

Abstract
BACKGROUND AND PURPOSE
Sauchinone, an antioxidant lignan, protects hepatocytes from iron-induced toxicity. This study investigated the protective effects of sauchinone against acetaminophen (APAP)-induced toxicity in the liver and the role of nuclear factor erythroid-2-related factor-2 (Nrf2) in this effect.
EXPERIMENTAL APPROACH
Blood biochemistry and histopathology were assessed in mice treated with APAP or APAP + sauchinone. The levels of mRNA and protein were measured using real-time PCR assays and immunoblottings.
KEY RESULTS
Sauchinone ameliorated liver injury caused by a high dose of APAP. This effect was prevented by a deficiency of Nrf2. Sauchinone treatment induced modifier subunit of glutamate-cysteine ligase, NAD(P)H:quinone oxidoreductase-1 (NQO1) and heat shock protein 32 in the liver, which was abolished by Nrf2 deficiency. In a hepatocyte model, sauchinone activated Nrf2, as evidenced by the increased nuclear accumulation of Nrf2, the induction of NQO1-antioxidant response element reporter gene, and glutamate-cysteine ligase and NQO1 protein induction, which contributed to the restoration of hepatic glutathione content. Consistently, treatment of sauchinone enhanced Nrf2 phosphorylation with a reciprocal decrease in its interaction with Kelch-like ECH-associated protein-1. Intriguingly, sauchinone activated protein kinase C-δ (PKCδ), which led to Nrf2 phosphorylation. In addition, it increased the inhibitory phosphorylation of glycogen synthase kinase-3β (GSK3β), derepressing Nrf2 activity, which was supported by the reversal of sauchinone's activation of Nrf2 by an activated mutant of GSK3β. Moreover, phosphorylation of GSK3β by sauchinone depended on PKCδ activation.
CONCLUSION AND IMPLICATIONS
Our results demonstrate that sauchinone protects the liver from APAP-induced toxicity by activating Nrf2, and this effect is mediated by PKCδ activation, which induces inhibitory phosphorylation of GSK3β.
Keywords: sauchinone, acetaminophen, Nrf2, PKCδ, GSK3β
Introduction
Saururus chinensis has been traditionally used for the treatment of fever, jaundice, oedema and inflammatory disease in oriental folk medicine (Chung and Shin, 1990). It has been shown that sauchinone, a bioactive lignan isolated from Saururus chinensis, protects hepatocytes and other types of cells (Sung and Kim, 2000; Song et al., 2003), and inhibits lipopolysaccharide-stimulated nitric oxide and cytokine production by suppressing nuclear factor-κB activation (Hwang et al., 2003; Lee et al., 2003). Previously, we found that sauchinone treatment protects the liver from iron-induced toxicity, which may result from LKB1-dependent activation of AMP-activated protein kinase (AMPK) (Kim et al., 2009). Oxidative stress causes injury of cells by generating highly reactive oxygen species (ROS) such as hydrogen peroxide, superoxide anions and hydroxyl radicals (Stadtman, 1992); excessive ROS induces damage to lipids, proteins and DNA in cells, and may also lead to cancer development and progression, cardiovascular diseases and neurodegenerative conditions. So, the ability of sauchinone to act as an antioxidant with an anti-inflammatory action renders this compound a prime candidate for cell and organ protection (Lee et al., 2003).
Tight control of ROS levels by antioxidant molecules and detoxifying enzymes is important to restore balance between oxidants and antioxidants in cells challenged with oxidative insults. Nuclear factor erythroid-2-related factor 2 (Nrf2), which is a member of the Cap'n'Collar family, activates the transcription of antioxidant genes such as glutamate-cysteine ligase (GCL) and NAD(P)H:quinone oxidoreductase-1 (NQO1) by binding to the antioxidant response elements (AREs). So, animals deficient in Nrf2 are highly susceptible to organ injury from toxic stimuli such as acetaminophen (APAP), benzo[a]pyrene, diesel and other oxidative stresses due to decreased antioxidant protection (Aoki et al., 2001; Enomoto et al., 2001; Ramos-Gomez et al., 2001). Similarly, mice with a hepatocyte-specific deletion of Kelch-like ECH-associated protein-1 (Keap1) that degrades Nrf2 exhibit an increase in the genes encoding for detoxifying enzymes and are resistant to APAP-induced toxicity (Okawa et al., 2006). Thus, Nrf2 may serve as an endogenous regulator by which cells combat oxidative stress.
Acetaminophen has been widely used as a hepatotoxicant in animal models. At higher doses, N-acetyl-p-benzoquinoneimine (NAPQI) is produced from APAP by CYP450 enzymes including CYP2E1. When NAPQI is not detoxified properly, it forms protein adducts in the liver and induces toxicity by depleting glutathione (GSH) (James et al., 2003). GCL is one of the major antioxidant enzymes critical for the homeostasis of the cellular redox system and detoxification of electrophiles, including NAPQI (Dalton et al., 2000), because it is a rate-limiting enzyme for the synthesis of GSH. Indeed, mice deficient in the modifier subunit of glutamate-cysteine ligase (GCLM) exhibited more extensive hepatic damage after APAP intoxication compared to wild-type (WT) animals, verifying the importance of GSH and GCL activity for liver protection (McConnachie et al., 2007). Another key antioxidant enzyme, NQO1 is capable of detoxifying NAPQI (Moffit et al., 2007), but at the same time, it is increased in the livers of patients who have died from an overdose of APAP (Aleksunes et al., 2006). Thus, an increase in the production of NQO1 may be an adaptive response to the toxicity of APAP. Because these Nrf2 target genes can detoxify highly reactive intermediates, chemical activators of Nrf2 might be utilized to protect hepatocytes from the toxic effects of APAP or other agents.
In view of the importance of free radical stress as a major contributor to the toxicity of APAP, in this study we investigated the potential of sauchinone to protect the liver from APAP-induced toxicity and used a hepatocyte model to determine the molecular basis of this action.
In the present study, we explored the effects of sauchinone on the expression of major antioxidant enzymes and the role of activating Nrf2 in this process, and identified a novel signalling pathway for regulation of Nrf2 activity stimulated by the antioxidant lignan. The results of this study using the Nrf2 knockout (KO) animal model clearly demonstrated that the ability of sauchinone to protect hepatocytes from APAP toxicity is mediated by activation of protein kinase C-δ (PKCδ) that leads to inhibitory phosphorylation of glycogen synthase kinase 3β (GSK3β).
Methods
Materials
Sauchinone was isolated from the n-hexane fraction of S. chinensis, as previously described, and its chemical structure was verified by a variety of spectroscopic analyses (Sung and Kim, 2000). Anti-GCL antibody was purchased from Neomarkers (Fremont, CA, USA), whereas anti-NQO1 antibody was supplied from Abcam (Cambridge, MA, USA). Antibodies directed against Nrf2, Keap1 and PKCδ were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-lamin A and anti-phospho-PKCδ antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Horseradish peroxidase-conjugated goat anti-rabbit and goat anti-mouse IgGs were supplied from Zymed Laboratories Inc. (San Francisco, CA, USA). Rottlerin was purchased from Calbiochem (San Diego, CA, USA). Non-targeting siRNA and siRNA directed against PKCδ were purchased from Dharmacon Research Inc. (Lafayette, CO, USA). Anti-β-actin, anti-phospho-serine, anti-phospho-GSK3β (Ser9), anti-GSK3β antibodies, APAP and other reagents in the molecular studies were obtained from Sigma Aldrich (St. Louis, MO, USA).
