

Abstract
Topoisomerase 1 (Top1) enzymes regulate DNA superhelicity by forming covalent cleavage complexes that undergo controlled rotation. Substitution of nucleoside analogs at the +1 position of the DNA duplex relative to the Top1 cleavage site inhibits DNA religation. The reduced efficiency for Top1-mediated religation contributes to the anticancer activity of widely used anticancer drugs including fluoropyrimidines and gemcitabine. In the present study, we report that mismatched base pairs at the +1 position destabilize the duplex DNA components for a model Top1 cleavage complex formation even though one duplex component does not directly include a mismatched base pair. Molecular dynamics simulations reveal G-dU and G-FdU mismatched base pairs, but not a G-T mismatched base pair, increase flexibility at the Top1 cleavage site, and affect coupling between the regions required for the religation reaction to occur. These results demonstrate that substitution of dT analogs into the +1 position of the non-scissile strand alters the stability and flexibility of DNA contributing to the reduced efficiency for Top1-mediated DNA religation. These effects are inherent in the DNA duplex and do not require formation of the Top1:DNA complex. These results provide a biophysical rationale for the inhibition of Top1-mediated DNA religation by nucleotide analog substitution.
1. Introduction
DNA topoisomerasesregulate the topological state of DNA as required to relieve superhelical density for important biological processes such as replication and transcription [1–3]. DNA topoisomerase 1 (Top1) is expressed at elevated levels during S-phase of the cell cycle and is the topoisomerase primarily responsible for relieving superhelical density generated in front of advancing replication forks in mammalian cells. Top1 preferentially binds superhelical DNA and forms a covalent complex as a result of nucleophilic attack by the hydroxyl of Tyr 723 on the phosphodiester backbone of the scissile strand of the DNA duplex. DNA superhelical density is reduced by controlled rotation of the scissile strand about the nonscissile strand in the cleavage complex [4, 5]. Following release of superhelical tension, the cleavage complex is dissociated by nucleophilic attack of the free 5′-OH of the scissile strand to reform the phosphodiester backbone. DNA sequences that have several A-tracts flanking a conserved DNA duplex motif are also substrates for DNA Top1 and serve as a model system for understanding DNA recognition and catalysis by Top1 [6].
Top1 is the sole target for the camptothecin (CPT) class of anticancer drugs. CPT forms a stable ternary complex upon binding to the Top1:DNA covalent cleavage complex. Stabilization of cleavage complexes by CPT converts Top1 into a cellular poison since collision of advancing replication forks with trapped Top1 cleavage complexes results in DNA double-strand breaks. Thus, CPT not only inhibits Top1 activity, but also converts the enzymatic activity into DNA damage that is potentially lethal to the cell. Over the last decade, it has been shown that a variety of nonnative nucleotide substitutions that may result from oxidative damage to DNA (e.g., 8-oxo-dG) or covalent modification of DNA nucleobases (e.g., benzpyrene adducts) also cause trapping of Top1 cleavage complexes and result in DNA DSB formation [7]. Work from our laboratory in collaboration with the Pommier lab has shown that misincorporation of deoxyribonucleotide analogs that have anticancer activity, such as FdU [8] and gemcitabine [9], into Top1 cleavage sites also causes trapping of Top1 cleavage complexes [10]. Poisoning of Top1 by FdU-substituted DNA contributes to the cytotoxicity and antitumor activities of fluoropyrimidines [8].
The structural basis for trapping of Top1 cleavage complexes by damaged nucleobases or misincorporation of nucleotide analogs into the nonscissile strand of DNA remains an area of investigation. Although the DNA sequence used in most model studies of Top1:DNA interactions contains several A-tracts [6], X-ray crystal structures do not reveal any bending of this DNA in either covalent or noncovalent complexes with DNA [5]. One question that remains unanswered is how introduction of nonnative nucleotides into the nonscissile strand of DNA inhibits the religation reaction. To investigate this issue, we have constructed a model Top1 cleavage site consisting of a 39 mer DNA hairpin consisting of 13 base pairs with a 10 mer single-stranded overhang (Figure 1). We have investigated the thermal stability of this DNA hairpin consisting of all native nucleotides and have compared the stability of the native sequence to sequences that contain a single C → dU, C → FdU (5-fluoro-2′-deoxyuridine), or C → T substitution. These substitutions result in a single mismatched base pair at the site corresponding to the +1 site relative to the site of Top1 cleavage. As expected, introduction of a mismatched base pair decreases the stability of the DNA hairpin by approximately 3°C. We also investigated the stability of DNA duplex formation between these DNA hairpins and a DNA 10 mer that is complementary to the 10-nucleotide overhang of the hairpin. The interaction of this DNA 10 mer with the hairpin provides a model system for the Top1 religation reaction. Unexpectedly, we find that the melting temperature for the formation of the 10 mer duplex that would be required for the Top1 religation reaction to occur is sensitive to the presence of DNA mismatched base pairs in the hairpin even though no mismatched base pairs are present in the 10 mer duplex region. Molecular dynamics simulations of this model system demonstrate that G-dU and G-FdU mismatched base pairs increase flexibility and affect coupling with the first 10 base pairs. We conclude that DNA mismatched base pairs adjacent to the Top1 cleavage site both decrease DNA stability and increase flexibility disfavoring formation of the DNA conformation required for Top1-mediated DNA religation.
