

Abstract
BACKGROUND AND PURPOSE
It is well established that cytochrome P450 2J (CYP2J) enzymes are expressed preferentially in the heart, and that ebastine is a substrate for CYP2J, but it is not known whether ebastine is metabolized in myocardium. Therefore, we investigated its pharmacokinetics in the rat isolated perfused heart.
EXPERIMENTAL APPROACH
Rat isolated hearts were perfused in the recirculating mode with ebastine for 130 min. The concentrations of ebastine and its metabolites, hydroxyebastine and carebastine, were measured using liquid chromatography with a tandem mass spectrometry. The data were analysed by a compartmental model. The time course of negative inotropic response was linked to ebastine concentration to determine the concentration–effect relationship.
KEY RESULTS
Ebastine was metabolized to an intermediate compound, hydroxyebastine, which was subsequently further metabolized to carebastine. No desalkylebastine was found. The kinetics of the sequential metabolism of ebastine was well described by the compartmental model. The EC50 of the negative inotropic effect of ebastine in rat isolated heart was much higher than free plasma concentrations in humans after clinical doses.
CONCLUSIONS AND IMPLICATIONS
The kinetics of ebastine conversion to carebastine via hydroxyebastine resembled that observed in human liver microsomes. The results may be of interest for functional characterization of CYP2J activity in rat heart.
Keywords: CYP2J, perfused rat heart, inotropy, myocardial metabolism of ebastine, pharmacokinetic/pharmacodynamic modelling
Introduction
The cytochrome P450 subfamily CYP2J is expressed in rat, mouse and human heart (Wu et al., 1996, 1997; Wang et al., 2002; DeLozier et al., 2007) where it metabolizes arachidonic acid to biologically active epoxyeicosatrienoic acids, which play an important role in the regulation of cardiac function (Chaudhary et al., 2009; Nithipatikom and Gross, 2010). In addition, CYP2J2 is mainly responsible for the hydroxylation of the H1-receptor antagonist ebastine in human liver and intestinal microsomes (Hashizume et al., 2002; Liu et al., 2006). The corresponding rat homologue CYP2J3 was found to be highly expressed in rat myocytes (Wu et al., 1997). In this study, we present the first kinetic data on the myocardial metabolism of ebastine. Based on the time course of perfusate concentrations and the heart tissue levels at the end of perfusion in a recirculating Langendorff perfusion apparatus, the kinetics of ebastine disposition and sequential formation of hydroxyebastine and carebastine was analysed using compartmental modelling. The estimated myocardial formation rate of hydroxyebastine served as a measure of CYP2J activity. In order to better understand the underlying uptake kinetics of ebastine into the heart, the myocardial concentration of ebastine was linked to the negative inotropic effect using pharmacokinetic–pharmacodynamic (PK–PD) modelling. Our approach allowed the estimation of concentration-response parameters for the cardiodepressive effect of ebastine.
Methods
Recirculating rat heart perfusion
All animal care and experimental procedures in this investigation conformed to the European Community guidelines for the use of experimental animals and were approved by the Animal Protection Body of the State of Sachsen-Anhalt, Germany. Isolated hearts from male Wistar rats (n = 6; weight range 294–328 g), were initially perfused (Langendorff method) with Krebs–Henseleit bicarbonate buffer containing 0.1% of bovine serum albumin at a constant flow of 9.5 mL·min−1 in the non-recirculating mode, as described previously (Weiss and Kang, 2002). A latex balloon was placed in the left ventricle and connected to a pressure transducer line. The balloon was inflated with 50% methanol to create a diastolic pressure of 5–6 mmHg. After 30 min stabilization, the hearts were beating spontaneously at an average rate of 255 beats/min. Coronary perfusion pressure, the left ventricular pressure and heart rate were measured continuously. Left ventricular developed pressure (LVDP) was calculated from systolic and end-diastolic pressures as LVDP = left ventricular systolic pressure – left ventricular end-diastolic pressure. Coronary resistance was obtained by dividing coronary perfusion pressure by flow. After stabilization, the perfusion system was switched to the recirculating mode. Twenty millilitres of the total recirculating perfusate (60 mL volume) was replaced by modified Krebs–Henseleit buffer containing ebastine in concentrations between 605.7 and 383.3 ng·mL−1. The first sample (0.5 mL) was taken after 5 min, when mixing in the recirculating system was complete, followed by further samples at 10 min and then every 10 min up to 130 min. At the end of the perfusion, hearts were weighed and quickly frozen in liquid nitrogen.
