

Abstract
A central question in the development of many branched tubular organs, including the Drosophila trachea, concerns the mechanisms and molecules that control the number and pattern of new branches arising from preexisting vessels. We report on a branching inhibitor, Fusion-6 (Fus-6) produced by specialized tracheal cells to prevent neighboring cells from branching. In Fus-6 mutants, cells that are normally quiescent acquire the branching fate and form an increased number of sprouts emanating from the primary branches. Fus-6 is identified as the headcase (hdc) gene and is expressed in a subset of the cells that extend fusion sprouts to interconnect the tracheal network. hdc expression is regulated by the transcription factor escargot (esg) because it is not expressed in the fusion cells of esg mutants and is ectopically activated in the trachea in response to esg misexpression. We show that the hdc mRNA encodes two overlapping protein products by an unusual suppression of translational termination mechanism. Translational readthrough is necessary for hdc function because rescue of the tracheal mutant phenotype requires the full-length hdc mRNA. In ectopic expression experiments with full-length and truncated hdc constructs, only the full-length cDNA encoding both proteins could inhibit terminal branching. We propose that hdc acts non-autonomously in an inhibitory signaling mechanism to determine the number of cells that will form unicellular sprouts in the trachea.
Keywords: Branching morphogenesis, inhibition, translational termination suppression
The sprouting of new branches from pre-existing vessels is a fundamental process in the development of many epithelial tube networks and results in elaborate patterns of emerging finer tubes in organs like the vertebrate circulatory system, lungs, and the Drosophila trachea. Understanding the mechanisms controlling branch sprouting and patterning is not only a challenging task to developmental biologists but is also of medical interest because of the emerging important role of deregulated angiogenesis in tumorigenesis and the pathogenesis of diseases such as rheumatoid arthritis and retinopathies (Folkman 1995). The cellular events accompanying the sprouting of new capillaries during angiogenesis include chemotactic migration and proliferation of endothelial cells, and formation of a capillary lumens (Risau and Flamme 1995). During the embryonic development of the Drosophila trachea, the sprouting of new capillaries initially generates a stereotyped pattern of fine branches, and later, during larval life, most of these sprouts ramify further to generate complex arrays of finer terminal branches or tracheoles (Manning and Krasnow 1993). Similar to endothelial capillary sprouting in vertebrates, tracheal branch sprouting occurs by the migration of single cells from the primary branches and the formation of unicellular tubules but does not involve cell division (Samakovlis et al. 1996a).
At the molecular level, several factors, including vascular endothelial growth factor (VEGF), an endothelial-specific growth and chemotactic factor, and members of the fibroblast growth factor (FGF) family, are thought to play a central activating role in capillary sprouting in vertebrate angiogenesis (Hanahan and Folkman 1996; Risau 1997). In flies, the number of cells of each branch that undergo terminal sprouting is regulated by the breathless (btl) gene encoding an FGF receptor homolog (Klämbt et al. 1992; Reichman-Fried and Shilo 1995; Lee et al. 1996) and branchless (bnl), a fly member of the FGF growth factor family (Sutherland et al. 1996). The sprouting process is associated with the activation of a set of marker genes (pantip markers) in the leading cells of each primary branch in response to FGF signaling (Samakovlis et al. 1996a; Sutherland et al. 1996). The expression pattern of these markers is dynamic and it gradually becomes restricted to the cells that form unicellular branches. Depending on which primary branch these unicellular sprouts originate from, they either retain the expression of pantip markers and further differentiate into terminal sprouts ramifying in response to respiratory demand or migrate and fuse to similar tubular extensions from adjacent tracheal metameres to interconnect the network. The processes of terminal branching and branch fusion are accompanied by the expression of separate classes of marker genes and they are controlled by different genes (Samakovlis et al. 1996a). Mutants in the pruned gene encoding a Drosophila serum response factor (DSRF) homolog abolish terminal branching (Guillemin et al. 1996), whereas the Fusion-1 (Fus-1) gene encoding the escargot (esg) transcription factor specifically affects branch fusion (Samakovlis et al. 1996b; Tanaka-Matakatsu et al. 1996). esg is an activator of the fusion program as well as a repressor of terminal branching that can drive ectopic tracheal fusion events and repress terminal branching when misexpressed in all tracheal cells (Samakovlis et al. 1996b). Both terminal and fusion genes are under the control of the pantip gene pointed (pnt) encoding an ets domain transcription factor. pnt is required for the transcriptional activation of pruned in the terminal cells and the repression of esg in the cells of the pantip group that do not acquire the fusion cell fate (Samakovlis et al. 1996a).
Endothelial sprouting is also controlled by secreted inhibitors that suppress the formation of new capillaries during tumor angiogenesis and it has been proposed that a balance of inductive and inhibitory signals determines whether endothelial cells of the blood vessels will remain quiescent or will grow new branches toward their targets (Hanahan and Folkman 1996). Little is known, however, of the cellular mechanisms and molecular components of this balance and how it normally regulates the number and pattern of new branches during organogenesis. In this study we have identified mutations in a tracheal gene, Fus-6, that increase the number of unicellular sprouts emanating from the dorsal primary branches. Fus-6 is identified as the headcase (hdc) gene which has been found previously to function in the development of the adult head structure (Weaver and White 1995). We show that hdc is expressed in a subset of the cells that extend fusion sprouts to interconnect the tracheal network and its expression is activated by the esg transcription factor. We provide evidence that hdc functions nonautonomously to suppress neighboring tracheal cells from adopting the branching fate in response to bnl and, thus, determines the number of unicellular sprouts emanating from the dorsal primary branches. We finally show that the hdc mRNA encodes two proteins by an unusual translational readthrough mechanism and that the termination suppression event is necessary for hdc gene function.
Results
lacZ expression and increased branching phenotypes in the Fus-6 mutants
One hundred of the ∼2000 cells of the tracheal epithelium express a set of fusion cell-specific marker genes and undergo a complex program of sprouting unicellular branches that fuse to each other and connect the independent metameric units of the trachea. In the Fus-6 marker strain, lacZ is expressed in a subset of the fusion cells of each tracheal metamere from stage 14 until the end of embryogenesis (Fig. 1; Samakovlis et al. 1996b). In Fus-6 embryos, the marker is selectively expressed in the fusion cells of the dorsal anastomoses in metameres 2–9 (Fig. 1A), and in the fusion cells of the lateral trunk (Fig. 1D) and ventral anastomoses (not shown). In these branches the fusion cells are in contact with terminal cells that sprout off to generate tracheoles. The fusion cells of the dorsal trunk and dorsal branches in metameres 1 and 10 that do not contact terminal cells do not express lacZ (Fig. 1F). Additional tissues expressing the marker during embryogenesis include clusters of cells in the CNS, the primordia of the imaginal tissues, and the imaginal tracheal cells of the spiracular branch. The original P-element insert was homozygous lethal and caused increased branching at the tips of the dorsal branches (Fig. 2). We generated 100 additional strains by P-element excision mutagenesis and screened them for lethality and tracheal phenotypes with the lumenal tracheal marker 2A12. We identified five new early pupal lethal alleles in the same complementation group as the original insert and several revertants of the lethal and tracheal phenotypes, suggesting that these phenotypes are associated with the transposon insertion. The Fus-650 and Fus-620 alleles showed the strongest tracheal phenotypes, and we selected Fus-650 for further analysis. All of the mutant embryos had additional fine branches emanating from the dorsal branches. These new branches were sprouting at the tips of the dorsal branches in a position where the fusion and terminal sprouts are found in the wild-type flies (Fig. 2). The effect of the mutation was sporadic in each embryo, on average 21% of the tracheal metameres (n = 96 metameres) had additional sprouts compared with 1% seen in the wild type (n = 96). Extra sprouting was also detected in the lateral trunk of the mutants, but there the frequency of increased branching was much lower, and we concentrated our phenotypic analysis on the dorsal branch. This incomplete penetrance of the phenotype is not the result of the hypomorphic nature of the alleles because neither Fus-6 RNA nor protein can be detected in embryos homozygous for the mutations (see below).
Figure 1.