Figure 1.
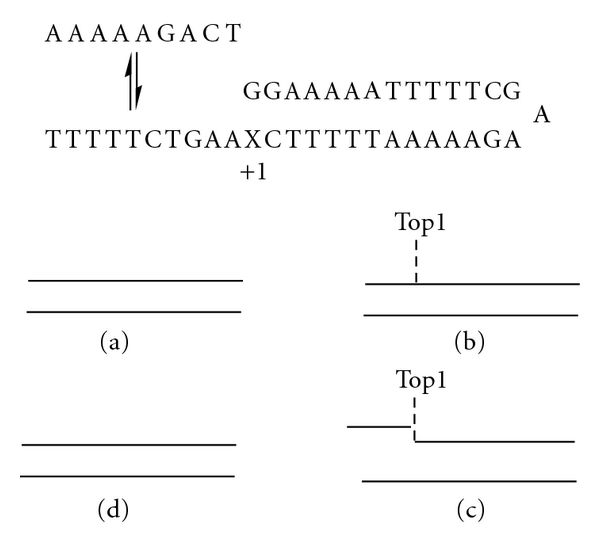
Model system used for evaluating the effects of mismatched base pairs on the stability of the intermediate required for Top1-mediated DNA religation. Top: depiction of the equilibrium between the 10 mer single-stranded DNA and the 39 mer DNA hairpin that constitutes a model system for Top1-mediated DNA religation. A G-X base pair is at the +1 position relative to the Top1 cleavage site. Bottom (a–d): depiction of the steps of Top1 binding, strand dissociation, strand reassociation, and religation.
2. Methods
2.1. Design and Synthesis of DNA Hairpin Sequences
A model Top1 cleavage site (Figure 1) was designed based upon a DNA sequence containing several A-tract motifs that had been previously demonstrated to be a suitable model substrate for the Top1 cleavage/religation reaction [6]. The model Top1 cleavage site was synthesized in two pieces, a 39 mer DNA hairpin containing the GAA sequence [11] that promotes hairpin formation and a 10-nucleotide single-strand DNA complementary to the 3′-terminus of the hairpin. Upon annealing, the two sequences from a duplex with a single nick in the phosphodiester backbone of one strand corresponding to the scissile strand of the Top1 cleavage complex (Figure 1). To investigate the effects of deoxynucleotide substitutions on the stability of the model Top1 cleavage site, the 39 mer was synthesized four times, once with dC as the putative base pairing partner for the 3′-terminal dG and also with T, dU, or FdU at this site. In this manner, the base pair at the +1 site of the model Top1 cleavage complex was either the native G-C base pair or was a G-T, G-FdU, or G-dU mismatched base pair. These mismatched base pairs occur at the junction corresponding to the site of cleavage for the Top1 complex. All DNA sequences were synthesized at the University of Calgary DNA core synthesis facility and purified by gel filtration chromatography.
2.2. Temperature-Dependent UV Spectroscopy (UV Melts)
Absorbance versus temperature profiles of each oligonucleotide in buffer were measured at 260 nm using a thermoelectrically controlled Aviv model 14DS UV-vis spectrophotometer (Lakewood, NJ). The temperature was scanned from 20°C to 95°C for the 39 mer DNA hairpins and from 1–100°C for the model Top1 cleavage sites at a heating rate of 0.6°C/min. DNA concentrations were 1.5–2.0 μM, and the buffer used was 10 mM sodium phosphate, pH 7.0 with 200 mM NaCl added for high-salt conditions. Shape analysis of the melting curves yielded Tm values using procedures reported earlier [12].
