

Abstract
The Notch signaling pathway is a conserved intercellular signaling mechanism that is essential for proper embryonic development in numerous metazoan organisms. We have examined the in vivo role of the Jagged2 (Jag2) gene, which encodes a ligand for the Notch family of transmembrane receptors, by making a targeted mutation that removes a domain of the Jagged2 protein required for receptor interaction. Mice homozygous for this deletion die perinatally because of defects in craniofacial morphogenesis. The mutant homozygotes exhibit cleft palate and fusion of the tongue with the palatal shelves. The mutant mice also exhibit syndactyly (digit fusions) of the fore- and hindlimbs. The apical ectodermal ridge (AER) of the limb buds of the mutant homozygotes is hyperplastic, and we observe an expanded domain of Fgf8 expression in the AER. In the foot plates of the mutant homozygotes, both Bmp2 and Bmp7 expression and apoptotic interdigital cell death are reduced. Mutant homozygotes also display defects in thymic development, exhibiting altered thymic morphology and impaired differentiation of γδ lineage T cells. These results demonstrate that Notch signaling mediated by Jag2 plays an essential role during limb, craniofacial, and thymic development in mice.
Keywords: Notch signaling, Jagged2, Serrate2, limb development, cleft palate, T cell differentiation
The Notch signaling pathway is an evolutionarily conserved signaling mechanism, and mutations in its components disrupt cell fate specification and embryonic development in organisms as diverse as insects, nematodes, and mammals (for recent reviews, see Artavanis-Tsakonas et al. 1995; Gridley 1997; Robey 1997; Weinmaster 1997). This signaling pathway was first identified and studied in Drosophila. The Notch gene of Drosophila encodes a large transmembrane receptor that, at the extracellular surface of a cell, interacts with membrane-bound ligands encoded by the Delta and Serrate genes. The signal induced by ligand binding is then transmitted at the intracellular surface in a process involving proteolysis of the receptor and interactions with several novel cytoplasmic and nuclear proteins (Fortini and Artavanis-Tsakonas 1994; Jarriault et al. 1995; Matsuno et al. 1995; Kopan et al. 1996; Blaumueller et al. 1997; Pan and Rubin 1997). Genes homologous to members of the Notch signaling pathway have been cloned from numerous vertebrate organisms, and many have been shown to be essential for normal embryonic development. In humans, the importance of Notch signaling for growth and development is underscored by the finding that mutations in genes encoding components of the Notch signaling pathway have been implicated in cancer and in two inherited disease syndromes (Ellisen et al. 1991; Joutel et al. 1996; Li et al. 1997; Oda et al. 1997).
Four genes encoding ligands for the Notch family of receptors have been cloned in mammals: Jagged1 (Jag1; Lindsell et al. 1995), Jagged2 (Jag2; Shawber et al. 1996; these genes are also referred to as Serrate1 and 2), Delta-like1 (Dll1; Bettenhausen et al. 1995), and Delta-like3 (Dll3; Dunwoodie et al. 1997). All of these Notch family ligands are transmembrane proteins that, in their extracellular domain, contain multiple EGF-like motifs as well as a second conserved motif termed the DSL domain (named after three invertebrate Notch ligands; Delta, Serrate, Lag-2). The DSL domain is required for interaction of ligands with Notch family receptors (Henderson et al. 1994; Muskavitch 1994). Two of these ligands, Jagged1 and Jagged2, have been shown to activate Notch1 in mammalian cells (Lindsell et al. 1995; Luo et al. 1997).
We have been studying the role of Notch signaling during embryonic development in mice. To study the biological role of the Jag2 gene, we made a targeted mutation that removes exons encoding the DSL domain of the Jagged2 protein. Mice homozygous for this deletion die at birth because of defects in craniofacial morphogenesis. The mutant mice have cleft palate, and dorsal portions of the tongue are fused to the unelevated palatal shelves. In addition, the mutant mice exhibit impaired differentiation of γδ lineage T cells and altered thymic morphology.
The Jag2 mutant homozygotes also exhibit syndactyly of the fore- and hindlimbs. Previously, we mapped the Jag2 gene to distal Chromosome 12 (Lan et al. 1997), near two mutations that exhibit limb and/or craniofacial defects, syndactylism (sm; Grüneberg 1956) and legless (lgl; Singh et al. 1991). Sidow et al. (1997) recently reported that the sm mutation is a missense mutation in the Jag2 gene. Our results suggest that sm is a hypomorphic allele of Jag2 because mice homozygous for our targeted DSL domain deletion exhibit completely penetrant perinatal lethality and syndactyly that is more severe than in sm homozygous mice, many of which survive to adulthood. These results demonstrate that Notch signaling mediated by Jag2 plays an essential role during limb, craniofacial, and thymic development in mice.
Results
Targeted disruption of the mouse Jag2 gene
Recently, we described the isolation of a gene, Jag2, which encodes a protein whose amino acid sequence and expression pattern during rat embryogenesis suggest that it functions as a ligand for the Notch family of receptors (Shawber et al. 1996). Mouse Jag2 cDNA and genomic clones were isolated by screening brain cDNA and genomic libraries (see Materials and Methods). To investigate the biological role of the Jag2 gene, we created a deletion allele by gene targeting. A Jag2 targeting vector was constructed that deleted genomic sequence encoding the DSL domain of the Jagged2 protein, which is required for interaction of ligands with Notch family receptors in both invertebrates (Henderson et al. 1994; Muskavitch 1994) and vertebrates (C. Lindsell and G. Weinmaster, unpubl.). Deletion of the DSL domain should create a null mutation in the Jag2 gene. We have named this mutant allele Jag2ΔDSL (Fig. 1A). The linearized targeting vector was electroporated into ES cells, and germ-line transmission of the Jag2ΔDSL mutant allele was obtained. Southern blot analyses confirmed the expected structure of the transmitted allele, and also confirmed that the fragment containing the exon encoding the DSL domain was deleted in the transmitted Jag2ΔDSL mutant allele (Fig. 1B). Mice heterozygous for the Jag2ΔDSL mutant allele appeared normal.
Figure 1.

Targeted disruption of the mouse Jag2 gene. (A) Targeting scheme. The top line shows the genomic organization of a portion of the Jag2 gene. Exons are indicated by black boxes. Additional uncharacterized exons are present 3′ of the exons indicated. The middle line represents the structure of the targeting vector. A 5.0-kb deletion was created that removes exons encoding the DSL domain and half of the first EGF repeat. Shown at the bottom is the predicted structure of the Jag2 locus following homologous recombination of the targeting vector. Probes used for Southern blot analysis are indicated. (E) EcoRI; (N) NotI; (S) SacI; (Sp) SpeI. (B) DNA isolated from embryos of the intercross of Jag2+/ΔDSL heterozygous mice was digested with EcoRI, blotted, and hybridized with the indicated probe. Genotypes of progeny are indicated at the top of the lane. Lack of hybridization of the DSL domain probe to DNA of the Jag2ΔDSL homozygotes confirms that the fragment containing the exon encoding the DSL domain is deleted in the transmitted Jag2ΔDSL allele.
Jag2ΔDSL mutant mice die at birth with cleft palate
To examine whether mice homozygous for the Jag2ΔDSL mutation were viable, heterozygous F1 animals were intercrossed, and the genotypes of F2 progeny were determined two to three weeks after birth. No mice homozygous for the mutation were found, indicating that the Jag2ΔDSL mutation was lethal prior to this stage. To determine when the Jag2ΔDSL homozygotes were dying, embryos from timed matings were isolated. This analysis revealed that Jag2ΔDSL homozygous mutants could complete embryogenesis, but died at or shortly after birth. Caesarean delivery at E18 of pups from heterozygous intercrosses indicated that the majority of the Jag2ΔDSL homozygotes were unable to breathe, became cyanotic, and died within a few minutes. A few of the Jag2ΔDSL homozygotes were able to breathe, but all of these died within a few hours. Autopsy of these Jag2ΔDSL homozygous neonates revealed that they contained large amounts of air in the stomach and intestines.
Further examination of the Jag2ΔDSL mutant neonates revealed a bilateral cleft of the secondary palate (Fig. 2). During normal palatal morphogenesis, the palatal shelves (consisting of the palatine and maxillary shelves) elevate and grow toward the midline of the embryo above the tongue. The shelves then undergo an epithelial-mesenchymal transition to fuse and form the secondary palate, which ossifies and forms the roof of the oral cavity. In Jag2ΔDSL homozygous embryos, the palatal shelves did not elevate, and the tongue became wedged between the shelves (Fig. 2A,B). In dorsal regions of the mutant embryos, the unelevated palatal shelves had undergone an epithelial-mesenchymal transition and fused on each side with the posterior portion of the tongue (Fig. 2C,D). Examination of stained skeletal preparations confirmed that the maxillary and palatine shelves were absent in the Jag2ΔDSL mutant neonates (Fig. 2E,F). The fusion of the unelevated palatal shelves with the tongue prevents proper formation of the oral cavity, and is the apparent cause of the breathing difficulties and perinatal lethality of the Jag2ΔDSL homozygous neonates. To determine if the palatal clefting in the Jag2ΔDSL homozygotes correlated with a domain of Jag2 expression, we analyzed craniofacial expression of Jag2 during embryogenesis in mice. High levels of expression were observed in the epithelial cell layer of the branchial arches and the area surrounding the nasal pits (Fig. 2G). Jag2 expression in craniofacial epithelia, including nasal, tongue, and palatal epithelia, was maintained throughout embryogenesis (Fig. 2H). A very similar pattern of Jag2 expression during mouse embryogenesis was recently reported by another group (Luo et al. 1997).
