

Abstract
There is an increasing role for positron emission tomography (PET) in oncology, particularly as a component of early phase clinical trials. As a non-invasive functional imaging modality, PET can be used to assess both pharmacokinetics and pharmacodynamics of novel therapeutics by utilizing radiolabelled compounds. These studies can provide crucial information early in the drug development process that may influence the further development of novel therapeutics. PET imaging probes can also be used as early biomarkers of clinical response and to predict clinical outcome prior to the administration of therapeutic agents. We discuss the role of PET imaging particularly as applied to phase 0 studies and discuss the regulations involved in the development and synthesis of novel radioligands. The review also discusses currently available tracers and their role in the assessment of pharmacokinetics and pharmacodynamics as applied to oncology.
LINKED ARTICLES
This article is part of a themed section on Imaging. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2011.163.issue-8BJP has previously published an Imaging in Pharmacology themed section, edited by A Davenport and C Daly. To view this section visit http://dx.doi.org/10.1111/bph.2010.159.issue-4
Keywords: positron emission tomography (PET), radioisotope, drug development, radiopharmaceutical
Introduction
Drug development is a lengthy, complex and expensive process. Currently the development of a typical investigational agent takes 10–15 years and costs approximately US $800 million. Despite the time and financial commitment, attrition rates are high with less than 10% of novel anticancer drugs entering the market from phase I studies (Dimasi, 2001). This low success rate has been ascribed to two major causes; the lack of predictive preclinical models and inadequate, complex trial designs (Bergstrom et al., 2003; Marchetti and Schellens, 2007). In parallel, the area of drug discovery and development is undergoing a rapid expansion in almost all therapeutic areas of medicine, particularly in oncology. This is in part fuelled by innovative technologies such as computer-aided drug design, combinatorial chemistry, and high-throughput methodologies for studying genomics and structural biology. The use of these tools has led to an increase in the identification of potentially druggable targets that drive pathological conditions. However, due to increasing costs and limited resources, it is not possible to develop all potential drug candidates and this has initiated discussions as to how to more effectively drive drug development.
It is generally accepted that early information on exposure of target tissue to investigational drug, modulation of the desired target, and biological effect will assist in the prompt translation of agents from the laboratory to the clinic (Workman et al., 2006). It is important to establish these measurements early in the drug development pathway, either prior to phase I studies (phase 0) or as part of phase I studies. To this end positron emission tomography (PET) can be used to obtain crucial pharmacokinetic (PK) and pharmacodynamic (PD) information non-invasively, and hence the role of PET imaging in drug development is increasing.
PET is a non-invasive imaging methodology that involves the administration of compounds labelled with positron-emitting radioisotopes (radiotracers or radioligands) that are formulated for intravenous injection (radiopharmaceuticals). Radiotracers are substrates of normal physiological pathways (activated probes) or localize to particular targets because of specific binding interactions (targeted probes). These radiolabelled probes are administered at tracer amounts so as to not perturb normal physiology but allows in vivo studies under physiological conditions. PET therefore allows the investigation of substrate-target interactions or physiological processes with the advantage of not having to administer an efficacious dose of a chemical compound as with classical pharmacological investigations. As radiotracers act as substrates for normal physiological processes they need to undergo extensive preclinical validation. The development of novel PET radiotracers is a complex process that requires a multidisciplinary research group including expertise in organic chemistry, radiochemistry, pharmacology, cell biology, physics, image analysis and oncology. In this review we initially describe the process by which novel cancer imaging tracers are developed, from conception to the clinic (Figure 1). The second part of the review will focus predominantly on PET for assessing PD end-points that illustrate the hallmarks of cancer, proliferation, apoptosis and angiogenesis studies in patients, and the utility of PET as an early biomarker of therapeutic response. We will also discuss the PET imaging in phase 0 and PK studies.
Figure 1.
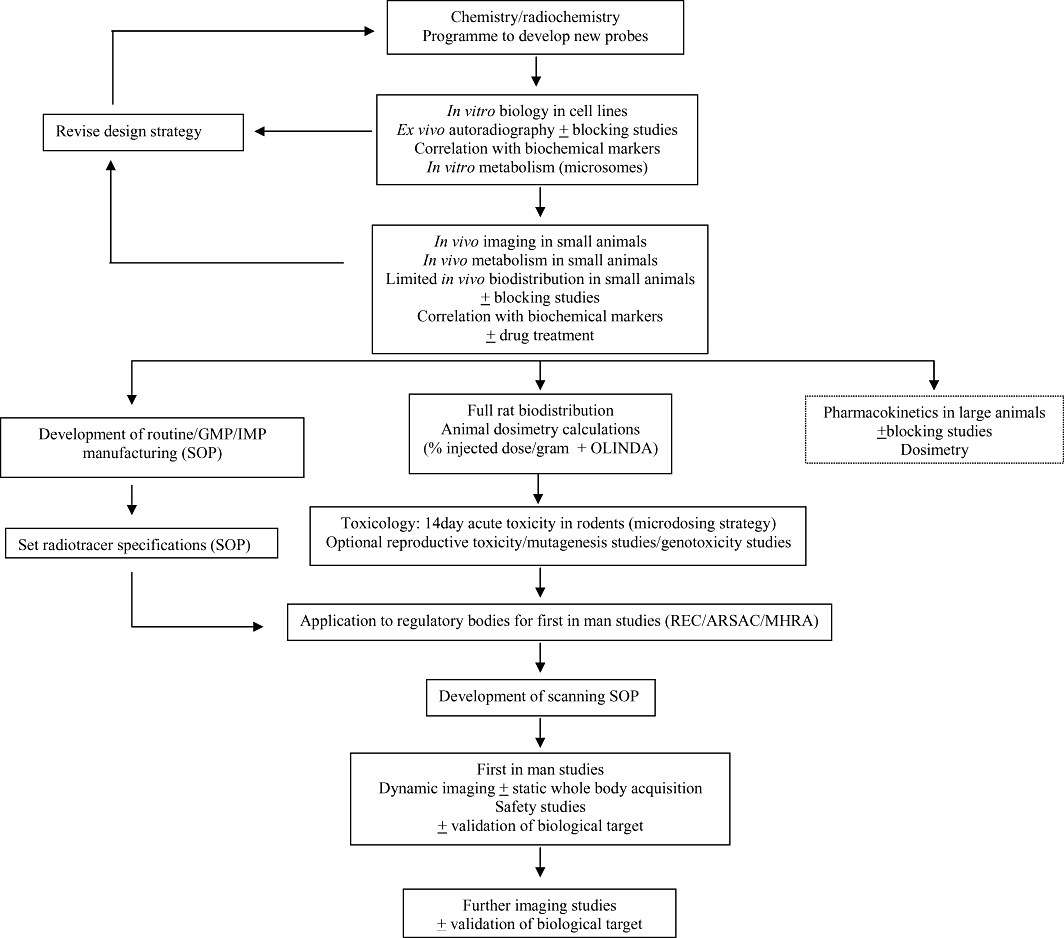
The developmental process involved in designing a new radiotracer for clinical use. OLINDA, organ level internal dose assessment.
Radiosynthesis
Any biological target that is present at increased or decreased levels in cancer cells can be visualized by PET. Ideally the target should be as specific as possible for the disease process, and consideration should also be given to the clinical information that might be gleaned from imaging the target or pathway. The development of a targeted radiotracer involves the synthesis of an extensive library of potential compounds for a particular target, with the expectation that only a few imaging agents will successfully progress to clinical PET studies. This library can contain several analogues of the parent compound that have a known affinity for a target and often may be based on previously known compounds. For example, in the design of a PET imaging tracer for the detection of αvβ3-integrin as a marker of angiogenesis, a focused library of peptides containing the arginine-glycine-aspartic acid (RGD) sequence were screened and [18F]Galacto-RGD, was taken forward into clinical development (Haubner et al., 2001). PET tracers are manufactured by automated synthesis modules the first of which was designed for the manufacture of [18F]fluorodeoxyglucose (FDG) (Iwata et al., 1984). Within this module the radiochemical hardware was interfaced with a microcomputer that controlled the 32 steps involved in the radiosynthesis of [18F]FDG (Fowler et al., 1981). More recently, the synthesis of [18F]FDG occurs via a high yield, one-pot sysnthesis reaction, which has become the synthetic pathway of most commercial [18F]FDG automation modules (Hamacher et al., 1986) (Figure 2).
Figure 2.
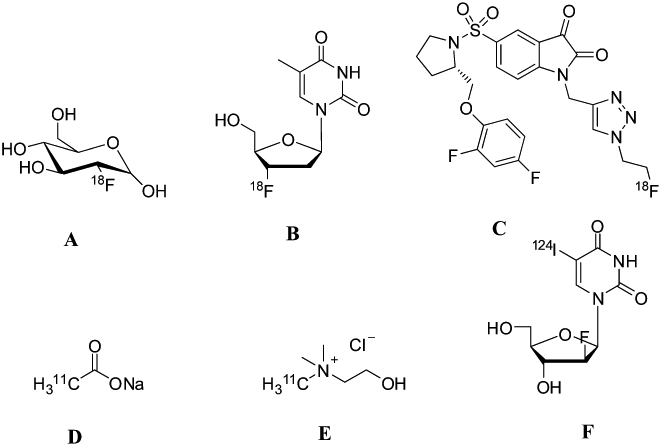
Chemical structures of common tracers involved in imaging. (A) [18F]FDG (B). [18F]FLT C isatin sulfonamide, [18F]ICMT-11, (D) [11C]acetate, (E) [11C]choline, (F) [124I]– 2′-fluoro-5-iodo-[beta]-D-arabinouridine (FIAU).
To have clinical utility, these potential tracers must have subnanomolar or nanomolar affinity for the physiological target so as to not interfere with normal biological function. Other desirable characteristics of radiotracers for somatic tumours include:
high specificity and/or selectivity
high plasma clearance and low plasma protein binding
neutral and hydrophilic to enhance elimination and reduce effective (radiation) dose
limited or measurable metabolism and amenable to kinetic modelling
low toxicity
good in vivo stability because of low metabolic clearance
Of central importance, the compound must have a suitable site for labelling with radioisotopes such as carbon-11 or fluorine-18, the most commonly used radioisotopes for PET in oncology. However, research utilizing other radioisotopes including copper-64, yttrium-86 and iodine-124 is increasing. When labelling a drug or molecule of choice, an element in the target molecule is substituted by its radioisotope. Problems arise however with the substitution of hydrogen as this does not have a positron emitting isotope. In this case hydrogen is substituted by fluorine-18, which may result in altered PK compared with the native molecule but these substitutions are conservative, and in the past have yielded useful probes such as [18F](FDG).
Radioisotopes are produced by charged particle nuclear reactions within a cyclotron where a target vessel containing a gas or fluid is bombarded by protons or deuterons. These radioisotopes are obtained from the cyclotron in simple chemical forms (Table 1). For example carbon-11 could be obtained as [11C]CO2 or [11C]CH4 and fluorine-18 as either [18F]fluoride or [18F]fluorine. Once generated, radiochemistry is then used to incorporate these radioactive substrates into the compounds of interest. This process involves a number of steps, culminating in the conversion of the radioisotope to a reactive intermediate that is then used to label the compound of interest. The radiolabelled compound is isolated, for instance by high-performance liquid chromatography (HPLC), to eliminate radiochemical and chemical by-products that may have arisen during synthesis. The product is then formulated and can be used for aseptic injection.
Table 1.
Characteristics of radionuclides commonly used in the development of PET tracers
| Radionuclide | Half-life (min) | Intermediate building blocks | Typical specific activity (GBq·µmol−1) |
|---|---|---|---|
| 11C | 20 | [11C]CH3I, [11C]HCN, [11C]CO, [11C]CO2, [11C]CH4 | 3.4 × 105 |
| 18F | 110 | [18F]F-, [18F]F2 | 6.3 × 104 |
| 76Br | 972 | [76Br]Br+,[76Br]Br- | 7.2 × 103 |
| 13N | 10 | [13N]NH4+,[13N]NH4-,[13N]NH2- | 7.0 × 105 |
| 15O | 2 | [15O]H2O | 3.4 × 106 |
| 124I | 40 (days) | [124I]I+, [124I]I- | 1.15 × 103 |
| 68Ga | 68 | [68Ga]Ga | 1.02 × 105 |
| 64Cu | 760 | [64Cu]CuCl2 | 2.2 × 108–1.85 × 109 |
Due to the short half-life of PET radioisotopes, the labelling of compounds involves considerable amounts of radioactivity, and therefore needs to be performed within remote-controlled lead-shielded ‘hot’ cells. The short half-life of radioisotopes also results in a time constraint for PET radiolabelling, and the introduction of the radioisotope at the latest possible stage in the synthesis is desirable. The development of novel synthetic procedures to improve this ‘late labelling’ is an active area of research.
Compounds labelled with carbon-11 or fluorine-18 are produced with high specific radioactivity, that is, these compounds have a high level of radioactivity and are associated with a very small amount of the stable compound or carrier. The amount of carrier present is usually sufficient to obtain a mass spectrum of the compound. To ascertain the position of the incorporated radioisotope, co-labelling experiments can be conducted. For example, for carbon-11 chemistry, very small amounts of carbon-13-labelled agents can be mixed with the carbon-11 equivalents and used in the radiosynthesis. The purified product is then examined by carbon-13 nuclear magnetic resonance and mass spectrometry.
The commonly used radionuclides in PET that are most appropriate for labelling of drugs are summarized in Table 1. Carbon-11 is an ideal nuclide for labelling molecules of biological interest, however, its short half-life (20.4 min), complex synthesis procedures and difficulties in obtaining high specific activities are limitations for routine manufacturing by PET centres. The metallic nuclides, gallium-68, copper-64 and yttrium-86 are useful for developing peptide- and protein-based radiotracers. Fluorine-18 is ideal for a number of reasons; it has a long half-life (110 min) allowing transportation from production to clinical PET imaging sites, it has acceptable dosimetry allowing multiple studies in a given patient, in terms of production it can be produced in large quantities in a cyclotron, has high labelling yields and finally, fluorine-18 has low positron energy compared with the other nuclides, which improves the resolution of images obtained (Vallabhajosula, 2007). Radionuclides are administered to patients, where they decay by positron emission, which results in the release of two gamma rays. Gamma emission is detected by detectors placed around the subject and images obtained showing the distribution of radionuclide in the body. Improvements in anatomical localization is achieved by combining images obtained by PET with concurrent computer tomographic (CT) images obtained prior to scanning.