2.3. Molecular Modeling and Molecular Dynamics Simulations
The simulation of the four hairpins is performed using NAMD [13] with the CHARMM27 force field [14, 15], with analysis performed using CHARMM [16]. A normal, matched DNA structure was built using PREDICTOR [17] to construct 23 base pairs using the same sequence as in the experiments. The GAA hairpin was added from a high-resolution NMR structure [18]. Coordinate manipulation commands within CHARMM were used to merge the two structural elements. The resulting overall structure was minimized in CHARMM with an r-dependent dielectric of 4r, and harmonic restraints to remove bad contacts. The minimization cycle was (1) 100 steps of steepest-descent minimization followed by 100 steps of conjugate gradient minimization both with best-fit harmonic constraints on the hairpin atoms with a mass-weighted force constant of 1 with the remaining bases fixed to relieve any bad contacts with the hairpin; (2) 100 steps of steepest-descent minimization followed by 100 steps of conjugate gradient minimization both with harmonic constraints on all atoms with a mass-weighted force constant of 10; (3) 100 steps of steepest-descent minimization followed by 100 steps of conjugate-gradient minimization both with harmonic constraints on all atoms with a mass-weighted force constant of 1. The other three hairpins were built by mutating the original matched structure and rebuilding the altered base within CHARMM followed by the same minimization protocol. Missing parameters for FdU were obtained from our previous quantum mechanical study [19] and the existing fluorine parameters within CHARMM27 supplemented by two dihedrals created using the parameters from our previous study [19] with force constants from the corresponding unperturbed dihedrals. The resulting structures were fully solvated and charge neutralized with TIP3P water in a cubic box using the visual molecular dynamics (VMD) package [20]. The simulation was performed in NAMD using [13] standard parameters: a 2.0 fs timestep using SHAKE on all bonds to hydrogen atoms, a 12 Å cutoff, Particle Mesh Ewald with a 1.0 Å grid determined by NAMD, Langevin constant pressure algorithm with a target pressure of 1.01325 bar, a piston period of 100 fs, a piston decay time of 50/ps, and a piston temperature of 300 K, all as implemented in NAMD. The simulation protocol consisted of 1000 steps of unconstrained steepest-descent minimization on the fully solvated and ionized system, followed by 250 ps of thermal equilibration to 300 K with temperature reassignment, followed by a 8–12 ns of production simulation; 10 different simulations with different random initial conditions were performed for each hairpin for a total of 40 simulations. Based on exchange between clusters after 4 ns all-atom clustering and leveling of the all-atom RMSDs (data not shown), the first 4 ns of each simulation was discarded as equilibration. The remaining 240 ns in total of simulation data was analyzed with structures saved every 2 ps. CHARMM's analysis routines were used to calculate the root-mean-square fluctuations, covariances, and for rmsd-based clustering with a 2.5 Å cutoff. Matlab was used to perform the clustering analysis of the data with single-linkage Euclidean distance measures and an inconsistent value of 0.001.
3. Results
3.1. UV Hyperchromicity Measurements
The effects of mismatched base pairs at the +1 position on the stability of the model Top1 cleavage site were investigated using UV hyperchromicity measurements. Initial studies focused on the stabilities of the four 39 mer DNA hairpins (Figure 2). The parental hairpin consisting of only Watson-Crick base pairs in the stem region had a Tm of 61.0°C. Introduction of a G-T mismatched base pair reduced the stability of the hairpin by 4.4°C. The G-dU and G-FdU mismatched base pairs also destabilized the hairpin decreasing the Tm by 3.0 and 3.5°C, respectively. The results demonstrate that introduction of a single mismatched base pair at the +1 site relative to Top1 cleavage destabilizes the duplex by 3–4.4°C. The results are consistent with previous studies indicating similar destabilization effects resulting from introduction of a single mismatched base pair.
Figure 2.
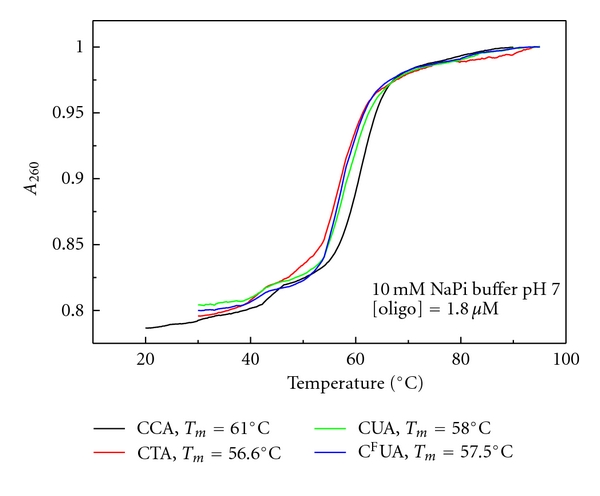
Stability of the 39 mer DNA hairpin corresponding to a model Top1 cleavage site. A single coordinated transition is observed corresponding to the thermal melting transition for the double-stranded region of the hairpin. As expected, the melting temperature is dependent upon the presence of a mismatched base pair at the terminus of the duplex region. The native sequence is most stable while C → T, C → dU, and C → FdU decrease the stability of the duplex region by 4.4°C, 3.0°C, and 3.5°C, respectively.
We next investigated the stability of the model Top1 cleavage complex using UV hyperchromicity measurements. The four 39 mer DNA hairpins consisting of 13 base pairs with a 10 mer single-stranded overhang were annealed to the 10 mer ssDNA complementary to the overhang region (Figure 1). The single mismatched base pair corresponding to the +1 site of a Top1 cleavage complex was at the stem terminus, such that annealing of the 10 mer to the overhang extended the stem, albeit with a nicked phosphodiester backbone. The UV hyperchromicity profiles for all four hairpins in the presence of the complementary 10 mer were biphasic, as expected, with release of the 10 mer occurring first followed by melting of the 39 mer hairpins (Figure 3). Under low salt conditions (10 mM phosphate buffer, pH 7), the melting temperatures for the two phases of the parental hairpin were 9.7°C and 61.1°C corresponding to release of the 10 mer and unfolding of the hairpin. The identical melting curve obtained in physiologically relevant salt conditions (10 mM phosphate buffer, pH 7, 200 mM NaCl) had a Tm of 33.1°C for release of the 10 mer and a Tm of 77.2°C for unfolding of the hairpin. Interestingly, the presence of the mismatched base pairs destabilized both components of the biphasic melting curves. For example, for the physiologically relevant salt conditions with the G-FdU base pair, the Tm for release of the 10 mer decreased from 33.1 to 30.1°C, and the Tm for unfolding of the hairpin decreased from 77.2 to 75.3°C. The decrease for the 39 mer hairpin is expected as the mismatched base pair is present in the stem of the hairpin, and the magnitude of destabilization is similar to that observed for the 39 mer hairpin alone. The destabilization of the 10 mer region is somewhat surprising as the sequence does not contain a mismatched base pair. The terminal base pair of the 10 mer, however, stacks upon the site of the mismatched base pair in the 39 mer hairpin, and the observed destabilization likely results from less efficient stacking interactions. The G-T and G-dU mismatched base pairs elicited similar degrees of destabilization as the G-FdU mismatch for both release of the 10 mer and melting of the DNA hairpin. The results indicate that the presence of a mismatched base pair decreases the stability of the model Top1 cleavage complex disfavoring adoption of the geometry required for religation to occur. The results are consistent with decreased stability of the duplex as contributing to the less favorable religation kinetics observed for model Top1 cleavage sites containing G-FdU or other mismatched base pairs [8].