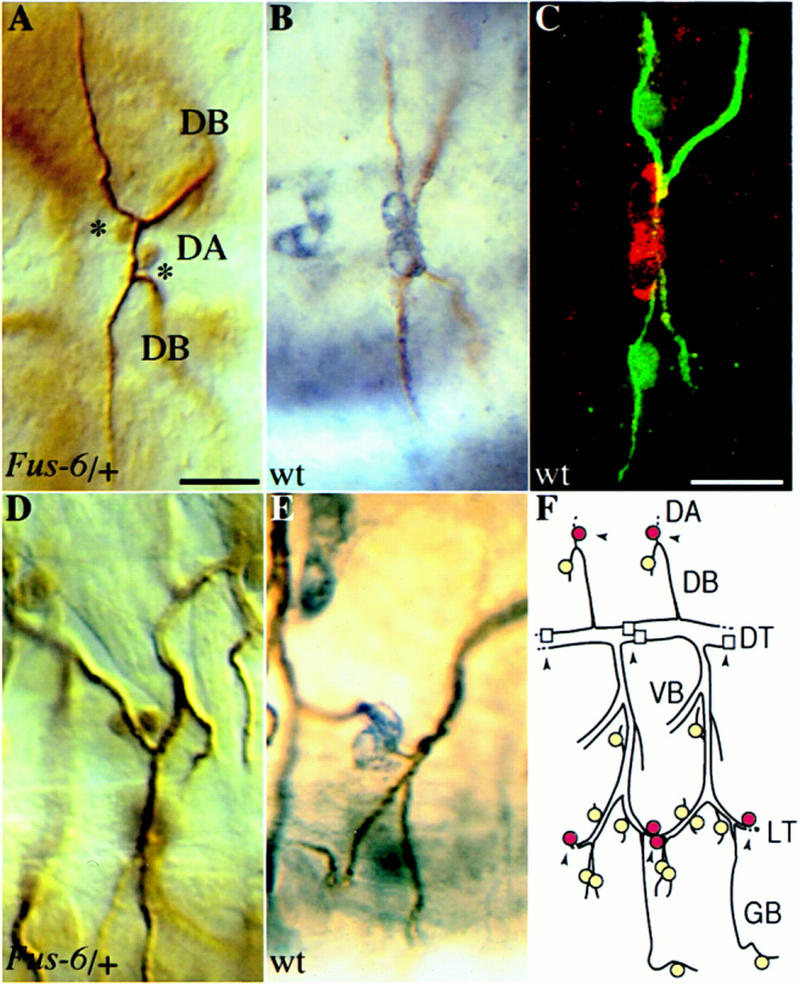
Expression of Fus-6 (hdc) in a subset of the tracheal fusion cells. The tracheal lacZ expression in the Fus-6 enhancer trap strain is identical to the expression of hdc. (A,D) Fus-6 embryos have been stained with antibodies against β-galactosidase and a tracheal lumenal antigen, (B,E) wild-type embryos were stained with antibodies against Hdc in purple and the tracheal lumen in brown. (A) Dorsal view (anterior left) focused at the dorsal anastomosis (DA). The nuclear lacZ marker is selectively expressed in the two fusion cells (indicated by asterisks) of the dorsal branches (DB) connecting two contralateral metameres. (B) hdc is expressed in the fusion cells forming the DA. lacZ (D) and hdc (E) expression in the fusion cells connecting the lateral trunk (lateral view, anterior left). (C) Hdc is cytoplasmic in the fusion cells of the dorsal anastomosis in contrast to the nuclear localization of DSRF in the adjacent terminal cells (dorsal view, anterior left). Embryos have been stained with antibodies against Hdc (red), DSRF (green), and tracheal lumen (green). (F) Schematic diagram of two central abdominal tracheal metameres (lateral view, anterior left) showing the cells expressing hdc as darker (red) circles and the neighboring cells expressing terminal markers as lighter (yellow) circles. Fusion points at the dorsal anastomosis (DA), lateral trunk (LT), and dorsal trunk (DT) are indicated by arrowheads. hdc is not expressed in the fusion cells of the DT (squares) that do not contact terminal cells. Bar in A and D, 10 μm; in B, C, and E, 10 μm.
Figure 2.
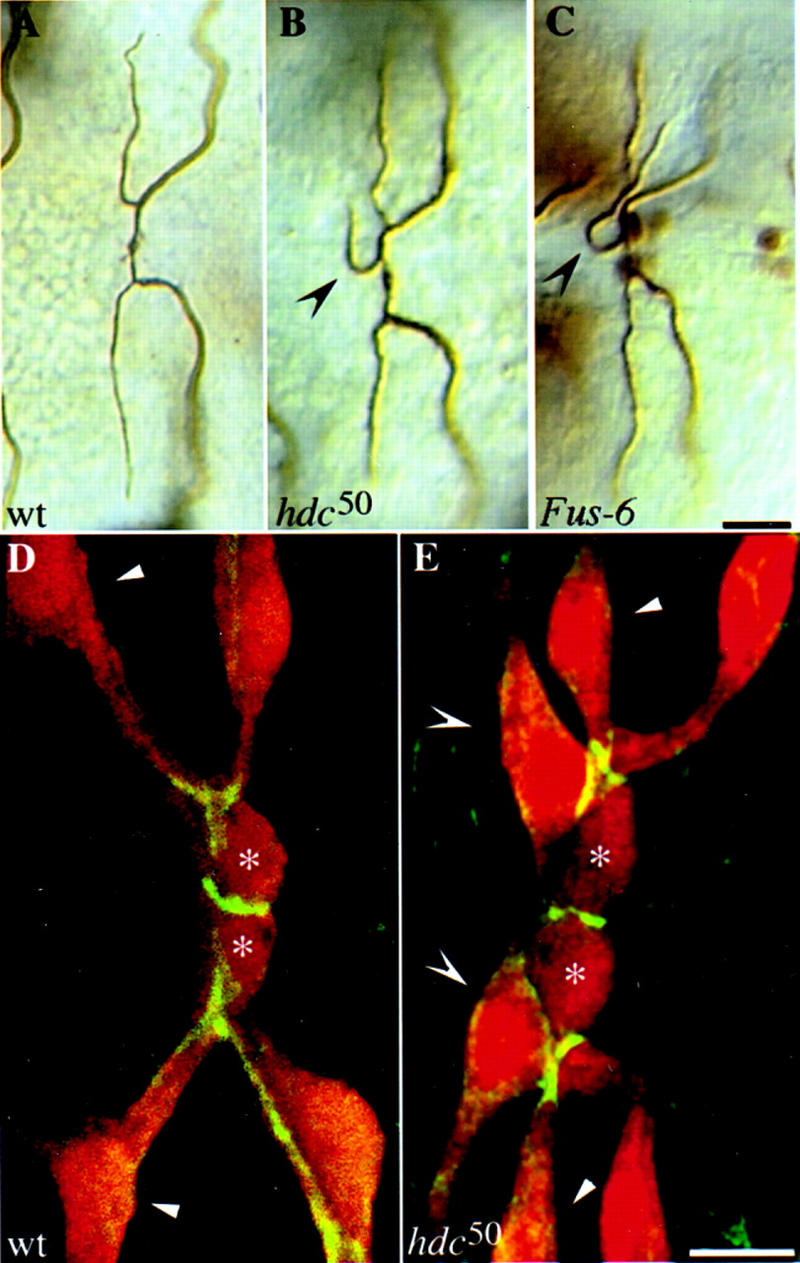
Tracheal phenotypes in Fus-6 (hdc) mutants. Additional sprouts emanate from the DBs in hdc and Fus-6 mutants. Dorsal views, anterior to the left, of wild-type and mutant embryos at stage 16. Wild-type (A), hdc50 homozygous (B), and Fus-6 homozygous (C) embryos stained with antibodies against the lumenal antigen and β-galactosidase. Extra branches are marked by concave arrowheads. Bar, 10 μm. Extra cells sprout off the DBs in hdc50 embryos. (D,E) Confocal images of the DB in wild-type (1-eve-1/+) and hdc50 (1-eve-1, hdc50/hdc50) embryos showing tracheal cells (anti-β-galactosidase in red) and cell junctions (anti-Coracle antiserum in green). The fusion cells are marked by asterisks, the terminal sprouts in the wild type by arrowheads, and the extra sprouting cells in the mutant by concave arrowheads. Bar, 5 μm.
More cells adopt the sprouting fate in Fus-6 mutants
The additional branches in Fus-650 mutants could derive from increased ramifications of the cells that normally branch or by additional tracheal cells acquiring the branching fate. Staining of mutant embryos for a lacZ marker expressed in all tracheal cells and an antibody against the cell junction protein, Coracle, showed that each additional sprout derived from a tracheal cell located at the tip of the dorsal branch contacting the cells that form the fusion and terminal cell sprouts in the wild-type embryos (Fig. 2D,E).