Figure 2.
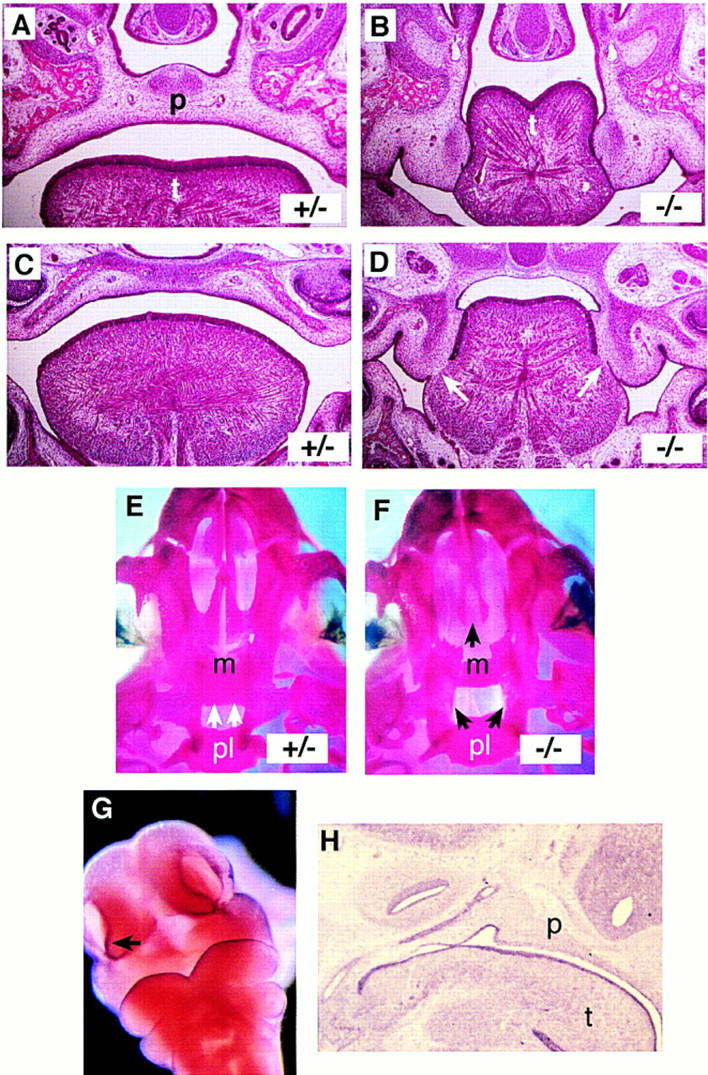
Cleft palate in Jag2ΔDSL homozygotes. (A–D) Frontal sections of E18 embryos. Sections A and B are more ventral than sections C and D. Genotypes are indicated in each panel. Note in the Jag2ΔDSL homozygote fusion of the tongue with the unelevated palatal shelves (white arrows in D). (E,F) Ventral view of stained skeletal preparations of neonatal skulls. The dorsal extent of the palatine shelves is indicated with white arrows in the heterozygote (E). In the Jag2ΔDSL homozygote (F), the palatine shelves have not grown toward and fused in the dorsal midline. The maxillary shelves are also more lateral in the Jag2ΔDSL homozygote. (G,H) Jag2 RNA expression in wild-type mouse embryos. In the head of a E10.5 mouse embryo (G), Jag2 is expressed in the epithelial cell layer of the branchial arches and the area surrounding the nasal pits. (H) Sagittal section of an E12.5 mouse embryo revealed high levels of Jag2 expression in the epithelium and muscles of the tongue, and epithelium of the palate and nasal pharynx. (m) Maxillary shelf; (p) palate; (pl) palatine shelf; (t) tongue.
Jag2ΔDSL mutant mice have syndactylous limbs and a hyperplastic apical ectodermal ridge
In the limb bud of wild-type embryos at E9.5, prior to formation of the apical ectodermal ridge (AER), Jag2 is expressed throughout the ectoderm of the developing limb bud (Fig. 3A). Once the AER forms at E10.5, Jag2 expression in the limb becomes restricted to the AER (Fig. 3B). Importantly, expression of the Notch1 gene has been observed in the AER in both chicks and rats (Myat et al. 1996; Shawber et al. 1996; Rodriguez-Esteban et al. 1997). Whole mount in situ hybridization of wild-type mouse embryos at E10.5 also revealed weak expression of Notch1 in the AER (Fig. 3C).
Figure 3.
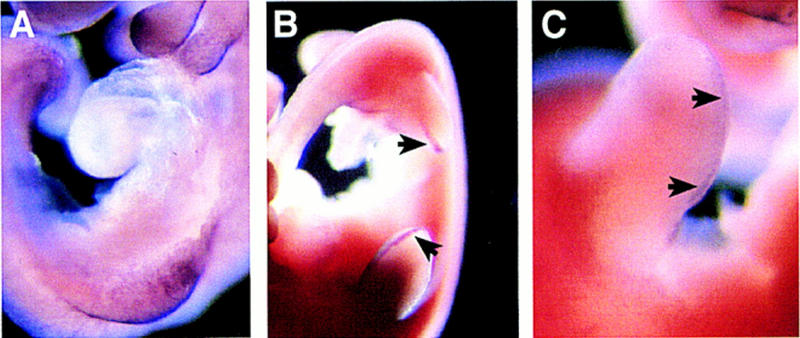
Expression of Jag2 and Notch1 in limb buds of wild-type embryos. Whole mount in situ hybridization of mouse embryos at E9.5 and E10.5. (A) Prior to formation of the AER at E9.5, Jag2 is expressed throughout the limb ectoderm. (B) After AER formation at E10.5, Jag2 expression becomes restricted to the AER (arrows). (C) Low levels of Notch1 expression are also detected in the AER at E10.5 (arrows).
Examination of the limbs of the Jag2ΔDSL homozygous neonates revealed that they exhibited syndactyly (digit fusions) of both the fore- and hindlimbs (Fig. 4B). The syndactyly affected digits 2, 3, and 4, and in many instances appeared to involve soft tissue fusions, although skeletal staining revealed that 5 of 13 mutant skeletons examined had primary chondrogenic or secondary osseous fusions of the distal phalanges, with the hindfeet more severely affected (Fig. 4C–F). Several of the mutant skeletons also exhibited splitting of the terminal phalanx of digit 2 of the hindfeet (Fig. 4E,F).
Figure 4.
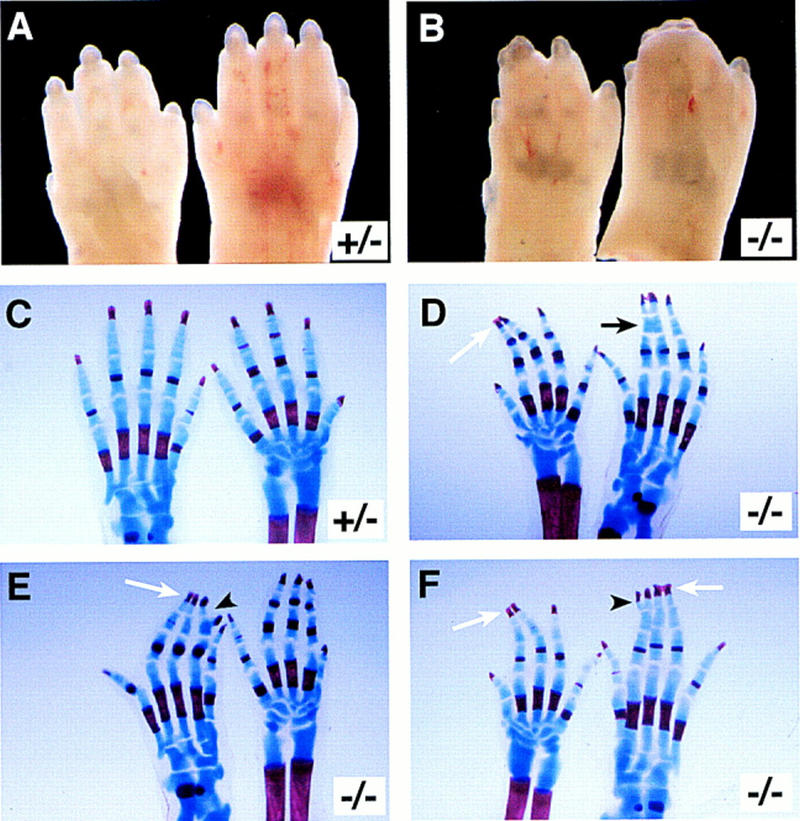
Limb defects in Jag2ΔDSL homozygotes. (A,B) Feet of neonatal mice. A forefoot (left) and hindfoot (right) is shown in each panel. Jag2ΔDSL homozygous neonates (B) exhibit syndactyly of both fore- and hindlimbs, although the hindlimbs are more severely affected. (C–F) Stained skeletal preparations of neonatal limbs of a heterozygous (C) and three Jag2ΔDSL homozygous mice (D–F). Both a fore- and a hindfoot are shown in each panel. The forefoot is on the right in C and E, and on the left in D and F. The homozygotes display several defects affecting distal phalanges, including secondary osseous fusions of both fore- and hindfeet (white arrows in D–F), primary chondrogenic fusions (black arrow in D), and terminal splitting of a distal phalanx (arrowheads in E,F).