For a radiotracer to be developed further, a number of characteristics need to be fulfilled during the radiosynthesis phase (Pimlott, 2005). It must be possible to:
Obtain sufficient yields (5–40% non-decay corrected, end of synthesis yields, depending on synthesis procedure) and rapid radiosynthesis. This optimization is particularly crucial in the development of a radiotracer for early clinical studies and may affect the cost of the radiotracer and logistics of these studies.
Obtain high radiochemical purity adequate for human imaging studies (preferably 95% or higher).
Obtain high specific radioactivity to allow injection of sufficient radioactivity into patients for imaging. The amount of compound present in a patient dose should be well below the amount that elicits a pharmacological response.
Obtain the radiopharmaceutical in a form that is acceptable to regulatory bodies with respect to impurity profile [European Medicines Agency (EMEA) or Food and Drug Administration (FDA)].
Once radiosynthesis has successfully been developed, a standard operating procedure (SOP) can be initiated for the synthesis of the radioligand, and to ensure that the radiopharmaceutical is sterile and endotoxin-free (Figure 1). The SOP will detail in full the synthesis and specifications for production and purity analysis of these tracers so that they are synthesized in a reproducible and reliable manner. The SOP will also indicate how scanning will be conducted. SOPs are central to both preclinical and clinical validation of a novel radiotracer before it is considered to be useful for imaging the target of interest. After synthesis, radiolabelled compounds must then undergo pharmacological characterization, initially in cell culture, and then in vivo to determine the biological action and affinity for the tumour target (Figure 1), a procedure that will be discussed in more detail later.
Production and quality control
The European Community Directive on Good Clinical Practice in clinical trials requires that the manufacture of investigational medicinal products be in compliance with Good Manufacturing Practice (GMP) standards within approved facilities, and that these facilities must be capable of meeting these standards. Within the UK, facilities involved in the production of radiopharmaceuticals must apply for a manufacturing license issued by the Medicines and Healthcare Products Regulatory Agency (MHRA), and these sites are regularly monitored. In the United States, the FDA acts as an equivalent regulatory body and is responsible for issuing guidance notes and regulations, and a summary of the regulations involved in radiotracer development is given in Table 2.
Table 2.
Regulatory guidelines pertaining to the manufacture and clinical use of radioligands
| Body | Locality | Regulation name | Process | Type of regulation |
|---|---|---|---|---|
| Medicines and Healthcare products Regulatory Agency (MHRA) | UK | Medicines Control Agency (MCA) ‘Specials’ License (MHRA, 2007) | Ensures manufacture of investigational medicinal products are in compliance with Good Manufacturing Practice (GMP) standards | Legislative and binding |
| Issue guidance notes and regulations | Non-binding | |||
| Investigational Medicinal Product ‘IMP’ or equivalent (UK ‘Specials’) (MHRA, 2010) | Ability to undertake human studies with medicinal products | Legislative and binding | ||
| Food and Drug Administration (FDA) | USA | Issue guidance notes and regulations | Non-binding | |
| Radioactive Drug Research Committee (RDRC) approval mechanism: Investigational new drug (IND) | Safety evaluation for entirely new radiolabelled compounds for which there has been no prior human exposure | Legislative and binding | ||
| Radioactive Drug Research Committee (RDRC) approval mechanism: Exploratory Investigational New Drug (eIND) | Safety evaluation for situations where there has been some prior human exposure to the radiolabelled compound | Legislative and binding | ||
| Critical Path Initiative | Action plan to assist investigators along the approval path towards IND or eIND | Encourage investigators to submit new tracers for approval. Non-binding | ||
| Radiopharmacy Committee of European Association of Nuclear Medicine (EANM) | EU (incl UK) | ‘Guidelines on Good Radiopharmacy Practice (GRPP)’ (Committee, 2007) | Production of radiopharmaceuticals, specific GMP issues such as compliance to GRPP, self-inspection, acceptance criteria and ensuring appropriate staff qualifications. | Non-binding |
| Administration of Radioactive Substances Advisory Committee (ARSAC) | UK | Certificate for the administration of radioactive medicinal products | Certification allows specific radiolabelled compounds of pre-defined effective dose to be administered to study subjects for a specified time interval | Legislative and binding |
It is not always feasible to perform detailed quality control tests prior to administration of radionuclides, particularly as the half-life of the majority of radionuclides is short. Therefore, an ongoing quality assurance system is often established prior to initial clinical studies to ensure that quality analysis occurs. For radioligands with a high specific radioactivity, it is recommended to evaluate the stability of the ligand during test runs for up to 4 h (for carbon-11 and fluorine-18) after radiosynthesis to ensure that changes in the chemical composition of the radioligand has not occurred.
A number of in-house quality control procedures need to be assessed prior to administration of the radionuclide to the patient. These include assessment of purity, nature of any impurities, sterility of the solution, absence of pyrogens and suitable pH. With regards to impurities generated during the radiosynthesis, it should be emphasized that the parent compound is given at only tracer doses, and hence exposure to minor impurities is negligible. Minor impurities, however, are still considered and limits set; (EMEA, 2009). Consideration should also be given to levels of the other components in the formulation, such as anti-oxidants and residual solvents. These concepts should all be outlined within the SOP, which are adhered to by the manufacturing unit and must be readily available for audit purposes. At many centres a simple checklist is used to ensure that the radiopharmaceutical released adheres to strict criteria.
Chemical identity and purity
Prior to the administration of the radiopharmaceutical to patients it is essential, as part of the developmental process, to provide evidence that the radiopharmaceutical prepared via the radiosynthetic route is identical to the non-radiolabelled reference compound. This may not be the case for example with fluorinated compounds such as [18F]FDG, where there are no known biological equivalent. If the radiosynthetic route differs from that of the non-radiolabelled compound then the radiosynthetic route should be documented and be available for review by an independent body. Following decay of the radiolabelled compound, the chemical identity should be determined by mass spectrometry and nuclear magnetic resonance, if possible. The purity of the radiochemical, radionuclide and radiopharmaceutical should be determined by HPLC-UV and HPLC-mass spectrometry. It is common to perform three to five runs of the radiosynthetic process once this has been determined to ensure the robustness and reproducibility of the manufacturing process and to ensure that the compound meets all clinical criteria. During this process batch numbers should be recorded including manufacturing records of the radiosynthesis and quality control analyses.
In vitro and in vivo studies
Prior to testing in human subjects, novel radiopharmaceuticals must undergo in vitro and in vivo validation in order to ascertain target specificity, biodistribution and toxicity. Radiotracers are initially assessed in vitro to assess target specificity, uptake and the impact on cellular responses. Previously, radioligands would then be transferred into animal models, now however, a number of authors have suggested the use of multicellular tumour spheroids (MTS) as a transitional model, as the cytology and the morphology of MTS resembles that of experimental tumours in mice (Fracasso and Colombatti, 2000; Kunz-Schughart et al., 2004). Therefore, the use of MTS would allow evaluation of dose and dosing regimen with less laboratory effort. Preclinical studies have shown MTS to be an appropriate in vitro system to investigate drug response to chemotherapy (Bates et al. 2000; Dubessy et al., 2000; Mueller-Klieser, 2000). A number of MTS of differing tumour types have been evaluated and shown to be good experimental models in which to investigate novel agents and PET imaging as a biomarker of response (Monazzam et al., 2006; 2007; 2009;). However, MTS does not substitute the animal systems.
Animal studies are performed as part of the development of novel radioligands to determine biodistribution, reproductive toxicology, carcinogenic toxicity and genotoxicity. An integral part of early radiotracer development is a biodistribution study. These studies are often the first conducted and are important in determining whether binding of the radioisotope is tumour specific or if it binds to other sites outside the tumour, that is, non-specific binding. High non-specific binding of a radiotracer is a common reason for the failure of a radiotracer to reach clinical studies. Furthermore, knowing the degree of non-specific binding will assist in calculating the absorbed dose by tissues (dosimetry). Toxicology and biodistribution studies assure that the radiopharmaceutical is non-toxic at doses to be administered to patients.
A key aspect of the in vivo assessment of radioligands for use in oncology is selection of appropriate tumour models. This is of particular importance with reference to biodistribution studies; if the tumour model does not express a receptor of interest for example then any proof of concept study will be negative. In a study by Ullrich and colleagues [18F]fluorothymidine ([18]FLT) was investigated as an early response marker to erlotinib, an epidermal growth factor receptor (EGFR) inhibitor in animal models of non-small cell lung cancer (Ullrich et al., 2008). The authors report a reduction in [18]FLT by PET in erlotinib-sensitive EGFR-dependent tumours; in contrast [18]FLT-PET changes were attenuated in tumours harbouring T790M secondary EGFR mutations that confer resistance to EGFR tyrosine kinase inhibitors. Similarly, in a study investigating [18]FLT-PET as a marker of response to mTOR inhibitor, rapamycin, reductions in [18]FLT-PET were not seen in PTEN wild-type resistant tumours but occurred in sensitive cell types (with PTEN loss), again highlighting the need for the target of interest to be present for proof of concept studies (Wei et al., 2008 p. 140).
Carcinogenicity studies are not required unless there is a specific concern. This is particularly true for tracers being developed for use in oncology where, in general, patients have limited life expectancy. In these patients, long-term repeat dose toxicology, genotoxicity and carcinogenicity studies are usually omitted. Similarly, reproductive toxicology studies are not required as long as patients of reproductive age will be advised to take appropriate precautions to avoid pregnancy, and pregnant and lactating patients will be excluded from any clinical studies.
In terms of toxicology studies, single-dose studies are performed in animals using the formulation to be used in patients. Single-dose toxicology studies are deemed appropriate in animals, as in the clinical setting, patients will usually receive only a single, low dose of the radioligand, and if repeated administrations are delivered, the doses are usually administered weeks apart and, from a toxicological standpoint, are considered equivalent to a single dose. All animal toxicology studies are performed under the highest quality standards possible [equivalent to Good Laboratory Practice (GLP)].
Initial dosimetry studies are performed in animals over a number of time points depending on the biologicalal and physical half-lives of the radioisotope. The radioactivity in blood, urine and tissues, expressed as a percentage of injected radioactivity, is determined to enable calculation of the predicted human effective radiation dose (mSv) to each tissue. Human dosimetry often tends to be the first human study of new radiotracers. The dose to critical organs, such as the bladder and liver, can limit the amount of radioactivity that can be injected in human studies and therefore has the potential to limit the success of the tracer.
PK studies, mainly in rodents but where possible in larger animals, play important roles in the prediction of human radiation organ dosimetry and toxicokinetics (Figure 3). This is an important concept as metabolism can affect not only the amount of tracer that is able to bind to the target, but the generation of radiolabelled metabolites may interfere with image analysis. Furthermore, rapid metabolism of the radiotracer during the imaging time frame can limit the potential usefulness of a tracer, and thus needs to be considered.
Figure 3.
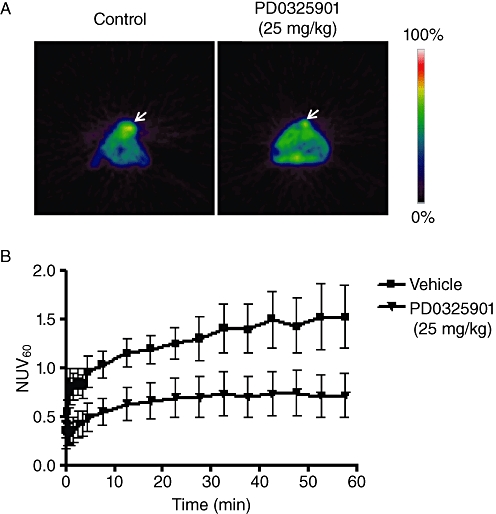
Use of [18F]FLT in positron emission tomography (PET) studies (A). Transverse [18F]FLT-PET images of mice bearing the SKMEL-28 human melanoma tumour, 10 days after inoculation with either vehicle or PD0325901 treatment. (B) [18F]FLT-PET time (min) versus radioactivity curve [as measured by normalized uptake value at 60 min postinjection (NUV60)] for the vehicle or PD0325901 treatment group (courtesy of Julius Leyton).
The potential immunogenicity of the radiopharmaceuticals is also of importance, particularly if the tracer is to be administered more than once to the same patient. This issue should be addressed by inspecting the structures of compounds for any possible alerts for immunogenicity by analogy with related compounds, for example, the presence of peptides or the presence of a penicilloic acid moiety.
Once animal toxicology and biodistribution studies are complete and the tracer is deemed suitable for clinical use from a scientific perspective, the radiotracer must undergo validation in the clinical setting. There are a number of regulatory bodies that manage the clinical development of radioligands to eusure that trials are run in a safe and consistent manner. These agencies tend to have similar remits in various countries, and the approval process is outlined in Table 2.
Role of imaging in early clinical trials
Pre-phase I or phase 0 studies
In 2003, the EMEA published a concept note followed by a Position Paper on the non-clinical safety studies required to support human clinical trials with a single dose of pharmacologically active compound using microdose techniques (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002941.pdf). These guidelines affect PET in two ways: firstly in terms of the development process of radioligands, but also use of PET as clinical trial end-points (Marchetti and Schellens, 2007). As sub-pharmacological single doses of the investigational agent are administered, ultrasensitive analytical methods such as PET are required to assess the drug distribution and PK. The paper raises the possibility of a reduced preclinical safety package for microdose studies and facilitates the development of novel radioligands (VanBrocklin, 2008).