What is the identity of the extra sprouting cells and when do they arise? We addressed the first question by using a set of tracheal cell-type specific markers. At embryonic stage 12, 3–4 cells at the tip of the dorsal branch express the pantip group of markers. In the dorsal and lateral trunk branches of each tracheal metameric unit, in which hdc is expressed, the expression of these marker genes becomes restricted in two steps. In the first step at stage 14, strong pantip marker expression is maintained in the two cells of the initial group that become selected to sprout, and in the second step, at stage 15, it ultimately becomes refined to one of the sprouting cells that will generate terminal branches (Fig. 3I) and (Samakovlis et al. 1996a). The first step in the restriction of the domain of pantip marker expression is preceded by the activation of separate classes of marker genes in a subset of the cells expressing pantip markers at stage 13. These cells either express terminal markers and undergo terminal branching or fusion markers and connect to similar fusion sprouts deriving from neighboring metameres (Fig. 3I; Samakovlis et al. 1996a). The remaining cells of the group, not expressing fusion or terminal markers, stop migrating, lose the expression of pantip markers, and acquire their position at the stalk of the dorsal branch. We found that the extra branching cells in the Fus-6 mutant expressed the two pantip and the two terminal marker genes tested (Fig. 3B,D; data not shown). In addition, during larval life these cells went on to form extensive networks of fine tracheoles (Fig. 3H). Thus, the extra branching cells arise from the group of cells expressing pantip markers and display the molecular and morphological features of terminal cells. These additional branching cells can first be detected in the mutant embryos at stage 14 with the appearance of an extra cell expressing pantip and terminal markers (Fig. 3I). The initiation of expression of the earliest terminal marker both in wild-type and mutant embryos occurs in a single cell of the dorsal branch at stage 13, arguing that the initial selection of a terminal cell from the pantip group is not affected in the mutants.
Figure 3.
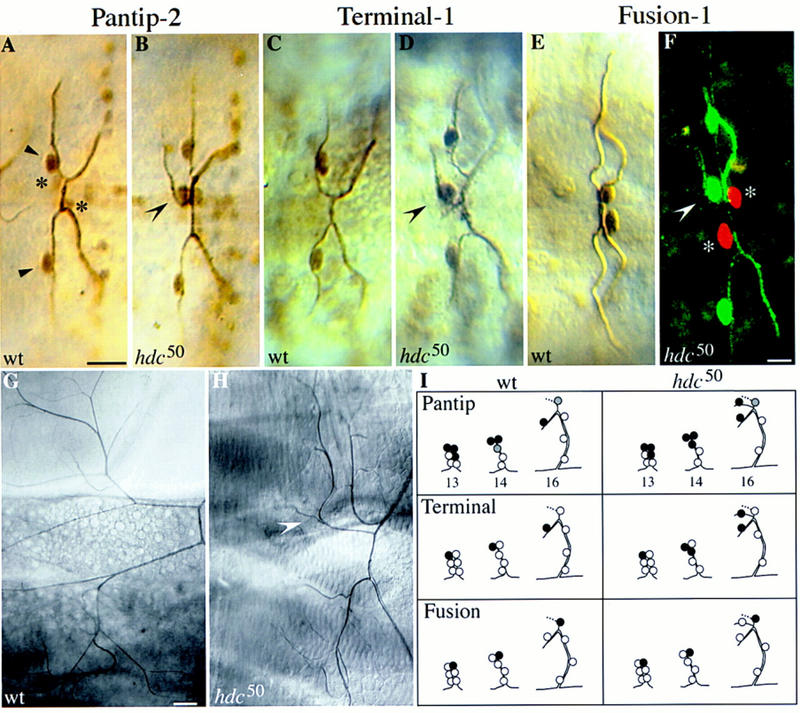
Expression of branching markers in hdc mutants. The extra sprouting cells express pantip and terminal but not fusion cell markers. Dorsal views, anterior to the left, of wild-type and hdc mutant embryos at stage 16. For the pantip and fusion markers, wild-type and hdc50 mutant embryos carrying one copy of the marker gene were double stained for β-galactosidase and the tracheal lumen. For the terminal marker panels, wild-type and hdc mutant embryos were triple stained with antibodies against the lumen, β-galactosidase, and DSRF. (A) A pair of contralateral DBs in a wild-type embryo, each expressing the pantip-2 marker strongly in the terminal (arrowheads) and weaker in the fusion (asterisks) cells. (B) Similar view of a hdc50 mutant embryo showing that the extra sprouting cell strongly expresses the pantip marker (concave arrowhead). (C,D) DBs of wild-type and hdc50 embryos stained for the DSRF terminal marker. The extra sprouting cell expressing DSRF is indicated by a concave arrowhead. (E,F) Expression of the Fus-1 marker in the fusion cells of wild-type DBs in E and hdc50 mutant in F. In the confocal micrograph in F, hdc50 embryos were stained both for the Fus-1 marker in red, and the terminal DSRF marker and tracheal lumen in green to show that the extra sprouting cell (concave arrowhead) expressed terminal but not fusion markers. Fusion cells are indicated by asterisks. Bar in A–E, 10 μm; in F, 5 μm. (G,H) Extra sprouts in hdc mutants ramify in to tracheoles during larval life. (G) Pair of DBs in a wild-type 3rd instar larva viewed by Nomarski optics, showing the extensive fine tracheoles emanating from each terminal branching cell. (H) Same view of a hdc50 mutant larva with an additional terminal branch extending tracheoles (concave arrowhead). Bar, 50 μm. (I) Schematic summary of pantip, terminal, and fusion marker expression in the wild-type and hdc mutants. The extra sprouting cells express pantip and terminal markers at stage 14.
The additional terminal sprouts in the mutant could arise by a change in the cell fate specification program of the fusion cell. This was not the case, because the expression of the three fusion markers tested was unchanged in the Fus-6 mutant (Fig. 3F; data not shown). Furthermore, the formation of the dorsal anastomosis was not affected in the mutants. The two fusion cells were able to migrate toward each other and were connected by intercellular junctions visualized with an antibody against the Coracle protein to form the dorsal anastomosis (Fig. 2E). Asymmetric cell division of the fusion cell to generate a terminal and a fusion sprout in the mutant was also excluded as a possible mechanism for the generation of new terminal branches because no cell divisions could be detected in the trachea of mutant embryos by BrdU labeling (see Materials and Methods). Suppression of cell death is also an unlikely mechanism to explain the presence of the extra branching cell because apoptosis does not occur during wild-type tracheal development (Samakovlis et al. 1996a).
What then is the cellular mechanism behind the increased branching in the mutant? Cell counts in the dorsal branches of wild-type and mutant embryos double stained with a general and a lumenal tracheal marker showed that, although the number of fine branches emanating from the dorsal branch increases by ∼20% in the mutants, the average number of cells in each branch remains the same (5.8 cells in the wild type, n = 119 metameres, compared with 6 cells in the mutant, n = 128 metameres). In addition, in the mutant branches, the number of cells in the stalk of the dorsal primary branch was consistently decreased by one cell in all of the metameres counted compared with the contralateral metameres that did not exhibit the extra sprouting phenotype (n = 13 metameres). Thus, the above results suggest that the extra branching cells arise by a cell fate transformation of a nonbranching cell adjacent to the terminal and fusion cells of the dorsal branch into a sprouting cell, and that the Fus-6 gene product acts nonautonomously to prevent neighboring tracheal cells from branching.
Fus-6 is allelic to hdc and is localized in the cytoplasm of the fusion cells
Genomic DNA flanking the site of insertion of the B9-3-52 P-element was recovered by plasmid rescue in Escherichia coli and used to initiate the molecular characterization of the Fus-6 locus. The recovered 1.4-kb genomic EcoRI fragment detected a hybridizing band of ∼4.5-kb on Northern blots and was also used as probe in RNA in situ hybridization experiments (data not shown). Hybridizing tissues included the central nervous system, the imaginal disc primordia, and the tracheal spiracular cells, in a pattern strongly reminiscent of the expression of the lacZ marker in the original enhancer trap strain. We used this fragment as a probe to screen two embryonic cDNA libraries and identified several hybridizing clones. Partial sequencing of the longest cDNA clone and database comparison of the sequence revealed that it was identical to the cDNA corresponding to the previously identified gene hdc (Weaver and White 1995) and homologous to two different Caenorhabditis elegans genes revealed by the worm genome project and a human EST sequence (Fig. 5, below). We established that the Fus-6 gene is allelic to hdc by the following criteria. First, using a monoclonal antibody against the hdc product, we showed that hdc is expressed in the tracheal fusion cells from the beginning of stage 14 until the end of embryogenesis, in a pattern identical to the Fus-6 lacZ marker (Fig. 1). The Hdc protein was detected by confocal microscopy in the cytoplasm of the fusion cells of the dorsal anastomoses in metameres 2–9, the lateral trunk and ventral anastomoses (Fig. 1). Second, alignment of the sequences from the recovered genomic fragment from the Fus-6 strain and the hdc cDNA revealed that in the mutant, the transposon is inserted in the hdc gene in a position corresponding to 220 bp upstream of the first nucleotide of the cDNA clone. Third, the lethality and tracheal phenotypes of our insertion and excision mutants are allelic to the phenotypes of the hdc43 loss-of-function allele (Weaver and White 1995). Finally, we have shown that Fus-650 is a strong loss-of-function allele of hdc, because we could not detect any hdc RNA or protein (Fig. 4; data not shown) in mutant embryos, and we could rescue the tracheal phenotype by hdc expression in the trachea (see below).