Figure 3.
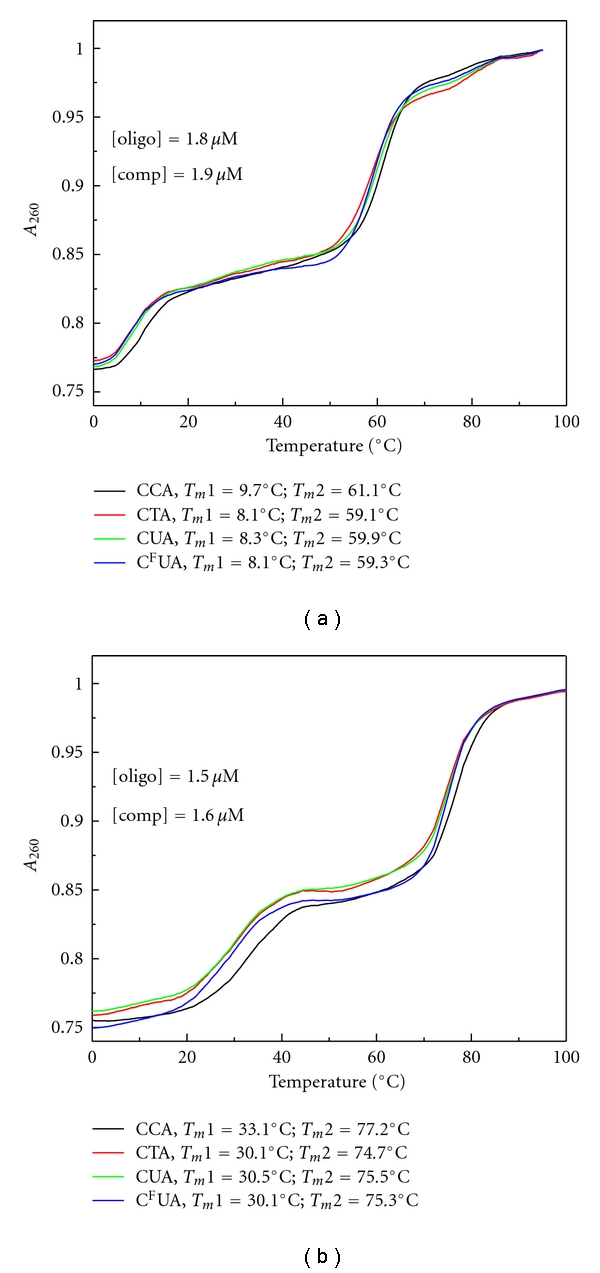
Thermal stability of the complex of the 10 mer ssDNA and the 39 mer DNA hairpin constituting a model Top1 religation complex. (a) Melting transition in 10 mM sodium phosphate buffer pH 7; (b) melting transition in the same buffer as A, but with 200 mM NaCl. A biphasic melting profile is observed in both instances. The lower melting temperature corresponds to the dissociation of the ssDNA from the 39 mer DNA hairpin, while the higher melting transition corresponds to the melting temperature for the duplex region of the hairpin. As was observed for the DNA hairpin alone (Figure 2), the melting temperature for the duplex region is sensitive to the presence of a mismatched base pair at the terminus. Surprisingly, the melting temperature for the complex between the single-stranded DNA 10 mer and the single-stranded region of the hairpin is also sensitive to the presence of a base pair mismatch in the duplex region even though the transition does not directly involve the mismatched base pair.