As there has been a significant decline in the number of new molecules submitted to the FDA over the past decade, the FDA responded to this by devising an action plan called the ‘Critical Path Initiative’ to facilitate the drug discovery and development process from inception to approval (FDA, 2004; 2010;). A recent initiative implemented by the FDA is the establishment of the Exploratory Investigational New Drug (eIND) application, a characteristic of which are microdosing/phase 0 studies. These studies involve the administration of a single ‘micro-dose’ or ‘tracer-dose’ of a novel agent or radioligand, either to healthy volunteers or patients. The aim of a phase 0 study is to obtain information on PK and/or distribution properties or receptor selectivity prior to phase I. The studies do not provide information about the safety or tolerability of the radiopharmaceutical. The definition of a microdose is ‘less than 1/100 of the dose of a test substance calculated (based on animal data) to yield a pharmacological effect of the test substance with a maximum dose of ≤100 µg or, in the case of biological agents, ≤30 nmol’ (CHMP, 2004). The translational research questions that justify a pre-phase I study include whether the agent distributes to the tumour, to what extent the agent distributes to normal tissues, and whether the metabolite profile in rodents and humans are similar. These studies are used to establish whether the radiopharmaceutical behaves in human subjects as expected from preclinical studies. Theoretically, microdose trials should allow an early discrimination between those lead compounds to take forward into further development and those to be discarded. The biodistribution of a compound is also important to determine the extent of non-specific binding. Pre-phase I studies can also yield information regarding PK modulation and scheduling. N-[2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA) is a DNA intercalating acridine derivative that was labelled with carbon-11 in order to determine the metabolic profile of DACA, the distribution to the tumour – as a predictive marker of response – and distribution to normal tissues – as a predictor of toxicity (Saleem et al., 2008). Twenty-four patients were enrolled in the study and [11C]DACA administered at 1/1000 of the phase I dose. This PK study illustrated that [11C]DACA undergoes extensive metabolism and its metabolites distributed well to the tumour. Low peak concentrations of [11C]DACA-derived radioactivity were obtained in the vertebrae and brain compared with other normal tissues suggesting that myelosuppression and neurotoxicity were unlikely to be dose-limiting toxicities. There was uptake within the myocardium suggesting the possibility of myocardial toxicity in the phase I study. In a similar study LY2181308, an antisense oligonucloetide to survivin, was labelled with carbon-11 and administered concurrently with therapeutic doses of unlabelled drug (Saleem et al., 2009). [11C]LY2181308 uptake in both normal tissue and tumour was assessed. The authors demonstrated the utility of microdosing experiments illustrating that active drug concentrations could be achieved within the tumour and that unlabelled LY2181308 saturated normal tissue kinetics resulting in increased tumour uptake. These studies establish the feasibility of performing PET studies with radiolabelled anticancer drugs in a phase 0 setting.
PET can also been utilized to provide proof of principle for mechanism of action of the labelled drug. The effect of eniluracil, a drug that inhibits dihydropyrimidine dehydrogenase, an enzyme responsible for the degradation of 5-fluorouracil (5-FU), was studied using PET (Ahmed et al., 1999; Saleem et al., 2000a,b;). The study illustrated that in eniluracil-naïve patients, [18F]FU-derived radioactivity localized more strongly to the normal liver than in metastases. In addition there was distinct localization of radioactivity in the gall bladder consistent with hepatobiliary clearance of [18F]FBAL that disappeared in the presence of eniluracil. In patients receiving eniluracil, there was increased plasma uracil and unmetabolized [18F]FU. There was also an increase in the half-life of [18F]FU-derived radioactivity in tumours, providing proof of principle for mechanism of action of eniluracil. More recently Rosso and collegues utilized a PET imaging approach to generate kinetics of temozolamide following administration of labelled drug to patients with gliomas. The authors illustrate that the utilization of impulse response function to predict drug exposures in both tumours and healthy tissue, assuming linear drug kinetics (Rosso et al., 2009).
Following these studies a novel radioligand will undergo formal clinical evaluation prior to licensing for clinical use. A list of PET tracers currently used in the clinical setting, either as research tools or for diagnostic purposes, is given in Table 3.
Table 3.
Common PET tracers used in oncology
| Isotope | Radiotracer | Target | Measured effect |
|---|---|---|---|
| 11C | [11C]choline | Phosphocholine | Choline kinase activity, membrane function |
| l-Methyl-[11C]methionine | Amino acid transporter | Protein synthesis, amino acid transport, tumour cell growth | |
| L-1-[11C]tyrosine | Amino acid uptake | Amino acid metabolism | |
| 2-[11C]thymidine | DNA | DNA synthesis. Tumour cell proliferation | |
| N-methyl-[11C]temozolamide, Carbonly-[11C]temozolamide, N-[2-(dimethylamino)ethyl]acridine-4-carboxamide, [11C]-1,3-bis-(2-chloroethyl)-1-nitrosourea | Drug pharmacokinetics | ||
| 18F | [18F]fluorodeoxyglucose | Glucose metabolism | Glucose transport. Aerobic and anaerobic glycolysis, glucose consumption or metabolism |
| 5-[18F]fluorouracil, [18F]fluorotamoxifen | Pharmacokinetics | ||
| [18F]fluoride | Incorporation in the hydroxyapatite crystals in bone | Bone metabolism; Bone blood flow and osteoblastic activity | |
| 1-[11C]acetate | Tricarboxylic acid cycle via acetyl coenzyme A | Lipid synthesis | |
| [18F]fluoroacetate | |||
| 3-[18F]fluorothymidine | Activity of thymidine kinase-1 | DNA synthesis. Tumour cells proliferation | |
| [18F]fluoroazomycin arabinoside, [18F]fluoromisonidazole | Hypoxic cells | Tumour hypoxia | |
| 16α-[18F]fluoro-17β-oestradiol | Oestrogen receptors in breast cancer | Receptor binding, response to anti-oestrogen therapy | |
| 16β-[18F]-5α-dihydrotestosterone (FDHT) | Androgen receptor (ARs) | Functional status of tumour ARs | |
| [18F]AH111585/fluciclatide, [18F]Galacto-RGD | Angiogenesis | Integrin receptors (αVβ3) on endothelial cells of neovasculature | |
| [18F]oligonucleotide | Gene expression | In vivo hybridization with mRNA | |
| 9-1-[18F]fluoro-3-hyroxy-3-propoxy- methylguanine | Gene expression | Substrate to herpes virus thymidine kinase | |
| 68Ga | [68Ga]DOTATOC, [68Ga]DOTANOC | Specific binding to somatostatin receptor SSTR-II; SSTR-II, III, V | Presence of SSTRII; SSTRII, III, IV |
| 124I | Na[124I] | Gene expression | Sodium iodide symporter expression |
| 15O | [15O]H2O | Perfusion | Blood flow/perfusion |
| [15O]CO | Blood volume | Antivascular response | |
| others | 124I-, 64Cu-, 86Y-Labeled antibodies | Binding to tumour antigens | Specific binding to tumour associated antigenic binding sites (such as CEA, PSMA, CD20 and CD22) |
| [124I]Annexin V, [64Cu]Annexin V, [18F]-Annexin V | Apoptosis | Specific binding to Phosphatidylserine (PS) on cell membrane |
Drug PKs
As discussed earlier, a successful imaging agent must be retained by the target but must not affect the target, nor can it be too toxic for use with other treatment agents. In contrast, therapeutic agents are designed to interact and affect the target of interest. Labelling drugs may therefore be useful in understanding the PK of an agent particularly as part of the drug development process (Figure 4). Parameters including drug absorption, distribution, metabolism and excretion, as well as the maximum plasma concentration and area under the plasma concentration versus time curve are major determinants of toxicity and efficacy of a drug, and can be achieved by labelling drugs of interest with radioisotopes such as carbon-11, fluorine-18 or nitrogen-13 (Osman et al., 2001; Saleem et al., 2001; 2008;). PK parameters are obtained through mathematical kinetic modelling, a process that involves comparing the concentrations of radioactivity in tissues with that in arterial plasma and calculating the plasma-tissue exchange rates to derive tissue PK parameters (Saleem et al., 2000a,b;). This allows the investigator to ascertain what happens to the tracer within the body and, in particular, within the target organ.
Figure 4.
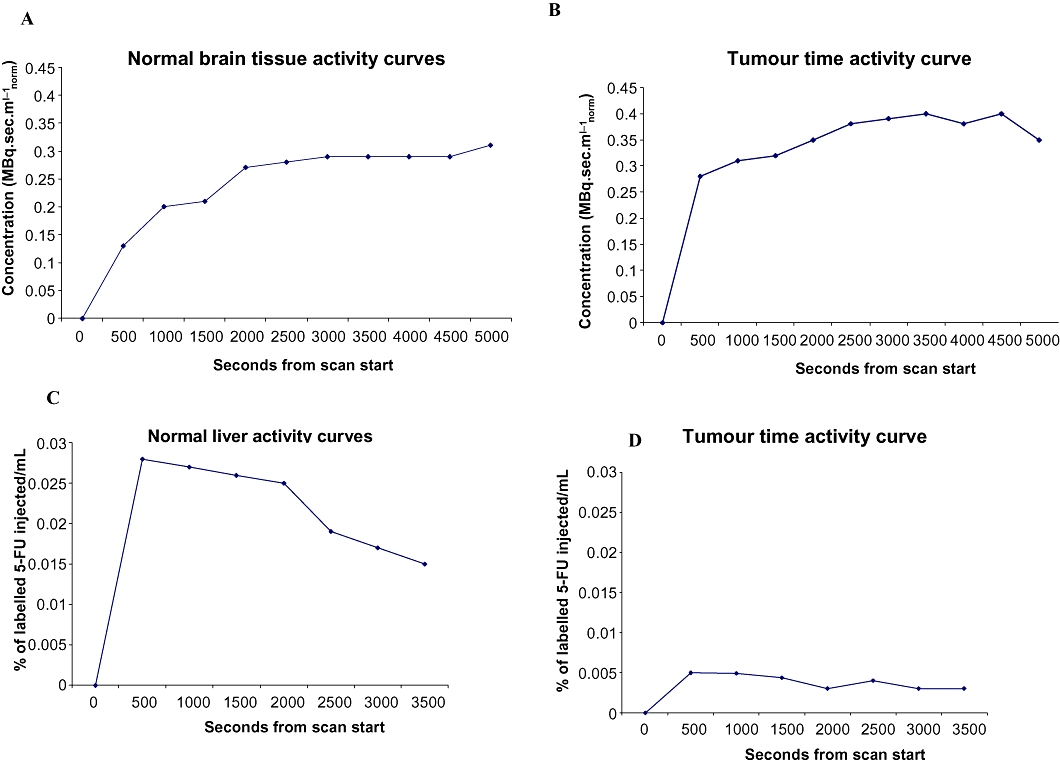
Uptake of tracers varies between normal and tumour tissue. (A, B) Time activity curves demonstrating normal brain and tumour uptake of [11Cmethyl]temozolomide. (C, D) Time activity curves demonstrating normal liver and tumour uptake of [18F]5-fluorouracil.
The interpatient variability in the PK of anticancer drugs is generally high and these drugs have a narrow therapeutic index. Steady state plasma PK often poorly reflect the levels of drug within normal or tumour tissues. PET PK studies may aid in determining intra-tumour drug exposure. For example when comparing uptake of [11C]-1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU) within the tumour after both intravenous and intra-arterial administration it was shown that intra-arterial administration resulted in 50 times higher intra-tumour concentrations (Tyler et al., 1986). Examples of drugs that have been evaluated by PET include [11C]temozolamide, [18F]5FU, [18F]paclitaxel, [13N]cisplatin and [11C]DACA (Ginos et al., 1987; Kissel et al., 1997; Dimitrakopoulou-Strauss et al., 1998; Saleem et al., 2001; 2003; Kurdziel et al., 2003). PK studies may also allow for the investigation of the mechanism of action of novel therapies prior to phase I. For example, the proposed mechanism of action of temozolamide was studied in patients using [11C]temozolmide tracers labelled at the methyl and carbonyl positions. The study demonstrated that temozolamide undergoes tissue-specific ring-opening in humans, although this conformational change was not tumour specific (Saleem et al., 2003).
On labelling the drug it is important to ensure that no modification has occurred to the molecule, as even a slight change can alter its physicochemical properties. Initial exploratory studies of hydrogen-3 (tritium) and carbon-14 labelled compounds in animals are often informative in determining if imaging in patients will be useful. For example, if a large number of degradation products are rapidly generated, imaging may add little value in determining response. On the other hand, imaging the distribution of a drug may provide valuable insight into further development and choice of tumours to be treated.
The PK of 5-FU has been successfully studied by labelling the drug with 18F, allowing enough time for kinetic studies to be performed with good reproducibility (Harte et al., 1998). Up to 80% of 5-FU is degraded through catabolism. A PK study in 13 patients with colorectal liver metastases showed a relationship between uptake of 5-FU and tumour response; the likelihood of disease stabilization was higher and survival was longer in patients whose tumours had a higher uptake of the radiolabelled drug (Moehler et al., 1998).
When used in a PK context, labelled macromolecules, for example, antibodies that target cell surface receptors may show specific targeting to receptors as well as significant non-selective distribution in intracellular and extracellular pools related in part to their slow systemic clearance (Verel et al., 2005). This characteristic of radiolabelled macromolecules poses a challenge when they are used in a PD context. In the case of IgG entities, this requires labelling with long-lived radioisotopes, for example, copper-64 and imaging typically at 24–48 h.
PDs
PD endpoints in PET imaging can be considered as either generic or specific end-points. Generic end-points image physiologic processes such as cellular proliferation, glucose metabolism, apoptosis, hypoxia and angiogenesis. Imaging of specific end-points provides proof of principle for a proposed mechanism of action such as the use of 16α-18F-fluroro-17β-oestradiol to assess oestrogen receptor status in breast cancer (Lee et al., 2009). We will discuss a number of these probes that are currently undergoing preclinical and clinical development or in routine clinical use.