Animals
Animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. Nrf2 KO mice were kindly supplied by Dr M Yamamoto (Tohoku University, Sendai, Japan), and bred at the College of Pharmacy, Seoul National University. WT and Nrf2 KO C57/BL6 mice at 8 weeks of age (∼25 g) were maintained in a clean room at the Animal Care Center, College of Pharmacy, Seoul National University. The animals were caged and supplied with filtered pathogen-free air, commercial chow (Purina, Korea) and water ad libitum at a temperature between 20 and 23°C with a 12 h light and dark cycle and relative humidity of 50%. Mice were treated with sauchinone (30 mg·kg−1, p.o., dissolved in 40% polyenthylene glycol and 0.5% methylcellulose) or vehicle (40% polyenthylene glycol and 0.5% methylcellulose) once daily for 3 days, and then were exposed to APAP (500 mg·kg−1, i.p., dissolved in 40% polyenthylene glycol) 1 h after last dose of sauchinone. Six hours after APAP administration, blood and liver tissue samples were taken for blood biochemical and histopathological analyses respectively.
Blood chemistry
Alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) in plasma were analysed using Spectrum®, an automatic blood chemistry analyser (Abbott Laboratories, Abbott Park, IL, USA).
Histopathology
Liver injury was assessed by light microscopy. The largest lobe of the liver was sliced, and tissue slices were fixed in 10% buffered-neutral formalin for 6 h. Fixed liver tissue slices were processed and embedded in a paraplast automatic tissue processor, Citadel 2000 (Shandon Scientific, Cheshire, UK). Sections of 4 µm in thickness were subjected to haematoxylin and eosin staining before being examined. A certified pathologist scored samples in a blinded fashion. An arbitrary scope was given to each microscopic field viewed at a magnification of 100× or 400× for the evaluation of central necrosis, hepatocyte degeneration and haemorrhage.
Determination of GSH content
Glutathione content in the liver tissue was quantified using a commercially available GSH determination kit (Oxis International, Portland, OR, USA). Briefly, the GSH-400 method was based on a chemical reaction, which proceeded in two steps. The first step led to the formation of substitution products (thioethers) between 4-chloro-1-methyl-7-trifluromethyl-quinolinum methylsulphate and all mercaptans, which were present in the sample. The second step included β-elimination reaction under alkaline conditions. This reaction was mediated by 30% NaOH, which specifically transformed the substituted product (thioether) obtained with GSH into a chromophoric thione.
Real-time PCR assays
Total RNA was isolated from cells using an RNeasy mini kit (QIAGEN, Valencia, CA, USA). The RNA (2 µg each) isolated was reverse-transcribed using oligo-d(T)16 primers to obtain cDNA. Real-time PCR was carried out according to the manufacturer's instructions (Light-Cycler 2.0; Roche, Mannheim, Germany). After PCR amplification, a melting curve of each amplicon was determined to verify its accuracy.
Immunoblot analyses
Sodium dodecyl sulphate polyacrylamide gel electrophoresis and immunoblot analyses were performed according to previously published procedures. Proteins were separated by gel electrophoresis and electrophoretically transferred to nitrocellulose paper. The nitrocellulose paper was incubated with antibodies overnight and was reacted with horseradish peroxidase-conjugated secondary antibody. Bands were developed using an ECL® chemiluminescence detection kit (Amersham Biosciences, Buckinghamshire, UK). The purity of nuclear fractions was confirmed by immunoblotting for lamin A/C (a nuclear marker). Equal loading of proteins was verified by immunoblottings for β-actin. Scanning densitometry of the immunoblots was performed with Image Scan & Analysis System (Alpha Innotech Corp., San Leandro, CA, USA). The area of each lane was integrated using the software AlphaEaseTM version 5.5, followed by background subtraction.
Cell culture
HepG2 cells, a human hepatocyte-derived cell line, were purchased from ATCC (Rockville, MD, USA). The cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 50 U·mL−1 penicillin and 50 µg·mL−1 streptomycin at 37°C in humidified atmosphere with 5% CO2. The cells were plated at a density of 5 × 106 cells per 10 cm diameter dish and preincubated for 24 h at 37°C. For all experiments, HepG2 cells were grown to 70–80% confluence and were subjected to no more than 20 cell passages. Protein samples were stored at −70°C until use.
Preparation of nuclear extracts
Nuclear extracts were prepared according to a previously published method. Briefly, HepG2 cells (1 × 107) in dishes were washed twice with ice-cold phosphate buffered saline (PBS) and then scraped from the dishes with 1 mL of PBS and transferred to microtubes. Cells were then centrifuged at 2000× g for 5 min. The supernatant was discarded, and the pellet was allowed to swell after the addition of 100 µL of hypotonic buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.5% Nonidet P-40, 1 mM dithiothreitiol and 0.5 mM phenylmethylsulphonyl fluoride. The lysates were incubated for 10 min on ice and then centrifuged at 7200× g for 5 min at 4°C. Pellets containing crude nuclei were resuspended in 50 µL of extraction buffer containing 20 mM HEPES (pH 7.9), 400 mM NaCl, 1 mM EDTA, 10 mM dithiothreitiol and 1 mM phenylmethylsulphonyl fluoride, and then were incubated for 30 min on ice. The samples were then centrifuged at 15 800× g for 10 min to obtain supernatants containing nuclear fractions. Nuclear fractions were stored at −70°C until use.
Transient transfection and luciferase reporter assay
Cells were plated at a density of 1 × 106 cells per well in 6-well dishes and transfected the following day. Briefly, the cells were incubated with 1 µg of pGL-1651 reporter plasmid and 3 µL of Lipofectamine® reagent (Life Technologies, Gaithersburg, MD, USA) in 1 mL of antibiotic-free minimum essential media medium for 3 h. The cells were incubated in a serum-free medium for 6 h and then exposed to medium containing FBS with or without 3–30 µM sauchinone. Luciferase activities were measured using a dual luciferase assay system (Promega, Madison, WI, USA).
Immunoprecipitation assays
To assess the interaction between Nrf2 and Keap1 or to measure the degree of Nrf2 phosphorylation, cell lysates were immunoprecipitated with anti-Nrf2 antibody overnight at 4°C. Cell lysates were incubated with anti-Nrf2 antibody overnight at 4°C. The antigen–antibody complex was immunoprecipitated after incubation with protein G-agarose for 2 h. Immune complexes were dissolved in 2× Laemmli buffer. Protein samples were resolved and immunoblotted with anti-Keap1 or anti-phospho-serine antibody.
Statistical analyses
One-way analysis of variance procedures were used to assess significant differences among treatment groups. For each significant effect of treatment, the Newman–Keuls test was used for comparisons of multiple group means. The criterion for statistical significance was set at P < 0.05 or P < 0.01.
Results
Protection of the liver from APAP-induced toxicity
First, the effects of pretreatment with sauchinone (p.o.) on APAP-induced liver injury were determined in WT and Nrf2 KO mice. The blood biochemistry showed that plasma ALT and AST levels were elevated 6 h after a single dose of 500 mg·kg−1 APAP treatment (i.p.). Pretreatment with sauchinone (30 mg·kg−1·day−1, p.o., for three consecutive days) significantly inhibited the increase in plasma ALT or AST activity in WT mice challenged with APAP (Figure 1, upper and middle). After APAP administration, the increases in ALT and AST activities were more in Nrf2 KO mice than in WT mice, consistent with results obtained previously (Enomoto et al., 2001), and the pretreatment with sauchinone failed to prevent this APAP-induced increase in plasma ALT and AST activities in these Nrf2 KO mice. Sauchinone treatment alone did not alter the blood biochemistry in Nrf2 KO mice. In addition, LDH was measured as another index of hepato-cellular injury. Consistently, sauchinone effectively prevented an increase in plasma LDH activity elicited by APAP in WT mice, but failed to do so in Nrf2 KO mice (Figure 1, lower).