Histological analysis revealed that the AER of Jag2ΔDSL homozygous embryos was hyperplastic. Whereas in wild-type and heterozygous littermates the AER was a relatively thin epithelial thickening at the distal end of the limb bud (Fig. 5A), in the Jag2ΔDSL homozygotes the AER was larger and protruded into the underlying mesenchyme of the progress zone (Fig. 5B). Hyperplasia of the AER was also revealed by whole mount in situ hybridization by use of marker genes expressed in the AER. These whole mount in situ studies also revealed that the shape of the foot plate differed between the Jag2ΔDSL homozygous embryos and their wild-type and heterozygous littermates. The foot plates of the Jag2ΔDSL homozygotes were rounder than those of their littermates, and did not exhibit the same degree of interdigital clefting as their littermates (see Figs. 5–7).
Figure 5.
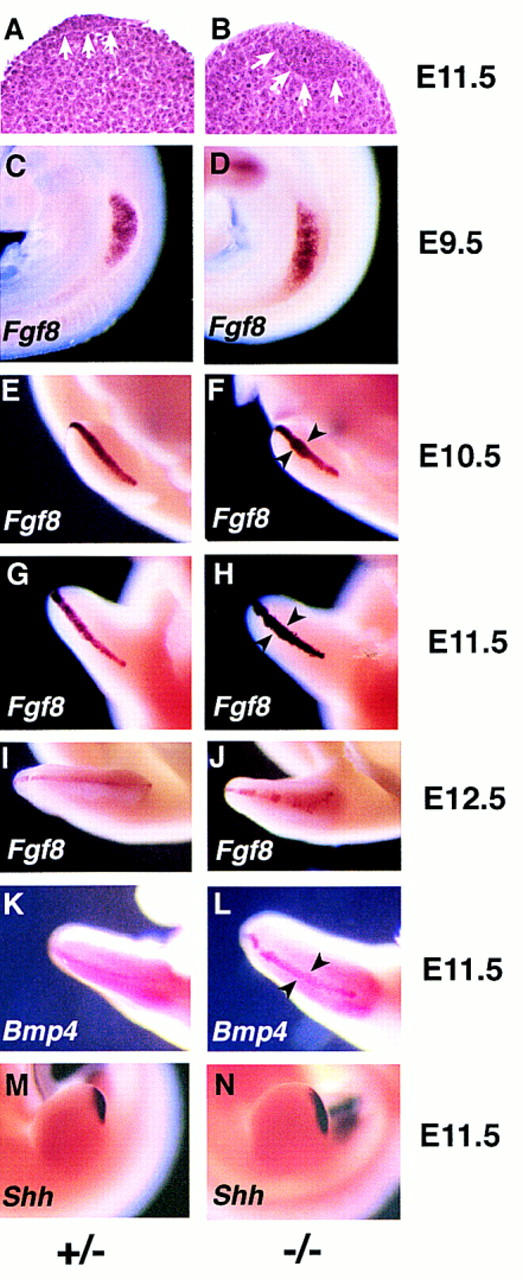
Jag2ΔDSL homozygotes have a hyperplastic AER and exhibit altered marker gene expression. (A,B) Histological analysis of forelimb buds of Jag2ΔDSL homozygous mutant embryo (B) and littermate control (A) at E11.5. The extent of the AER is indicated by the white arrows. (C–J) Time course of Fgf8 expression in the limb bud. (C,D) At E9.5, Fgf8 is expressed in a broad band in the forelimb bud ectoderm of both the Jag2ΔDSL homozygote and the littermate control. (D,E) At E10.5, the Fgf8 expression domain in the AER of the forelimb is broader in the some of the Jag2ΔDSL homozygotes than in wild-type or heterozygous littermates. (G,H) At E11.5, the Fgf8 expression domain is expanded in the AER of both fore- and hindlimbs of all Jag2ΔDSL homozygotes. (I,J) At E12.5, the Fgf8 expression remains broader in the mutants, and is also more irregular along the anterior margin of the limb bud. (K,L) Bmp4 expression at E11.5. (M,N) Shh expression at E11.5.
Figure 7.
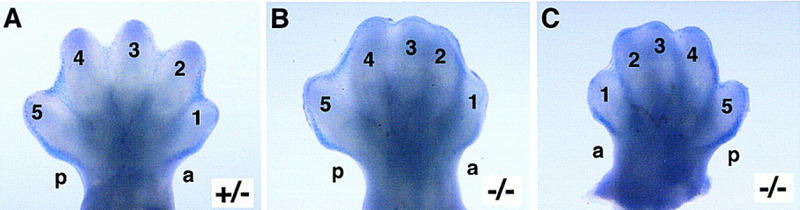
Visualization of interdigital cell death by Nile Blue Sulphate vital staining. Limb buds were isolated at E13.5. Plantar (ventral) apects of hindfeet are shown. Apoptotic cells that stain blue with Nile Blue Sulphate are observed between all forming digits of wild-type and heterozygous littermates, as well as in the anterior (a) and posterior (p) necrotic zones (A) In Jag2ΔDSL homozygotes (B,C), the numbers of staining cells are greatly reduced in the regions where digits 2, 3, and 4 would normally form. There is no apparent reduction of staining cells in the region between digits 1 and 2, and in the anterior and posterior necrotic zones.
Fgf8 (fibroblast growth factor 8) is both an excellent marker for the AER and is one of the molecules required for AER function (Niswander and Martin 1993; Fallon et al. 1994; Laufer et al. 1994; Mahmood et al. 1995; Crossley et al. 1996; Vogel et al. 1996). We examined Fgf8 expression in the limb buds of Jag2ΔDSL homozygotes and littermate controls from E9.5 to E12.5. At E9.5 (prior to AER formation), Fgf8 was expressed as a broad stripe at the dorsal/ventral boundary of the forelimb bud and throughout the ectoderm of the hindlimb bud. This pattern of Fgf8 expression is the same in both Jag2ΔDSL homozygotes and littermate controls (Fig. 5C,D). At E10, the AER forms first on the forelimb bud and then on the hindlimb bud. When examined at E10.5, the Fgf8 expression domain in the newly formed AER of the hindlimb bud was the same in both Jag2ΔDSL homozygotes and littermate controls. However, the Fgf8 expression domain is expanded in some of the mutant forelimb buds (Fig. 5E,F). At E11.5, all six mutant embryos examined exhibited expanded domains of Fgf8 expression in both fore- and hindlimb buds (Fig. 5G,H). By E12.5, the Fgf8 expression domain in the Jag2ΔDSL homozygotes was still broader but was also more irregular, particularly along the anterior margin of the limb bud, than in control littermates (Fig. 5I,J).
We also examined expression of other genes expressed in the AER to determine whether they would show expanded expression domains similar to Fgf8. We examined expression of the genes encoding the bone morphogenetic proteins Bmp2, Bmp4, and Bmp7. At E10.5, all of these genes were expressed in the AER, and no differences in their domains of expression were detected between Jag2ΔDSL homozygotes and littermate controls (data not shown). Other sites of expression of these genes in the limb bud (e.g., expression of Bmp2 in the zone of polarizing activity and expression of Bmp7 in the anterior and posterior mesenchyme) were also unchanged in the mutants. By E11.5, expression of both Bmp2 and Bmp7 was downregulated in the AER, whereas Bmp4 was still expressed in the AER. This Bmp4 expression domain is expanded in the Jag2ΔDSL homozygotes (Fig. 5K,L). These data are consistent with the time course of the AER hypertrophy that we had observed histologically and by Fgf8 expression.
Previous work has demonstrated a positive feedback loop in the developing limb between the AER and the zone of polarizing activity (ZPA), the organizing center responsible for anterior–posterior patterning of the limb (Laufer et al. 1994; Niswander et al. 1994; for review, see Johnson and Tabin 1997). The ZPA exerts its activity through the production of the morphogen Sonic hedgehog (Shh). Because of this positive feedback loop between the AER and the ZPA, one consequence of a hyperplastic AER might be an expansion of the ZPA. Therefore, we examined Shh expression in limb buds of Jag2ΔDSL homozygotes and littermate controls, and found that the domain of Shh expression was expanded in the Jag2ΔDSL homozygous mutants (Fig. 5M,N).