Tumour metabolism and proliferation
[18F]FDG is the commonest clinically available tracer and assesses the generic end-point of glucose utilization, as a measure of metabolism. Despite the increasing number of potential radiotracers, PET is generally still synonymous with [18F]FDG. Metabolic imaging with [18F]FDG relies on differential utilization of glucose in cancer cells compared with normal cells (Kelloff et al., 2005). [18F]FDG enters the cell and is phosphorylated by hexokinase to [18F]FDG-6-phosphate. [18F]FDG-6-phosphate cannot undergo any further metabolism and is trapped within the cell, with the rate of accumulation of [18F]FDG-6-phosphate being proportional to the rate of glucose utilization. Therefore, the more metabolically active the cell the more glucose is utilized. The major limitation with [18F]FDG is the false positivity seen within areas of inflammation (Subhas et al. 2005; Rosenbaum et al., 2006). Furthermore, as [18F]FDG relies on increased cellular proliferation compared with surrounding tissues, it has low sensitivity in slow growing, well-differentiated and less metabolically active tumours (Shreve et al. 1999; Burton et al., 2004). Moreover, [18F]FDG distributes within the renal collecting system obscuring pelvic lesions.
[18F]FDG has been extensively studied both for the staging of tumours and response to cytotoxic therapy having an overall average sensitivity of 84% and a specificity of 88% in cancer (Gambhir et al., 2001). More recently [18F]FDG has been used to monitor responses to targeted therapies particular those that affect glucose homeostasis such as the mTOR and Akt inhibitors (Contractor and Aboagye, 2009) (Table 4). The most impressive results with [18F]FDG were seen in the early trials of imatinib mesylate, a c-kit inhibitor used in the management of gastrointestinal stromal tumours (GISTs). In these tumours, response on [18F]FDG-PET was seen as early as 24 h after therapy, with the response on PET imaging correlated with progression-free survival (Stroobants et al., 2003). This is of particular importance in early phase clinical trials as standard radiological assessment of these tumours often fails to show any change in tumour size. Currently [18F]FDG is approved for the staging and response assessment of a number of tumour types. A number of guidelines have been developed for the assessment of response including the European Organization for Research and Treatment of Cancer (EORTC) and the National Cancer Institute guidelines (Young et al. 1999; Shankar et al., 2006). More recently the PERCIST guidelines were suggested by Wahl and colleagues but remain to be adopted (Wahl et al., 2009).
Table 4.
Selected clinical studies of the use of [18F]FDG for monitoring therapy response
| Tumour type | Therapy | Change in FDG uptake with therapy | Reference |
|---|---|---|---|
| Lymphoma | Reduction after 7 days correlates with progression free survival and overall survival | Romer et al., 1998 | |
| Reduction after two cycles correlation with complete response, progression free survival and overall survival | Haioun et al., 2005 | ||
| Gastrointestinal stromal tumour | Imatinib | Reduction after 7 days; correlated with response | van Oosterom et al., 2001 |
| Imatinib | Reduction at 1 month correlated with response | Holdsworth et al., 2007 | |
| Biliary tract cancer | Bevacizumab, gemcitabine and oxaliplatin | Reduction at 6 weeks; correlation with progression-free survival and overall survival | Zhu et al., 2010 |
| Non-small cell lung cancer | Gefitinib | Reduction on day 2, associated with improved progression free survival | Sunaga et al., 2008 |
| Breast cancer | Cyclophosphamide, doxorubicin, vincristine, and prednisolone or docetaxel | Reduction after first cycle predictive of response | Smith et al., 2000 |
| Epirubicin, cyclophosphamide or epirubicin, paclitaxel | Reduction after first cycle predictive of response | Schelling et al., 2000 | |
| Lapatinib | Reduction after 1 month correlated with response | Kawada et al., 2007 | |
| Prostate cancer | Goserelin | Reduction 1 after−5 months of therapy | Oyama et al., 2001 |
| Gastric cancer | Cetuximab, irinotecan, 5-fluorouracil | Reduction correlated with response, time to progression and overall survival | Di Fabio et al., 2007 |
| Gastro-oesophageal cancer | Epirubicin, cisplatin and 5-fluorouracil | Reduction at 14 days correlates with response and overall survival | Ott et al., 2006 |
Proliferation
Proliferation is one of the hallmarks of malignancy (Hanahan and Weinberg, 2000) and the majority of chemotherapeutic agents act to inhibit proliferation of cancer cells. It has been shown in a number of tumour types that the rate of proliferation, as indicated by Ki-67 and PCNA expression and S-phase fraction of tumour samples, is predictive of both survival and prognosis (Stuart-Harris et al., 2008; Yerushalmi et al., 2010) There are inherent difficulties in patient acceptability of routine biopsies, particularly if performed serially. Furthermore, there may be issues of tumour accessibility and, because of tumour heterogeneity, the biopsy sample may not be representative of the proliferative activity of the whole tumour. There is a need therefore to assess tumour proliferation, non-invasively.
Thymidine is a nucleoside utilized in DNA replication in proliferating cells, and both thymidine and its analogues have been extensively studied as potential tracers of cellular proliferation. Thymidine was first labelled with tritium, which is still used for in vitro work. During the process of development, thymidine was labelled with carbon-11. However, [11C]thymidine undergoes rapid metabolism resulting in significant levels of labelled metabolites within the blood (Shields et al., 1992). The fluorinated analogue, [18F]FLT, has a suitable nuclide half-life (109.8 min) for clinical use and stability against phosphorylase-mediated degradation in vivo and is now extensively used as a marker of proliferation (Shields et al., 1998).
After injection, [18F]FLT enters the cell by both Na+-dependent active nucleoside transporters and by passive diffusion. [18F]FLT follows the salvage pathway of DNA synthesis and like thymidine undergoes phosphorylation by thymidine kinase–1 (TK1) to [18F]FLT-monophosphate (Seitz et al., 2002). [18F]FLT is a selective substrate for TK1 whereas thymidine is also phosphorylated by TK2. TK1 is virtually absent in quiescent cells but is increased in proliferating cells (Sherley and Kelly, 1988; Munch-Petersen et al., 1995). Phosphorylated [18F]FLT is not significantly incorporated into DNA and is trapped within the cytosol. The rate limiting step for [18F]FLT accumulation is the initial phosphorylation by TK1; it is also the rate limiting step in the salvage pathway of DNA synthesis, therefore the handling of [18F]FLT reflects cellular proliferation (Shields et al., 1998).
In a paper by Barthel and colleagues the authors investigated the ability of [18F]FLT-PET to monitor response to chemotherapy in mice bearing sarcomas injected with 5-FU (Barthel et al., 2003). The authors reported reduced uptake of [18F]FLT following treatment that correlated with the PCNA labelling index within the tumour and a reduction in tumour volume. The authors then considered the role of TK1 in [18F]FLT-PET imaging by contrasting the uptake of [18F]FLT in TK1 knock-out and functional heterozygote lymphoma models (Barthel et al., 2005). They reported higher uptake in TK1 heterozygote lymphoma compared with the knock-out model, which correlated with higher TK1 protein expression within the tumours and accumulation of [18F]FLT. These studies were then reproduced in a clinical setting. In the seminal paper by Kenny and colleagues, [18F]FLT-PET was reported as being an early response marker in patients receiving chemotherapy for breast cancer when compared with RECIST assessment (Figure 5) (Kenny et al., 2005; 2007;). Furthermore, the [18F]FLT uptake parameters within the breast tumours correlated with Ki-67 expression within surgical resection specimens lending further support of [18F]FLT as a PD marker for early clinical response.
Figure 5.
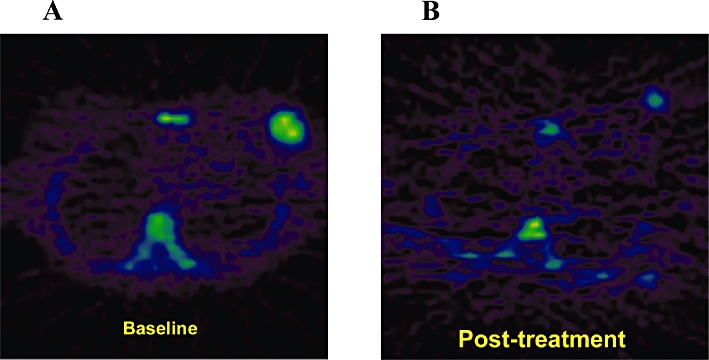
[18F]FLT-positron emission tomography images to assess early response (1 week) to epirubicin, cyclophosphamide and 5-fluorouracil chemotherapy in a patient with T3N0M0 breast cancer. (A) Pretreatment uptake is seen in the right breast and (B) post-treatment response is observable as a reduction of [18F]FLT uptake (courtesy of Dr. Laura Kenny).
However, [18F]FLT has several limitations (Barwick et al., 2009). Firstly, [18F]FLT uptake in tumours depends on TK1 activity, including therapy-induced activation of the salvage pathways (Perumal et al., 2006; Pillai et al., 2008; Kenny et al., 2009). Therefore, agents that only inhibit de novo DNA synthesis such as fluoropyrimidines may increase FLT uptake. Other therapeutic agents inhibit both pathways and subsequently reduce [18F]FLT uptake. Moreover, tumours with low TK1 expression or low proliferative activity may not be readily identified (Bading and Shields, 2008). High physiological uptake of [18F]FLT in liver and bone marrow also limits the assessment of disease in these organs. However, the recent introduction of kinetic filtering has led to significant signal reduction from the liver, improving visualization (Gray et al., 2010). There is evidence to suggest that membrane localization of nucleoside transporters can be enhanced in tumours in response to a number of chemotherapy agents, which may result in a ‘flare’ response with [18F]FLT. Perumal and colleagues investigated the role of [18F]FLT-PET in monitoring intra-tumour inhibition of thymidylate synthase in vivo 1–2 h following 5-FU injection into mice bearing RIF-1 fibrosarcomas (Perumal et al., 2006). The authors reported an increase in the 60 min [18F]FLT tumour/heart radioactivity ratio in drug treated mice compared with vehicle controls. Plasma and tumour deoxyuridine levels increased significantly, but TK1 expression and ATP remained unchanged. Cellular assays for membrane uptake suggested a functional role of type-1 equilibrative nucleoside transporter (ENT-1). The authors hypothesized that [18F]FLT uptake is mediated by redistribution of ENT-1 transporters to the plasma membrane 1–2 h after treatment. This was utilized clinically by Kenny and colleagues in breast cancer patients receiving capecitabine therapy where the authors suggested that PET retention parameters obtained following capecitabine therapy could be utilized as a PD marker for TS inhibitors (Kenny et al., 2009). There have been a number of studies considering the uptake of [18F]FLT in both the preclinical (Table 5) and clinical (Table 6) setting, with differing results according to the type of therapy utilized and at differing time points, both of which need to be considered in the utilization of [18F]FLT as a response marker.
Table 5.
Preclinical studies of the use of [18F]FLT for monitoring therapy response
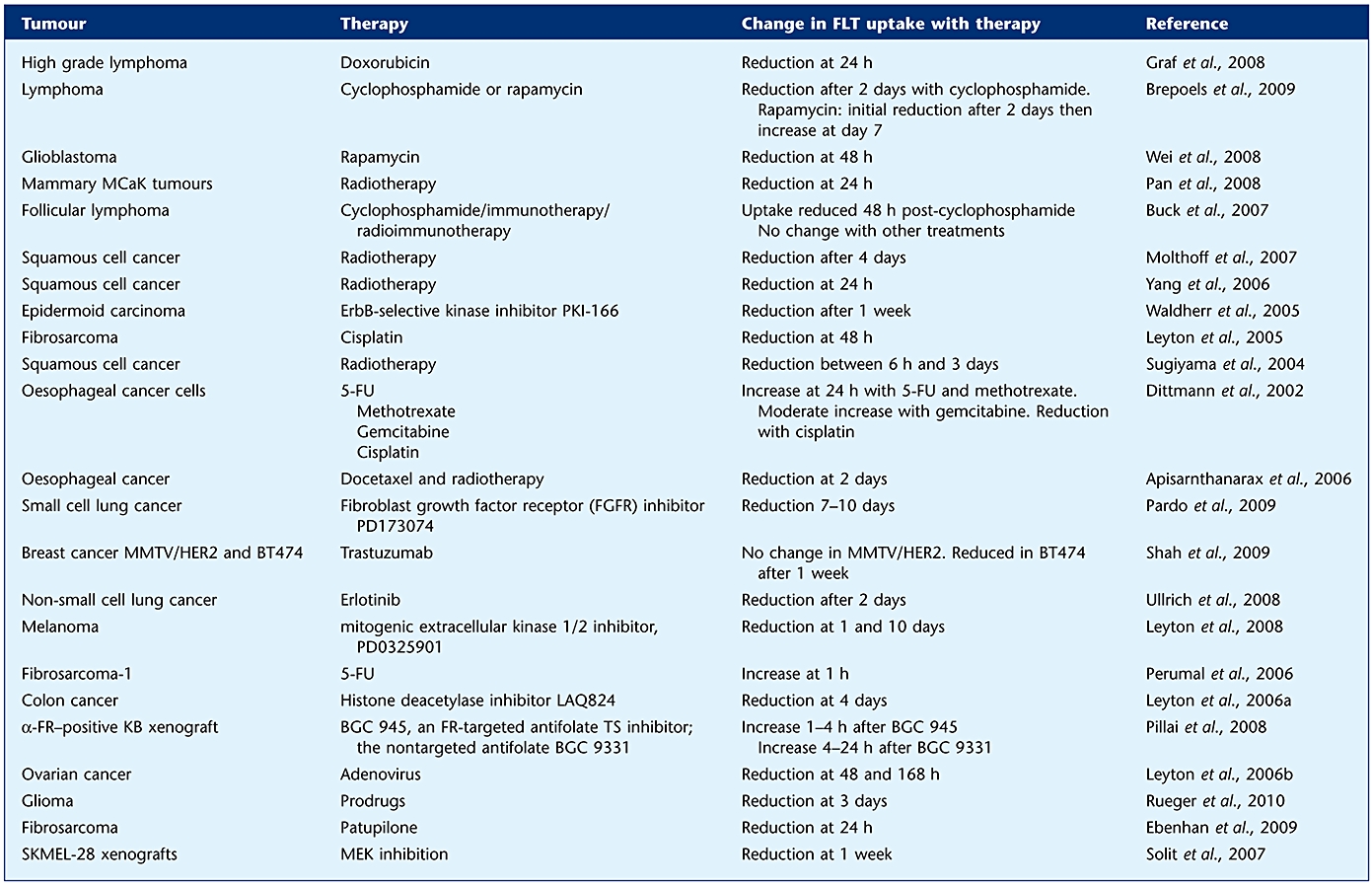 |
Table 6.