Figure 1.
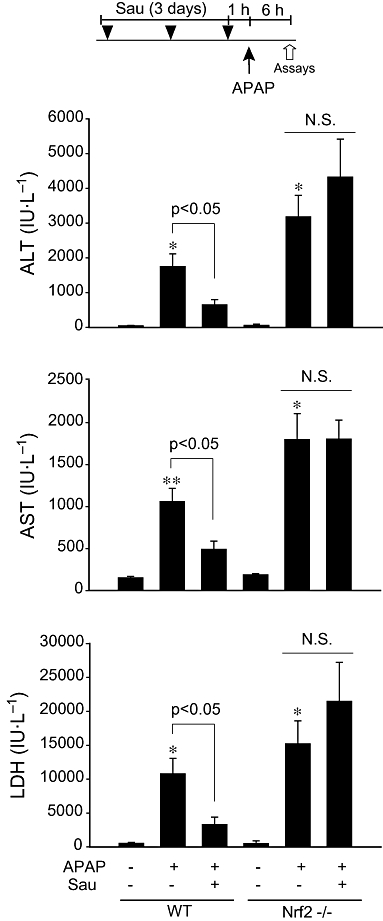
The effect of sauchinone (Sau) on acetaminophen (APAP)-induced liver injury. Male wild-type (WT) or Nrf2 knockout mice that had been treated with Sau (30 mg·kg−1·day−1, p.o., for 3 days) were administered a single dose of APAP (500 mg·kg−1, i.p.). The animals were killed 6 h after APAP treatment. The activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were measured in the plasma samples. N.S., not significant.
To verify the protective effects of sauchinone against APAP-induced hepatotoxicity, we examined the extent of liver injury histopathologically. In WT mice, the vehicle produced no apparent abnormalities but APAP caused haemorrhage in the central area of the liver, as usually seen after APAP intoxication, and pretreatment of these mice with sauchinone resulted in an obvious protective effect against this APAP-induced toxicity (Figure 2, left). As expected, there were no observable changes in the liver tissue of Nrf2 KO mice treated with vehicle. In contrast, treatment of Nrf2 KO mice with APAP resulted in significantly increased haemorrhage and necrosis in the central area, which was more severe than that in WT mice exposed to APAP. Consistent with the blood biochemical results, sauchinone pretreatment of Nrf2 KO mice failed to prevent central liver necrosis elicited by APAP (Figure 2, right).
Figure 2.
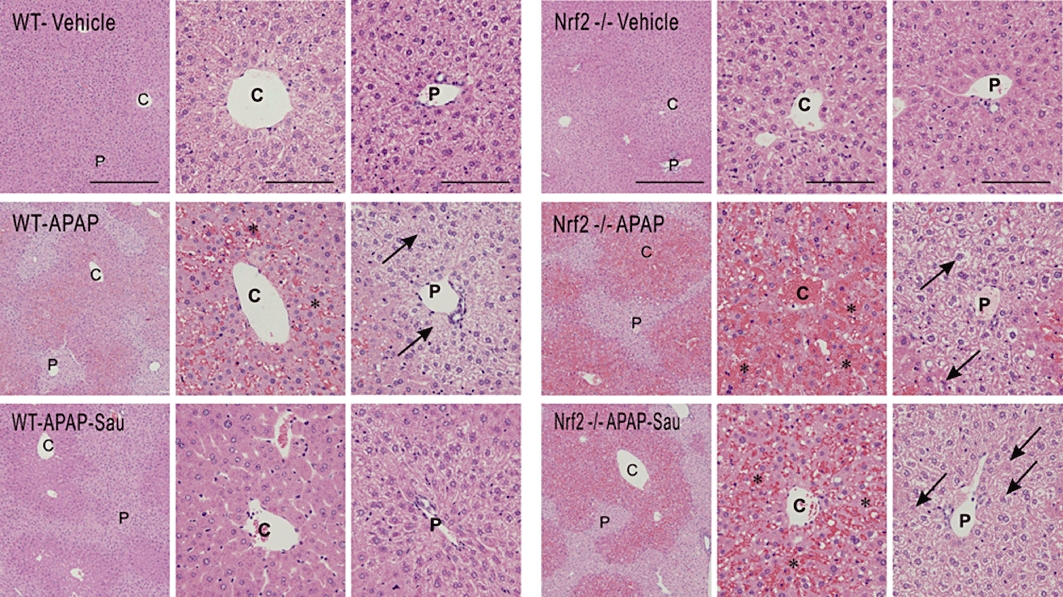
Haematoxylin and eosin staining of the liver. Mice were treated with acetaminophen (APAP) or APAP + sauchinone (Sau), as described in the legend to Figure 1 (n = 6 each). Arrows indicate degeneration and necrosis of the hepatocytes. C, central vein; P, portal space; *, haemorrhage; scale bars = 200 µm in left column and 50 µm in middle and right columns; Low-power view (left column) and high-power view of the central and portal area (right column). Nrf2, nuclear factor erythroid-2-related factor-2; WT, wild-type.
Restoration of GSH by induction of GCL and other antioxidant enzymes
Glutathione has an important role in protecting hepatocytes from the toxic effects of APAP; hence, we determined the hepatic GSH levels to monitor the actual improvement in the liver function. As expected, sauchinone pretreatment almost completely prevented the decrease in the GSH caused by APAP (Figure 3A). Next, we determined the effects of sauchinone on the induction of Nrf2 target genes in WT and Nrf2 KO mice. Because GCL is a redox-sensitive rate-limiting enzyme that catalyses GSH synthesis, the effect of sauchinone on the expression of GCLM mRNA was assessed in the liver. As expected, sauchinone significantly increased the level of GCLM mRNA in WT mice and this was not observed in Nrf2 KO animals (Figure 3B). Moreover, it effectively elevated the mRNA levels of other Nrf2 target genes [i.e. NQO1 and heat shock protein 32 (HSP32, haeme oxygenase-1) ]. Again, the ability of sauchinone to increase the mRNA of these target genes was severely impaired in the Nrf2 KO group (Figure 3B). Immunoblots of these proteins confirmed similar changes (Figure 3C). Additionally, we examined the levels of nuclear Nrf2 in the livers of WT and KO mice treated with sauchinone alone or in combination with APAP. Either sauchinone or APAP caused an increase in nuclear Nrf2 content. Moreover, simultaneous treatment of WT mice with both agents resulted in an enhanced increase in nuclear Nrf2 (Figure 3D). As expected, these increases in nuclear Nrf2 content were not observed in the Nrf2 KO group. All of these data indicate that the activation of Nrf2 by sauchinone has an important role in the induction of these genes as well as protecting the liver from APAP intoxication.
Figure 3.