3.2. Molecular Dynamics Simulations
Comparing molecular dynamics simulations for the four different 49 mer DNA hairpins (39 mer DNA hairpin extended without a nick in the phosphodiester backbone) demonstrates that all the mismatches indeed do produce profound effects on the initial 10 base pairs that form the recognition sequence and its complement. Due to the observed effects on the thermal stability and the novel influence of different mismatches occurring outside the recognition sequence, three different atomic measures of structural fluctuations within these first 10 base pairs are used to quantify this influence. First, atomic root-mean-square fluctuations (RMSFs) are calculated for all heavy atoms and averaged on a per-base level. This quantifies the extent to which each atom fluctuates about its equilibrium position and averages these fluctuations at the base-level for comparisons to determine how the different mismatches affect both the overall atomic fluctuations of the first 10 base pairs and the pattern of fluctuations. Second, correlated fluctuations are calculated. These determine the extent to which atomic fluctuations, regardless of magnitude, are correlated or anticorrelated and are averaged at the base level for comparison among the four different simulation types. This measure, referred to as the covariance matrix, determines how the different mismatches affect the coupling within the first 10 base pairs. This method has been used by multiple research groups to analyze communication within proteins [21] and the effects of various perturbations on communication [22]. Performing the same analysis on DNA can provide similar information. The third and final measure used is cluster analysis, in which the different structural snapshots across all the simulations are clustered together to find the conformations accessed and their populations in the different hairpins.
The first noticeable effect of mismatch formation is on the RMSFs of the G-dU and G-FdU hairpins; the average RMSF over the first 10 base pairs increases by 13.5% and 12.0% for the G-dU and G-FdU hairpins, respectively, relative to the average RMSF of the matched hairpin (Table 1). Surprisingly, the G-T mismatch shows almost no overall increase in flexibility (<1%). However, all the hairpins show delocalized changes in flexibility in the first 10 bases relative to the matched hairpin (Figure 4).
Table 1.
DNA RMSFs (Å)*.
| Matched | 2.136 |
| G-T | 2.120 |
| G-dU | 2.423 |
| G-FdU | 2.391 |
*The root-mean-square fluctuations in Å for the first 10 base pairs of each sequence are averaged.
Figure 4.
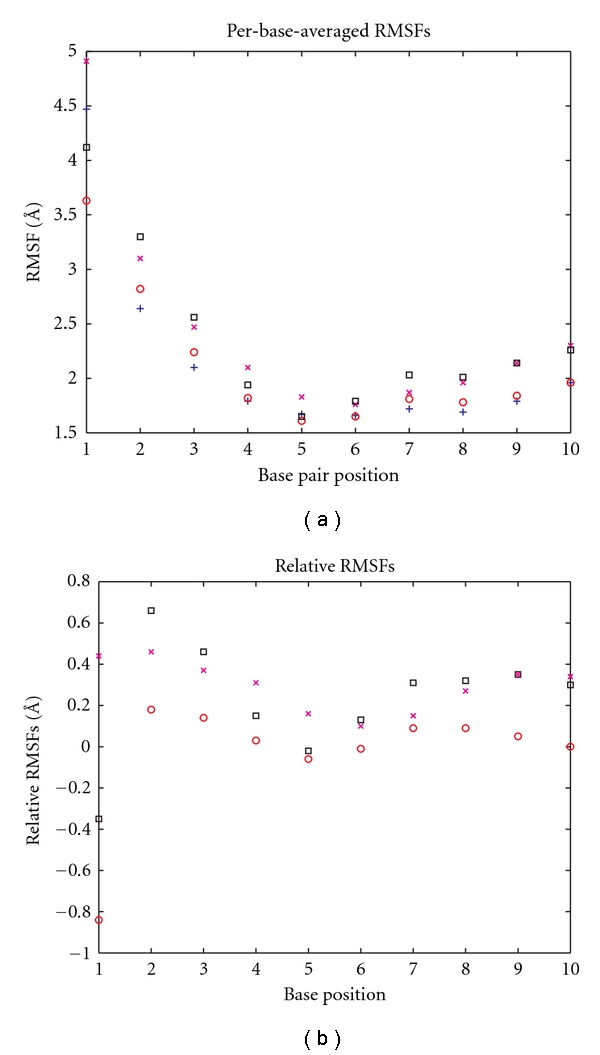
(a) Per-base-averaged RMSFs. The atomic root-mean-square fluctuations for the first ten bases for each of the DNA sequences are depicted: matched DNA as blue +s, T mismatched as red °s, dU mismatched as magenta ×s, and FdU mismatched as black squares. (b) Per-base-averaged RMSFs relative to matched DNA as (a), but with the matched DNA rmsf values subtracted.
The covariance matrices (Figure 5) also exhibit delocalized changes in dynamics, as measured by correlated fluctuations, mostly concentrated within the chain where the mismatches occur (bases 1–10). The largest such changes occur with the FdU mismatch (Figure 5(c)), although more pairs exhibit large perturbations with the dU mismatch (Figure 5(d)), including bases in the opposite chain near the mismatch.
Figure 5.