Clinical studies of the use of [18F]FLT for monitoring therapy response
| Tumour type | Therapy | Change in FLT uptake with therapy | Reference |
|---|---|---|---|
| Lung | Gefitinib | Reduction after 7 days; correlated with response | Sohn et al., 2008 |
| Chemo/radiotherapy | Reduction at day 2, 8 and 29 | Everitt et al., 2009 | |
| Gliomas | Bevacizumab and irinotecan | Reduction at 6 weeks; correlation with progression-free survival | Chen et al., 2007 |
| Temozolamide | Correlation with tumour response | Galldiks et al., 2010 | |
| Germ cell tumours | Combination chemotherapy | Reduction; no association with response | Pfannenberg et al., 2010 |
| Non-Hodgkin's lymphoma | CHOP, R-CHOP, rituximab | Reduction after 2 days; correlated with response. No change with rituximab | Herrmann et al., 2007 |
| Breast cancer | Various chemotherapy and hormone therapy | Reduction at 2 weeks; correlated with response | Pio et al., 2006 |
| Combination 5-FU, epirubicin, cyclophosphamide | Reduction at 1 week; correlated with response | Kenny et al., 2007 | |
| Colorectal | Combination 5-FU and radiotherapy | Reduction at 2 weeks; no correlation with response | Wieder et al., 2007 |
| Oropharyngeal cancer | Radiotherapy | Reduction at 1 week | Troost et al., 2010 |
| Radiotherapy | Increase 2–3 months compared with baseline; correlation with presence of disease | Been et al., 2009 | |
| Melanoma | CTLA4 blockade | No change in tumour uptake | Ribas et al., 2010 |
| Colon (COLO205 cells) | Checkpoint kinase 1 (Chk1)inhibitor and docetaxel | Reduction with docetaxel and combination treatment at day 2 + 16 | Zhang et al., 2009 |
The choline pathway can also be imaged as a marker of cell growth, and clinical response biomarker in malignancy. Phosphocholine is a biosynthetic product of choline kinase and is a precursor for membrane phosphatidylcholines, a class of phospholipids and a major component of biological membranes. The expression and activity of phosphocholine is up-regulated in a number of tumour types including prostate, lung, breast and colon cancer (Ramirez de Molina et al., 2002; 2007; Glunde et al., 2005). This increase in phosphocholine levels seen in malignancy is largely attributable to the up-regulation of choline kinase that occurs with malignant transformation. However, increased phosphocholine levels may also be due to increased breakdown via phospholipase C (Glunde et al., 2004). Choline can be radiolabelled with either carbon-11 ([11C]choline) or flurorine-18 ([18F]fluorocholine, or [18F]FCH). As [11C]Choline has reduced excretion by the urinary collecting system it has been used clinically in the imaging of pelvic tumours.
[11C]Choline has been most extensively used in the assessment of nodal disease in prostate cancer (Hara et al., 1998; de Jong et al., 2003; Reske et al., 2006; Scher et al., 2007). However, the utility of [11C]choline as an early response biomarker for the management of prostate cancer is yet to be established. Promising preclinical work by Krause and colleagues with [11C]choline showed utility for early therapeutic response to docetaxel in a prostate cancer xenograft model (Krause et al. 2010). Kenny and colleagues report on a subgroup of patients treated with trastuzumab who had early responses measured by [11C]choline-PET (Kenny et al., 2010). These findings require further investigation. [18F]-fluoromethyl-choline (FCH) has the advantage of longer half-life compared with [11C]choline, but these tracers undergo oxidation, with metabolites being detected within the plasma soon after administration. Thus the relative contribution of both the parent compound and the metabolites are difficult to ascertain when a late imaging protocol is used. Leyton and colleagues have recently reported data with a deuterated analogue,[18F]-fluoromethyl-[1,2-2H4]-choline (D4-[18F]FCH), which exhibits greater in vivo stability, and sensitivity particularly for late tumour imaging. Moreover, the authors report that D4-[18F]FCH was a biomarker of response in mice treated with the mitogenic inhibitor of the extracellular signal-regulated kinase, PD0325901 (Leyton et al., 2009).
Apoptosis
Apoptosis, or programmed cell death, is the likely mechanism behind the tumourcidal effects of both standard chemotherapeutic agents and many novel agents (Hanahan and Weinberg, 2000). An early event in apoptosis involves phosphatidylserine, which is normally confined to the cytoplasmic leaflet of the cell translocating to the external leaflet of the cell membrane. This has been exploited for imaging by labelling annexin V, an endogenous protein that has a high affinity for membrane bound phosphatidylderine, for imaging of apoptosis in vivo (Blankenberg et al., 1998). Annexin V was initially labelled with technetium-99 m and utilized as a SPECT radionuclide, to detect cardiac thrombosis in patients with atrial fibrillation (Blankenberg et al., 1999; Hofstra et al., 2000). SPECT imaging differs from PET imaging in that it involves the administration of a gamma-emitting tracer. An improved labelling methodology resulted in the development of [99mTc]-HYNIC-annexin V, which has been used in the assessment of lung cancer, where a patient responding to therapy had an increased uptake of the radiotracer (Blankenberg, 2008). Other annexin V-based radioisotopes with longer half-lives have been developed including iodine-124 with the specific advantage that its long half-life allows the characterization of longer-term biologicalal processes, and [18F]-annexin V (Glaser et al., 2003; Yagle et al., 2005).
There is still considerable interest in imaging apoptosis, as this is the predominant outcome from many cytotoxic and mew mechanism-based anticancer agents. [18F]-labelled 5-fluoropentyl-2-methyl-malonic acid ([18F]-ML10) is a 18F radiotracer which is a member of the ApoSense family of small-molecule apoptosis probes that utilize the γ-carboxyglutamic-acid (Gla)-domain protein as a potential critical structural moiety, enabling binding to cells with anionic phospholipid surfaces (Cohen et al., 2009). Early studies reported that induction of apoptosis is associated with cellular uptake of the probe, which is also caspase-dependent, and is blocked by caspase inhibition. Uptake of the tracer occurs in parallel with binding of Annexin-V and disruption of mitochondrial membrane potential. Uptake and accumulation of ([18F]-ML10 is lost upon membrane disruption and therefore may be utilized to differentiate apoptosis versus necrosis (Cohen et al., 2009). Whilst there are currently no published studies of the utility of [18F]-ML10 in the imaging of malignancies, a recent study has validated the use of an analogue of ML-10, ML-9. In this study [3H]-ML-9 was used to detect apoptosis induced by doxorubicin in in vivo models of malignancy (Grimberg et al., 2009). The authors report marked uptake of [3H]-ML-9 following treatment which correlated with both caspase activation and DNA fragmentation. Clinical validation of [18F]-ML10 as a marker of apoptosis is underway.
The process of cell death involves the family of caspases that execute cell death. Given the role of these enzymes in cell death there is considerable interest in imaging caspases. Isatin sulphonamides are novel agents in development that bind selectively to the active site of caspase-3 (and -7). Work by our group illustrates the utility of a specific [18F]isatin sulfonamide, [18F]ICMT-11, which binds to a range of drug-induced apoptotic cancer cells in vitro and to murine lymphoma xenografts (Smith et al., 2008; Nguyen et al., 2009). Furthermore, the increased signal intensity in tumours after drug treatment was associated with increased apoptosis. This tracer is currently undergoing further validation.
Angiogenesis
Angiogenesis is an essential process that supports tumour growth and metastasis through neovascularization that is driven by growth factors including vascular endothelial growth factor (VEGF) (Shibuya, 2008). There are two main strategies for the targeting of angiogenesis for therapeutic benefit: direct inhibition of angiogenesis growth factors and their receptors, for example, the anti-VEGF inhibitor bevacizumab, and indirect inhibition of blood flow through targeting of established vasculature, for example, the tubulin binding drug combretastatin A4-phosphate. Despite the introduction of these agents into clinical practice, there are few validated imaging biomarkers that can currently be used to predict or assess response to these therapies. Naturally there is considerable interest in this area
There have been a number of PET methodologies developed for imaging angiogenesis. Direct imaging of VEGF and its receptor has been studied by a number of investigators with IgG radiolabelled with long-lived isotopes but these are yet to be used routinely in the clinic. In a study by Collingridge and colleagues, the free and membrane bound VEGF in tumours was imaged directly in mice through utilization of an anti-VEGF antibody labelled with iodine-124 ([124I]-SHPP-VG76e) (Collingridge et al., 2002). This study highlighted the need to evaluate antibody diagnostics following labelling to assure that they retain immunoreactivity and the use of control antibodies to assess blood pooling. Specific imaging of VEGF at 24 h after injection of the radiotracer was demonstrated in tumours expressing different levels of VEGF, as well as following presaturation of VEGF binding sites by unlabelled VG76e. In a phase I study, HuMV833, a humanized anti-VEGF monoclonal antibody, was labelled with iodine-124 and the distribution of the antibody, as well as its clearance, was assessed (Jayson et al., 2002). The results indicated heterogenous uptake both within individual tumours and between individual patients. Recently, bevacizumab was labelled with zirconium-89 and assessed in an ovarian cancer model in vivo (Nagengast et al., 2007). The study reported higher uptake of radiolabeled bevacizumab compared with control radiolabelled IgG in the human xenografts. However, compared with other studies investigating radiolabelled bevacizumab the tumour uptake was lower. This may have been as there is no clear evidence that VEGF is up-regulated in this tumour model, which again underscores the importance of appropriate tumour models in proof of concept studies. With respect to the targeting of the VEGF receptor, Cai and colleagues recently published an in vivo study of [64Cu]-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-VEGF121 (Cai et al., 2006). The report suggests rapid and specific uptake of [64Cu]-DOTA-VEGF121 in tumours that express high levels of VEGFR-2. Furthermore, the authors have developed a VEGFR-2-specific PET tracer based on mutated VEGF121 that has lower binding affinity for VEGFR-1 without compromising VEGFR-2 binding affinity (Wang et al., 2007). Given the angiogenesis inducing properties of VEGF isoforms, it will be interesting to see how VEGFR-targeting probes are transferred into clinical use.
There is increasing interest in imaging integrin αvβ3. Integrins are a family of cell-adhesion receptors that allow the interaction of tumour cells and tumour endothelial cells with the extracellular matrix (ECM). The integrin αvβ3 is highly expressed on activated endothelial cells during angiogenesis. The tripeptide amino acid sequence, RGD, is present in a number of ECM proteins including vitronectin, fibronectin, and thrombospondin. Cyclic peptide ligands possessing the RGD sequence have high affinity for these integrins and, when labelled appropriately with positron- or γ-emitting radioisotopes, can be used for imaging αvβ3 receptor by PET. Haubner and colleagues synthesized [18F]Galacto-RGD for evaluation of tumour angiogenesis in a number of tumour types (Haubner et al., 2001). In these studies it was noted that tracer uptake corresponded to expression of αvβ3 and tracer uptake could be blocked by excess unlabelled compound. [18F]Galacto-RGD has been evaluated clinically in a number of tumour types illustrating good biodistribution, low background uptake and heterogeneous uptake within tumours that correlated with expression of αvβ3 within tumour samples. More recently a new pegylated RGD-based radioligand for αvβ3 ([18F]AH111585/[18F]fluciclatide) was developed with high tumour retention. In a recent phase I study, Kenny and colleagues reported that [18F]fluciclatide is retained in tumour tissues and detects primary and metastatic breast cancer by PET (Kenny et al., 2008 #125). A number of other clinical studies are currently underway utilizing this tracer as a response marker and results are awaited.
Visualization of blood flow offers another strategy for evaluating the vascular phenotype. Clinically, [15O]H2O has been extensively used to measure tissue perfusion in response to anti-vascular therapy. [15O]H2O is freely diffusible into and out of tissue water and is biologically inert, having the desired qualities for a tracer of perfusion. In a seminal study in phase I patients, [15O]H2O-PET was performed in patients receiving the anti-vascular agent combrestatin A4. The authors reported a 30–60% reduction in tumour flow at higher dose levels. This trial was important in delineating the role of PET imaging in phase I drug development. In particular, results from the PET study confirmed the mechanism of action of combrestatin A4 and allowed for the selection of dosing schedule to be taken forward into phase II (Collins, 2003).
Conclusion
In summary, the development of a novel radiotracer is a complicated process involving an experienced multi-disciplinary research team. Described here are the initial steps in identifying and developing a potential radioligand for clinical use. As described, this involves extensive in vitro and in vivo characterization. A number of oncology drugs have been radiolabelled for PET imaging of PK and various drugs have been evaluated for target modulation and response using PET. Imaging of the tumours and normal tissues preclude extrapolation of plasma PK-PD to tissue PK-PD and the inherent assumptions associated with that as these are measured directly by the imaging method. As oncology continues to move towards the concept of personalized medicine, the role of PET imaging in achieving this aim will develop further. PET imaging allows a non-invasive, functional assessment of pathophysiological processes occurring within a patient, and is becoming an essential tool in the assessment of disease diagnosis, progression and response to therapy. Furthermore, as the cost of drug development increases, PET may be useful in demonstrating proof of concept for the proposed molecular and biological mechanisms of action for a drug, as well as being a useful biomarker. This is an area where reduced regulatory burden on investigators will boost innovation and rapid transition of research technology into patient tools.
Acknowledgments
We thank Kaiyumars Contractor, Lynn Maslen and Graham Smith for their help with this MS and Laura Kenny and Julius Leyton for allowing us to use unpublished images.
This work was supported by Cancer Research UK/Engineering and Physical Sciences Research Council/Medical Research Council/National Institute for Health Research Grant C2536/A10337.