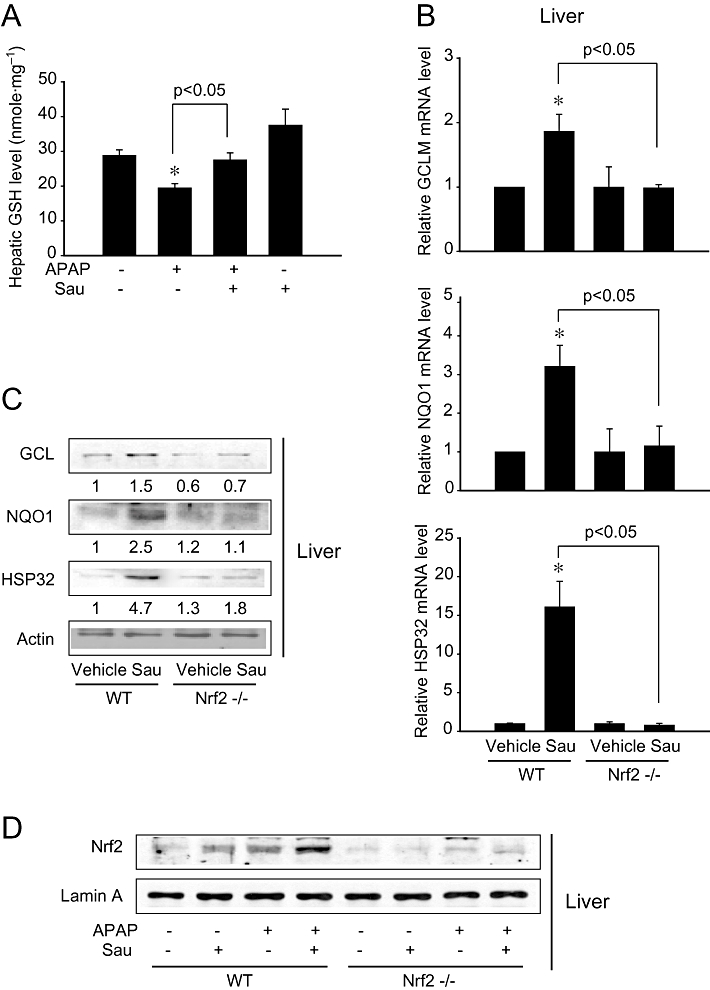
The effects of sauchinone (Sau) on hepatic glutathione (GSH) contents and glutamate-cysteine ligase (GCL), NAD(P)H:quinone oxidoreductase-1 (NQO1) and heat shock protein 32 (HSP32) expression. (A) Hepatic GSH contents. Mice were treated with acetaminophen (APAP) or APAP + Sau, as described in the legend to Figure 1 (n = 6 each). (B) Real-time PCR assays for modifier subunit of glutamate-cysteine ligase (GCLM), NQO1 and HSP32. Sau (30 mg·kg−1·day−1) was administered to wild-type (WT) or nuclear factor erythroid-2-related factor-2 (Nrf2) knockout mice for 3 days, and the liver samples were collected 6 h after last injection of Sau (n = 6 each). (C) Immunoblottings for GCL, NQO1 and HSP32 in the liver homogenates. Values indicate scanning densitometric results. Representative data from six samples. (D) Immunoblottings for Nrf2 in the nuclear fractions of liver homogenates. Representative data from six samples.
Next, an experimental cell model was used to explore the effects of sauchinone on the levels of GCL and NQO1. Real-time PCR assays revealed that treatment of HepG2 cells with 30 µM sauchinone notably increased the mRNA levels of the catalytic subunit of GCL, GCLM and NQO1 12 h after treatment (Figure 4A). Immunoblot analysis verified that sauchinone increased the levels of GCL and NQO1 (Figure 4B). Thus, treatment of hepatocyte-derived cells with sauchinone induced an increase in the levels of antioxidant and cytoprotective enzymes. Overall, our results demonstrate that an Nrf2-mediated increase in antioxidant enzymes induced by sauchinone may contribute to the protection of hepatocytes from oxidative and electrophilic stress.
Figure 4.
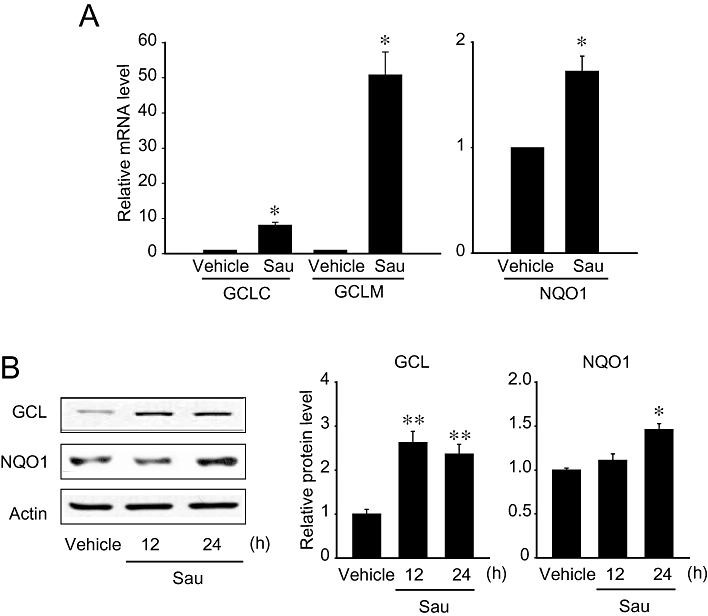
Induction of glutamate-cysteine ligase (GCL) and NAD(P)H:quinone oxidoreductase-1 (NQO1) by sauchinone. (A) Real-time PCR assays. The expression of nuclear factor erythroid-2-related factor-2 (Nrf2) target genes was monitored in HepG2 cells using real-time PCR assays, in which the mRNA level of GAPDH was used as a reference for data normalization. Cells were treated with vehicle or 30 µM sauchinone (Sau) for 12 h and subjected to the preparation of mRNA, from which cDNA was synthesized by reverse transcriptase. Fold changes were calculated by correlation coefficients of crossing point (Cp) for triplicate PCR results. (B) Immunoblottings for GCL and NQO1. The proteins of interest were immunoblotted in the lysates of HepG2 cells that had been treated with vehicle or 30 µM sauchinone for 24 h. GCLC, catalytic subunit of glutamate-cysteine ligase; GCLM, modifier subunit of glutamate-cysteine ligase.
Nrf2-dependent ARE gene transactivation
Nuclear factor erythroid-2-related factor 2 is sequestered by Keap1 in the cytoplasm under normal conditions, but translocates into the nucleus after stimulation. To assess the effect of sauchinone treatment on the activation of Nrf2-ARE, we performed a dose– and time–response study in HepG2 cells. Treatment of the cells with 3–30 µM sauchinone for 3 h markedly increased the nuclear Nrf2 content in a concentration-dependent manner (Figure 5A left). Moreover, nuclear accumulation of Nrf2 occurred as early as 1 h after sauchinone treatment, reached a maximum at 3 h, and this was maintained up to at least 12 h after treatment (Figure 5A, right).
Figure 5.
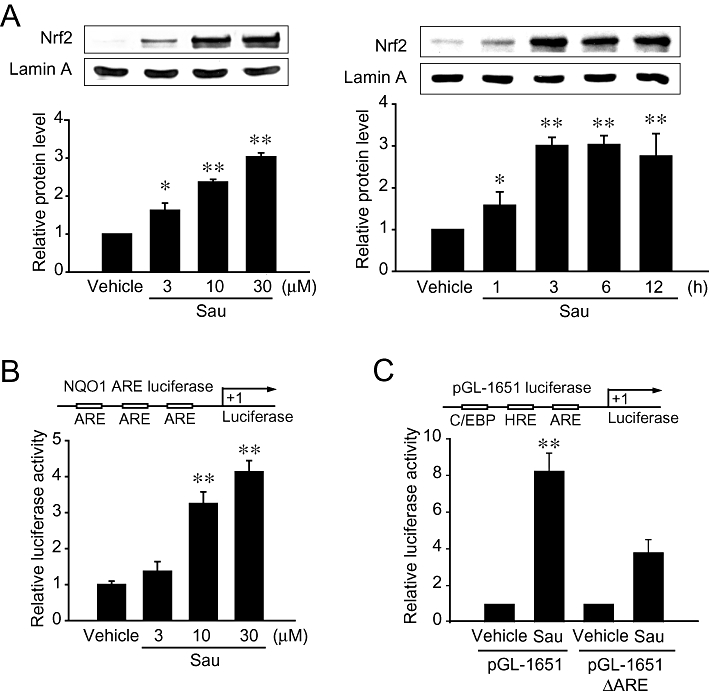
The effects of sauchinone (Sau) on nuclear factor erythroid-2-related factor-2 (Nrf2) activation and antioxidant response element (ARE) reporter gene induction. (A) Nuclear accumulation of Nrf2. Nrf2 contents were measured in the nuclear fractions of HepG2 cells treated with vehicle or Sau as indicated above. (B) NAD(P)H:quinone oxidoreductase-1 (NQO1) ARE reporter assays. Luciferase activity was measured on the lysates of cells treated with 3–30 µM sauchinone for 24 h following transfection with a NQO1-ARE construct. (C) The effect of Sau on the induction of luciferase activity from pGL-1651 or pGL-1651-ΔARE. Luciferase activity was measured in cells that had been transfected with pGL-1651 or pGL-1651-ΔARE, and exposed to vehicle or 30 µM Sau for 24 h. The values represent the mean ± SE of four separate experiments (significant compared with control, **P < 0.01; control = 1). C/EBP, CCAAT/enhancer-binding protein; HRE, HNF response element.