It is apparent from the histological analyses and marker gene expression data described above that the AER forms normally in the Jag2ΔDSL homozygous mutants and becomes hyperplastic between E10.5 and E11.5. We examined whether there might be decreased programmed cell death in the AER of the Jag2ΔDSL homozygous mutants by analyzing in situ detection of terminal transferase-catalyzed digoxigenin-dUTP labeling (TUNEL assay) on sections of E10.5 limb buds. Very few apoptotic cells were detected in the AER, and the numbers of labeled cells did not differ significantly between Jag2ΔDSL homozygotes and littermate controls (data not shown).
Jag2ΔDSL mutant mice exhibit reduced Bmp2 and Bmp7 expression and decreased interdigital cell death
In amniote vertebrates, the formation of digits occurs by the elimination of mesenchymal cells in the interdigital regions through programmed cell death. Apoptotic cell death in the interdigital regions is regulated by BMP-mediated signaling (Macias et al. 1996, 1997; Yokouchi et al. 1996; Zou and Niswander 1996). Therefore, we examined Jag2ΔDSL homozygotes for expression of Bmp2, Bmp4, and Bmp7 at later stages than were used to examine hypertrophy of the AER. At E13.5 in wild-type embryos, Bmp2 was strongly expressed in the interdigital mesenchyme and along the margins of the foot plate in the anterior and posterior necrotic zones (Fig. 6A). In the Jag2ΔDSL homozygotes, expression of Bmp2 was reduced between developing digits 2 and 3 (the digits most severely affected by the syndactylous phenotype; Fig. 6B). Bmp7 was also expressed in the interdigital mesenchyme of wild-type embryos at E13.5, although it was not expressed as strongly as Bmp2 (Fig. 6C). In the Jag2ΔDSL homozygotes, Bmp7 expression was reduced, particularly in the distal portions of the interdigital region (Fig. 6D). Bmp4 was expressed at E13.5 in the distal mesenchyme of the limb bud (Fig. 6E; also see Dunn et al. 1997). Bmp4 expression was unaffected in the Jag2ΔDSL homozygotes (Fig. 6F).
Figure 6.
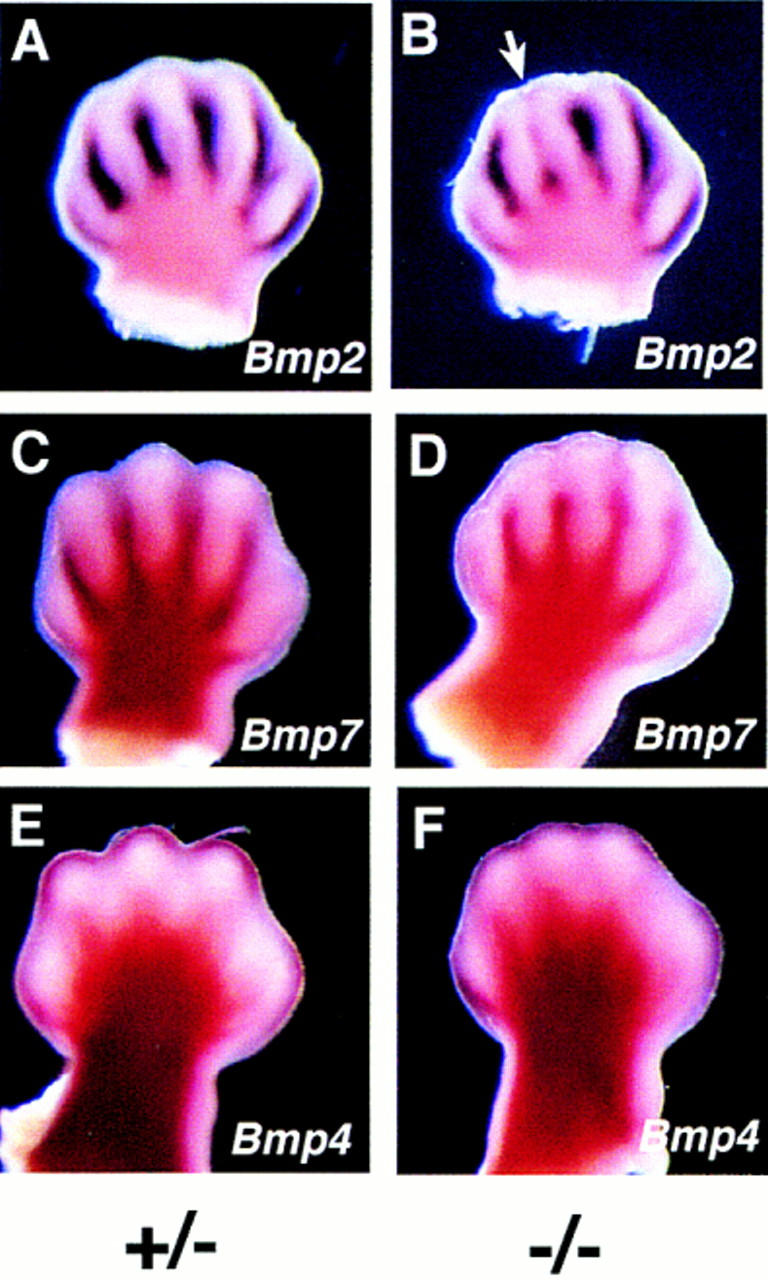
Expression of Bmp2 and Bmp7 is reduced in limbs of Jag2ΔDSL homozygotes. (A,B) Bmp2 expression. Bmp2 expression is reduced in Jag2ΔDSL homozygous mutants between the developing digits 2 and 3 (white arrow). (C,D) Bmp7 expression. Bmp7 expression is reduced in distal regions of the interdigital mesenchyme in the Jag2ΔDSL mutants. (E,F) Bmp4 expression. Bmp4 expression in distal mesenchyme is unaffected in the Jag2ΔDSL mutants. Note the altered morphology of the foot plate in the Jag2ΔDSL homozygous mutants (B,D,F). All embryos shown are at E13.5.
We also directly visualized apoptotic cells in the limb bud by vital staining with Nile Blue Sulphate (Mori et al. 1995). This analysis revealed a reduction of interdigital cell death in the mutants in the region where digits 2, 3, and 4 were forming, whereas the region between digits 1 and 2 and the anterior and posterior necrotic zones (in which anterior and posterior marginal mesoderm of the limb bud is removed) were unaffected (Fig. 7A–C). Thus, the syndactyly of the Jag2ΔDSL homozygotes is accompanied by hyperplasia of the AER, altered morphology of the foot plate, an expanded domain of Fgf8 expression, and reductions in expression of Bmp2 and Bmp7, and a reduction of interdigital cell death.
Jag2ΔDSL mutant mice exhibit impaired differentiation of γδ T cells
Another major site of Jag2 expression during embryogenesis is the thymus (Shawber et al. 1996; Luo et al. 1997). Both Jag2 and Notch1 are coexpressed in the fetal thymus (Luo et al. 1997), suggesting a role for the Notch signaling pathway during thymic development. Indeed, Robey and colleagues have shown previously that perturbations in Notch signaling mediated by the Notch1 gene can affect the development of particular T cell lineages (Robey et al. 1996; Washburn et al. 1997). To examine whether loss of Jag2 gene function leads to similar alterations in T cell development, we performed flow cytometric analyses on thymocytes isolated from Jag2ΔDSL homozygous mutant embryos and control littermates at E18 (∼1 day prior to birth).
The vast majority (>95%) of mature murine T cells express either the CD4 or CD8 marker and utilize antigen-specific T cell receptor (TCR) molecules consisting of an α and β chain heterodimer (αβ lineage T cells). During their course of differentiation in the thymus, TCRαβ expression is first observed in CD4+/CD8+ (double positive) T cells. However, a subset of murine T cells utilize an antigen-specific TCR molecule comprised of a γδ chain heterodimer (γδ lineage T cells). In the thymus, TCRγδ expression is generally restricted to a subset of CD4−/CD8− (double negative) cells whose appearance precedes that of double positive cells. In fetal thymuses from Jag2ΔDSL homozygotes, the percentage of total γδ T cells, as well as those among the CD4−/CD8− double negative subset, was approximately one-half that observed in wild-type and heterozygous littermate controls (representative data shown in Fig. 8A,B; data from all embryos analyzed is summarized in Table 1). In contrast, fetal thymuses from Jag2ΔDSL homozygotes and littermate controls did not differ in the proportion of CD4/CD8 double positive cells that expressed rearranged TCRαβ molecules. Similarly, fetal thymuses from Jag2ΔDSL homozygotes and littermate controls did not differ in proportions of total double negative, double positive, CD4 single positive, or CD8 single positive cells.