Glossary
Abbreviations
- 5-FU
5-fluorouracil
- ARSAC
Administration of Radioactive Substances Advisory Committee
- BCNU
1,3-bis-(2-chloroethyl)-1-nitrosourea
- CT
computer tomography
- DACA
N-[2-(dimethylamino)ethyl]acridine-4-carboamine
- DOTA
1,4,7,10-tetraazacyclododecane-N,N′,N″,N′″-tetraacetic acid
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- eIND
Exploratory Investigational New Drug
- EMEA
European Medicines Agency
- ENT
equilibrative nucleoside transporter
- EORTC
European Organisation for Research and Treatment of Cancer
- FDA
Food and Drug Administration
- GLP
Good Laboratory Practice
- GMP
Good Manufacturing Practice
- GRPP
Guidelines on Good Radiopharmacy Practice
- IMP
investigational medicinal product
- MCA
Medicines Control Agency
- MHRA
Medicines Healthcare products Regulatory Agency
- ML-10
5-fluoropentyl-2-methyl-malonic acid
- MTS
multicellular tumour spheroids
- PD
pharmacodynamics
- PET
positron emission tomography
- PK
pharmacokinetics
- RDRC
Radioactive Drug Research Committee
- RGD
arginine-glycine-aspartic acid
- SOP
standard operating procedure
- TK
thymidine kinase
- VEGF
vascular endothelial growth factor
Conflicts of interest
The authors have no conflict of interest to declare.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Ahmed FY, Johnston SJ, Cassidy J, O'Kelly T, Binnie N, Murray GI, et al. Eniluracil treatment completely inactivates dihydropyrimidine dehydrogenase in colorectal tumors. J Clin Oncol. 1999;17:2439–2445. doi: 10.1200/JCO.1999.17.8.2439. [DOI] [PubMed] [Google Scholar]
- Apisarnthanarax S, Alauddin MM, Mourtada F, Ariga H, Raju U, Mawlawi O, et al. Early detection of chemoradioresponse in esophageal carcinoma by 3′-deoxy-3′-3H-fluorothymidine using preclinical tumor models. Clin Cancer Res. 2006;12:4590–4597. doi: 10.1158/1078-0432.CCR-05-2720. [DOI] [PubMed] [Google Scholar]
- Bading JR, Shields AF. Imaging of cell proliferation: status and prospects. J Nucl Med. 2008;49(Suppl 2):64S–80S. doi: 10.2967/jnumed.107.046391. [DOI] [PubMed] [Google Scholar]
- Barthel H, Cleij MC, Collingridge DR, Hutchinson OC, Osman S, He Q, et al. 3′-deoxy-3′-[18F]fluorothymidine as a new marker for monitoring tumor response to antiproliferative therapy in vivo with positron emission tomography. Cancer Res. 2003;63:3791–3798. [PubMed] [Google Scholar]
- Barthel H, Perumal M, Latigo J, He Q, Brady F, Luthra SK, et al. The uptake of 3′-deoxy-3′-[18F]fluorothymidine into L5178Y tumours in vivo is dependent on thymidine kinase 1 protein levels. Eur J Nucl Med Mol Imaging. 2005;32:257–263. doi: 10.1007/s00259-004-1611-0. [DOI] [PubMed] [Google Scholar]
- Barwick T, Bencherif B, Mountz JM, Avril N. Molecular PET and PET/CT imaging of tumour cell proliferation using F-18 fluoro-L-thymidine: a comprehensive evaluation. Nucl Med Commun. 2009;30:908–917. doi: 10.1097/MNM.0b013e32832ee93b. [DOI] [PubMed] [Google Scholar]
- Bates RC, Edwards NS, Yates JD. Spheroids and cell survival. Crit Rev Oncol Hematol. 2000;36:61–74. doi: 10.1016/s1040-8428(00)00077-9. [DOI] [PubMed] [Google Scholar]
- Been LB, Hoekstra HJ, Suurmeijer AJ, Jager PL, van der Laan BF, Elsinga PH. [18F]FLT-PET and [18F]FDG-Pet in the evaluation of radiotherapy for laryngeal cancer. Oral Oncol. 2009;45:e211–e215. doi: 10.1016/j.oraloncology.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Bergstrom M, Grahnen A, Langstrom B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur J Clin Pharmacol. 2003;59:357–366. doi: 10.1007/s00228-003-0643-x. [DOI] [PubMed] [Google Scholar]
- Blankenberg FG. In vivo detection of apoptosis. J Nucl Med. 2008;49(Suppl 2):81S–95S. doi: 10.2967/jnumed.107.045898. [DOI] [PubMed] [Google Scholar]
- Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, et al. In vivo detection and imaging of phosphatidylserine expression during programmed cell death. Proc Natl Acad Sci U S A. 1998;95:6349–6354. doi: 10.1073/pnas.95.11.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, et al. Imaging of apoptosis (programmed cell death) with 99mTc annexin V. J Nucl Med. 1999;40:184–191. [PubMed] [Google Scholar]
- Brepoels L, Stroobants S, Verhoef G, De Groot T, Mortelmans L, De Wolf-Peeters C. 18)F-FDG and (18)F-FLT uptake early after cyclophosphamide and mTOR inhibition in an experimental lymphoma model. J Nucl Med. 2009;50:1102–1109. doi: 10.2967/jnumed.109.062208. [DOI] [PubMed] [Google Scholar]
- Buck AK, Kratochwil C, Glatting G, Juweid M, Bommer M, Tepsic D, et al. Early assessment of therapy response in malignant lymphoma with the thymidine analogue [18F]FLT. Eur J Nucl Med Mol Imaging. 2007;34:1775–1782. doi: 10.1007/s00259-007-0452-z. [DOI] [PubMed] [Google Scholar]
- Burton C, Ell P, Linch D. The role of PET imaging in lymphoma. Br J Haematol. 2004;126:772–784. doi: 10.1111/j.1365-2141.2004.05069.x. [DOI] [PubMed] [Google Scholar]
- Cai W, Wu Y, Chen K, Cao Q, Tice DA, Chen X. In vitro and in vivo characterization of 64Cu-labeled Abegrin, a humanized monoclonal antibody against integrin alpha v beta 3. Cancer Res. 2006;66:9673–9681. doi: 10.1158/0008-5472.CAN-06-1480. [DOI] [PubMed] [Google Scholar]
- Chen W, Delaloye S, Silverman DH, Geist C, Czernin J, Sayre J, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot study. J Clin Oncol. 2007;25:4714–4721. doi: 10.1200/JCO.2006.10.5825. [DOI] [PubMed] [Google Scholar]
- Agency EM, editor. CHMP. Position Paper on Non-Clinical Safety Studies to Support Clinical Trials with A Single Microdose. London: European Medicines Agency; 2004. C.f.M.P.f.H.U. [Google Scholar]
- Cohen A, Shirvan A, Levin G, Grimberg H, Reshef A, Ziv I. From the Gla domain to a novel small-molecule detector of apoptosis. Cell Res. 2009;19:625–637. doi: 10.1038/cr.2009.17. [DOI] [PubMed] [Google Scholar]
- Collingridge DR, Carroll VA, Glaser M, Aboagye EO, Osman S, Hutchinson OC, et al. The development of [(124)I]iodinated-VG76e: a novel tracer for imaging vascular endothelial growth factor in vivo using positron emission tomography. Cancer Res. 2002;62:5912–5919. [PubMed] [Google Scholar]
- Collins JM. Functional imaging in phase I studies: decorations or decision making? J Clin Oncol. 2003;21:2807–2809. doi: 10.1200/JCO.2003.05.100. [DOI] [PubMed] [Google Scholar]
- Contractor KB, Aboagye EO. Monitoring predominantly cytostatic treatment response with 18F-FDG PET. J Nucl Med. 2009;50(Suppl 1):97S–105S. doi: 10.2967/jnumed.108.057273. [DOI] [PubMed] [Google Scholar]
- Di Fabio F, Pinto C, Rojas Llimpe FL, Fanti S, Castellucci P, Longobardi C, et al. The predictive value of 18F-FDG-PET early evaluation in patients with metastatic gastric adenocarcinoma treated with chemotherapy plus cetuximab. Gastric Cancer. 2007;10:221–227. doi: 10.1007/s10120-007-0438-3. [DOI] [PubMed] [Google Scholar]
- Dimasi JA. Risks in new drug development: approval success rates for investigational drugs. Clin Pharmacol Ther. 2001;69:297–307. doi: 10.1067/mcp.2001.115446. [DOI] [PubMed] [Google Scholar]
- Dimitrakopoulou-Strauss A, Strauss LG, Schlag P, Hohenberger P, Irngartinger G, Oberdorfer F, et al. Intravenous and intra-arterial oxygen-15-labeled water and fluorine-18-labeled fluorouracil in patients with liver metastases from colorectal carcinoma. J Nucl Med. 1998;39:465–473. [PubMed] [Google Scholar]
- Dittmann H, Dohmen BM, Kehlbach R, Bartusek G, Pritzkow M, Sarbia M, et al. Early changes in [18F]FLT uptake after chemotherapy: an experimental study. Eur J Nucl Med Mol Imaging. 2002;29:1462–1469. doi: 10.1007/s00259-002-0925-z. [DOI] [PubMed] [Google Scholar]
- Dubessy C, Merlin JM, Marchal C, Guillemin F. Spheroids in radiobiology and photodynamic therapy. Crit Rev Oncol Hematol. 2000;36:179–192. doi: 10.1016/s1040-8428(00)00085-8. [DOI] [PubMed] [Google Scholar]
- EANM Radiopharmacy Committee. 2007. Guidelines on Current Good Radiopharmacy Practice (cGRPP) in the Preparation of Radiopharmaceuticals.
- Ebenhan T, Honer M, Ametamey SM, Schubiger PA, Becquet M, Ferretti S, et al. Comparison of [18F]-tracers in various experimental tumor models by PET imaging and identification of an early response biomarker for the novel microtubule stabilizer patupilone. Mol Imaging Biol. 2009;11:308–321. doi: 10.1007/s11307-009-0216-1. [DOI] [PubMed] [Google Scholar]
- Agency TEME, editor. EMEA. Questions & Answers on the CHMP Guideline on the Limits of Genotoxic Impurities (EMA/CHMP/SWP/431994/2007/Revision 2) London: 2009. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002907.pdf. [Google Scholar]
- Everitt S, Hicks RJ, Ball D, Kron T, Schneider-Kolsky M, Walter T, et al. Imaging cellular proliferation during chemo-radiotherapy: a pilot study of serial 18F-FLT positron emission tomography/computed tomography imaging for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2009;75:1098–1104. doi: 10.1016/j.ijrobp.2008.12.039. [DOI] [PubMed] [Google Scholar]
- FDA. Innovation or stagnation: challenge and opportunity on the critical path to new medical products. 2004.
- FDA. Critical Path 2010 Update. 2010. U.S. Department of Health & Human Services.