Figure 5.

(A) Deduced amino acid sequence of the hdc longer product. The nucleotide sequence derives from the cDNA sequence in Weaver and White (1995) and our longest, partially sequenced, cDNA clone. Start and stop codons are underlined and X is used as a symbol for the unknown amino acid incorporated at the internal UAA stop codon. The different regions homologous to the two C. elegans ORFs are highlighted. (B) BLASTP amino acid sequence comparisons of the hdc products to the two C. elegans ORFs and the human EST sequence. The C. elegans ORF 1763999 is homologous (34% identity, 60% similarity) to the region between amino acids 453–587 in the Hdc sequence shared by both products (highlighted in gray in A). The C. elegans ORF 2315488 and the ORF deriving from the human EST are homologous to a region between amino acids 85 and 172 (38% identity, 63% homology) in the Hdc sequence shared by both products (highlighted in a gray box in A). The same C. elegans ORF is also homologous (36% identity, 56% homology) to a region between amino acids 1042 and 1067 unique to the carboxyl terminus of the longer product (highlighted in a gray box in A).
Figure 4.
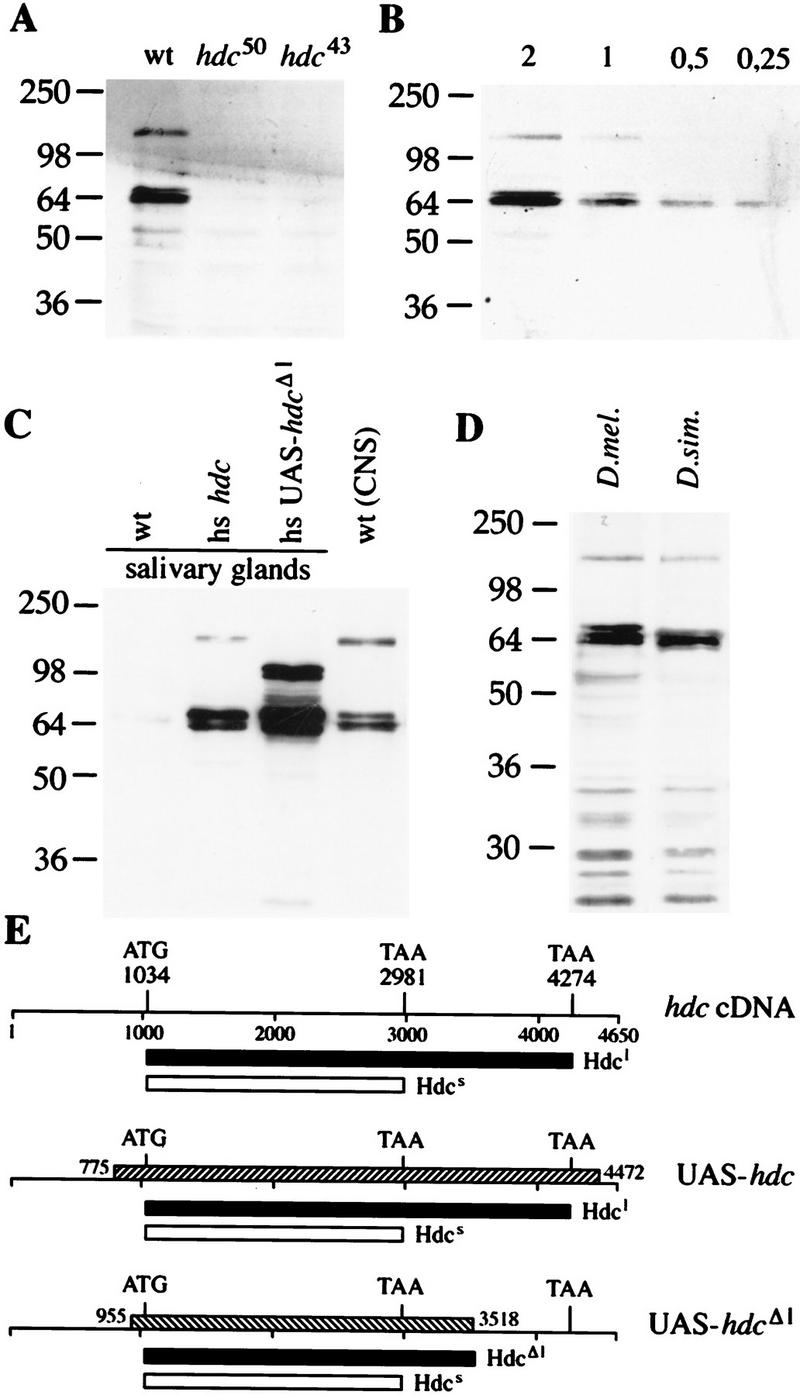
Translational readthrough generates two hdc products. Western blots probed with the antibody against the hdc products (A). The antibody recognizes two proteins in CNS and imaginal disk extracts from wild-type larvae. Both bands are absent in extracts from larvae homozygous for hdc50 and hdc43. (B) Quantitation of the amount of the two products relative to each other. Serial dilutions of a CNS and imaginal disk extract were loaded. The shorter product is ∼fourfold more abundant than the longer one. (C) Full-length cDNA is necessary to generate a product with the same size as the longer endogenous protein. Extracts of salivary glands from transgenic larvae carrying the full-length or a truncated version of the hdc cDNA under heat shock control were compared with CNS and imaginal disk extracts from wild type. (D) Two Hdc proteins are detected in extracts from D. simulans larval CNS and disks. (E) Drawings of the hdc cDNA clone and the fragments used to generate the transgenic strains shown as hatched bars. Start and stop codons are indicated and the predicted products are shown as black or white bars under each construct. Hdcl designates the longer product, and Hdcs the shorter one.
hdc encodes two protein products by suppression of translational termination
An unusual feature of the hdc cDNA sequence is its potential to encode more than one polypeptide. There is a 3241 nucleotide long ORF starting with an AUG flanked by a conserved Drosophila translational initiation site (Cavener and Ray 1991). The sequence of this putative ORF is interrupted by an internal UAA stop codon at position 2981 and continues in frame until it is interrupted again by another UAA stop codon followed by a polyadenylation signal (Weaver and White 1995; Fig. 5). This intriguing structure suggests that the hdc mRNA has the potential to encode at least two proteins: One with a predicted molecular mass of 70 kD generated by termination at the first UAA codon, and one with a molecular mass of 125 kD generated by suppression of translational termination. To investigate this possibility, we first sequenced the region containing the first UAA codon from 10 independent genomic clones deriving from genomic DNA PCR amplification, and then from 20 independent RT–PCR derived embryonic cDNA clones. The UAA codon was present in all clones excluding the possibility that the original clone was a cloning artifact. These results also argue against an RNA editing mechanism that would modify the UAA codon in the mature mRNA population.
Then, we used a monoclonal antibody generated against the amino-terminal part that is common to both putative translational products (Weaver and White 1995) and attempted to detect them in extracts of wild-type and mutant larvae by Western blots. The antibody recognized one strong band of ∼70 kD and a weaker band of ∼130 kD. Both signals were absent in the extracts made from hdc50 and hdc43 mutant larvae. The presence of more than one band at the 70-kD range indicates either posttranslational modification of the shorter product or possibly inefficient termination at the UAA stop codon. Quantitation of the signal from lanes containing serial dilutions of the wild-type extract showed that the shorter protein is approximately four times more abundant than the longer product (Fig. 4B).
Thus, the hdc gene encodes two different polypeptides, and their molecular weights suggest that they derive from suppression of translational termination. To establish that the longer protein is not the product of an as yet unidentified splice form or derives from extensive post-translational modifications of the shorter protein, we ran Western blots of protein extracts from transgenic fly strains expressing either the full length hdc cDNA or a deletion derivative that lacks most of the carboxy-terminal part of the longer isoform, both under the control of a heat inducible promoter (Fig. 4C). In these experiments we used protein extracts from dissected CNS and larval imaginal discs from wild-type flies and dissected salivary glands, a tissue that normally expresses hdc at very low levels, from the transgenic strains. We could detect the two isoforms of the expected molecular mass only in the extracts prepared from CNS of wild-type larvae and the strain expressing the full-length cDNA in salivary glands after heat shock. The strain expressing the deletion construct produced the shorter isoform and a protein of higher molecular mass corresponding to the expected readthrough product from the construct (Fig. 4C). Thus, our full-length cDNA clone containing the internal UAA codon accounts for both products observed in wild-type flies and the longer product is not the product of posttranslational modifications of the short isoform. Our results strongly argue for a translational termination suppression mechanism involved in generating the two protein products from the hdc mRNA and this mechanism appears to be conserved in flies because we could detect two proteins on Western blots of extracts prepared from the closely related Drosophila simulans (Fig. 4D). Interestingly, one of the C. elegans homologs identified by BLAST database searches is homologous to the amino acids 440–574 shared by the two products, whereas the other is homologous to an 84-amino acid amino-terminal region common to both products and a region of 24 amino acids at the carboxy-terminal end, unique to the longer product (Fig. 5). The presence of two C. elegans genes with homologies to distinct domains of the two hdc products suggested that both products may be functional. Attempts to detect the proteins in more distant Drosophila species and C. elegans failed, probably because the epitope recognized by our monoclonal antibody is not conserved.