Determination of ebastine and its metabolites
Ebastine and its three metabolites in perfusate and in heart tissue were measured by a previously reported method with slight modifications (Kang et al., 2004). Ebastine, desalkylebastine, hydroxyebastine and carebastine were kindly donated by Almirall Prodesfarma, SA (Barcelona, Spain). The concentrations of ebastine and its three metabolites were quantified using liquid chromatography/mass spectrometry with a API 4000 liquid chromatography with a tandem mass spectrometry (LC/MS/MS) System (Applied Biosystems, Foster City, CA) equipped with an electrospray ionization interface used to generate positive ions [M+ H]+. The compounds were separated on a reversed-phase column (Kinetex C18, 2.1 × 50 mm, 2.6 µm particle size; Phenomenex, Torrance, CA) with an isocratic mobile phase consisting of acetonitrile and 0.1% formic acid (60:40%, v/v). The mobile phase was eluted using an Agilent 1100 series pump (Agilent, Wilmington, DE) at 0.3 mL·min−1. Quantitation was performed by multiple reaction monitoring (MRM) of the protonated precursor ion and the related product ion for ebastine and its three metabolites. The mass transitions used for ebastine, hydroxyebastine, carebastine and methaqualone (internal standard) were m/z 470.2 → 167.1, 486.4 → 167.1, 500.2 → 167.1 and 251.4 → 131.9, respectively; that for desalkylebastine was m/z 268.3 → 167.1. Calibration graphs in perfusate and in heart tissue were derived from the peak area ratio of ebastine and three metabolites to the internal standard with a linear regression respectively.
Methaqualone (900 µL, 10 ng·mL−1 in acetonitrile) was added to perfusate (300 µL) and mixed vigorously for 3 min. After centrifugation (15 700× g, 10 min), 200 µL of the supernatant was transferred into a vial; 5 µL was injected into the LC/MS/MS system.
All hearts were divided randomly into four pieces and weighed. Each piece was homogenized in three times its volume of phosphate buffer solution (0.5 M, pH 7.4) on ice with a tissue homogenizer (Ultra-Turrax T 25, IKA-Labortechnik, Staufen, Germany). Acetonitrile including methaqulaone was added to precipitate proteins in the homogenate. After vortexing for 1 min and centrifuging at 15 700× g for 10 min, the supernatant was injected onto the analytical column (Kang and Weiss, 2001).
Modelling and data analysis
Pharmacokinetics
The model of cardiac disposition and sequential metabolism of ebastine (Figure 1) describes drug uptake from the reservoir with volume Vres into the tissue compartment with an uptake rate constant kin,e and an efflux rate constant kout,e. Because no information was available on ebastine metabolism in rat heart, the model relies on the pathway of sequential ebastine metabolism in human liver microsomes (Liu et al., 2006), taking the observation into account that enzymes of the CYP3A subfamily are either absent from or minimally expressed in, human and rat myocardium (Thum and Borlak, 2000a,b; DeLozier et al., 2007). Thus, no dealkylation of ebastine and hydroxyebastine was assumed. The formation of hydroxyebastine and its subsequent metabolism to carebastine are denoted by rate constants keh and khc, respectively, and the metabolism of carebastine by kcm. Hydroxyebastine and carebastine formed in myocardium are transported into the reservoir (kout,h and kout,c) and undergo reuptake (kin,h and kin,c). The time courses of the concentrations of ebastine, Ceb(t), hydroxyebastine, Chy(t), and carebastine, Cca(t) in reservoir, obtained by solving the differential equations corresponding to the compartmental model (Figure 1), are fitted to the data to estimate the model parameters. However, in order to estimate the parameters keh, khc and kcm, we need additional information because metabolism of ebastine and hydroxyebastine by other enzymes cannot be excluded, a priori. This information comes from the tissue concentrations of the substances measured in the heart at the end of perfusion (Aend). First, the ebastine concentration, Ceb(t), and amount, Aeb,end, was fitted to estimate kin,e, kout,e. and keh. Second, holding these parameters constant, the hydroxyebastine data, Chy(t), and the amount in the heart, Ahy,end, were fitted to estimate kout,h, kin,h and khc, and third, in the same way, the carebastine data, Cca(t), were fitted to get kin,c, kout,c and kcm.