- Fowler JS, MacGregor RR, Wolf AP, Farrell AA, Karlstrom KI, Ruth TJ. A shielded synthesis system for production of 2-deoxy-2-[18F]fluoro-D-glucose. J Nucl Med. 1981;22:376–380. [PubMed] [Google Scholar]
- Fracasso G, Colombatti M. Effect of therapeutic macromolecules in spheroids. Crit Rev Oncol Hematol. 2000;36:159–178. doi: 10.1016/s1040-8428(00)00084-6. [DOI] [PubMed] [Google Scholar]
- Galldiks N, Kracht LW, Burghaus L, Ullrich RT, Backes H, Brunn A, et al. Patient-tailored, imaging-guided, long-term temozolomide chemotherapy in patients with glioblastoma. Mol Imaging. 2010;9:40–46. [PubMed] [Google Scholar]
- Gambhir SS, Czernin J, Schwimmer J, Silverman DH, Coleman RE, Phelps ME. A tabulated summary of the FDG PET literature. J Nucl Med. 2001;42:1S–93S. [PubMed] [Google Scholar]
- Ginos JZ, Cooper AJ, Dhawan V, Lai JC, Strother SC, Alcock N, et al. [13N]cisplatin PET to assess pharmacokinetics of intra-arterial versus intravenous chemotherapy for malignant brain tumors. J Nucl Med. 1987;28:1844–1852. [PubMed] [Google Scholar]
- Glaser M, Collingridge DR, Aboagye EO, Bouchier-Hayes L, Hutchinson OC, Martin SJ, et al. Iodine-124 labelled annexin-V as a potential radiotracer to study apoptosis using positron emission tomography. Appl Radiat Isot. 2003;58:55–62. doi: 10.1016/s0969-8043(02)00239-7. [DOI] [PubMed] [Google Scholar]
- Glunde K, Jie C, Bhujwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res. 2004;64:4270–4276. doi: 10.1158/0008-5472.CAN-03-3829. [DOI] [PubMed] [Google Scholar]
- Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 2005;65:11034–11043. doi: 10.1158/0008-5472.CAN-05-1807. [DOI] [PubMed] [Google Scholar]
- Graf N, Herrmann K, den Hollander J, Fend F, Schuster T, Wester HJ, et al. Imaging proliferation to monitor early response of lymphoma to cytotoxic treatment. Mol Imaging Biol. 2008;10:349–355. doi: 10.1007/s11307-008-0162-3. [DOI] [PubMed] [Google Scholar]
- Gray KR, Contractor KB, Kenny LM, Al-Nahhas A, Shousha S, Stebbing J, et al. Kinetic filtering of [(18)F]Fluorothymidine in positron emission tomography studies. Phys Med Biol. 2010;55:695–709. doi: 10.1088/0031-9155/55/3/010. [DOI] [PubMed] [Google Scholar]
- Grimberg H, Levin G, Shirvan A, Cohen A, Yogev-Falach M, Reshef A, et al. Monitoring of tumour response to chemotherapy in vivo by a novel small-molecule detector of apoptosis. Apoptosis. 2009;14:257–267. doi: 10.1007/s10495-008-0293-7. [DOI] [PubMed] [Google Scholar]
- Haioun C, Itti E, Rahmouni A, Brice P, Rain JD, Belhadj K, et al. [18F]fluoro-2-deoxy-D-glucose positron emission tomography (FDG-PET) in aggressive lymphoma: an early prognostic tool for predicting patient outcome. Blood. 2005;106:1376–1381. doi: 10.1182/blood-2005-01-0272. [DOI] [PubMed] [Google Scholar]
- Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27:235–238. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hara T, Kosaka N, Kishi H. PET imaging of prostate cancer using carbon-11-choline. J Nucl Med. 1998;39:990–995. [PubMed] [Google Scholar]
- Harte RJ, Matthews JC, O'Reilly SM, Price PM. Sources of error in tissue and tumor measurements of 5-[18F]fluorouracil. J Nucl Med. 1998;39:1370–1376. [PubMed] [Google Scholar]
- Haubner R, Wester HJ, Weber WA, Mang C, Ziegler SI, Goodman SL, et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–1785. [PubMed] [Google Scholar]
- Herrmann K, Wieder HA, Buck AK, Schoffel M, Krause BJ, Fend F, et al. Early response assessment using 3′-deoxy-3′-[18F]fluorothymidine-positron emission tomography in high-grade non-Hodgkin's lymphoma. Clin Cancer Res. 2007;13:3552–3558. doi: 10.1158/1078-0432.CCR-06-3025. [DOI] [PubMed] [Google Scholar]
- Hofstra L, Liem IH, Dumont EA, Boersma HH, van Heerde WL, Doevendans PA, et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet. 2000;356:209–212. doi: 10.1016/s0140-6736(00)02482-x. [DOI] [PubMed] [Google Scholar]
- Holdsworth CH, Badawi RD, Manola JB, Kijewski MF, Israel DA, Demetri GD, et al. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol. 2007;189:W324–W330. doi: 10.2214/AJR.07.2496. [DOI] [PubMed] [Google Scholar]
- Iwata R, Ido T, Takahashi T, Monma M. Automated synthesis system for productino of 2-deoxy-[18F]fluoro-D-glucose with computer control. Appl Radiat Isot. 1984;35:445–454. [Google Scholar]
- Jayson GC, Zweit J, Jackson A, Mulatero C, Julyan P, Ranson M, et al. Molecular imaging and biological evaluation of HuMV833 anti-VEGF antibody: implications for trial design of antiangiogenic antibodies. J Natl Cancer Inst. 2002;94:1484–1493. doi: 10.1093/jnci/94.19.1484. [DOI] [PubMed] [Google Scholar]
- de Jong IJ, Pruim J, Elsinga PH, Vaalburg W, Mensink HJ. 11C-choline positron emission tomography for the evaluation after treatment of localized prostate cancer. Eur Urol. 2003;44:32–38. doi: 10.1016/s0302-2838(03)00207-0. discussion 38–39. [DOI] [PubMed] [Google Scholar]
- Kawada K, Murakami K, Sato T, Kojima Y, Ebi H, Mukai H, et al. Prospective study of positron emission tomography for evaluation of the activity of lapatinib, a dual inhibitor of the ErbB1 and ErbB2 tyrosine kinases, in patients with advanced tumors. Jpn J Clin Oncol. 2007;37:44–48. doi: 10.1093/jjco/hyl116. [DOI] [PubMed] [Google Scholar]
- Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11:2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- Kenny LM, Vigushin DM, Al-Nahhas A, Osman S, Luthra SK, Shousha S, et al. Quantification of cellular proliferation in tumor and normal tissues of patients with breast cancer by [18F]fluorothymidine-positron emission tomography imaging: evaluation of analytical methods. Cancer Res. 2005;65:10104–10112. doi: 10.1158/0008-5472.CAN-04-4297. [DOI] [PubMed] [Google Scholar]
- Kenny L, Coombes RC, Vigushin DM, Al-Nahhas A, Shousha S, Aboagye EO. Imaging early changes in proliferation at 1 week post chemotherapy: a pilot study in breast cancer patients with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Eur J Nucl Med Mol Imaging. 2007;34:1339–1347. doi: 10.1007/s00259-007-0379-4. [DOI] [PubMed] [Google Scholar]
- Kenny LM, Coombes RC, Oulie I, Contractor KB, Miller M, Spinks TJ, et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients. J Nucl Med. 2008;49:879–886. doi: 10.2967/jnumed.107.049452. [DOI] [PubMed] [Google Scholar]
- Kenny LM, Contractor KB, Stebbing J, Al-Nahhas A, Palmieri C, Shousha S, et al. Altered tissue 3′-deoxy-3′-[18F]fluorothymidine pharmacokinetics in human breast cancer following capecitabine treatment detected by positron emission tomography. Clin Cancer Res. 2009;15:6649–6657. doi: 10.1158/1078-0432.CCR-09-1213. [DOI] [PubMed] [Google Scholar]
- Kenny LM, Contractor KB, Hinz R, Stebbing J, Palmieri C, Jiang J, et al. Reproducibility of [11C]choline-positron emission tomography and effect of trastuzumab. Clin Cancer Res. 2010;16:4236–4245. doi: 10.1158/1078-0432.CCR-10-0468. [DOI] [PubMed] [Google Scholar]
- Kissel J, Brix G, Bellemann ME, Strauss LG, Dimitrakopoulou-Strauss A, Port R, et al. Pharmacokinetic analysis of 5-[18F]fluorouracil tissue concentrations measured with positron emission tomography in patients with liver metastases from colorectal adenocarcinoma. Cancer Res. 1997;57:3415–3423. [PubMed] [Google Scholar]
- Krause BJ, Souvatzoglou M, Herrmann K, Weber AW, Schuster T, Buck AK, et al. [11C]Choline as pharmacodynamic marker for therapy response assessment in a prostate cancer xenograft model. Eur J Nucl Med Mol Imaging. 2010;37:1861–1868. doi: 10.1007/s00259-010-1493-2. [DOI] [PubMed] [Google Scholar]
- Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J Biomol Screen. 2004;9:273–285. doi: 10.1177/1087057104265040. [DOI] [PubMed] [Google Scholar]
- Kurdziel KA, Kiesewetter DO, Carson RE, Eckelman WC, Herscovitch P. Biodistribution, radiation dose estimates, and in vivo Pgp modulation studies of 18F-paclitaxel in nonhuman primates. J Nucl Med. 2003;44:1330–1339. [PubMed] [Google Scholar]
- Lee JH, Rosen EL, Mankoff DA. The role of radiotracer imaging in the diagnosis and management of patients with breast cancer: part 1 – overview, detection, and staging. J Nucl Med. 2009;50:569–581. doi: 10.2967/jnumed.108.053512. [DOI] [PubMed] [Google Scholar]
- Leyton J, Latigo JR, Perumal M, Dhaliwal H, He Q, Aboagye EO. Early detection of tumor response to chemotherapy by 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography: the effect of cisplatin on a fibrosarcoma tumor model in vivo. Cancer Res. 2005;65:4202–4210. doi: 10.1158/0008-5472.CAN-04-4008. [DOI] [PubMed] [Google Scholar]
- Leyton J, Alao JP, Da Costa M, Stavropoulou AV, Latigo JR, Perumal M, et al. In vivo biological activity of the histone deacetylase inhibitor LAQ824 is detectable with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Cancer Res. 2006a;66:7621–7629. doi: 10.1158/0008-5472.CAN-05-3962. [DOI] [PubMed] [Google Scholar]
- Leyton J, Lockley M, Aerts JL, Baird SK, Aboagye EO, Lemoine NR, et al. Quantiflying the activity of adenoviral E1A CR2 deletion mutants using renilla luciferase bioluminescence and 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography imaging. Cancer Res. 2006b;66:9178–9185. doi: 10.1158/0008-5472.CAN-06-1539. [DOI] [PubMed] [Google Scholar]
- Leyton J, Smith G, Lees M, Perumal M, Nguyen QD, Aigbirhio FI, et al. Noninvasive imaging of cell proliferation following mitogenic extracellular kinase inhibition by PD0325901. Mol Cancer Ther. 2008;7:3112–3121. doi: 10.1158/1535-7163.MCT-08-0264. [DOI] [PubMed] [Google Scholar]
- Leyton J, Smith G, Zhao Y, Perumal M, Nguyen QD, Robins E, et al. [18F]fluoromethyl-[1,2-2H4]-choline: a novel radiotracer for imaging choline metabolism in tumors by positron emission tomography. Cancer Res. 2009;69:7721–7728. doi: 10.1158/0008-5472.CAN-09-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti S, Schellens JH. The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. Br J Cancer. 2007;97:577–581. doi: 10.1038/sj.bjc.6603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MHRA. 2007. Rules and Guidance for Pharmaceutical Manufacturers and Distributors 2007.
- MHRA. 2010. Applying to Conduct A Clinical Trial: Additional Information, Vol. 2010. Medicines and Healthcare products Regulatory Agency (MHRA) http://www.mhra.gov.uk/Howweregulate/Medicines/Licensingofmedicines/Clinicaltrials/Applyingforaclinicaltrialauthorisation/Additionalinformation/index.htm. [Google Scholar]
- Moehler M, Dimitrakopoulou-Strauss A, Gutzler F, Raeth U, Strauss LG, Stremmel W. 18F-labeled fluorouracil positron emission tomography and the prognoses of colorectal carcinoma patients with metastases to the liver treated with 5-fluorouracil. Cancer. 1998;83:245–253. [PubMed] [Google Scholar]
- Molthoff CF, Klabbers BM, Berkhof J, Felten JT, van Gelder M, Windhorst AD, et al. Monitoring response to radiotherapy in human squamous cell cancer bearing nude mice: comparison of 2′-deoxy-2′-[18F]fluoro-D-glucose (FDG) and 3′-[18F]fluoro-3′-deoxythymidine (FLT) Mol Imaging Biol. 2007;9:340–347. doi: 10.1007/s11307-007-0104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monazzam A, Razifar P, Simonsson M, Qvarnstrom F, Josephsson R, Blomqvist C, et al. Multicellular tumour spheroid as a model for evaluation of [18F]FDG as biomarker for breast cancer treatment monitoring. Cancer Cell Int. 2006;6:6. doi: 10.1186/1475-2867-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monazzam A, Josephsson R, Blomqvist C, Carlsson J, Langstrom B, Bergstrom M. Application of the multicellular tumour spheroid model to screen PET tracers for analysis of early response of chemotherapy in breast cancer. Breast Cancer Res. 2007;9:R45. doi: 10.1186/bcr1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monazzam A, Razifar P, Ide S, Rugaard Jensen M, Josephsson R, Blomqvist C, et al. Evaluation of the Hsp90 inhibitor NVP-AUY922 in multicellular tumour spheroids with respect to effects on growth and PET tracer uptake. Nucl Med Biol. 2009;36:335–342. doi: 10.1016/j.nucmedbio.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Mueller-Klieser W. Tumor biology and experimental therapeutics. Crit Rev Oncol Hematol. 2000;36:123–139. doi: 10.1016/s1040-8428(00)00082-2. [DOI] [PubMed] [Google Scholar]
- Munch-Petersen B, Cloos L, Jensen HK, Tyrsted G. Human thymidine kinase 1. Regulation in normal and malignant cells. Adv Enzyme Regul. 1995;35:69–89. doi: 10.1016/0065-2571(94)00014-t. [DOI] [PubMed] [Google Scholar]
- Nagengast WB, de Vries EG, Hospers GA, Mulder NH, de Jong JR, Hollema H, et al. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med. 2007;48:1313–1319. doi: 10.2967/jnumed.107.041301. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO. Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci U S A. 2009;106:16375–16380. doi: 10.1073/pnas.0901310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358:1421–1423. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- Osman S, Rowlinson-Busza G, Luthra SK, Aboagye EO, Brown GD, Brady F, et al. Comparative biodistribution and metabolism of carbon-11-labeled N-[2-(dimethylamino)ethyl]acridine-4-carboxamide and DNA-intercalating analogues. Cancer Res. 2001;61:2935–2944. [PubMed] [Google Scholar]
- Ott K, Weber WA, Lordick F, Becker K, Busch R, Herrmann K, et al. Metabolic imaging predicts response, survival, and recurrence in adenocarcinomas of the esophagogastric junction. J Clin Oncol. 2006;24:4692–4698. doi: 10.1200/JCO.2006.06.7801. [DOI] [PubMed] [Google Scholar]
- Oyama N, Akino H, Suzuki Y, Kanamaru H, Ishida H, Tanase K, et al. FDG PET for evaluating the change of glucose metabolism in prostate cancer after androgen ablation. Nuclear Med Commun. 2001;22:963–969. doi: 10.1097/00006231-200109000-00004. [DOI] [PubMed] [Google Scholar]