The longer protein is necessary for hdc function
To assess the significance of the unusual translational termination suppression event in the hdc mRNA, we tested whether the shorter, more abundant protein was sufficient for rescue of the tracheal mutant phenotype or whether the longer product was necessary for the full function of the gene. Using the UAS–GAL4 system (Brand and Perrimon 1993), we expressed the full-length cDNA or the truncated version missing the carboxyl terminus of the longer form in the tracheal fusion cells of homozygous mutant embryos. The two constructs were expressed under the control of the same fusion cell-specific driver strain and the results of these experiments are summarized in Table 1. Rescue of the extra branching phenotype required the longer construct encoding both proteins, the shorter form alone is unable to fully rescue the mutant phenotype. Western blots of protein extracts and in situ antibody stainings to control for expression levels of the transgenes revealed that both constructs produced the expected products and that the truncated construct was even expressed at higher levels than the full-length construct (Fig. 4C; data not shown). These results show that the translational termination suppression event generating the longer protein is not an unusually frequent termination error but is necessary to produce the functional gene product.
Table 1.
Rescue of hdc50 null mutant tracheal phenotype with the full-length or the truncated hdc construct
| Genotype
|
Affected dorsal branchesa (%)
|
|---|---|
| OregonR (wild type) | 1 (n = 96) |
| hdc50/hdc50 | 21 (n = 192) |
| p127–Gal4/+;UAS–hdc hdc50/hdc50 | 0 (n = 80) |
| UAS–hdcΔ1/p127–Gal4; hdc50/hdc50 | 12.5 (n = 48) |
Extra branches and additional cells expressing DSRF.
Misexpression of hdc can suppress terminal branching
The analysis of the loss-of-function mutants and the expression of the hdc proteins exclusively in a subset of fusion cells in wild-type embryos suggested that hdc functions nonautonomously to inhibit neighboring cells from adopting the terminal cell fate and undergo further branching. Consistently, generalized misexpression of the full-length hdc cDNA in all tracheal cells by use of the UAS–GAL4 system suppresses terminal branching and the expression of the terminal cell marker DSRF in ∼8.5% of the dorsal branches (Fig. 6B,E, n = 816). Suppression of terminal branching and DSRF expression could also be detected at similar levels in the ganglionic branches in which hdc is not normally expressed (data not shown). Generalized misexpression of the truncated form under the control of the same driver strains was found to have a marginal effect on terminal branching and the expression of terminal markers, arguing again for the requirement of the longer ORF for full hdc function (Fig. 6E).
Figure 6.
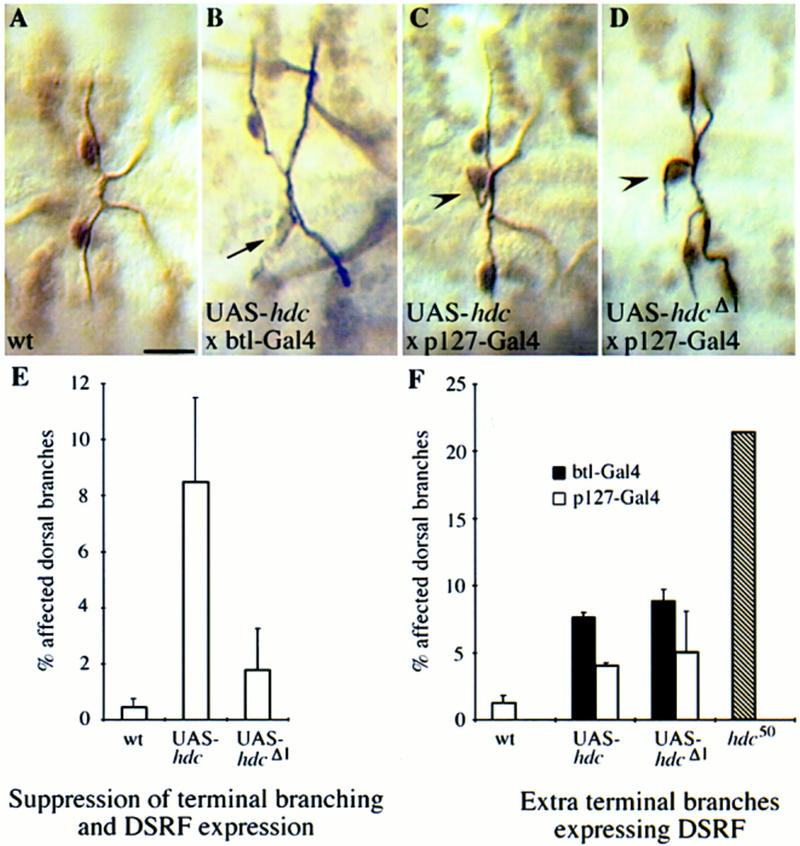
Misexpression of hdc in all tracheal cells can suppress terminal branching. The GAL4 expression system was used to express wild-type and mutant Hdc proteins throughout the developing tracheal system (B,E,F) and selectively in the fusion cells (C,D,F) of hdc+ embryos. Expressing UAS–hdc in all tracheal cells with the pantracheal btl–GAL4 driver results in suppression of terminal branching and expression of the terminal marker DSRF (arrow in B). (E) Comparison of the terminal branching suppression phenotype of UAS–hdc and UAS–hdcΔl. The full-length construct is necessary for suppression. (C) Expression of UAS–hdc with the p127-GAL4 fusion cell-specific driver or the pantracheal btl–GAL4 driver causes extra branching phenotypes similar to the hdc50 loss-of-function mutant (arrowheads). Expression of the UAS–hdcΔ1 construct with the same drivers results in a similar phenotype (D,F). Bar in A–D, 10 μm.
An additional, unexpected, phenotype in the same embryos expressing the full length hdc construct in all tracheal cells was the presence of extra terminal sprouts in ∼5% of the dorsal branches (n = 816) which resembled the tracheal defects observed in the hdc null mutants (Fig. 6F). This extra branching phenotype was also detected when we overexpressed hdc under the control of a fusion cell-specific driver strain (Fig. 6C,F). Because the same UAS construct expressed under the control of the same fusion cell-specific driver strain is able to rescue the tracheal phenotype in hdc null mutants, this result argues that an imbalance in the concentration of the hdc products in the fusion cell can cause the same phenotypes as the loss-of-function mutations. We explored this phenotype to ask whether hdc function is sensitive to the dosage of the longer or the shorter protein product. We find that overexpression of the truncated form in wild-type flies alone also caused increased branching (Fig. 6D,F).
hdc expression in the trachea is regulated by esg
Branch fusion in the Drosophila trachea is a complex process involving two specialized cells at the tip of each fusing branch that undergo a series of morphological changes to generate a bicellular anastomosis and connect the two tracheal branches. We have proposed that esg acts as a regulator in a genetic hierarchy that controls fusion cell identity and function (Samakovlis et al. 1996b). In esg mutants, the fusion cells of the dorsal branches fail to undergo the fusion process and express later fusion markers, instead, they express terminal markers and ramify into tracheoles during larval life. Ectopic expression of esg in all tracheal cells is sufficient to induce ectopic branch fusions and suppress terminal branching and expression of terminal genes. Having identified Fus-6 as hdc, we asked whether its expression in the trachea is regulated by esg. First, we established that hdc expression was absent in the fusion cells of esg mutants (Fig. 7B). Then, we misexpressed esg in all tracheal cells using the UAS–GAL4 system and found that this was sufficient to induce hdc expression in one to two additional tracheal cells at the tips of the dorsal and lateral trunk branches (Fig. 7C; data not shown). Thus, esg is not only necessary for hdc expression in the fusion cells, but it is also sufficient to induce hdc expression in the cells of the pantip group of the dorsal and lateral branches. Because hdc acts nonautonomously as a branching inhibitor the above results predict that in esg mutants not only the fusion cell, but also one of the stalk cells may acquire the terminal cell fate. We analyzed the extra terminal branching phenotype of esg strong loss-of-function mutants and we find a third cell extending a unicellular branch and expressing the terminal marker DSRF in ∼16% of the 176 dorsal branches analyzed (Fig. 7D). These results show that esg can suppress terminal cell fate in the fusion cells by repressing terminal genes like pruned, and in neighboring cells by the activation of hdc and perhaps other fusion genes that act non-cell autonomously as branching inhibitors.
Figure 7.