Figure 8.
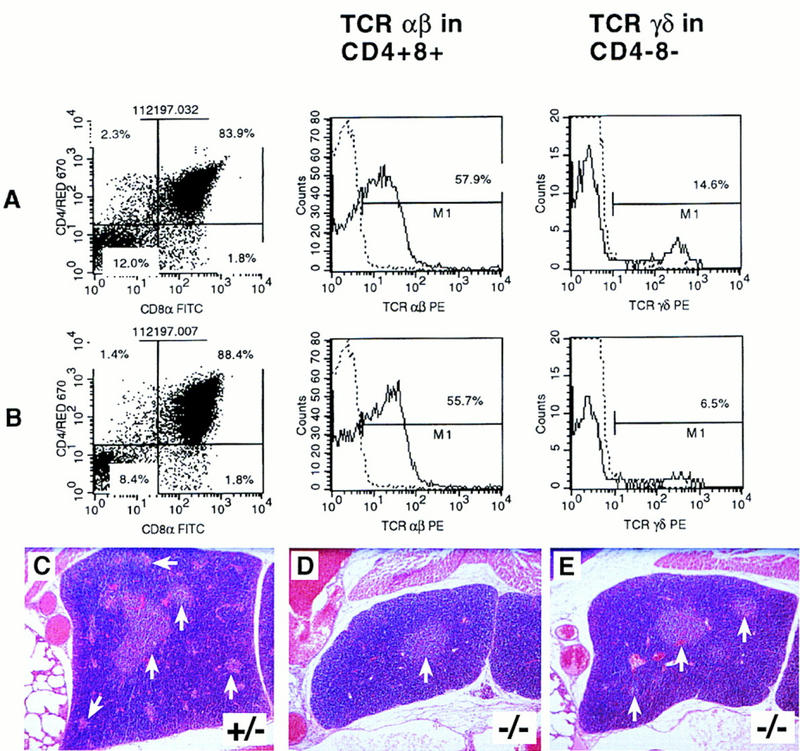
Jag2ΔDSL homozygotes exhibit impaired differentiation of γδ T cells and altered thymic morphology. (A,B) FACS profiles of fetal thymocytes from a representative control littermate (A) and a Jag2ΔDSL homozygous mutant (B) assessed for CD4 vs. CD8 expression, proportion of TCRαβ+ cells in the CD4+CD8+ population, and TCRγδ+ cells in the CD4−CD8− population. Broken lines denote the background staining levels that were used to establish gates for respectively assessing labeling of CD4+CD8+ and CD4−CD8− cells by phycoerythrin-conjugated TCR αβ- or γδ-specific monoclonal antibodies. Annotated numbers denote the percent positive cells of the indicated phenotype. (C–E) Altered morphology of the fetal thymus in Jag2ΔDSL homozygotes at E18. (C) Thymus section from heterozygous littermate. (D,E) Thymus sections from two Jag2ΔDSL homozygotes. In the heterozygote, both darker-stained cortical regions and multiple lighter-stained medullary regions (arrows) can be observed. In the Jag2ΔDSL homozygotes, both the number and the size of the developing medullary regions are reduced.
Table 1.
Summary of flow cytometry analyses of fetal thymocytes from Jag2ΔDSLmutant embryos and littermate controls
| Thymic subset analyzed |
Jag2 genotype
|
|
|---|---|---|
| +/+, +/−
|
−/−
|
|
| CD4−CD8− | 7.9 ± 0.9 (n = 14) | 8.0 ± 1.1 (n = 11) |
| CD4+CD8+ | 83.6 ± 1.2 (n = 14) | 85.6 ± 1.4 (n = 11) |
| CD4+CD8− | 3.2 ± 0.3 (n = 14) | 3.3 ± 0.3 (n = 11) |
| CD4−CD8+ | 4.3 ± 0.9 (n = 14) | 3.3 ± 0.9 (n = 11) |
| TCRαβ+ among CD4+CD8+ cells |
52.1 ± 1.0 (n = 19) | 50.5 ± 1.1 (n = 15) |
| TCRγδ+ among total thymocytes | 2.6 ± 0.1 (n = 19) | 1.3 ± 0.1 (n = 15)a |
| TCRγδ+ among CD4−CD8− cells |
19.6 ± 1.3 (n = 19) | 11.8 ± 1.0 (n = 15)a |
Total cells isolated per thymic lobe did not differ between mutants and controls. No differences were noted between +/+ and +/− littermate controls, so data from these two genotypes were combined. Data are presented as the mean % ± s.e.m. of cells expressing the indicated differentiation markers. Numbers of embryos tested are given in parentheses.
Significantly less (P < 0.001, Student’s t-test) than in +/+ and +/− littermate controls.
Histological analyses also revealed alterations in thymic morphology in the Jag2ΔDSL homozygous embryos. Examination of hematoxylin and eosin stained sections of fetal thymuses from wild-type and Jag2ΔDSL heterozygous littermate controls revealed darker-stained cortical regions and multiple lighter-stained medullary regions (Fig. 8C). In sections of fetal thymuses from Jag2ΔDSL homozygotes, the amount and number of the lighter-staining medullary regions appeared reduced (Fig. 8D,E). Thus, Jag2ΔDSL homozygous mutant embryos exhibit both impaired differentiation of γδ lineage T cells and altered thymic morphology.
Discussion
To determine the biological role of the Jag2 gene in mice, we constructed and analyzed animals homozygous for a deletion that removed the exons encoding the DSL domain of the Jagged2 protein. The phenotype of the Jag2ΔDSL homozygotes demonstrates that Jag2-mediated Notch signaling is essential for proper craniofacial morphogenesis, as well as for proper thymic and limb development. These phenotypes correlate well with domains of Jag2 expression in the tongue and palatal epithelium, in the embryonic thymus, and in the AER of the limb bud.
Cleft palate in Jag2ΔDSL mutant mice
Cleft palate is among the most common birth defects in humans, and is also commonly observed in both spontaneous and targeted mouse mutants and in mouse embryos exposed to teratogenic agents during embryogenesis (e.g., Lohnes et al. 1994; Satokata and Maas 1994; Kaartinen et al. 1995; Proetzel et al. 1995; Orioli et al. 1996; Mo et al. 1997, and references therein). This suggests that disruptions in multiple developmental pathways can lead to palatal clefting. Much less commonly observed, however, is completely penetrant cleft palate, which we have seen in the Jag2ΔDSL homozygous mutants. This suggests that the Jag2 gene may play a highly specific role during palatal morphogenesis, and that the palatal clefting observed in the Jag2ΔDSL homozygotes is not a secondary defect.
During normal palatal morphogenesis, the palatal shelves must elevate, then grow toward the midline of the embryo above the tongue. When the two palatal shelves from either side of the embryo come into proximity, they undergo an epithelial–mesenchymal transition to fuse and form the secondary palate, which ossifies and forms the roof of the oral cavity (Ferguson 1988). Mutations have been found that block different stages of this process. In some mutants that exhibit cleft palate, the shelves elevate and come into proximity, but are unable to undergo the epithelial–mesenchymal transformation and fail to fuse along the midline of the embryo (e.g., Kaartinen et al. 1995; Proetzel et al. 1995). In other mutants, the palatal shelves elevate but do not grow toward the midline of the embryo, possibly because of a deficiency of craniofacial mesenchyme (e.g., Satokata and Maas 1994). The type of cleft palate exhibited in the Jag2ΔDSL homozygotes represents a third category, in which the palatal shelves fail to elevate. Lack of palatal shelf elevation is also observed in mouse embryos homozygous for a particular deletion in the piebald complex (O’Brien et al. 1996). However, the fusions between the unelevated palatal shelves and the tongue that we observe in the Jag2ΔDSL homozygotes appears to be quite unusual.
Thymic development in Jag2ΔDSL mutant mice
Alterations in Notch signaling mediated by the Notch1 gene have been shown previously to affect development of particular T cell lineages (Robey et al. 1996; Washburn et al. 1997). The Jag2 gene is expressed at high levels in the embryonic thymus (Shawber et al. 1996; Luo et al. 1997), which suggests that Jagged2 protein may function as the ligand for the Notch1 protein during thymic development. Jag2ΔDSL homozygous mutant embryos at E18 exhibit both altered thymic morphology and a reduction in the number of γδ T cells. These results demonstrate that Notch signaling mediated through the Jag2 gene is essential for normal thymic development and for the differentiation of normal numbers of γδ T cells.