Figure 1.
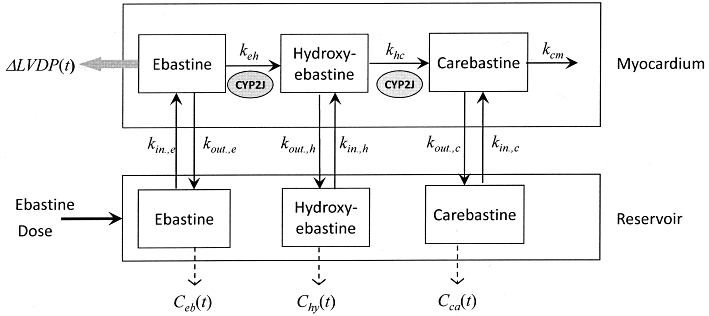
Working compartmental model used to evaluate the sequential cardiac metabolism of ebastine in the rat isolated heart with recirculating perfusate.
The data were analysed by a population approach with maximum likelihood estimation via the EM algorithm implemented in the software ADAPT 5 (D'Argenio et al., 2009). The MLEM program provides estimates of the population mean and inter-subject variability as well as of the individual subject parameters (conditional means). We assumed log-normally distributed model parameters, and that the measurement error has a standard deviation that is a linear function of the measured quantity.
Pharmacodynamics
The amount of ebastine in myocardium, Aeb(t), predicted by the model was directly linked to the inotropic effect, E(t),
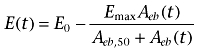 |
(1) |
where Emax, and Aeb,50 are the maximal response and the amount required to produce 50% maximal response respectively. As a measure of inotropic response E(t), we used the change in left ventricular developed pressure LVDP(t), that is the decrease in LVDP with respect to the baseline (pre-drug) value E0 = LVDP0. Since the maximal achievable reduction in LVDP is just E0, we used this as Emax value in Equation 1. The pharmacodynamic parameters E0, and Aeb,50 were estimated fitting Equation 1 to the observed LVDP(t) data up to the time before the re-increase is stopped (and an apparent plateau value is reached). The EC50 of the cardiodepressive effect of ebastine was then calculated according to the distribution at steady state
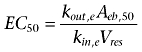 |
(2) |
Results
Hydroxyebastine and carebastine were measured together with ebastine in the perfusate, while desalkylebastine was below the quantification limit (1 ng·mL−1) at all time points measured. Figure 2 shows the average concentration–time profiles of ebastine, hydroxyebastine and carebastine in the reservoir after adding doses ranging between 7.8 and 12.1 mg ebastine (10.1 ± 1.5 mg; mean ± SD) at time t = 0. The amount of ebastine, hydroxyebastine and carebastine in the left ventricle at the end of perfusion were 22.3 ± 4.3%, 3.6 ± 0.9% and 19.1 ± 5.4% (means ± SD) of the dose respectively. No desalkylebastine was found in myocardium. The observed and model predicted perfusate concentrations [Ceb(t), Chy(t) and Cca(t)] and amounts in heart [Aeb(t), Ahy(t) and Aca(t)] are illustrated in Figure 3A–C for one heart. The experiment with an AIC value of the ebastine disposition data that was closest to the group median value was selected as the example. Parameter estimates with inter-individual variability in parentheses are listed in Table 1. Ebastine decreased LVDP to 57.3 ± 4.9% of baseline level with maximum effect at 10 min. Figure 4 shows the fit obtained by using Eq. 1 of the time course of the negative inotropic response corresponding to the time course of ebastine amount in heart, Aeb(t), shown in Figure 3A. The population estimates of the pharmacodynamic parameters (mean ± SD) are E0 = 107 ± 5.4 mmHg and EC50 = 8.9 ± 0.5 ng·mL−1. No significant changes in heart rate and coronary resistance were observed.
Figure 2.