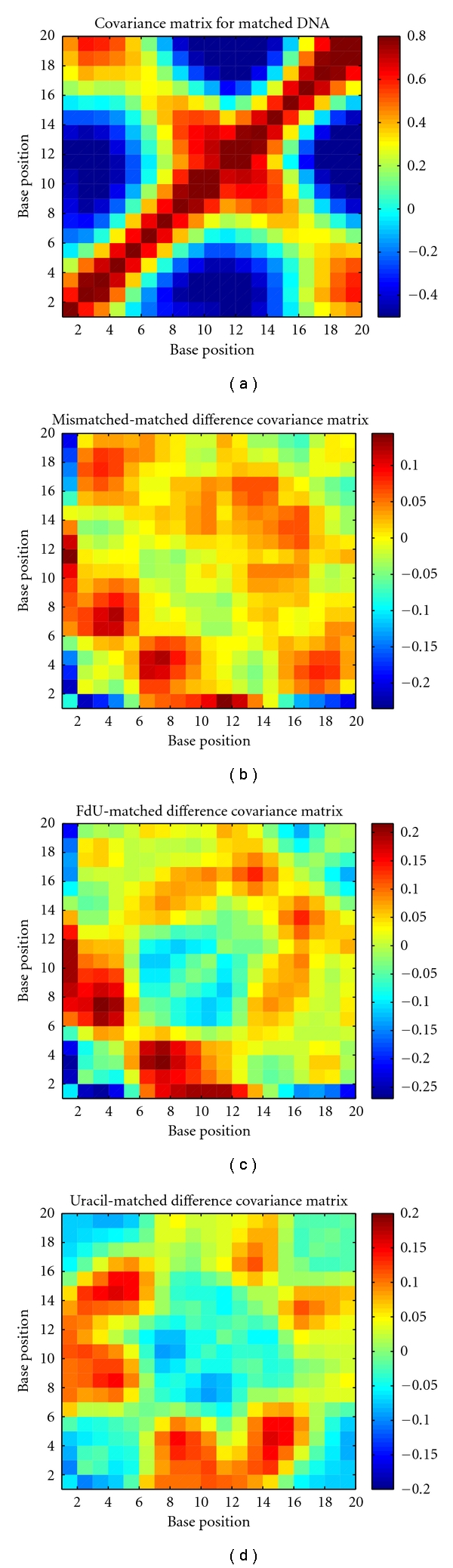
(a) Matched covariance matrix. The per-base-averaged covariance matrix plotted as a color map with red indicating high correlations of fluctuations blue indicating high anticorrelation of fluctuations, and yellow indicat uncorrelated. (b) Mismatched-matched difference covariance matrix. The matched covariance matrix (a) subtracted from the mismatched covariance matrix (not shown). (c) FdU-matched difference covariance matrix. The matched covariance matrix (a) subtracted from the FdU-mismatch covariance matrix (not shown). (d) Uracil-matched difference covariance matrix. The matched covariance matrix (a) subtracted from the uracil-mismatch covariance matrix (not shown).
Both the RMSFs and covariances demonstrate that there are delocalized changes in atomic covariances and their correlations in the recognition sequences despite the mismatch occurring outside this sequence; clustering analysis sheds light on the population shifts and conformational changes that may give rise to these variations, and that may perturb the recognition and binding by Top1.
Clustering analysis on the first 10 base pairs shows that there are four different conformations that are accessible to each of the four DNA sequences. The difference lies in their populations (Table 2). The different mismatches shift population among the different conformations, with the dominate change being increasing population in the rarest conformation found in the normal, matched DNA simulation. The population of this conformation increases in the different mismatched from T, to dU and to FdU.
Table 2.
Cluster populations (%).
| Cluster no./Sequence | Matched | G-T | G-dU | G-FdU |
|---|---|---|---|---|
| 1 | 29.0% | 28.5% | 14.2% | 32.9% |
| 2 | 37.4% | 30.1% | 32.7% | 20.2% |
| 3 | 24.8% | 22.6% | 27.8% | 20.4% |
| 4 | 8.8% | 18.9% | 25.3% | 26.4% |
The actual structural rearrangements that occur during these conformational changes are actually quite modest (Figure 6), as is expected in a system as structured as duplex DNA. The different mismatches induce subtle shifts in the base backbone and the base interactions these shifts are especially large at the position just downstream from the mismatch (Figure 6(b)), and especially so in the conformation that is rarest in the matched DNA and dominate in the FdU mismatch (Figure 6(c)).
Figure 6.