It is informative to compare our experiments with those of Robey and colleagues (Washburn et al. 1997). In the experiments described in this report, we directly analyzed thymocytes of Jag2ΔDSL homozygotes and littermate controls just before birth (we could not analyze them after birth, because of the completely penetrant neonatal lethality associated with the craniofacial defects of the Jag2ΔDSL homozygotes). However, thymocytes from Notch1-deficient mice cannot be analyzed directly because homozygous Notch1-deficient embryos die during embryogenesis, prior to differentiation of the thymus (Swiatek et al. 1994; Conlon et al. 1995). Therefore, Washburn et al. (1997) examined T cell differentiation in irradiated rag1 mutant mice reconstituted with a mixture of equal portions of fetal liver or bone marrow cells from Notch1+/+ and Notch1+/− donor mice (Notch1−/− stem cells could not be examined, because Notch1-deficient embryos die prior to differentiation of fetal liver cells). Under these conditions, they found that T cells derived from Notch1+/− stem cells are less likely than T cells derived from Notch1+/+ stem cells to differentiate as αβ T cells. This effect was observed only in chimeras reconstituted with a mixture of Notch1+/+ and Notch1+/− stem cells. In either intact Notch1+/− mice, or in same genotype chimeras (i.e., chimeras reconstituted with only Notch1+/+ or Notch1+/− stem cells, but not a mixture of the two), no effect on differentiation of αβ T cells was observed. Washburn et al. (1997) also found that T cells derived from Notch1+/− stem cells are less likely than T cells derived from Notch1+/+ stem cells to differentiate as γδ T cells, and observed that the ratio of γδ to αβ T cells was higher in T cells derived from Notch1+/− stem cells. Therefore, they suggested that reduced Notch signaling mediated by the Notch1 gene favored differentiation of the γδ lineage over the αβ lineage.
Our analysis of T cell differentiation in Jag2ΔDSL homozygous mutant embryos provides further evidence that perturbations of the Notch signaling pathway can influence the differentiation of particular T cell subsets. However, in the Jag2ΔDSL homozygotes, only differentiation of γδ lineage T cells is impaired, whereas differentiation of αβ T cells is unaffected. This may suggest that another ligand interacts with the Notch1 protein during differentiation of the αβ T cell lineage. A more detailed analysis of the role of the Jag2 gene during T cell differentiation will require chimera studies similar to those performed to analyze Notch1 function (Washburn et al. 1997).
Limb defects in mice mutant for two different Jag2 mutant alleles
Previously, we mapped the Jag2 gene to distal Chromosome 12 (Lan et al. 1997), near the spontaneous mouse mutation sm. Sidow et al. (1997) have shown recently that the sm mutation is a missense mutation in the Jag2 gene that causes a glycine to serine substitution in the first EGF repeat. Therefore, we will refer to the mutant allele present in the sm mutants as the Jag2sm allele. The Jag2sm mutation arose spontaneously in the A/Fa inbred strain (Grüneberg 1956). In Jag2sm homozygotes, digits of the forefeet were joined by soft tissues only, whereas fusions of cartilage or bone were often observed on the hindfeet. Considerable postnatal mortality of Jag2sm homozygotes was also observed, although the cause of this lethality was not studied further (Grüneberg 1956).
Morphological and histological examination of the limb buds of Jag2sm homozygotes indicates that the AER is hyperplastic (Grüneberg 1960; Milaire 1967; Sidow et al. 1997). We show here that the AER of the Jag2ΔDSL homozygotes is also hyperplastic (Fig. 5A,B). The authors of these earlier studies suggested that the syndactyly of the Jag2sm homozygotes was the result of mechanical constraints caused by the hyperplastic AER (Grüneberg 1960; Milaire 1967). However, recent work has established that the onset of interdigital cell death in the foot plate is concomitant with the cessation of AER function, and that local administration of fibroblast growth factors (FGFs) in the interdigital region of chick limb buds can inhibit interdigital cell death and cause the formation of soft-tissue syndactyly (Macias et al. 1996). This phenotype is strikingly similar to the phenotypes of the Jag2ΔDSL and Jag2sm mutants. We demonstrate here that in the Jag2ΔDSL homozygotes, Bmp2 and Bmp7 expression and interdigital cell death are reduced. Our model is that the hyperplastic AER of the Jag2ΔDSL (and Jag2sm) homozygotes produces excess FGF activity that inhibits expression of Bmp2 and Bmp7 in the foot plate, with a consequent reduction of interdigital cell death. The expansion of the Shh expression domain in the Jag2ΔDSL homozygotes (presumably via the positive feedback loop between the ZPA and the AER) supports our idea that the hyperplastic AER of the Jag2ΔDSL mutants produces excess FGF activity. Antagonistic interactions between the FGF and BMP signaling pathways have been reported previously for both limb and mandibular development (Niswander and Martin 1993; Neübuser et al. 1997). However, the altered morphology of the foot plate observed in the Jag2ΔDSL and Jag2sm homozygotes most likely also contributes to the syndactylous phenotype, particularly the secondary osseous fusions of the distal phalanges.
Our model that excess FGF activity produced by the hyperplastic AER inhibits BMP expression in the limb mesenchyme and thus causes a reduction of interdigital cell death is consistent with the hypothesis that BMPs are important regulators of programmed cell death in the limb bud (e.g., Macias et al. 1996, 1997; Yokouchi et al. 1996; Zou and Niswander 1996; Dunn et al. 1997; Hofman et al. 1997). However, given the gross morphological change of the AER and the expansion of the Fgf8 expression domain throughout the anterior-posterior axis of the Jag2ΔDSL mutant AER, it is striking that only interdigital cell death, but not cell death in the anterior and posterior necrotic zones, are affected in the Jag2ΔDSL homozygotes. It has been shown that overexpression of a dominant-negative form of BMP2/4 receptor Ia in the chick limb inhibited programmed cell death in both the interdigital mesenchyme and the anterior and posterior necrotic zones (Yokouchi et al. 1996). This indicates that, at least in the chick, BMPs are regulators of programmed cell death in all regions of the limb mesenchyme. In Jag2ΔDSL mutant limb buds, both Bmp2 and Bmp7 are downregulated only in the interdigital regions, leaving the BMP-mediated apoptotic signaling pathway intact in the anterior and posterior necrotic zones. One possible explanation for the specific reduction of Bmp2/7 expression only in the interdigital region of the Jag2ΔDSL mutant limb bud might be an asymmetric distribution of a positive regulator(s) of Bmp2/7 transcription in the limb bud. If such a positive regulator were expressed at higher levels in the anterior and posterior regions of the limb mesenchyme, it could counteract the inhibitory FGF activity and result in unaltered expression of Bmp2 and Bmp7 in the anterior and posterior mesenchyme.
Amorphic and hypomorphic alleles of Jag2
The Jag2sm missense mutation changes an invariant glycine to serine at codon 267 in the first EGF repeat of the Jagged2 protein, which Sidow et al. (1997) suggest creates a hypomorphic Jag2 mutant allele. The more severe phenotype of the Jag2ΔDSL homozygotes supports the idea that Jag2sm is a hypomorphic allele, whereas Jag2ΔDSL is an amorphic (null) allele. The limb phenotype of Jag2ΔDSL homozygotes, although similar to that of Jag2sm homozygotes, appears more severe. Neither secondary osseous fusions of the forefeet nor terminal splitting of distal phalanges (both of which are present in Jag2ΔDSL homozygotes) were reported in Jag2sm homozygotes (Grüneberg 1956, 1960). In addition, no Jag2ΔDSL homozygote has survived for more than a few hours after birth, whereas many Jag2sm homozygotes are viable. Although postnatal lethality was noted in the original description of the Jag2sm mutation (Grüneberg 1956), its cause was not determined. One obvious implication from our work is that the postnatal lethality originally described for the Jag2sm mutants may be caused by incompletely penetrant craniofacial defects, such as the cleft palate and tongue fusions observed in the Jag2ΔDSL homozygotes. Interestingly, a genome scan in mice for teratogen-induced cleft palate and cleft lip susceptibility loci identified several chromosomal regions associated with clefting susceptibility (Diehl and Erickson 1997). One of the identified regions maps to distal chromosome 12 and includes the map postition of the Jag2 gene. This result suggests Jag2 as a possible candidate gene involved in teratogen-induced clefting susceptibility in mice.
Recent results in the chick have suggested that Jag2 acts downstream of the Radical Fringe gene in regulating formation of the AER (Laufer et al. 1997; Rodriguez-Esteban et al. 1997). Whether this regulatory pathway is conserved in mice is open to question, because expression of the Radical Fringe gene cannot be detected in mouse limb buds by whole mount in situ hybridization (Johnston et al. 1997; N. Zhang and T. Gridley, unpubl.). Our results demonstrate that Jag2 is not required in mice for AER formation, and instead suggest that Jag2 may act to negatively regulate growth and function of the AER.