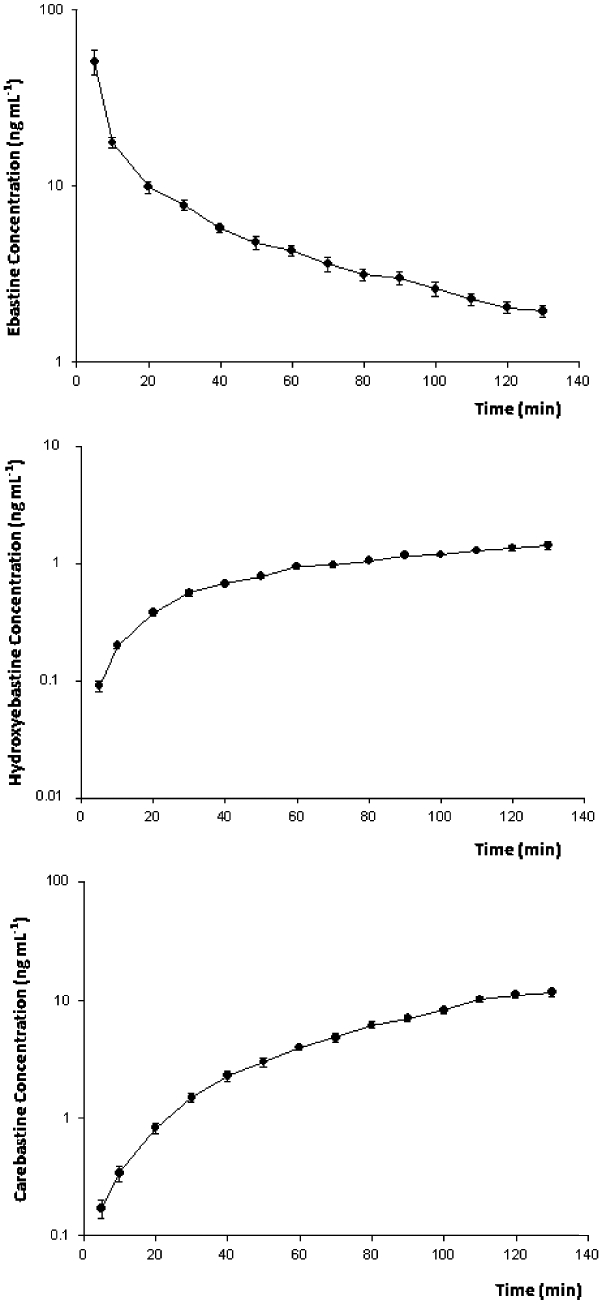
Time course of the reservoir concentration of ebastine (A), hydroxyebastine (B) and carebastine (C). The results represent mean ± SEM of raw data from six hearts.
Figure 3.
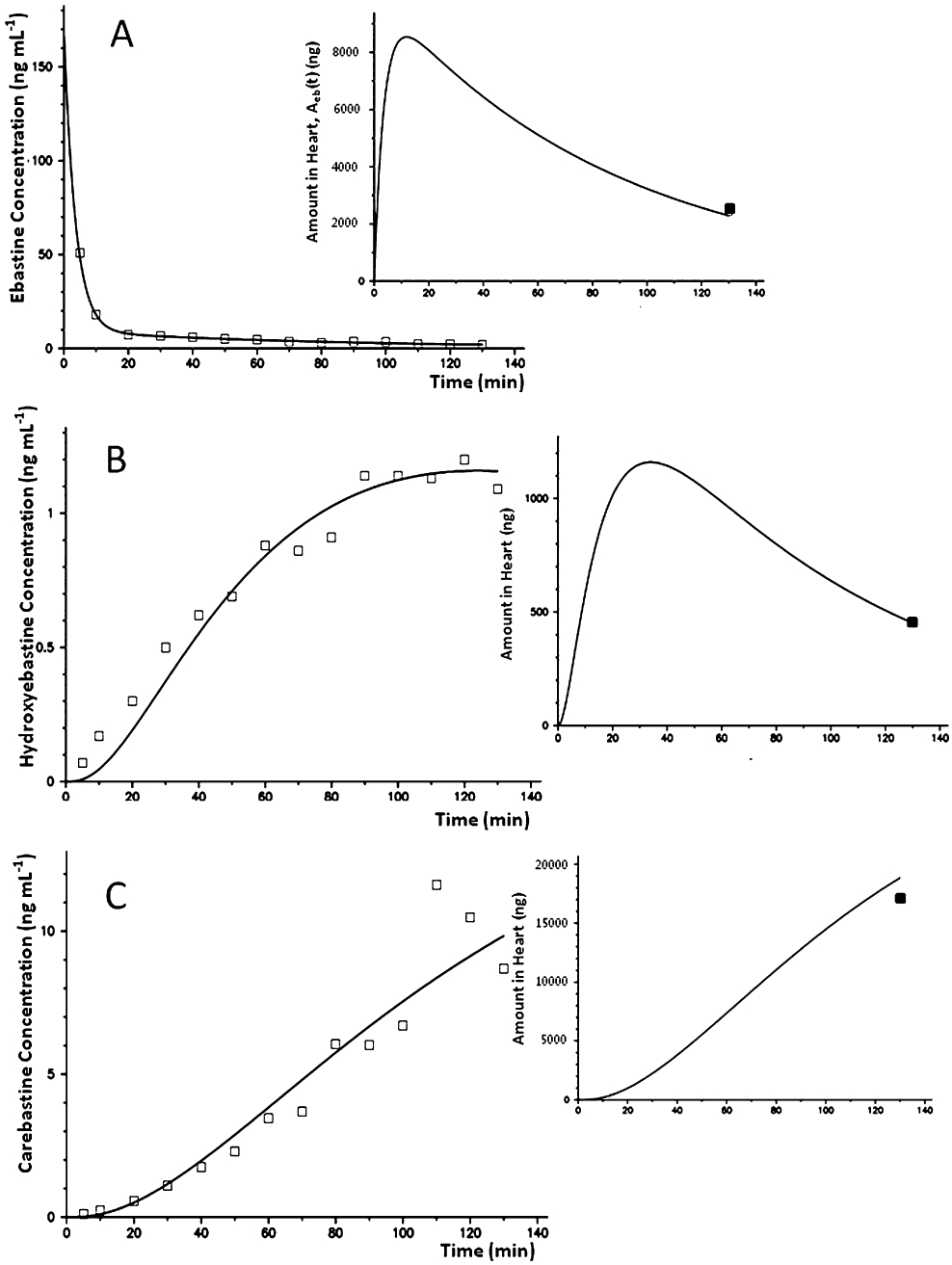
Model fit of the reservoir concentration of ebastine (A), hydroxyebastine (B) and carebastine (C) in one heart. The inserts show the time courses of amount in the heart as predicted by the model to fit the amount measured at the end of perfusion.
Table 1.
Parameters for the model of cardiac uptake and metabolism kinetics of ebastine and formation of hydroxyebastine and carebastine, respectively, estimated in an rat isolated heart preparation