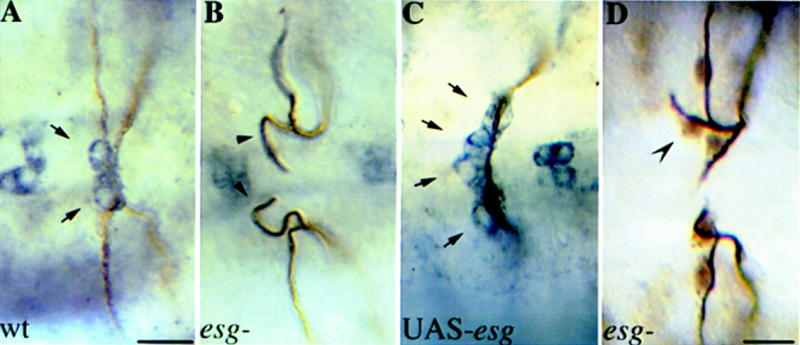
hdc expression is regulated by esg. (A–C) Embryos double stained with antibodies against the tracheal lumen in brown and Hdc in blue. (A) Dorsal view focusing on hdc expression in the dorsal branches (DBs) of wild type, (B) esg mutant, and (C) C38–GAL4/UAS-esg embryos. (Arrows in A) hdc expression in the two fusion cells of wild-type embryos. hdc expression was not detectable in esg mutants (Arrowheads in B), whereas in UAS-esg embryos, hdc is expressed in four cells at the tip of the DBs (Arrows in C). Bar in A–C, 10 μm. (D) Dorsal view of an esg mutant embryo stained with antibodies against tracheal lumen and DSRF. A third branching cell expressing DSRF is indicated by a concave arrowhead. Bar, 10 μm.
Discussion
hdc is a novel branching inhibitor
In hdc mutants, additional cells acquire the branching fate and sprout off to form terminal branches. hdc is expressed in the fusion cells of the growing dorsal primary branches and is required for turning off the expression of pantip and terminal markers in neighboring cells. Thus, it acts nonautonomously to inhibit the cells of the initial group expressing pantip markers that have not been selected to express terminal genes at stage 13 from continuing migrating and acquiring the terminal cell fate. In the wild type, during the early stages of cell migration and primary branching, a group of tracheal cells located at the growing tip of each bud express a set of pantip marker genes (Samakovlis et al. 1996a). The number of cells that will initially express the pantip markers is regulated by the activation of Breathless, an FGF receptor homolog in flies, and Branchless, a secreted FGF homolog produced by tissues surrounding the tracheal epithelium (Lee et al. 1996; Sutherland et al. 1996). The expression of pantip genes is dynamically regulated as it later becomes restricted to the cells that will continue migrating and express either fusion or terminal markers (Samakovlis et al. 1996a) and ultimately to the cells expressing terminal markers. Thus, the pantip markers define a group of cells at the tip of each branch among which the terminal and fusion cells will be selected. The remaining cells of the pantip group will cease migrating, intercalate, and acquire their position at the stalk of the primary branch. We propose that hdc participates in an inhibitory signaling mechanism to restrict the domain of bnl responsive cells (Fig. 8). This inhibitory signaling does not affect the cell identity of the terminal and fusion cells because their fate is already established at stage 13, before hdc is expressed in the fusion cell, but acts to prevent the rest of the cells in the pantip group from migrating so that they retain their position surrounding the stalk of the primary branch. The identification of hdc as a branching inhibitor produced by a subset of the cells that undergo sprouting to inhibit their neighboring cells from branching uncovers a new mechanism involved in patterning the sprouting of new branches. The decision whether endothelial cells will sprout during angiogenesis in vertebrates is also thought to be controlled by a balance of activating and inhibiting signals (Hanahan and Folkman 1996), and it will be interesting to see whether any of the known inhibitors of angiogenesis or the human homolog of hdc are produced from the sprouting cells in response to branching signals and act in a similar mechanism to determine the pattern of endothelial sprouting in vertebrates.
Figure 8.
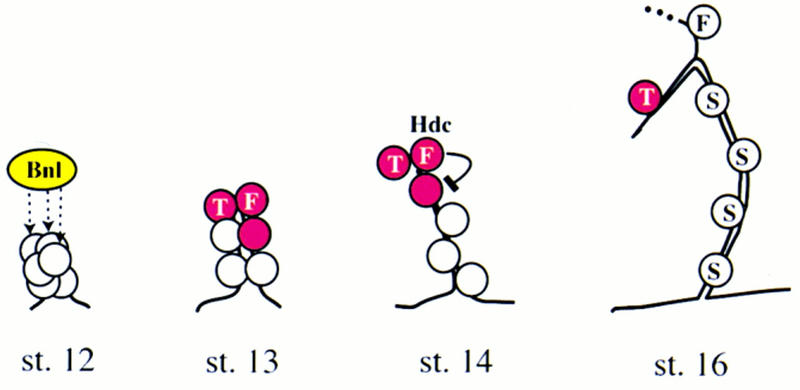
Model of the inhibitory action of hdc on sprouting. Secreted Bnl protein (dashed arrows) activates the expression of pantip marker genes (red) in the tip cells of the dorsal primary branch closest to the source of the signal. Single cells of the pantip group are selected to express terminal (T) or fusion (F) cell-specific markers. The fusion cell expresses hdc to inhibit the Bnl mediated induction of terminal branching in the remaining cell of the pantip group, which later becomes a stalk cell (S).
What is the inhibitory signal?
The expression of hdc exclusively in the fusion cells and its function to prevent neighboring cells from sprouting is consistent with the hypothesis that hdc might be the inhibitory signal itself. The localization of the hdc protein products in the cytoplasm, however, suggests that hdc may play a role in the fusion cell to facilitate signaling through another molecule secreted to the cell surface. Then, where does the signal come from? One possibility is that it derives exclusively from the hdc expressing fusion cell to inhibit the rest of the cells expressing pantip markers to further differentiate into terminal cells. In this scenario, hdc would be acting to enhance this fusion cell-specific signal. This hypothesis assumes different signals for restricting the number of sprouting cells in the tracheal branches in which hdc is not expressed. Another possibility is that a common inhibitory signal is operating in the initial group of cells expressing pantip markers in all primary branches to determine which cells in the group will retain the expression of pantip genes and continue sprouting. All tracheal primary branches express the pantip markers in a group of cells at their growing end at stage 12 in response to the activating bnl signal and a subset of these cells are selected to acquire either the fusion or the terminal cell fate at stage 13 and sprout unicellular branches. The rest of the cells in the group lose pantip gene expression and do not sprout, they instead become stalk cells suggesting that an inhibitory signal is produced by the terminal and fusion cells of all primary tracheal branches to restrict the domain of pantip gene expression. The phenotypes of hdc loss-of-function mutants and the expression of the gene only in the branches that send off both terminal and fusion sprouts suggest that hdc may be necessary to potentiate a more general inhibitory signal in the dorsal and lateral branches in which the cells expressing pantip markers will give rise to two different sprouting cells and stalk cells. The ability of hdc to suppress terminal branching and DSRF expression after ectopic expression in the ganglionic branches that do not extend fusion sprouts is consistent with the hypothesis that it can enhance an inhibitory signal operating on the pantip cells of all primary branches. The characterization of two tracheal genes, in addition to hdc, that increase the number of unicellular sprouts (M.A. Krasnow, pers. comm.) should help to understand the interplay between these molecules during sprouting of new branches.
hdc expression is regulated by esc
Previously, we have shown that the transcription factor encoded by the esg gene is a key regulator of the fusion cell fate. In esg mutants, the fusion cells do not express later fusion marker genes including the P-element insert in hdc and they inappropriately express terminal markers. Furthermore, ectopic expression of esg in all tracheal cells was sufficient to suppress terminal branching and to induce ectopic fusions (Samakovlis et al. 1996b). With the hdc antibody at hand, we have shown that esg misexpression in all tracheal cells is sufficient to induce hdc expression in additional tracheal cells. Interestingly, this activation was not evident in all cells of the tracheal epithelium; it was localized to the tip cells of the primary branches that send off both terminal and fusion cells suggesting these cells contain a cofactor necessary for hdc activation by esg. The loss-of-function phenotypes in the dorsal branches of esg and hdc embryos are similar, in both mutants the number of terminal branches is increased. The cellular processes by which the extra terminal branches arise are, however, different. In the case of esg mutants, the fusion cell partly acquires the terminal cell identity, it does not undergo branch fusion, and is also unable to inhibit neighboring cells that normally become stalk cells from sprouting. In hdc mutants, the fusion process appears normal, the defect is caused by aberrant signaling to the cells expressing pantip markers. Thus, hdc is an esg downstream gene in the trachea and acts after esg and the specification of the fusion cell fate.