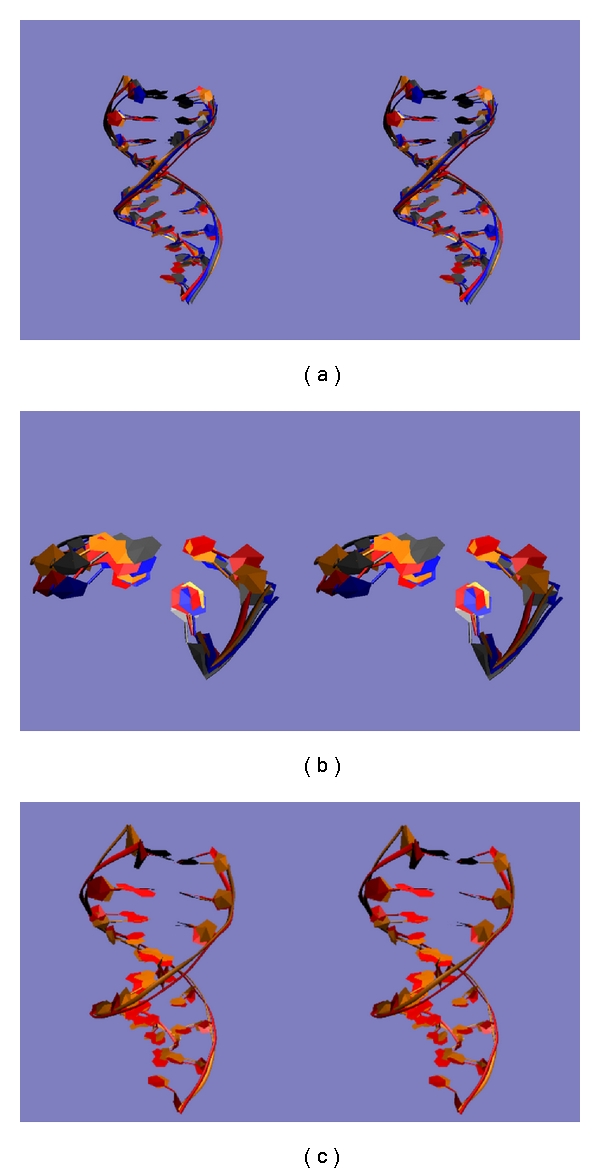
(a) Overall conformations adopted in the four clusters. The first ten base pairs of the centroids of each of the four clusters are shown in VMD's NewRibbon format. Cluster 1 (see Table 2 for numbering) is blue, cluster 2 is red, cluster 3 is gray, and cluster 4 is orange. (b) Conformations near the mismatched site in the four clusters. The two base pairs of the centroids of each of the four clusters downstream from the mismatched site are shown in VMD's New Ribbon format. Cluster 1 (see Table 2 for numbering) is blue, cluster 2 is red, cluster 3 is gray, and cluster 4 is orange. (c) Overall conformations of cluster 2 and cluster 4. The first ten base pairs of the centroids of each of the most common and the rarest clusters for matched DNA are shown in VMD's NewRibbon format. Cluster 2 (see Table 2 for numbering) is red and cluster 4 is orange.
One issue that can also be addressed is that of the classification of the different DNA sequences, which ones are more similar, and which are more different. With three different measures, the simplest approach to this problem is to cluster the four sequences based on each of these three measures (Table 3). By RMSF, two clusters emerge, one consisting of matched and mismatched, and one of FdU and dU; by cluster population, three clusters emerge, one consisting of matched and mismatched, one of FdU, and one with dU. However, when clustering is based on covariance matrices, although three clusters emerge, the mismatched and FdU sequences are in one cluster with the other two as singleton clusters. These results suggest that the mismatched and matched sequences are most similar, although with the mismatched more similar to FdU in one dynamical measure (covariances), and FdU is most similar to either dU or mismatched depending on the measure used.
Table 3.
Clustering of simulations via different measures.
| Measure/Sequence | Matched | G-T | G-dU | G-FdU |
|---|---|---|---|---|
| RMSF | 1 | 1 | 2 | 2 |
| Covariance matrix | 1 | 1 | 2 | 3 |
| Cluster population | 1 | 2 | 3 | 2 |
Cluster number for each simulation for each measure.
4. Discussion
DNA mismatched base pairs occur at high levels in cells both as a consequence of base damage (e.g., cytidine deamination) [23] as well as errors during DNA replication. Cancer cells frequently have defects in DNA repair processes that may result in greater levels of DNA mismatched base pairs being present in the replicated genome. Further, treatment with fluoropyrimidine drugs such as 5FU or FdUMP [10] results in thymineless conditions and imbalanced deoxynucleotide pools resulting in greater preponderance of mismatched base pairs, including G-FdU mismatched base pairs, introduced during replication and subsequent attempted repair processes. The introduction of such mismatched base pairs has been demonstrated to decrease the stability of duplex DNA [23, 24]. The present work demonstrates that mismatched base pairs have a novel effect on the DNA component of a Top1 cleavage complex by destabilizing the putative religation intermediate that consists of a DNA duplex with a nicked phosphodiester backbone.
A G-dU (or G-FdU) base pair in wobble geometry has two hydrogen bonds as does an A-T Watson-Crick base pair and thus is a relatively conservative substitution although at elevated pH an ionized G-FdU base pair may form [24]. G-FdU and other mismatched base pairs not repaired prior to initiation of DNA replication interfere with the religation step of Topoisomerase 1 activity when the mismatched base pair is proximal to the site of Top1 cleavage [8]. The structural and thermodynamic basis for the decreased Top1-mediated DNA religation due to mismatched base pairs remains incompletely understood. While direct interactions between DNA and Top1 protein may play a major role, the inherent stability of the DNA duplex likely also is a contributing factor. The present results demonstrate that G-FdU and other mismatched base pairs destabilize the interaction of the scissile strand with the nonscissile strand of a model Top1 cleavage complex. The extent of this destabilization is greater under high salt conditions similar to that which occurs in eukaryotic cells.