| Parameter (h−1) | Uptake | Metabolism | |
|---|---|---|---|
| Ebastine | kin,e | kout,e | keh |
| 16.3 (16) | 0.846 (32) | 0.834 (9) | |
| Hydroxyebastine | kin,h | kout,h | khc |
| 0.067 (7) | 0.307 (49) | 4.82 (22) | |
| Carebastine | kin,c | kout,c | kcm |
| 13.7 (28) | 492 (17) | 1.31 (5) | |
Mean values and inter-individual variability as relative standard error (%) in brackets, n = 6.
Figure 4.
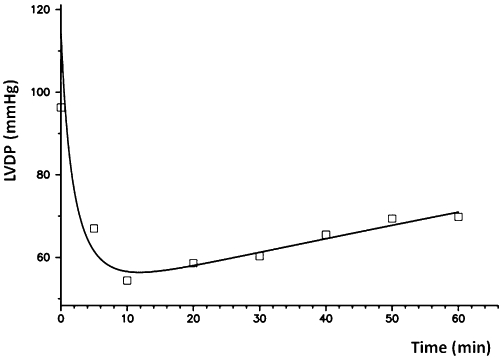
Fit of the time course of the negative inotropic response as function of the amount of ebastine in heart, Aeb(t), shown in Figure 3A.