Translational readthrough is necessary for hdc function
We have demonstrated that the hdc gene produces two proteins by suppression of the UAA stop codon found in its mRNA. Furthermore, translational termination suppression is necessary for the function of the gene because the truncated form missing the carboxy-terminal part, unique to the larger protein, could not rescue the tracheal mutant phenotype nor suppress terminal branching when expressed in all tracheal cells. Genes with internal stop codons can be found in organisms ranging from bacteria to mammals and several retroviruses. The best characterized example of suppression of in-frame stop codons in bacteria and eucaryotes is the suppression of UGA codons by incorporation of a selenocysteine (Böck et al. 1991). In Drosophila, two transcripts have been identified with a single in-frame UGA stop codon. One derives from the kelch gene involved in oogenesis that produces large 160-kD and shorter 76-kD overlapping proteins (Xue and Cooley 1993), and the other from the out at first (oaf) gene whose protein products have not been characterized (Bergstrom et al. 1995). The relative ratio of the two Kelch proteins is developmentally regulated and the protein encoded by the shorter ORF is sufficient to rescue the ring canal phenotype of kelch mutants. Mutant analysis of substitutions of the UGA stop codon in the longer product suggested that it is generated by tRNA suppression (Robinson and Cooley 1997).
In retroviruses such as the moloney murine leukemia virus, suppression of UAG stop codon is necessary for the production of the gag—pol fusion protein and viral propagation. UAA stop codons can also be suppressed in the same context both in infected mammalian cells and reticulocyte lysates (Feng et al. 1989; Li and Rice 1989). This termination codon suppression in heterologous cell culture and in vitro systems suggests that the necessary machinery for readthrough exists in mammals provided the cis-acting elements are also present.
The translational readthrough in the hdc mRNA is the first example of UAA suppression in a natural gene. We determined the relative ratio between the longer and the shorter products to be ∼1:4. This is an unusually high ratio for the longer form; in the retroviral case, the shorter product is 20 times more abundant than the longer one. Also, we have shown by ectopic expression experiments that all the necessary components for the suppression event are present in all tissues of the animal tested, suggesting that the regulation of such readthrough events should rely on cis-acting sequences present in the mRNA. UAA is the most common termination codon in invertebrates and in 12% of the Drosophila genes analyzed is followed by a C, just like the internal stop codon in hdc (Cavener and Ray 1991). Because both the hdc termination codon and its immediate context are used in many genes, it is unlikely that the two hdc products are caused by high leakiness of termination. The mechanisms of translational termination suppression not involving selenocysteine incorporation are largely unknown. The proposed mechanism of readthrough in retroviruses involves potential pseudoknot structures made by the sequences 3′ of the UAG codon (Feng et al. 1992). Secondary structure predictions on the hdc mRNA sequence did not reveal any potential for the formation of a significant pseudoknot structure although it identified a potential large stem loop structure composed of 79 nucleotides located 3 nucleotides downstream of the UAA codon. Such a structure may cause ribosomes to pause, thereby facilitating ribosome slippage or allowing suppressing tRNAs to successfully compete with release factor at the readthrough site. Site directed mutagenesis and sequence swap experiments should provide informative evidence as to whether the mechanism of the two suppression events is similar. Our present analysis of hdc function does not exclude that the shorter protein form might have a function together with the longer readthrough product. The presence of two C. elegans genes, one homologous to a region unique to the larger product and the other to a common region, indicates that both hdc products may be functional. In addition, the phenotypes generated by overexpression of both the full-length and the truncated constructs in the fusion cells suggest that hdc function is sensitive to imbalances in the total amounts of both proteins as well as to the relative ratio between them.
Materials and methods
Drosophila strains
The enhancer trap markers 1-eve-1 (Tracheal-1), Pantip-2 and Pantip-4, Fus-1 (esg) and Fus-2, Terminal-1(pruned/DSRF) and Terminal-3 have been described (Perrimon et al. 1991; Samakovlis et al. 1996a). The Fus-6, B9-3-52 enhancer trap allele was generated in the laboratory of Y.N. Jan (Hartenstein and Jan 1992), and the rest of the markers used in the study in the laboratory of A. Spradling (Spradling et al. 1995). The Fus-6 marker is a P[lacZ] insertion at cytological position 99 F. P-element excision mutagenesis from Fus-6 resulted in a recovery of five early pupal lethals. One of these, hdc50, with the strongest tracheal phenotypes (visualized by mAb 2A12 staining), was selected and further characterized. hdc43 is a null hdc allele and hshdc87.1 is a P-element transgenic strain containing the full-length coding region under the control of the hsp70 promoter (Weaver and White 1995). For analysis of embryonic phenotypes, mutants were balanced over CyO or TM3 balancers carrying lacZ transposons to identify homozygous mutant embryos. To identify homozygous hdc50 larvae, the mutant chromosome was kept over the TM6b balancer. The esg mutant strains used were the null esg allele G66 (Whiteley et al. 1992) and the strong EMS-induced allele VS2 (Ashburner et al. 1990; Hayashi et al. 1993).
Embryo fixation and staining
Embryo fixation, staining, and light and confocal fluorescence microscopy were as described (Samakovlis et al. 1996b). The lumen-specific antibody used was mAb2A12 diluted 1:5. The mouse monoclonal anti-Hdc antibody mAbU33.7 (Weaver and White 1995) was used at 1:1 for in situ stainings. The anti-DSRF monoclonal antibody was mAb 2-161 (1:1000) from M. Gilman (Ariad Corporation, Boston, MA) and the anti-Coracle guinea pig polyclonal antiserum (1:1000) was from R. Fehon (Fehon et al. 1994). Biotin-, Cy2-, FITC-, and Cy3-conjugated secondary antibodies (Jackson) were used at 1:300 dilution. For the BrdU incorporation experiment, embryos were collected in 25°C for 2 hr and were then treated in two different ways according to Bodmer et al. (1989): (1) Embryos were aged 8–10 hr followed by exposure to BrdU for 30 min and then again aged for 4–6 hr before fixation; or (2) the embryos were aged for 11–15 hr followed by BrdU exposure for 30 min before fixation. Staining procedures of the BrdU labeled embryos was as before but with the following modifications. After incubation with mAb2A12 and anti-β-galactosidase antibody to detect homozygous mutants, embryos were fixed for 10 min in 4% formaldehyde in PBS before anti-BrdU antibody was added. Of the five hdc mutant embryos (80 metameres) with BrdU incorporation in the nervous system inspected, none showed incorporation in the trachea. Embryo staging was according to Campos-Ortega and Hartenstein (1985). For the rescue experiments, embryos of the cross (males) p127–Gal4/CyOftzlacZ; hdc50/TM3UbxlacZ × (females) p127–Gal4/CyOftzlacZ; UAS–hdc/hdc50 or p127–Gal4/CyOftzlacZ; hdc50/TM3UbxlacZ × UAS–hdcΔl;hdc50/TM3UbxlacZ were stained against Hdc, DSRF, and the tracheal lumen to score extra branching and DSRF expression and identify mutant embryos expressing Hdc under the control of the GAL4 driver strain.
Molecular biology
Cloning and sequencing of the hdc cDNA clones was done according to Sambrook et al. (1989). The longest cDNA clone was from an embryonic cDNA library (from P. Hurban) that extended from nucleotide 775 to nucleotide 4472 in the hdc sequence (Weaver and White 1995). This clone was partially sequenced and used in all subsequent experiments. RNA in situ hybridizations on embryos were according to Lehmann and Tautz (1994). Amplification of genomic DNA from the hdc gene was carried out by PCR with primers corresponding to positions 2844–2860 and 3349–3366 in the hdc sequence (Weaver and White 1995). The same primers were used for RT–PCR after first strand cDNA synthesis with an oligo dT primer and AMV reverse transcriptase. The genomic EcoRI rescued fragment was sequenced with a primer complementary to the P-element transposon inverted repeat.
UAS constructs and GAL4 strains for ectopic expression of hdc and esg
For the UAS–hdc transgenic fly strains, a 3.7-kb KpnI–NotI fragment from BSSK-hdc cDNA vector was isolated and cloned into pUAST vector (Brand and Perrimon 1993). The construct contains the start codon ATG at 1034bp and the two TAA termination codons and produces two proteins as the full-length hdc mRNA. The truncated form, UAS-hdcΔ1 was constructed with a 2.6-kb XhoI fragment from BSSk-hdc cDNA plasmid cloned into pUAST vector. This construct contains the start codon ATG and the first TAA termination codon (Fig. 5). Embryos from the w118 strain were injected according to Spradling (1986) and two independent strains were characterized for each construct. They were tested for tracheal phenotypes and protein expression and gave similar results. The UAS–esg strain has been described (Fuse et al. 1994). The following driver strains were used: btl–GAL4 (Shiga et al. 1996) expressing GAL4 in all tracheal cells from stage 11, TrGAL4, C38–GAL4 expressing GAL4 in most tracheal cells beginning at stage 13 (Guillemin et al. 1996), and p127–GAL4 that expresses GAL4 selectively in the tracheal fusion cells from stage 13. Embryos carrying one copy of the GAL4 driver and the UAS constructs were collected at 20°C for 8 hr and then were transferred to 29°C for 10 hr to maximize GAL4 activity. Then, embryos were fixed and stained to examine tracheal phenotypes.