To assess the functional role of sauchinone-induced activation of Nrf2 for ARE-driven reporter gene expression (Figure 5B, upper), we investigated the ability of sauchinone to increase NQO1-ARE luciferase gene transactivation. Treatment of NQO1-ARE-transfected cells with sauchinone resulted in a threefold to fourfold increase in luciferase activity (Figure 5B, lower). Luciferase activity assays were additionally performed in HepG2 cells transfected with pGL-1651 that contained the luciferase gene downstream of the −1.65 kb GSTA2 promoter region (Figure 5C, upper). Sauchinone also increased luciferase induction from the construct. In addition, the ability of sauchinone to induce luciferase from pGL-1651 was prevented by the ARE deletion mutation (i.e. pGL-1651-ΔARE) (Figure 5C, lower). These results support the concept that sauchinone-induced activation of Nrf2 contributes to the ARE-driven gene transactivation. The important consequence of this finding is that sauchinone notably activates Nrf2, which serves as a key molecule in the activation of ARE for target gene transactivation.
Sauchinone activates PKCδ that phosphorylates Nrf2
Because Nrf2 activation requires phosphorylation, we further assessed Nrf2 phosphorylation using immunoprecipitation assays. Sauchinone enhanced Nrf2 phosphorylation at the serine residue in HepG2 cells, as did tert-butylhydroquinone (Figure 6A, upper). The phosphorylation of Nrf2 may be accompanied by the release of Nrf2 from Keap1, an inhibitory protein. Immunoprecipitation and immunoblot analyses showed that sauchinone treatment notably decreased the Nrf2-Keap1 interaction, the degree of which was comparable to that caused by tert-butylhydroquinone (Figure 6A, lower). These results indicate that sauchinone increases Nrf2 phosphorylation and reciprocally decreases the Nrf2-Keap1 interaction.
Figure 6.
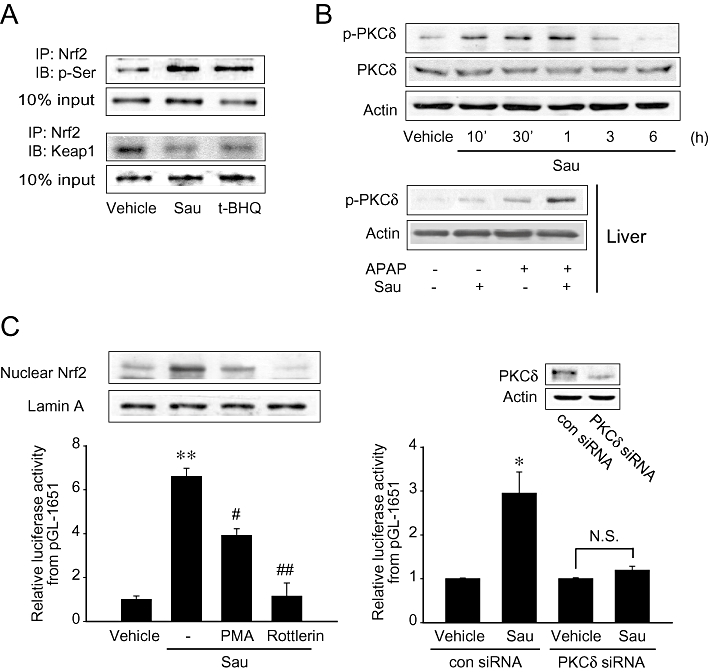
Sauchinone (Sau) activation of protein kinase C δ (PKCδ) and PKCδ-dependent nuclear factor erythroid-2-related factor-2 (Nrf2) activity. (A) Nrf2 phosphorylation with a reciprocal decrease in the interaction between Nrf2 and Kelch-like ECH-associated protein-1 (Keap1). HepG2 cells were treated with vehicle or Sau for 3 h. Nrf2 immunoprecipitates were subjected to immunoblottings for phosphorylated serine and Keap1. Results were confirmed by repeated experiments. tert-butylhydroquinone, t-BHQ. (B) The effect of Sau treatment on the phosphorylation of PKCδ. Immunoblottings were performed on the lysates of HepG2 cells treated with 30 µM Sau for 10 min–6 h (upper), or the livers of mice treated with 30 mg·kg−1 Sau and/or 500 mg·kg−1 APAP for 6 h (lower). (C) Inhibition by phorbol 12-myristate 13-acetate (PMA) or rottlerin of the ability of Sau to activate Nrf2. Nrf2 was immunoblotted in the nuclear fractions of cells treated with 30 µM Sau alone or in combination with 1 µM PMA or 2 µM rottlerin for 3 h. Luciferase activity was measured on the lysates of pGL-1651-transfected cells exposed to Sau alone or in combination with PMA or rottlerin for 24 h (left). In addition, luciferase activity was similarly determined after siRNA transfection (right). Data represent the mean ± SE of 4 separate experiments (significant compared with the respective control, *P < 0.05, **P < 0.01).
Protein kinase C δ catalyses Nrf2 phosphorylation at serine 40 residue, which is required for Nrf2 activation (Habelhah et al., 2004). In an attempt to identify the upstream effector necessary for Nrf2 phosphorylation by sauchinone, we further examined the effect of sauchinone on PKCδ activation. Immunoblottings revealed that PKCδ was phosphorylated in HepG2 cells from 10 min–1 h after sauchinone treatment, indicating PKCδ activation (Figure 6B, upper). Consistent with this finding, treatment of mice with either sauchinone (30 mg·kg−1·day−1, for 3 days) or APAP (500 mg·kg−1, for 6 h) also facilitated PKCδ phosphorylation in the liver (Figure 6B, lower). Moreover, simultaneous treatment with both sauchinone and APAP induced a greater increase in the phosphorylation. To link PKCδ activation and Nrf2 phosphorylation, we examined the degree of Nrf2 activation after PKCδ inhibition with either phorbol 12-myristate 13-acetate or rottlerin (a specific PKCδ inhibitor). Pretreatment with either phorbol 12-myristate 13-acetate or rottlerin prevented the ability of sauchinone to increase pGL-1651 gene transactivation (Figure 6C, left). In accord with this, PKCδ knockdown using siRNA resulted in a similar effect (Figure 6C, right). These results demonstrate that activated PKCδ is responsible for Nrf2 activation by sauchinone.