Discussion
To our knowledge, this is the first study showing that ebastine is metabolized in the heart. The use of ebastine as a test substrate for CYP2J2 activity was recently suggested by Liu et al. (2006) based on results obtained for human liver microsomes. Although the rat heart CYP2J3 isoform is 70% homologous to human heart CYP2J2 (Wu et al., 1997), it is not clear yet which CYP450 enzymes are responsible for hydroxylation of ebastine in rat heart. This limitation notwithstanding, the results provide new insight on the kinetics of sequential metabolism of ebastine in rat heart and the relative activities of the CYP450 enzymes that could be involved. With the exception that no desalkylebastine was found, the picture of consecutive metabolism is quite similar to that reported for human liver microsomes (Liu et al., 2006). Thus, the rate constant of hydroxyebastine carboxylation (khc) is much higher than that of ebastine hydroxylation; consequently, the concentration of hydroxyebastine formed from ebastine remains relatively low. On the other hand, the elimination rate constant of carebastine kcm is much lower than its formation rate constant (khc) and carebastine becomes one of the major, relatively stable metabolites of ebastine. The good fit of the amounts of ebastine, hydroxyebastine and carebastine in the heart suggests that no metabolism of ebastine and hydroxyebastine by other pathways (e.g. dealkylation) is required to achieve mass balance. This is in accordance with the finding that CYP3A isoforms are either not present or expressed at very low levels, in rat myocardium (Thum and Borlak, 2000b). It is not clear, however, which enzyme is responsible for the metabolism of carebastine (kcm). In a clinical pharmacokinetic study, it was recently suggested that metabolism of carebastine is not solely by CYP3A4 (Shon et al., 2010). Interestingly, the plasma concentration–time courses of ebastine and its metabolites hydroxyebastine and carebastine observed in vivo (Kang et al., 2004) resemble those observed here for ex vivo Langendorff-perfused rat hearts in the recirculation mode. The question arises here whether cardiac metabolism of ebastine contributes to total ebastine clearance in vivo. Although there was an extraction of about 90% in the rat perfused heart, cardiac extraction may be much lower in vivo due to the high degree of plasma protein binding but the possibility that part of the ebastine clearance in vivo is due to metabolism in the heart cannot be excluded. Note that a recirculating heart perfusion was chosen because it allows for the accumulation of the metabolites formed. The population modelling approach where in fitting simultaneously the concentration–time data from all hearts, using information from all instead of only one heart, has proven successful when parameters are poorly estimated using individual fits (Krudys et al., 2006).
In order to compare our estimate for the EC50 of the negative inotropic effect of ebastine with the situation in vivo, an unbound fraction of <0.2% measured in rats (Fujii et al., 1997) would result in a value of 3.2 µg·mL−1 that is fourfold higher than the EC50 for QT prolongation in rats (Ohtani et al., 1999). However, because these ebastine concentrations are much higher than the clinically achievable peak plasma concentrations in man (<5 ng·mL−1) (Moss and Morganroth, 1999), our results are not in contradiction to the fact that ebastine has no clinically deleterious cardiac effects (Moss and Morganroth, 1999). The mechanism underlying the negative inotropic effect of ebastine at higher concentrations is not yet understood. Besides the hypothesis that ebastine causes release of histamine (which reduces ventricular contractility in rats) (Llenas et al., 1999), it would be interesting to know whether an inhibition of CYP2J per se could have a negative inotropic effect.
In summary, kinetic modelling of the sequential metabolism of ebastine in rat heart suggests that CYP2J plays a major role and revealed similarities to metabolism patterns observed in human liver microsomes and the intact rat. The results may add to our understanding of the functional properties of rat CYP2J and suggest that ebastine could be used as a test substrate to assess myocardial CYP2J activity.
Acknowledgments
We are grateful to Petra Arendt for assistance in the experiment. This work was supported by a grant from the Deutsche Forschungsgemeinschaft, Bonn, Germany (WE 2190/5–1).
Glossary
Abbreviations
- CYP
cytochrome P450
- LVDP
left ventricular developed pressure
- k
rate constant
Conflicts of interest
The authors declare no conflicts of interest.
Supporting Information
Teaching Materials; Figs 1–4 as PowerPoint slide.