- Pan MH, Huang SC, Liao YP, Schaue D, Wang CC, Stout DB, et al. FLT-PET imaging of radiation responses in murine tumors. Mol Imaging Biol. 2008;10:325–334. doi: 10.1007/s11307-008-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B, et al. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009;69:8645–8651. doi: 10.1158/0008-5472.CAN-09-1576. [DOI] [PubMed] [Google Scholar]
- Perumal M, Pillai RG, Barthel H, Leyton J, Latigo JR, Forster M, et al. Redistribution of nucleoside transporters to the cell membrane provides a novel approach for imaging thymidylate synthase inhibition by positron emission tomography. Cancer Res. 2006;66:8558–8564. doi: 10.1158/0008-5472.CAN-06-0898. [DOI] [PubMed] [Google Scholar]
- Pfannenberg C, Aschoff P, Dittmann H, Mayer F, Reischl G, von Weyhern C, et al. PET/CT with 18F-FLT: does it improve the therapeutic management of metastatic germ cell tumors? J Nucl Med. 2010;51:845–853. doi: 10.2967/jnumed.109.070425. [DOI] [PubMed] [Google Scholar]
- Pillai RG, Forster M, Perumal M, Mitchell F, Leyton J, Aibgirhio FI, et al. Imaging pharmacodynamics of the alpha-folate receptor-targeted thymidylate synthase inhibitor BGC 945. Cancer Res. 2008;68:3827–3834. doi: 10.1158/0008-5472.CAN-08-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimlott SL. Radiotracer development in psychiatry. Nucl Med Commun. 2005;26:183–188. doi: 10.1097/00006231-200503000-00002. [DOI] [PubMed] [Google Scholar]
- Pio BS, Park CK, Pietras R, Hsueh WA, Satyamurthy N, Pegram MD, et al. Usefulness of 3′-[F-18]fluoro-3′-deoxythymidine with positron emission tomography in predicting breast cancer response to therapy. Mol Imaging Biol. 2006;8:36–42. doi: 10.1007/s11307-005-0029-9. [DOI] [PubMed] [Google Scholar]
- Ramirez de Molina A, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez J, Bonilla F, et al. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biophys Res Commun. 2002;296:580–583. doi: 10.1016/s0006-291x(02)00920-8. [DOI] [PubMed] [Google Scholar]
- Ramirez de Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Taron M, Ramirez de Molina V, Cejas P, et al. Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: a retrospective study. Lancet Oncol. 2007;8:889–897. doi: 10.1016/S1470-2045(07)70279-6. [DOI] [PubMed] [Google Scholar]
- Reske SN, Blumstein NM, Neumaier B, Gottfried HW, Finsterbusch F, Kocot D, et al. Imaging prostate cancer with 11C-choline PET/CT. J Nucl Med. 2006;47:1249–1254. [PubMed] [Google Scholar]
- Ribas A, Benz MR, Allen-Auerbach MS, Radu C, Chmielowski B, Seja E, et al. Imaging of CTLA4 blockade-induced cell replication with (18)F-FLT PET in patients with advanced melanoma treated with tremelimumad. J Nucl Med. 2010;51:340–346. doi: 10.2967/jnumed.109.070946. [DOI] [PubMed] [Google Scholar]
- Romer W, Hanauske AR, Ziegler S, Thodtmann R, Weber W, Fuchs C, et al. Positron emission tomography in non-Hodgkin's lymphoma: assessment of chemotherapy with fluorodeoxyglucose. Blood. 1998;91:4464–4471. [PubMed] [Google Scholar]
- Rosenbaum SJ, Lind T, Antoch G, Bockisch A. False-positive FDG PET uptake – the role of PET/CT. Eur Radiol. 2006;16:1054–1065. doi: 10.1007/s00330-005-0088-y. [DOI] [PubMed] [Google Scholar]
- Rosso L, Brock CS, Gallo JM, Saleem A, Price PM, Turkheimer FE, et al. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009;69:120–127. doi: 10.1158/0008-5472.CAN-08-2356. [DOI] [PubMed] [Google Scholar]
- Rueger MA, Ameli M, Li H, Winkeler A, Rueckriem B, Vollmar S, et al. 18)F]FLT PET for non-invasive monitoring of early response to gene therapy in experimental gliomas. Mol Imaging Biol. 2010;13:547–557. doi: 10.1007/s11307-010-0361-6. [DOI] [PubMed] [Google Scholar]
- Saleem A, Aboagye EO, Price PM. In vivo monitoring of drugs using radiotracer techniques. Adv Drug Deliv Rev. 2000a;41:21–39. doi: 10.1016/s0169-409x(99)00054-x. [DOI] [PubMed] [Google Scholar]
- Saleem A, Yap J, Osman S, Brady F, Suttle B, Lucas SV, et al. Modulation of fluorouracil tissue pharmacokinetics by eniluracil: in-vivo imaging of drug action. Lancet. 2000b;355:2125–2131. doi: 10.1016/s0140-6736(00)02380-1. [DOI] [PubMed] [Google Scholar]
- Saleem A, Harte RJ, Matthews JC, Osman S, Brady F, Luthra SK, et al. Pharmacokinetic evaluation of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide in patients by positron emission tomography. J Clin Oncol. 2001;19:1421–1429. doi: 10.1200/JCO.2001.19.5.1421. [DOI] [PubMed] [Google Scholar]
- Saleem A, Brown GD, Brady F, Aboagye EO, Osman S, Luthra SK, et al. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003;63:2409–2415. [PubMed] [Google Scholar]
- Saleem A, Aboagye EO, Matthews JC, Price PM. Plasma pharmacokinetic evaluation of cytotoxic agents radiolabelled with positron emitting radioisotopes. Cancer Chemother Pharmacol. 2008;61:865–873. doi: 10.1007/s00280-007-0552-2. [DOI] [PubMed] [Google Scholar]
- Saleem A, Ranson M, Callies S, Lahn M, Prenant C, Brown GD, et al. Microdosing imaging pharmacokinetic (PK) study of the antisense oligonucleotide (ASO) to survivin (LY2181308) using positron emission tomography (PET): a novel paradigm in clinical drug development. J Clin Oncol. 2009;27:15s. suppl; abstr 3578. [Google Scholar]
- Schelling M, Avril N, Nahrig J, Kuhn W, Romer W, Sattler D, et al. Positron emission tomography using [(18)F]Fluorodeoxyglucose for monitoring primary chemotherapy in breast cancer. J Clin Oncol. 2000;18:1689–1695. doi: 10.1200/JCO.2000.18.8.1689. [DOI] [PubMed] [Google Scholar]
- Scher B, Seitz M, Albinger W, Tiling R, Scherr M, Becker HC, et al. Value of 11C-choline PET and PET/CT in patients with suspected prostate cancer. Eur J Nucl Med Mol Imaging. 2007;34:45–53. doi: 10.1007/s00259-006-0190-7. [DOI] [PubMed] [Google Scholar]
- Seitz U, Wagner M, Neumaier B, Wawra E, Glatting G, Leder G, et al. Evaluation of pyrimidine metabolising enzymes and in vitro uptake of 3′-[(18)F]fluoro-3′-deoxythymidine ([(18)F]FLT) in pancreatic cancer cell lines. Eur J Nucl Med Mol Imaging. 2002;29:1174–1181. doi: 10.1007/s00259-002-0851-0. [DOI] [PubMed] [Google Scholar]
- Shah C, Miller TW, Wyatt SK, McKinley ET, Olivares MG, Sanchez V, et al. Imaging biomarkers predict response to anti-HER2 (ErbB2) therapy in preclinical models of breast cancer. Clin Cancer Res. 2009;15:4712–4721. doi: 10.1158/1078-0432.CCR-08-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar LK, Hoffman JM, Bacharach S, Graham MM, Karp J, Lammertsma AA, et al. Consensus recommendations for the use of 18F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute Trials. J Nucl Med. 2006;47:1059–1066. [PubMed] [Google Scholar]
- Sherley JL, Kelly TJ. Regulation of human thymidine kinase during the cell cycle. J Biol Chem. 1988;263:8350–8358. [PubMed] [Google Scholar]
- Shibuya M. Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. BMB Rep. 2008;41:278–286. doi: 10.5483/bmbrep.2008.41.4.278. [DOI] [PubMed] [Google Scholar]
- Shields AF, Graham MM, Kozawa SM, Kozell LB, Link JM, Swenson ER, et al. Contribution of labeled carbon dioxide to PET imaging of carbon-11-labeled compounds. J Nucl Med. 1992;33:581–584. [PubMed] [Google Scholar]
- Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- Shreve PD, Anzai Y, Wahl RL. Pitfalls in oncologic diagnosis with FDG PET imaging: physiologic and benign variants. Radiographics. 1999;19:61–77. doi: 10.1148/radiographics.19.1.g99ja0761. quiz 150–151. [DOI] [PubMed] [Google Scholar]
- Smith IC, Welch AE, Hutcheon AW, Miller ID, Payne S, Chilcott F, et al. Positron emission tomography using [(18)F]-fluorodeoxy-D-glucose to predict the pathologic response of breast cancer to primary chemotherapy. J Clin Oncol. 2000;18:1676–1688. doi: 10.1200/JCO.2000.18.8.1676. [DOI] [PubMed] [Google Scholar]
- Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Arstad E, et al. Design, synthesis, and biological characterization of a caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J Med Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- Sohn HJ, Yang YJ, Ryu JS, Oh SJ, Im KC, Moon DH, et al. [18F]Fluorothymidine positron emission tomography before and 7 days after gefitinib treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin Cancer Res. 2008;14:7423–7429. doi: 10.1158/1078-0432.CCR-08-0312. [DOI] [PubMed] [Google Scholar]
- Solit DB, Santos E, Pratilas CA, Lobo J, Moroz M, Cai S, et al. 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography is a sensitive method for imaging the response of BRAF-dependent tumors to MEK inhibition. Cancer Res. 2007;67:11463–11469. doi: 10.1158/0008-5472.CAN-07-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroobants S, Goeminne J, Seegers M, Dimitrijevic S, Dupont P, Nuyts J, et al. 18FDG-Positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec) Eur J Cancer. 2003;39:2012–2020. doi: 10.1016/s0959-8049(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Stuart-Harris R, Caldas C, Pinder SE, Pharoah P. Proliferation markers and survival in early breast cancer: a systematic review and meta-analysis of 85 studies in 32,825 patients. Breast. 2008;17:323–334. doi: 10.1016/j.breast.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Subhas N, Patel PV, Pannu HK, Jacene HA, Fishman EK, Wahl RL. Imaging of pelvic malignancies with in-line FDG PET-CT: case examples and common pitfalls of FDG PET. Radiographics. 2005;25:1031–1043. doi: 10.1148/rg.254045155. [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Sakahara H, Sato K, Harada N, Fukumoto D, Kakiuchi T, et al. Evaluation of 3′-deoxy-3′-18F-fluorothymidine for monitoring tumor response to radiotherapy and photodynamic therapy in mice. J Nucl Med. 2004;45:1754–1758. [PubMed] [Google Scholar]
- Sunaga N, Oriuchi N, Kaira K, Yanagitani N, Tomizawa Y, Hisada T, et al. Usefulness of FDG-PET for early prediction of the response to gefitinib in non-small cell lung cancer. Lung Cancer. 2008;59:203–210. doi: 10.1016/j.lungcan.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Troost EG, Bussink J, Hoffmann AL, Boerman OC, Oyen WJ, Kaanders JH. 18F-FLT PET/CT for early response monitoring and dose escalation in oropharyngeal tumors. J Nucl Med. 2010;51:866–874. doi: 10.2967/jnumed.109.069310. [DOI] [PubMed] [Google Scholar]
- Tyler JL, Yamamoto YL, Diksic M, Theron J, Villemure JG, Worthington C, et al. Pharmacokinetics of superselective intra-arterial and intravenous [11C]BCNU evaluated by PET. J Nucl Med. 1986;27:775–780. [PubMed] [Google Scholar]
- Ullrich RT, Zander T, Neumaier B, Koker M, Shimamura T, Waerzeggers Y, et al. Early detection of erlotinib treatment response in NSCLC by 3′-deoxy-3′-[F]-fluoro-L-thymidine ([F]FLT) positron emission tomography (PET) Plos One. 2008;3:e3908. doi: 10.1371/journal.pone.0003908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhajosula S. (18)F-labeled positron emission tomographic radiopharmaceuticals in oncology: an overview of radiochemistry and mechanisms of tumor localization. Semin Nucl Med. 2007;37:400–419. doi: 10.1053/j.semnuclmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- VanBrocklin HF. Radiopharmaceuticals for drug development: United States regulatory perspective. Current Radiopharmaceuticals. 2008;1:2–6. [Google Scholar]
- Verel I, Visser GW, Van Dongen GA. The promise of immuno-PET in radioimmunotherapy. J Nucl Med. 2005;46(Suppl 1):164S–171S. [PubMed] [Google Scholar]
- Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50(Suppl 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldherr C, Mellinghoff IK, Tran C, Halpern BS, Rozengurt N, Safaei A, et al. Monitoring antiproliferative responses to kinase inhibitor therapy in mice with 3′-deoxy-3′-18F-fluorothymidine PET. J Nucl Med. 2005;46:114–120. [PubMed] [Google Scholar]
- Wang H, Cai W, Chen K, Li ZB, Kashefi A, He L, et al. A new PET tracer specific for vascular endothelial growth factor receptor 2. Eur J Nucl Med Mol Imaging. 2007;34:2001–2010. doi: 10.1007/s00259-007-0524-0. [DOI] [PubMed] [Google Scholar]
- Wei LH, Su H, Hildebrandt IJ, Phelps ME, Czernin J, Weber WA. Changes in tumor metabolism as readout for Mammalian target of rapamycin kinase inhibition by rapamycin in glioblastoma. Clin Cancer Res. 2008;14:3416–3426. doi: 10.1158/1078-0432.CCR-07-1824. [DOI] [PubMed] [Google Scholar]
- Wieder HA, Geinitz H, Rosenberg R, Lordick F, Becker K, Stahl A, et al. PET imaging with [18F]3′-deoxy-3′-fluorothymidine for prediction of response to neoadjuvant treatment in patients with rectal cancer. Eur J Nucl Med Mol Imaging. 2007;34:878–883. doi: 10.1007/s00259-006-0292-2. [DOI] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Chung YL, Griffiths JR, Hart R, Leach MO, et al. Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J Natl Cancer Inst. 2006;98:580–598. doi: 10.1093/jnci/djj162. [DOI] [PubMed] [Google Scholar]
- Yagle KJ, Eary JF, Tait JF, Grierson JR, Link JM, Lewellen B, et al. Evaluation of 18F-annexin V as a PET imaging agent in an animal model of apoptosis. J Nucl Med. 2005;46:658–666. [PubMed] [Google Scholar]
- Yang YJ, Ryu JS, Kim SY, Oh SJ, Im KC, Lee H, et al. Use of 3′-deoxy-3′-[18F]fluorothymidine PET to monitor early responses to radiation therapy in murine SCCVII tumors. Eur J Nucl Med Mol Imaging. 2006;33:412–419. doi: 10.1007/s00259-005-0011-4. [DOI] [PubMed] [Google Scholar]
- Yerushalmi R, Woods R, Ravdin PM, Hayes MM, Gelmon KA. Ki67 in breast cancer: prognostic and predictive potential. Lancet Oncol. 2010;11:174–183. doi: 10.1016/S1470-2045(09)70262-1. [DOI] [PubMed] [Google Scholar]
- Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–1782. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- Zhang C, Yan Z, Painter CL, Zhang Q, Chen E, Arango ME, et al. PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin Cancer Res. 2009;15:4630–4640. doi: 10.1158/1078-0432.CCR-08-3272. [DOI] [PubMed] [Google Scholar]
- Zhu AX, Meyerhardt JA, Blaszkowsky LS, Kambadakone AR, Muzikansky A, Zheng H, et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol. 2010;11:48–54. doi: 10.1016/S1470-2045(09)70333-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.