Protein extractions and Western blots
Protein extracts from whole larvae or dissected salivary glands, CNS, and imaginal disc, electrophoresis, and Western blots were performed according to Sambrook et al. (1989). The anti-Hdc antibody was diluted 1:3 for Western blots and hybridizing bands were detected with the Amersham ECL reagent according to the manufacturer.
Acknowledgments
We thank S. Cumberledge, S. Hayashi, R. Fehon, P. Hurban, M. Gilman and the Umeå Stock Center for strains, antibodies, and cDNA libraries. We also thank J. Hemphälä for assistance in cloning and sequencing. We thank G. Beitel, G. Björk, T. Edlund, H. Semb, S. Tuck, and the members of our laboratory for their comments on the manuscript and M.A. Krasnow for stimulating discussions. P.S. is supported by a predoctoral fellowship from the Umeå Biotechnology Fund. This work was supported by a grant from the Swedish Research Council (NFR) to C.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL christos@ucmp.umu.se; FAX 46-90-778007.
References
- Ashburner M, Thompson P, Roote J, Lasko PF, Grau Y, El Messal M, Roth S, Simpson P. The genetics of a small autosomal region of Drosophila melanogaster containing the structural gene for alcohol dehydrogenase. VII. Characterization of the region around the snail and cactus loci. Genetics. 1990;126:679–694. doi: 10.1093/genetics/126.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom D, Merli C, Cygan J, Shelby R, Blackman R. Regulatory autonomy and molecular characterization of the Drosophila out at first gene. Genetics. 1995;139:1331–1346. doi: 10.1093/genetics/139.3.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böck A, Forchhammer K, Heider J, Baron C. Selenoprotein synthesis: An expansion of the genetic code. Trends Biochem Sci. 1991;16:463–467. doi: 10.1016/0968-0004(91)90180-4. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Carretto R, Jan Y. Neurogenesis of the peripheral nervous system in Drosophila embryos: DNA replication patterns and cell lineages. Neuron. 1989;3:21–32. doi: 10.1016/0896-6273(89)90112-8. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega AJ, Hartenstein V. The embryonic development of Drosophila melanogaster. New York: Springer-Verlag; 1985. [Google Scholar]
- Cavener D, Ray S. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991;19:3185–3192. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon R, Dawson I, Artavanis-Tsakonas S. A Drosophila homologue of membrane-skeleton protein 4.1 is associated with septate junctions and is encoded by the coracle gene. Development. 1994;120:545–557. doi: 10.1242/dev.120.3.545. [DOI] [PubMed] [Google Scholar]
- Feng Y, Levin J, Hatfield D, Schaefer T, Gorelick R, Rein A. Suppression of UAA and UGA termination codons in mutant murine leukemia viruses. J Virol. 1989;63:2870–2873. doi: 10.1128/jvi.63.6.2870-2873.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Yuan H, Rein A, Levin J. Bipartite signal for read-through suppression in murine leukemia virus mRNA: An eight-nucleotide purine-rich sequence immediately downstream of the gag termination codon followed by an RNA pseudoknot. Virology. 1992;66:5127–5132. doi: 10.1128/jvi.66.8.5127-5132.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatioid and other disease. Nature Medicine. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Fuse N, Hirose S, Hayashi S. Diploidy of Drosophila imaginal cells is maintained by a transcriptional repressor encoded by escargot. Genes & Dev. 1994;8:2270–2281. doi: 10.1101/gad.8.19.2270. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Groppe J, Ducker K, Treisman R, Hafen E, Affolter M, Krasnow MA. The pruned gene encodes the Drosophila serum response factor and regulates cytoplasmic outgrowth during terminal branching of the tracheal system. Development. 1996;122:1353–1362. doi: 10.1242/dev.122.5.1353. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Jan YN. Studying Drosophila embryogenesis with P-lacZ enhancer trap lines. Roux’s Arch Dev Biol. 1992;201:194–220. doi: 10.1007/BF00188752. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Hirose S, Metcalfe T, Shirras AD. Control of imaginal cell development by the escargot gene of Drosophila. Development. 1993;118:105–115. doi: 10.1242/dev.118.1.105. [DOI] [PubMed] [Google Scholar]
- Klämbt C, Glazer L, Shilo B-Z. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes & Dev. 1992;6:1668–1678. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- Lee T, Hacohen N, Krasnow M, Montell DJ. Regulated Breathless receptor tyrosine kinase activity required to pattern cell migration and branching in the Drosophila tracheal system. Genes & Dev. 1996;10:2912–2921. doi: 10.1101/gad.10.22.2912. [DOI] [PubMed] [Google Scholar]
- Lehmann R, Tautz D. In Situ Hybridization to RNA. In: Goldstein LSB, Fyrberg EA, editors. Methods in cell biology: Drosophila melanogaster—Practical uses in cell and molecular biology. San Diego, CA: Academic Press; 1994. pp. 575–596. [DOI] [PubMed] [Google Scholar]
- Li G, Rice C. Mutagenesis of the in-frame opal termination codon preceding nsP4 of Sindbis virus: Studies of translational readthrough and its effect on virus replication. J Virol. 1989;63:1326–1337. doi: 10.1128/jvi.63.3.1326-1337.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Krasnow MA. Development of the Drosophila tracheal system. In: Bate M, Arias AM, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 609–685. [Google Scholar]
- Perrimon N, Noll E, McCall K, Brand A. Generating lineage-specific markers to study Drosophila development. Dev Genet. 1991;12:238–252. doi: 10.1002/dvg.1020120309. [DOI] [PubMed] [Google Scholar]
- Reichman-Fried M, Shilo BZ. Breathless, a Drosophila FGF receptor homolog, is required for the onset of tracheal cell migration and tracheole formation. Mech Dev. 1995;52:265–273. doi: 10.1016/0925-4773(95)00407-r. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- Robinson DN, Cooley L. Examination of the function of two kelch proteins generated by stop codon suppression. Development. 1997;124:1405–1417. doi: 10.1242/dev.124.7.1405. [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Hacohen N, Manning G, Sutherland D, Guillemin K, Krasnow MA. Branching morphogenesis of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development. 1996a;122:1395–1407. doi: 10.1242/dev.122.5.1395. [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Manning G, Steneberg P, Hacohen N, Cantera R, Krasnow MA. Genetic control of epithelial tube fusion during Drosophila tracheal development. Development. 1996b;122:3531–3536. doi: 10.1242/dev.122.11.3531. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Shiga Y, Tanaka-Matakatsu M, Hayashi S. A nuclear GFP/β-galactosidase fusion protein as a marker for morphogenesis in living Drosophila. Dev Growth Differ. 1996;38:99–106. [Google Scholar]
- Spradling AC. P element-mediated transformation. In: Roberts DB, editor. Drosophila—a practical approach. Oxford, UK: IRL Press Limited; 1986. pp. 188–197. [Google Scholar]
- Spradling AC, Stern DM, Kiss I, Roote J, Laverty T, Rubin G. Gene disruptions using P transposable elements: An integral component of the Drosophila genome project. Proc Natl Acad Sci. 1995;92:10824–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland D, Samakovlis C, Krasnow MA. branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87:1091–1101. doi: 10.1016/s0092-8674(00)81803-6. [DOI] [PubMed] [Google Scholar]
- Tanaka-Matakatsu M, Uemura T, Oda H, Takeichi M, Hayashi S. Cadherin-mediated cell adhesion and cell motility in Drosophila trachea regulated by the transcription factor escargot. Development. 1996;122:3697–3705. doi: 10.1242/dev.122.12.3697. [DOI] [PubMed] [Google Scholar]
- Weaver T, White R. headcase, an imaginal specific gene required for adult morphogenesis in Drosophila melanogaster. Development. 1995;121:4149–4160. doi: 10.1242/dev.121.12.4149. [DOI] [PubMed] [Google Scholar]
- Whiteley M, Noguchi PD, Sensabaugh SM, Odenwald WF, Kassis JA. The Drosophila gene escargot encodes a zinc finger motif found in snail-related genes. Mech Dev. 1992;36:117–127. doi: 10.1016/0925-4773(92)90063-p. [DOI] [PubMed] [Google Scholar]
- Xue F, Cooley L. kelch encodes a component of intercellular bridges in Drosophila egg chambers. Cell. 1993;72:681–693. doi: 10.1016/0092-8674(93)90397-9. [DOI] [PubMed] [Google Scholar]