References
- Chaudhary KR, Batchu SN, Seubert JM. Cytochrome P450 enzymes and the heart. IUBMB Life. 2009;61:954–960. doi: 10.1002/iub.241. [DOI] [PubMed] [Google Scholar]
- D'Argenio DZ, Schumitzky A, Wang X. ADAPT 5 User's Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. Los Angeles, CA: Biomedical Simulations Resource; 2009. [Google Scholar]
- DeLozier TC, Kissling GE, Coulter SJ, Dai D, Foley JF, Bradbury JA, et al. Detection of human CYP2C8, CYP2C9, and CYP2J2 in cardiovascular tissues. Drug Metab Dispos. 2007;35:682–688. doi: 10.1124/dmd.106.012823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Matsumoto S, Hatoyama T, Miyazaki H. Studies on the first-pass metabolism of ebastine in rats. Arzneimittelforschung. 1997;47:949–953. [PubMed] [Google Scholar]
- Hashizume T, Imaoka S, Mise M, Terauchi Y, Fujii T, Miyazaki H, et al. Involvement of CYP2J2 and CYP4F12 in the metabolism of ebastine in human intestinal microsomes. J Pharmacol Exp Ther. 2002;300:298–304. doi: 10.1124/jpet.300.1.298. [DOI] [PubMed] [Google Scholar]
- Kang W, Liu KH, Ryu JY, Shin JG. Simultaneous determination of ebastine and its three metabolites in plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;813:75–80. doi: 10.1016/j.jchromb.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Kang W, Weiss M. Influence of P-glycoprotein modulators on cardiac uptake, metabolism, and effects of idarubicin. Pharm Res. 2001;18:1535–1541. doi: 10.1023/a:1013022212738. [DOI] [PubMed] [Google Scholar]
- Krudys KM, Kahn SE, Vicini P. Population approaches to estimate minimal model indexes of insulin sensitivity and glucose effectiveness using full and reduced sampling schedules. Am J Physiol Endocrinol Metab. 2006;291:E716–E723. doi: 10.1152/ajpendo.00346.2005. [DOI] [PubMed] [Google Scholar]
- Liu KH, Kim MG, Lee DJ, Yoon YJ, Kim MJ, Shon JH, et al. Characterization of ebastine, hydroxyebastine, and carebastine metabolism by human liver microsomes and expressed cytochrome P450 enzymes: major roles for CYP2J2 and CYP3A. Drug Metab Dispos. 2006;34:1793–1797. doi: 10.1124/dmd.106.010488. [DOI] [PubMed] [Google Scholar]
- Llenas J, Cardelus I, Heredia A, de Mora F, Gristwood RW. Cardiotoxicity of histamine and the possible role of histamine in the arrhythmogenesis produced by certain antihistamines. Drug Saf. 1999;21:33–38. doi: 10.2165/00002018-199921001-00005. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Morganroth J. Cardiac effects of ebastine and other antihistamines in humans. Drug Saf. 1999;21:69–80. doi: 10.2165/00002018-199921001-00009. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Gross GJ. Epoxyeicosatrienoic acids: novel mediators of cardioprotection. J Cardiovasc Pharmacol Ther. 2010;15:112–119. doi: 10.1177/1074248409358408. [DOI] [PubMed] [Google Scholar]
- Ohtani H, Sato H, Iga T, Kotaki H, Sawada Y. Pharmacokinetic-pharmacodynamic analysis of the arrhythmogenic potency of a novel antiallergic agent, ebastine, in rats. Biopharm Drug Dispos. 1999;20:101–106. doi: 10.1002/(sici)1099-081x(199903)20:2<101::aid-bdd160>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Shon JH, Yeo CW, Liu KH, Lee SS, Cha IJ, Shin JG. Itraconazole and rifampin alter significantly the disposition and antihistamine effect of ebastine and its metabolites in healthy participants. J Clin Pharmacol. 2010;50:195–204. doi: 10.1177/0091270009348974. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Gene expression in distinct regions of the heart. Lancet. 2000a;355:979–983. doi: 10.1016/S0140-6736(00)99016-0. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Cytochrome P450 mono-oxygenase gene expression and protein activity in cultures of adult cardiomyocytes of the rat. Br J Pharmacol. 2000b;130:1745–1752. doi: 10.1038/sj.bjp.0703465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Yang Y, Sullivan MF, Min J, Cai J, Zeldin DC, et al. Induction of cardiac cytochrome p450 in cocaine-treated mice. Exp Biol Med. 2002;227:182–188. doi: 10.1177/153537020222700305. [DOI] [PubMed] [Google Scholar]
- Weiss M, Kang W. P-glycoprotein inhibitors enhance saturable uptake of idarubicin in rat heart: pharmacokinetic/pharmacodynamic modeling. J Pharmacol Exp Ther. 2002;300:688–694. doi: 10.1124/jpet.300.2.688. [DOI] [PubMed] [Google Scholar]
- Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, et al. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J Biol Chem. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.