Materials and methods
Isolation of genomic and cDNA clones and targeting vector construction
To isolate mouse Jagged2 cDNA clones, the dBEST expressed sequence tag database was searched with the rat Jagged2 cDNA sequence (Shawber et al. 1996). The insert from one of the homologous EST clones identified in this search (dBEST no. 336371) was used as a probe to screen a mouse brain cDNA library. One clone isolated in this screen (CJ4a) had a 1.4-kb insert that exhibited >85% nucleotide sequence identity to the rat Jagged2 cDNA (the mouse gene was given the symbol Jag2 by the International Committee on Standardized Genetic Nomenclature for Mice). The sequence of this partial cDNA clone has been submitted to GenBank (accession number AF010137). Genomic clones were isolated from a Lambda DASH-II mouse genomic library (Stratagene), with a 1.2-kb insert encoding part of the EGF repeat region of the rat Jagged2 cDNA as a probe. The genomic organization of a portion of the mouse Jag2 locus was determined by restriction enzyme mapping, blot hybridization, and nucleotide sequencing.
To construct the targeting vector, a 2.2-kb SacI fragment of the Jag2 gene was subcloned upstream of a PGK–neo expression cassette (Soriano et al. 1991), and a 4.0-kb SpeI–NotI Jag2 fragment was subcloned downstream of the PGK–neo cassette. This resulted in the deletion of a 5.0-kb genomic fragment containing the exons encoding the DSL domain and half of the first EGF repeat of the Jagged2 protein. We refer to this allele as Jag2ΔDSL. An HSV–tk cassette (Mansour et al. 1988) was also introduced to allow negative selection against random integration of the targeting vector.
Electroporation, selection and screening of ES cells, and mouse and embryo genotyping
CJ7 ES cells were electroporated with 25 μg of linearized targeting vector, selected, screened, and injected into blastocysts from C57BL/6J mice, and the resulting chimeras bred with C57Bl/6J females as described previously (Swiatek et al. 1994). F1 animals heterozygous for the Jag2ΔDSL allele were intercrossed for analysis. Mice and embryos were genotyped by Southern blot analysis or by allele-specific PCR. PCR primers for the wild-type Jag2 allele were DSL1 (5′-ACTACAGTGCCACCTGCAAC-3′) and DSL2 (5′-CTTCGTGGCCTACTAAAGCC-3′), giving a 374-bp amplification product. The primers for the Jag2ΔDSL allele were J2KO2 (5′-GCACGAGACTAGTGAGACGTG-3′), located in the neo cassette and J2KO3 (5′-GAGTGAGAGTGTTCATGCTGAG-3′), giving a 530-bp amplification product.
Histology and in situ hybridization
Embryos were dissected and DNA was prepared from the yolk sacs or tails for genotyping by PCR or by Southern blot analysis. Embryos for histological analysis were fixed in Bouin’s fixative. Fixed embryos were dehydrated through graded alcohols, embedded in paraffin, sectioned at 6 μm, and stained with hematoxylin and eosin. Embryos for in situ hybridization were fixed overnight at 4°C in 4% paraformaldehyde in PBS. Whole mount in situ hybridization and hybridization of cryostat-sectioned embryos with nonradioactive probes were performed as described previously (Lindsell et al. 1996).
Detection of apoptotic cell death and skeletal analyses
Apoptotic cell death in the AER of Jag2ΔDSL homozygotes and littermate controls at E10.5 was assessed by TUNEL labeling. E10.5 embryos were fixed in 3.7% paraformaldehyde in PBS overnight at 4°C, embedded in paraffin, and sectioned. Apoptotic cells were detected by use of the ApopTag in situ apoptosis detection kit (Oncor, Gaithersburg, MD) following the manufacturer’s instructions. Apoptotic cell death in limb buds at E13.5 was assessed by whole mount vital dye staining with Nile Blue sulfate (Mori et al. 1995). Embryos from Jag2ΔDSL heterozygous intercrosses were dissected and were stained in a 1:50,000 (wt/vol) solution of Nile Blue sulfate (Fluka) in Hank’s balanced salt solution for 60–90 min at 37°C. After staining, limb buds were rinsed in PBS and photographed. Alizarin-red-stained skeletal preparations were performed as described (Martin et al. 1995).
Flow cytometry
Multicolor flow cytometric (FACScan, Becton Dickinson, Palo Alto, CA) analysis was used to compare T cell differentiation in fetal thymuses from Jag2ΔDSL homozygous mutant embryos and heterozygous and wild-type littermates. Single cell suspensions were prepared from thymuses of individual embryos at E18 and suspended at 2 × 107/ml in FACS buffer (Ca2+ and Mg2+ free PBS containing 0.1% sodium azide with 2% FBS). Aliquots of 106 thymocytes (50 μl) were stained simultaneously with monoclonal antibodies specific for the T cell differentiation markers CD4 and CD8. CD8 expression was detected with the monoclonal antibody 53-6.72 conjugated to a green fluorescent FITC tag. CD4 expression was detected with the biotin conjugated monoclonal antibody GK1.5 that was developed subsequently with a red fluorescent streptavidin-Red670 tag (Pharmingen, San Diego CA). Separate aliquots of thymocytes stained for both CD4 and CD8 expression were assessed respectively for the presence of rearranged T cell receptor (TCR) αβ or γδ complexes with the monoclonal antibodies H57-597 and GL3 conjugated to a phycoerythrin (PE) tag, whose red fluoresence intensity could be readiliy distinguished from that of Red670. Viable cells, identified by their forward versus side scatter profiles, were analyzed for staining with the antibodies described by use of the Cell Quest 3.0 data reduction program. Data are presented as the mean % ± s.e.m. of cells expressing the indicated array of differentiation markers.
Acknowledgments
We thank A. Sidow and E. Lander for communicating results prior to publication, C. Hicks and B. Griffeth for technical assistance, N. Zhang for help with the phage library screening, B. Hogan, E. Robertson, and G. Martin for probes, and A. Gossler and T. O’Brien for comments on the manuscript. This work was supported by grants from the National Institutes of Health (NS36437, HD34883 to T.G.; NS31885 to G.W.; DK46266, DK51090, AI41469 to D.V.S.) and the March of Dimes Foundation (1-FY97-0193 to T.G.; 5-FY94-0757 to G.W.). R.J. was supported by a National Research Service Award postdoctoral fellowship. This work was also supported by training grants from the National Cancer Institute to The Jackson Laboratory (Y.L.) and UCLA (C.S.), and by a Core grant from the National Cancer Institute to The Jackson Laboratory.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gridley@jax.org; FAX (207) 288-6077.
References
- Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- Bettenhausen B, Hrabe de Angelis M, Simon D, Guenet J-L, Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development. 1995;121:2407–2418. doi: 10.1242/dev.121.8.2407. [DOI] [PubMed] [Google Scholar]
- Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90:281–291. doi: 10.1016/s0092-8674(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Monowada G, MacArthur CA, Martin GR. Roles for FGF8 in the induction, initiation, and maintenance of chick limb development. Cell. 1996;84:127–136. doi: 10.1016/s0092-8674(00)80999-x. [DOI] [PubMed] [Google Scholar]
- Diehl SR, Erickson RP. Genome scan for teratogen-induced clefting susceptibility loci in the mouse: Evidence of both allelic and locus heterogeneity distinguishing cleft lip and cleft palate. Proc Natl Acad Sci. 1997;94:5231–5236. doi: 10.1073/pnas.94.10.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn NR, Winnier GE, Hargett LK, Schrick JJ, Fogo AB, Hogan BLM. Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev Biol. 1997;188:235–247. doi: 10.1006/dbio.1997.8664. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL, Henrique D, Harrison SM, Beddington RSP. Mouse Dll3: A novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development. 1997;124:3065–3076. doi: 10.1242/dev.124.16.3065. [DOI] [PubMed] [Google Scholar]
- Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, Sklar J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- Fallon JF, Lopez A, Ros MA, Savage MP, Olwin BB, Simandl BK. FGF-2: Apical ectodermal ridge growth signal for chick limb development. Science. 1994;264:104–107. doi: 10.1126/science.7908145. [DOI] [PubMed] [Google Scholar]
- Ferguson, M.W.J. 1998. Palate development. Development (Suppl.) 103: 41–60. [DOI] [PubMed]
- Fortini ME, Artavanis-Tsakonas S. The Suppressor of Hairless protein participates in Notch receptor signaling. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- Gridley T. Notch signaling during vertebrate development and disease. Mol Cell Neurosci. 1997;9:103–108. doi: 10.1006/mcne.1997.0610. [DOI] [PubMed] [Google Scholar]
- Grüneberg H. Genetical studies on the skeleton of the mouse. XVIII. Three genes for syndactylism. J Genet. 1956;54:113–145. [Google Scholar]
- Grüneberg H. Genetical studies on the skeleton of the mouse. XXV. The development of syndactylism. Genet Res. 1960;1:196–213. [Google Scholar]