PKCδ-dependent GSK3β phosphorylation is involved in the activation of Nrf2
Glycogen synthase kinase 3β is an inhibitory upstream component that regulates Nrf2 phosphorylation and subcellular localization (Salazar et al., 2006); Nrf2 phosphorylation by GSK3β facilitates the exclusion of Nrf2 from the nucleus, which may be associated with its pro-apoptotic effect. As part of an effort to understand the molecular basis underlying Nrf2 activation by sauchinone, we tested the possibility that sauchinone treatment increases the phosphorylation of GSK3β. Sauchinone increased GSK3β phosphorylation at the serine 9 residue 30 min or 1 h after treatment (Figure 7A, upper). Similarly, sauchinone treatment markedly enhanced the phosphorylation of GSK3β in the liver tissue (Figure 7A, lower). APAP had a weak effect. As a functional study, we examined whether an alteration in GSK3β activity affected the ability of sauchinone to activate Nrf2. Transfection with a constitutively active genetic variant of GSK3β (ΔSer9GSK3β, CA-GSK3β) completely inhibited the nuclear accumulation of Nrf2 induced by sauchinone (Figure 7B, upper). Consistent with this result, transfection with ΔSer9GSK3β prevented the ability of sauchinone to promote pGL-1651 gene transactivation (Figure 7B, lower), showing that this response is regulated by activated GSK3β. Our results indicate that increased phosphorylation of GSK3β by sauchinone is involved in Nrf2 activation.
Figure 7.
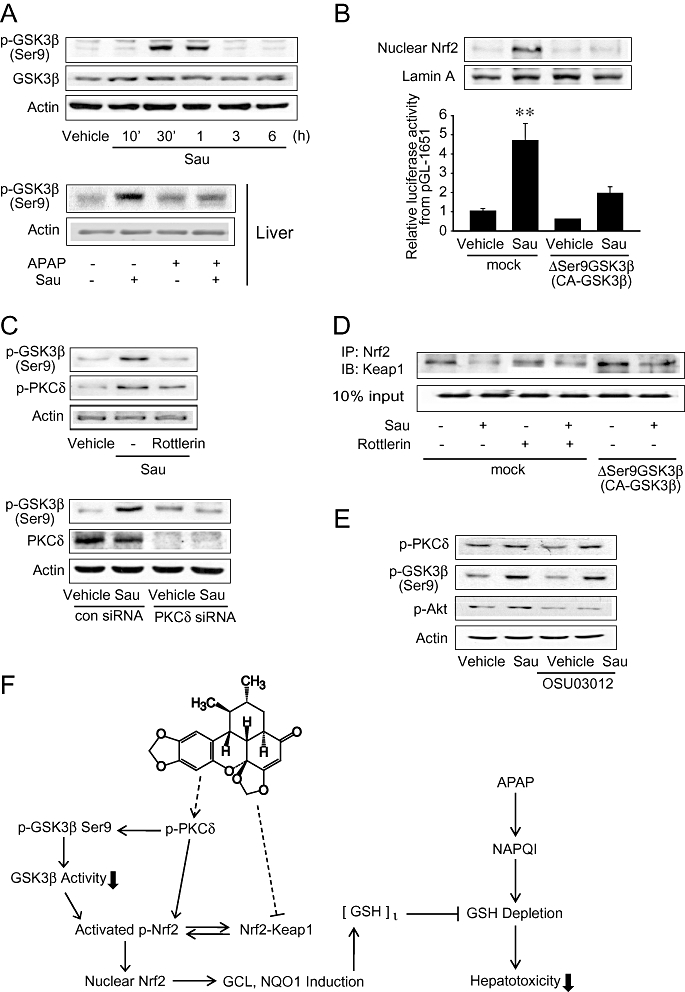
Increase in GSK3β phosphorylation by sauchinone (Sau). (A) Immunoblottings for glycogen synthase kinase 3β (GSK3β). Ser9-phosphorylated or total GSK3β was immunochemically measured on the lysates of HepG2 cells treated with 30 µM Sau for 10 min–6 h (upper), or on the homogenates of livers of mice treated with 30 mg·kg−1 Sau and/or 500 mg·kg−1 acetaminophen (APAP) for 6 h (lower). Results were confirmed in three separate experiments. (B) Reversal by ΔSer9GSK3β (CA-GSK3β) of the ability of Sau to activate nuclear factor erythroid-2-related factor-2 (Nrf2). Nrf2 was immunoblotted in the lysates of mock- or ΔSer9GSK3β-transfected cells treated with vehicle or Sau for 3 h. Lamin A immunoblotting verified equal loading and the purity of nuclear proteins (upper). Luciferase activity was measured on the lysates of cells that had been transfected with pGL-1651 in combination with ΔSer9GSK3β for 24 h, followed by Sau treatment (lower). (C) Inhibition of Sau-induced GSK3β phosphorylation by protein kinase C δ (PKCδ) inhibition. The proteins of interest were immunoblotted on the lysates of cells treated with vehicle, Sau, Sau + rottlerin for 1 h. Similarly, the proteins were assessed after transfection with non-targeting siRNA (con siRNA) or siRNA directed against PKCδ. Results were confirmed by three repeated experiments. (D) Immunoprecipitation and immunoblot assays. Interaction between Kelch-like ECH-associated protein-1 (Keap1) and Nrf2 was measured in mock- or CA-GSK3β-transfected HepG2 cells that had been treated with 5 µM rottlerin for 1 h and continuously exposed to vehicle or Sau for 3 h. Nrf2 immunoprecipitates were subjected to immunoblottings for Keap1. Results were confirmed by repeated experiments. (E) Immunoblottings for p-PKCδ or p-GSK3β (Ser9) on the lysates of cells treated with 1 µM OSU03012 for 1 h and continuously incubated with Sau for 1 h. (F) A schematic diagram illustrating the proposed mechanism by which sauchinone protects the liver against APAP-induced toxicity. GCL, glutamate-cysteine ligase; GSH, glutathione; NAPQI, N-acetyl-p-benzoquinoneimine; NQO1, NAD(P)H:quinone oxidoreductase-1.
To understand in more depth the molecular mechanism underlying Nrf2 activation, we attempted to link PKCδ activation and GSK3β phosphorylation by sauchinone. Interestingly, inhibition of PKCδ using either rottlerin or PKCδ siRNA, completely abolished the ability of sauchinone to increase GSK3β phosphorylation (Figure 7C). So, sauchinone-induced activation of PKCδ may lead to the phosphorylation of GSK3β. As a continuing effort to understand the basis of Nrf2 activation, we examined the effect of modulating PKCδ or GSK3β activity on the attenuation of the Nrf2 and Keap1 interaction by sauchinone. Inhibition of Keap1 binding was not reversed by either PKCδ inhibition or ΔSer9GSK3β transfection, suggesting that the dissociation of Nrf2 from Keap1 is independent of the PKCδ-GSK3β pathway (Figure 7D). 3-Phosphoinositide-dependent kinase 1 (PDK1) may activate PKC isoforms (Feldman et al., 2005). Treatment of HepG2 cells with OSU03012 (a PDK1 inhibitor) did not affect the phosphorylation of PKCδ or GSK3β induced by sauchinone (Figure 7E). In addition, the activation of PKCδ was not affected by U73122 treatment (a PLC inhibitor) (data not shown). Our data suggest that neither PDK1 nor PLC is involved in the activation of PKCδ by sauchinone.
Collectively, these findings indicate that PKCδ activated by sauchinone in conjunction with its inhibitory GSK3β phosphorylation may coordinately regulate the activating phosphorylation of Nrf2. Hence, the phosphorylation of Nrf2 coupled with the inhibition of Keap1 activity by sauchinone promotes Nrf2-ARE activity and this may contribute to the protection of the liver from APAP-induced toxicity (Figure 7F).