Molecular dynamics simulations demonstrate that the mismatched base pairs increase the flexibility of the duplex. The extent of this increase in flexibility is dependent upon the type of mismatch with G-dU and G-FdU mismatches displaying the greatest increased flexibility. These calculations reveal that the mismatched base pair causes increased atomic fluctuations up to 10 base pairs removed from the site of the mismatch. This increased flexibility makes it less likely that the scissile strand will adopt the correct conformation required for the religation reaction. Thus, the thermodynamic measurements obtained from the UV hyperchromicity data demonstrate that formation of the complex required for religation is disfavored by all DNA mismatched base pairs at the +1 site of the religation complex while the molecular dynamics simulations reveal that the G-dU and G-FdU mismatched pairs are especially potent at increasing conformational flexibility and decreasing the likelihood religation will occur. Overall, our results provide new insights into the structural and dynamic process of Top1-mediated DNA religation and the influence of mismatched base pairs, particularly G-dU and G-FdU mismatched base pairs, at disfavoring this process.
Acknowledgment
This work was supported by Grants nos. NIH CA-102532 (W. H. Gmeiner) and NIH P30 CA-12197.
References
- 1.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chemistry and Biology. 2010;17(5):421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pommier Y. DNA topoisomerase I Inhibitors: chemistry, biology, and interfacial inhibition. Chemical Reviews. 2009;109(7):2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen AY, Liu LF. DNA topoisomerases: essential enzymes and lethal targets. Annual Review of Pharmacology and Toxicology. 1994;34:191–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 4.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434(7033):671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 5.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279(5356):1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 6.Krogh S, Mortensen UH, Westergaard O, Bonven BJ. Eukaryotic topoisomerase I-DNA interaction is stabilized by helix curvature. Nucleic Acids Research. 1991;19(6):1235–1241. doi: 10.1093/nar/19.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Advances in Cancer Research. 2001;80:188–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 8.Liao ZY, Sordet O, Zhang HL, et al. A novel polypyrimidine antitumor agent FdUMP[10] induces thymineless death with topoisomerase I-DNA complexes. Cancer Research. 2005;65(11):4844–4851. doi: 10.1158/0008-5472.CAN-04-1302. [DOI] [PubMed] [Google Scholar]
- 9.Pourquier P, Gioffre C, Kohlhagen G, et al. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clinical Cancer Research. 2002;8(8):2499–2504. [PubMed] [Google Scholar]
- 10.Gmeiner WH, Yu S, Pon RT, Pourquier P, Pommier Y. Structural basis for topoisomerase I inhibition by nucleoside analogs. Nucleosides, Nucleotides and Nucleic Acids. 2003;22(5-8):653–658. doi: 10.1081/NCN-120022604. [DOI] [PubMed] [Google Scholar]
- 11.Hirao I, Kawai G, Yoshizawa S, et al. Most compact hairpin-turn structure exerted by a short DNA fragment, d(GCGMGC) in solution: an extraordinarily stable structure resistant to nucleases and heat. Nucleic Acids Research. 1994;22(4):576–582. doi: 10.1093/nar/22.4.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marky LA, Breslauer KJ. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers. 1987;26(9):1601–1620. doi: 10.1002/bip.360260911. [DOI] [PubMed] [Google Scholar]
- 13.Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 15.MacKerell AD, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56(4):257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 16.Brooks BR, Bruccoleri RD, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. Journal of Computational Chemistry. 1983;4:187–217. [Google Scholar]
- 17.Farwer J, Packer MJ, Hunter CA. PREDICTOR: a web-based tool for the prediction of atomic structure from sequence for double helical DNA with up to 150 base pairs. In Silico Biology. 2007;7(6):595–600. [PubMed] [Google Scholar]
- 18.Ulyanov NB, Bauer WR, James TL. High-resolution NMR structure of an AT-rich DNA sequence. Journal of Biomolecular NMR. 2002;22(3):265–280. doi: 10.1023/a:1014987532546. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh S, Salsbury FR, Jr., Horita DA, Gmeiner WH. Zn2+ selectively stabilizes FdU-substituted DNA through a unique major groove binding motif. Nucleic Acids Research. 2011;39(10):4490–4498. doi: 10.1093/nar/gkr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Cook M, Karplus M. Protein motions at zero-total angular momentum: the importance of long-range correlations. Biophysical Journal. 2000;79(6):2902–2908. doi: 10.1016/S0006-3495(00)76527-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salsbury FR, Crowder MW, Kingsmore SF, Huntley JJA. Molecular dynamic simulations of the metallo-beta-lactamase from Bacteroides fragilis in the presence and absence of a tight-binding inhibitor. Journal of Molecular Modeling. 2009;15(2):133–145. doi: 10.1007/s00894-008-0410-0. [DOI] [PubMed] [Google Scholar]
- 23.Ullman JS, McCarthy BJ. The relationship between mismatched base pairs and the thermal stability of DNA duplexes. II. Effects of deamination of cytosmine. Biochimica et Biophysica Acta. 1973;294(3):416–424. doi: 10.1016/0005-2787(73)90096-8. [DOI] [PubMed] [Google Scholar]
- 24.Sowers LC, Eritja R, Kaplan B, Goodman MF, Fazakerly GV. Equilibrium between a wobble and ionized base pair formed between fluorouracil and guanine in DNA as studied by proton and fluorine NMR. Journal of Biological Chemistry. 1988;263(29):14794–14801. [PubMed] [Google Scholar]