- Henderson ST, Gao D, Lambie EJ, Kimble J. lag-2 may encode a signaling ligand for the GLP-1 and LIN-12 receptors of C. elegans. Development. 1994;120:2913–2924. doi: 10.1242/dev.120.10.2913. [DOI] [PubMed] [Google Scholar]
- Hofman C, Luo G, Balling R, Karsenty G. Analysis of limb patterning in BMP-7-deficient mice. Dev Genet. 1996;19:43–50. doi: 10.1002/(SICI)1520-6408(1996)19:1<43::AID-DVG5>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signaling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Tabin CJ. Molecular models for vertebrate limb development. Cell. 1997;90:979–990. doi: 10.1016/s0092-8674(00)80364-5. [DOI] [PubMed] [Google Scholar]
- Johnston SH, Rauskolb C, Wilson R, Prabhakaran B, Irvine KD, Vogt TF. A family of mammalian Fringe genes implicated in boundary determination and the Notch pathway. Development. 1997;124:2245–2254. doi: 10.1242/dev.124.11.2245. [DOI] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Maréchal E, Maciazek J, Vayssiére C, Cruaud C, Cabanis E-A, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-βb3 indicates defects of epithelial-mesenchymal interaction. Nature Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: Importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Jiang R, Shawber C, Weinmaster G, Gridley T. The Jagged2 gene maps to Chromosome 12 and is a candidate for the lgl and sm mutations. Mamm Genome. 1997;8:875–876. doi: 10.1007/s003359900642. [DOI] [PubMed] [Google Scholar]
- Laufer E, Nelson CE, Johnson RL, Morgan BA, Tabin C. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell. 1994;79:993–1003. doi: 10.1016/0092-8674(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Laufer E, Dahn R, Orozco OE, Yeo C-Y, Pisenti J, Henrique D, Abbott UK, Fallon JF, Tabin C. Expression of Radical fringe in limb-bud ectoderm regulates apical ectodermal ridge formation. Nature. 1997;386:366–373. doi: 10.1038/386366a0. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: A mammalian ligand that activates Notch1. Cell. 1995;80:909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, Dollé P, Dierich A, Gorry P, Gansmuller A, Chambon P. Function of the retinoic acid receptors (RARs) during development. (I) Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120:2723–2748. doi: 10.1242/dev.120.10.2723. [DOI] [PubMed] [Google Scholar]
- Luo B, Aster JC, Hasserjian RP, Kuo F, Sklar J. Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Mol Cell Biol. 1997;17:6057–6067. doi: 10.1128/mcb.17.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias D, Gañan Y, Ros MA, Hurle JM. In vivo inhibition of programmed cell death by local administration of FGF-2 and FGF-4 in the interdigital areas of the embryonic chick leg bud. Anat Embryol. 1996;193:533–541. doi: 10.1007/BF00187925. [DOI] [PubMed] [Google Scholar]
- Macias D, Gañan Y, Sampath TK, Piedra ME, Ros MA, Hurle JM. Role of BMP-2 and OP-1 (BMP-7) in programmed cell death and skeletogenesis during chick limb development. Development. 1997;124:1109–1117. doi: 10.1242/dev.124.6.1109. [DOI] [PubMed] [Google Scholar]
- Mahmood R, Bresnick J, Hornbruch A, Mahony C, Morton N, Colquhoun K, Martin P, Lumsden A, Dickson C, Mason I. A role for FGF-8 in the initiation and maintenance of vertebrate limb bud outgrowth. Curr Biol. 1995;5:797–806. doi: 10.1016/s0960-9822(95)00157-6. [DOI] [PubMed] [Google Scholar]
- Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes & Dev. 1995;9:1237–1249. doi: 10.1101/gad.9.10.1237. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Diederich RJ, Go MJ, Blaumueller CM, Artavanis-Tsakonas S. Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development. 1995;121:2633–2644. doi: 10.1242/dev.121.8.2633. [DOI] [PubMed] [Google Scholar]
- Milaire J. Histochemical observations on the developing foot of normal, oligosyndactylous (Os/+) and syndactylous (sm/sm) mouse embryos. . Arch Biol (Liege) 1967;78:223–288. [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi X-M, Tsui L-C, Cheng SH, Joyner AL, Hui C-c. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- Mori C, Nakamura N, Kimura S, Irie H, Takigawa T, Shiota K. Programmed cell death in the interdigital tissue of the fetal mouse limb is apoptosis with DNA fragmentation. Anat Rec. 1995;242:103–110. doi: 10.1002/ar.1092420114. [DOI] [PubMed] [Google Scholar]
- Muskavitch MA. Delta-Notch signaling and Drosophila cell fate choice. Dev Biol. 1994;166:415–430. doi: 10.1006/dbio.1994.1326. [DOI] [PubMed] [Google Scholar]
- Myat A, Henrique D, Ish-Horowicz D, Lewis J. A chick homologue of Serrate and its relationship with Notch and Delta homologues during central neurogenesis. Dev Biol. 1996;174:233–247. doi: 10.1006/dbio.1996.0069. [DOI] [PubMed] [Google Scholar]
- Neubüser A, Peters H, Balling R, Martin GR. Antagonistic interactions between FGF and BMP signaling pathways: A mechanism for positioning the sites of tooth formation. Cell. 1997;90:247–255. doi: 10.1016/s0092-8674(00)80333-5. [DOI] [PubMed] [Google Scholar]
- Niswander L, Martin GR. FGF-4 and BMP-2 have opposite effects on limb growth. Nature. 1993;361:68–71. doi: 10.1038/361068a0. [DOI] [PubMed] [Google Scholar]
- Niswander L, Jeffrey S, Martin GR, Tickle C. Positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature. 1994;371:609–612. doi: 10.1038/371609a0. [DOI] [PubMed] [Google Scholar]
- O’Brien TP, Metallinos DL, Chen H, Shin MK, Tilghman SM. Complementation mapping of skeletal and central nervous system abnormalities in mice of the piebald deletion complex. Genetics. 1996;143:447–461. doi: 10.1093/genetics/143.1.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nature Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- Orioli D, Henkemeyer M, Lemke G, Klein R, Pawson T. Sek4 and Nuk receptors cooperate in guidance of commissural axons and in palate formation. EMBO J. 1996;15:6035–6049. [PMC free article] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MWJ, Doetschman T. Transforming growth factor-β3 is required for secondary palate fusion. Nature Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey E. Notch in vertebrates. Curr Opin Gen Dev. 1997;7:551–557. doi: 10.1016/s0959-437x(97)80085-8. [DOI] [PubMed] [Google Scholar]
- Robey E, Chang D, Itano A, Cado D, Alexander H, Lans D, Weinmaster G, Salmon P. An activated form of Notch influences the choice between CD4 and CD8 T cell lineages. Cell. 1996;87:483–492. doi: 10.1016/s0092-8674(00)81368-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Esteban C, Schwabe JW, De La Peña J, Izpisúa-Belmonte JC. Radical fringe positions the apical ectodermal ridge at the dorsoventral boundary of the vertebrate limb. Nature. 1997;386:360–366. doi: 10.1038/386360a0. [DOI] [PubMed] [Google Scholar]
- Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nature Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- Shawber C, Boulter J, Lindsell CE, Weinmaster G. Jagged2: A Serrate-like gene expressed during rat embryogenesis. Dev Biol. 1996;180:370–376. doi: 10.1006/dbio.1996.0310. [DOI] [PubMed] [Google Scholar]
- Sidow A, Bulotsky MS, Kerrebrock AW, Bronson RT, Daly MJ, Reeve MP, Hawkins TL, Birren BW, Jaenisch R, Lander ES. Serrate2 is disrupted in the mouse limb development mutant syndactylism. Nature. 1997;389:722–725. doi: 10.1038/39587. [DOI] [PubMed] [Google Scholar]
- Singh G, Supp DM, Schreiner C, McNeish J, Merker H-J, Copeland NG, Jenkins NA, Potter SS, Scott W. legless insertional mutation: Morphological, molecular, and genetic characterization. Genes & Dev. 1991;5:2245–2255. doi: 10.1101/gad.5.12a.2245. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- Swiatek PJ, Lindsell CE, Franco del Amo F, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes & Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- Vogel A, Rodriguez C, Izpisúa-Belmonte JC. Involvement of FGF-8 in initiation, outgrowth and patterning of the vertebrate limb. Development. 1996;122:1737–1750. doi: 10.1242/dev.122.6.1737. [DOI] [PubMed] [Google Scholar]
- Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes BJ, Cado D, Robey E. Notch activity influences the αβ versus γδ T cell lineage decision. Cell. 1997;88:833–843. doi: 10.1016/s0092-8674(00)81929-7. [DOI] [PubMed] [Google Scholar]
- Weinmaster G. The ins and outs of Notch signaling. Mol Cell Neurosci. 1997;9:91–102. doi: 10.1006/mcne.1997.0612. [DOI] [PubMed] [Google Scholar]
- Yokouchi Y, Sakiyama J-i, Kameda T, Iba H, Suzuki A, Ueno N, Kuroiwa A. BMP-2/-4 mediate programmed cell death in chicken limb buds. Development. 1996;122:3725–3734. doi: 10.1242/dev.122.12.3725. [DOI] [PubMed] [Google Scholar]
- Zou H, Niswander L. Requirement for BMP signaling in interdigital apoptosis and scale formation. Science. 1996;272:738–741. doi: 10.1126/science.272.5262.738. [DOI] [PubMed] [Google Scholar]