Discussion
Acetaminophen is one of the safest and most widely used medications for minor illness and pain. Despite a good safety profile when used at recommended doses, high doses of APAP may result in major morbidity and mortality; intoxication from APAP overdose is mostly secondary to liver failure, which results from massive hepatic necrosis (Cohen and Khairallah, 1997). In addition to its clinical relevance, APAP is widely used as a model toxicant to evaluate liver protection due to the reproducible hepatotoxicity it induces in laboratory animals by well-characterized metabolic pathways. APAP is primarily metabolized in the liver by UDP-glucuronosyltransferase-mediated glucuronidation. NAPQI, a reactive metabolic intermediate of APAP, is usually detoxified in vivo by GSH conjugation. However, when high doses are administered or under conditions of low cellular GSH levels, a larger fraction of APAP is metabolized to NAPQI by cytochrome P450 (Sarich et al., 1997). NAPQI then covalently binds to macromolecules and induces massive hepatic necrosis (Parkinson and Ogilvie, 2008). Currently, no other agents except N-acetylcysteine, a GSH precursor, and its analogues are available as antidotes for APAP poisoning.
A number of experimental studies have shown that sauchinone may serve as a possible antioxidant; sauchinone inhibits iron-induced oxidative stress, which depends on AMPK activation. Moreover, it prevents the induction of inducible nitric oxide synthase in macrophages by suppressing nuclear factor-κB activity (Hwang et al., 2003; Kim et al., 2009). Sauchinone has also been shown to inhibit receptor activator of nuclear factor κB ligand-induced osteoclastogenesis by reducing ROS generation (Han et al., 2007), supporting its potential use for the prevention of bone resorption. In addition, sauchinone induces the activation of LKB1, which leads to AMPK activation: these events contribute to cell survival, protecting the liver from the toxicity induced by iron accumulation (Kim et al., 2009). Also, sauchinone has the ability to inhibit liver X receptor α-mediated sterol regulatory element binding protein-1c induction and sterol regulatory element binding protein-1c-dependent hepatic lipogenesis, thereby protecting hepatocytes from oxidative stress elicited by fat accumulation (Kim et al., 2010). During the course of the present study, it was found that sauchinone increases HSP32 by activating Nrf2 (Jeong et al., 2010). Nonetheless, the molecular basis of the phase II antioxidant enzyme induction by sauchinone is unclear. More importantly, our study is the first in vivo report demonstrating that sauchinone is a bona fide Nrf2 activator and has a protective effect on the liver.
Activation of Nrf2 contributes to cellular defence mechanism through the up-regulation of genes containing ARE(s) in their promoter regions (Itoh et al., 1997). Proteins encoded from Nrf2 target genes exert their effects by decreasing the amount of oxidative stress and restoring the balance between oxidants and antioxidants in the cell. The importance of these enzymes in cytoprotection is highlighted by the observations that mice deficient of Nrf2 were considerably more sensitive to the acute toxicities of APAP, butylated hydroxytoluene and hyperoxia (Chan and Kan, 1999; Enomoto et al., 2001; Cho et al., 2002). Moreover, Nrf2 KO mice are more susceptible to the tumourigenicity of benzo[a]pyrene (Ramos-Gomez et al., 2001). However, controversially, it has also been found that mice with glutathione peroxidase-1 overexpression are more susceptible to APAP-induced liver injuries (Mirochnitchenko et al., 1999).
In the present study, we characterized the hepatoprotective effect of sauchinone against APAP intoxication; sauchinone protected mice from APAP-induced hepatic injury, as determined by the changes in plasma ALT, AST, LDH and histopathology. In addition, our finding that sauchinone treatment failed to protect the liver from APAP-induced injury during the absence of Nrf2 strongly supports the key role of this transcription factor in liver protection. Indeed, sauchinone facilitated Nrf2 translocation to the nucleus, which correlated well with increases in nuclear Nrf2 content and Nrf2-driven gene transactivation. Furthermore, our data demonstrated the induction of Nrf2 target genes including the catalytic subunit of GCL, GCLM and NQO1 by sauchinone both in vitro and in vivo. As expected, the induction of hepatic GCLM, NQO1 and HSP32 by sauchinone was not observed in Nrf2-null mice. These results indicate that both the hepatoprotective effect and the induction of cytoprotective genes induced by sauchinone are mediated by activation of Nrf2.
Several kinase pathways, including PKC, mitogen-activated protein kinase, phosphatidylinositol 3-kinase and PKR-like endoplasmic reticulum kinase, have been shown to influence Keap1-Nrf2-ARE signalling flow through posttranscriptional modification (Yu et al., 2000; Lee et al., 2001; Huang et al., 2002; Cullinan and Diehl, 2004). Among the kinases implicated in the activation of Nrf2, PKCδ phosphorylates Nrf2 at serine 40 upon its dissociation from Keap1. Our results presented here provide compelling evidence that sauchinone activates PKCδ phosphorylation, the inhibition of which attenuates the ability of sauchinone to activate Nrf2. Moreover, sauchinone-induced PKCδ phosphorylation preceded the nuclear accumulation of Nrf2 (10 min–1 h vs. 1 h–12 h). Therefore, it is apparent that sauchinone activation of PKCδ indeed contributes to the promotion of Nrf2 activity. Our results that chemical inhibition of PDK1 or PLC did not change PKCδ phosphorylation by sauchinone rule out the possibility that PDK1 or PLC lies upstream of PKCδ.
It is also noteworthy that GSK3β mediates the antioxidant cell response by phosphorylating Nrf2 to exclude Nrf2 from the nucleus and thereby inhibiting the ARE (Salazar et al., 2006). Interestingly, sauchinone treatment decreased GSK3β activity, as determined by its Ser9 phosphorylation. This contention is strengthened by our observation that the activation of Nrf2 by sauchinone was inhibited by CA-GSK3β overexpression. Another important finding of this study is that PKCδ acts as an upstream kinase of GSK3β, as supported by the observation that PKCδ inhibition by rottlerin or siRNA prevented the ability of sauchinone to induce GSK3β phosphorylation. Moreover, the time-course response of sauchinone-induced PKCδ phosphorylation matched well with that of GSK3β phosphorylation (i.e. 10 min–1 h vs. 30 min–1 h). It was recently reported that APAP treatment causes GSK3β activation and facilitates its translocation to mitochondria (Shinohara et al., 2010), implying that GSK3β may be a mediator of APAP-induced liver injury. The silencing of GSK3β protected mice from APAP-induced liver toxicity by decreasing the loss of hepatic GCL and promoting GSH recovery is in line with the present findings. Our finding that APAP treatment alone increased the phosphorylations of PKCδ and GSK3β suggests that the PKCδ-GSK3β pathway may be part of the adaptive responses to APAP.
Kelch-like ECH-associated protein-1 is an inhibitory protein responsible for Nrf2 ubiquitination and proteasomal degradation (Huang et al., 2002). In the absence of electrophiles or oxidants, Keap1 binds tightly with Nrf2 to repress the activity of Nrf2. Upon exposure to the inducers, the Keap1-Nrf2 complex is disrupted, allowing Nrf2 to translocate to the nucleus and transactivate target genes (Wakabayashi et al., 2004). Our results that sauchinone inhibited Keap1 binding with Nrf2, an effect that was not reversed by PKCδ inhibition or GSK3β activation, support the notion that Keap1 dissociation from Nrf2 by sauchinone may be independent of PKCδ activation. The direct target of sauchinone may be responsible for these events and remains to be identified.
Recently, it has been shown that variants in CD44 contribute to APAP-induced liver toxicity in an animal model and in human volunteers (Harrill et al., 2009). CD44 high T cell subsets increased the levels of Nrf2 and its target gene transcripts (Kim and Nel, 2005). So, APAP-induced hepatotoxicity and immunological changes involving CD44 may activate Nrf2 as an adaptive response. The direct relationship between CD44 and Nrf2 activation and the molecular basis should be studied.
In conclusion, our results demonstrate for the first time that sauchinone protects the liver from APAP-induced intoxication by activating Nrf2, and provide important information on the pharmacological mechanism and the potential application of sauchinone for the prevention and/treatment of liver intoxication. More importantly, our findings identify a novel signalling pathway activated by a unique plant lignan for the activation of Nrf2 and Nrf2-dependent target genes, as its effects are mediated by PKCδ activation in conjunction with its inhibition of GSK3β.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant funded by the Korea government (MEST) (No. 2009-0063233).
Glossary
Abbreviations
- ALT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- APAP
acetaminophen
- ARE
antioxidant response element
- AST
aspartate aminotransferase
- GCL
glutamate-cysteine ligase
- GCLM
modifier subunit of glutamate-cysteine ligase
- GSH
glutathione
- GSK3β
glycogen synthase kinase 3β
- HSP32
heat shock protein 32
- Keap1
Kelch-like ECH-associated protein-1
- KO
knockout
- LDH
lactate dehydrogenase
- NAPQI
N-acetyl-p-benzoquinoneimine
- NQO1
NAD(P)H:quinone oxidoreductase-1
- Nrf2
nuclear factor erythroid-2-related factor-2
- PKCδ
protein kinase C δ
- PMA
phorbol 12-myristate 13-acetate
- ROS
reactive oxygen species
- WT
wild-type
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Aleksunes LM, Goedken M, Manautou JE. Up-regulation of NAD(P)H quinone oxidoreductase 1 during human liver injury. World J Gastroenterol. 2006;12:1937–1940. doi: 10.3748/wjg.v12.i12.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Zhang LY, Kensler TW, Kleeberger SR. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am J Respir Cell Mol Biol. 2002;26:42–51. doi: 10.1165/ajrcmb.26.1.4536. [DOI] [PubMed] [Google Scholar]
- Chung BS, Shin MG. Dictionary of Korean Folk Medicine. Seoul: Young Lim Sa; 1990. pp. 813–814. [Google Scholar]
- Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 1997;29:59–77. doi: 10.3109/03602539709037573. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O'Connor T, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2005;280:19867–19874. doi: 10.1074/jbc.M501367200. [DOI] [PubMed] [Google Scholar]
- Habelhah H, Takahashi S, Cho SG, Kadoya T, Watanabe T, Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. EMBO J. 2004;23:322–332. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KY, Yang D, Chang EJ, Lee Y, Huang H, Sung SH, et al. Inhibition of osteoclast differentiation and bone resorption by sauchinone. Biochem Pharmacol. 2007;74:911–923. doi: 10.1016/j.bcp.2007.06.044. [DOI] [PubMed] [Google Scholar]
- Harrill AH, Watkins PB, Su S, Ross PK, Harbourt DE, Stylianou IM, et al. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res. 2009;19:1507–1515. doi: 10.1101/gr.090241.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- Hwang BY, Lee JH, Jung HS, Kim KS, Nam JB, Hong YS, et al. Sauchinone, a lignan from Saururus chinensis, suppresses iNOS expression through the inhibition of transactivation activity of RelA of NF-kappaB. Planta Med. 2003;69:1096–1101. doi: 10.1055/s-2003-45189. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- Jeong GS, Lee DS, Li B, Byun E, Kwon DY, Park H, et al. Protective effect of sauchinone by upregulating heme oxygenase-1 via the p38 MAPK and Nrf2/ARE pathways in HepG2 cells. Planta Med. 2010;76:41–47. doi: 10.1055/s-0029-1185906. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Nel AE. The role of phase II antioxidant enzymes in protecting memory T cells from spontaneous apoptosis in young and old mice. J Immunol. 2005;175:2948–2959. doi: 10.4049/jimmunol.175.5.2948. [DOI] [PubMed] [Google Scholar]
- Kim YW, Lee SM, Shin SM, Hwang SJ, Brooks JS, Kang HE, et al. Efficacy of sauchinone as a novel AMPK-activating lignan for preventing iron-induced oxidative stress and liver injury. Free Radic Biol Med. 2009;47:1082–1092. doi: 10.1016/j.freeradbiomed.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Kim YW, Kim YM, Yang YM, Kim TH, Hwang SJ, Lee JR, et al. Inhibition of SREBP-1c-mediated hepatic steatosis and oxidative stress by sauchinone, an AMPK-activating lignan in Saururus chinensis. Free Radic Biol Med. 2010;48:567–578. doi: 10.1016/j.freeradbiomed.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Lee AK, Sung SH, Kim YC, Kim SG. Inhibition of lipopolysaccharide-inducible nitric oxide synthase, TNF-alpha and COX-2 expression by sauchinone effects on I-kappaBalpha phosphorylation, C/EBP and AP-1 activation. Br J Pharmacol. 2003;139:11–20. doi: 10.1038/sj.bjp.0705231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Hanson JM, Chu WA, Johnson JA. Phosphatidylinositol 3-kinase, not extracellular signal-regulated kinase, regulates activation of the antioxidant-responsive element in IMR-32 human neuroblastoma cells. J Biol Chem. 2001;276:20011–20016. doi: 10.1074/jbc.M100734200. [DOI] [PubMed] [Google Scholar]
- McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, et al. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99:628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- Mirochnitchenko O, Weisbrot-Lefkowitz M, Reuhl K, Chen L, Yang C, Inouye M. Acetaminophen toxicity. Opposite effects of two forms of glutathione peroxidase. J Biol Chem. 1999;274:10349–10355. doi: 10.1074/jbc.274.15.10349. [DOI] [PubMed] [Google Scholar]
- Moffit JS, Aleksunes LM, Kardas MJ, Slitt AL, Klaassen CD, Manautou JE. Role of NAD(P)H:quinone oxidoreductase 1 in clofibrate-mediated hepatoprotection from acetaminophen. Toxicology. 2007;230:197–206. doi: 10.1016/j.tox.2006.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- Parkinson A, Ogilvie B. Biotransformation. In: Klaassen CD, editor. Casarett and Doull's Toxicology: The Basic Science of Poisons. New York: McGraw-Hill; 2008. pp. 161–304. [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- Sarich T, Kalhorn T, Magee S, al-Sayegh F, Adams S, Slattery J, et al. The effect of omeprazole pretreatment on acetaminophen metabolism in rapid and slow metabolizers of S-mephenytoin. Clin Pharmacol Ther. 1997;62:21–28. doi: 10.1016/S0009-9236(97)90148-X. [DOI] [PubMed] [Google Scholar]
- Shinohara M, Ybanez MD, Win S, Than TA, Jain S, Gaarde WA, et al. Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. J Biol Chem. 2010;285:8244–8255. doi: 10.1074/jbc.M109.054999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Kim YC, Moon A. Sauchinone, a lignan from Saururus chinensis, inhibits staurosporine-induced apoptosis in C6 rat glioma cells. Biol Pharm Bull. 2003;26:1428–1430. doi: 10.1248/bpb.26.1428. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- Sung SH, Kim YC. Hepatoprotective diastereomeric lignans from Saururus chinensis herbs. J Nat Prod. 2000;63:1019–1021. doi: 10.1021/np990499e. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Chen C, Mo YY, Hebbar V, Owuor ED, Tan TH, et al. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem. 2000;275:39907–39913. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.