

Abstract
Herein we report the discovery, synthesis and evaluation of a series of N-(4-acetamido)-phenylpicolinamides as positive allosteric modulators of mGlu4.a Compounds from the series show submicromolar potency at both human and rat mGlu4. In addition, pharmacokinetic studies utilizing subcutaneous dosing demonstrated good brain exposure in rats.
Introduction
G-Protein coupled receptors (GPCRs) comprise a large protein family of transmembrane receptors and have historically been the most studied class of drug targets. GPCRs are activated by orthosteric ligands binding which induce macromolecular protein changes and stimulation of signal transduction pathways and cellular responses inside the cell. The GPCR superfamily is grouped into three classes (class A (rhodopsin-like), B (secretin family) and C (metabotropic glutamate)).1 The mGlus belong to the Class C GPCR subfamily and are further subdivided into three classes.2 Group I consists of mGlu1 and mGlu5, Group II consists of mGlu2 and mGlu3, and Group III consists of mGlu4, mGlu6, mGlu7 and mGlu8. Due to the lack of selectivity and physicochemical properties of orthosteric ligands of the mGlus, significant effort has been initiated on identifying compounds that act via allosteric sites (i.e., sites different from the orthosteric site) on the receptors.3, 4 This approach has been successful for the Group I and II glutamate receptors5 (mGlu1,6, 7 mGlu5,8, 9 and mGlu28) with multiple tool compounds associated with each receptor; however, in contrast, the Group III family has been studied in much less detail due to the paucity of selective ligands – including allosteric modulators.
One subtype that has received much attention of late due to the discovery of selective allosteric ligands and its implication in a number of disease states, such as Parkinson's disease (PD), epilepsy, and anxiety, is mGlu4. The interest in targeting this receptor using allosteric modulation was initially sparked via studies with the compound N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide, PHCCC10, 11, 1, a partially selective mGlu4 positive allosteric modulator (PAM). With the exception of partial antagonist activity at mGlu1, 1 was shown to be selective versus the other mGlus. Compound 1 has been an invaluable tool compound for the study of mGlu4 and has been shown to be active in acute rodent models of Parkinson's disease (PD) when administered either via intracerebroventricular (icv) injection or systemically in a 50% DMSO vehicle. Although 1 has been a breakthrough compound for the study of mGlu4, it has substantial drawbacks due to its poor pharmacokinetic properties and relatively flat structure-activity relationship. Over the past three years, our laboratories and others have added to the list of selective and novel tool compounds for the study of mGlu4 via positive allosteric modulation (Figure 1). Thus far, only two disclosed mGlu4 PAMs (3 (VU0361737)12 and 413) have been shown to be CNS penetrant when dosed systemically in a nontoxic vehicle. In this communication, we herein disclose a second series of N-(4-acetamido)-phenylpicolinamides that act as mGlu4 positive allosteric modulators and show that representative compounds are also CNS penetrant.
Figure 1.

Structures of 1 and other recently reported mGlu4 PAMs.
Starting from a functional high-throughput screening (HTS) initiated at Vanderbilt University, a number of N-(4-acetamido)-phenylcarboxamide compounds were discovered, as exemplified by compound 5 (Figure 2, Pubchem Compound ID (CID): 1314024; ChemBridge ID: 7637085). This compound represented a large class of hits that were identified by our HTS and signaled a starting point for a lead identification program in our laboratories.
Figure 2.

Structure of initial HTS hit.
Results
The compounds were prepared via a common intermediate such that the phenyl core and the 2-chlorophenyl amide could be held constant while the furyl amide portion was evaluated (Scheme 1). To this end, the nitro aniline, 6, was acylated with 2-chlorophenyl acid chloride to give 8. Hydrogenation of the nitro group (Ra-Ni, H2) followed by amide formation led to compounds 9a–k and 10a–d. An alternative approach was employed for compounds 12a–n. First, the aniline of 6 was di-Boc protected (Boc2O, DMAP), followed by nitro reduction (H2 (1 atm), Pd/C) and picolinamide formation (picolinyl acid chloride, diisopropyl ethyl amine) leading to 12. Finally, Boc deprotection (4 M HCl, dioxane) followed by amide formation yielded 12a–n.
Scheme 1.

Synthesis of N-(4-acetamido)-phenylcarboxamides, 9, 10, and 12. All library compounds were purified by mass-directed prep LC where required.14, 15
Discussion
The initial SAR was centered around the furyl amide portion (9a–k) in order to replace the 5-bromo-2-furyl amide portion as this is a high molecular weight compound (MW = 449.7). Several heterocyclic replacements, as well as aryl and carbocycles, were evaluated. The two pharmacological assays we utilized to assess mGlu4 PAM potency (1. Chinese Hamster Ovary cells expressing human mGlu4 and the chimeric G protein Gqi5 to induce calcium mobilization and 2. HEK cells expressing rat mGlu4 in conjunction with G protein regulated Inwardly Rectifying Potassium (K) channels (GIRK) to induce thallium flux16) show some day-to-day variability in the maximal PAM response; for this reason, all data have been normalized to the % response of the control PAM, 1, to compare relative efficacy.17, 18 Initial removal of the bromine resulted in a more potent compound (9a, furyl, 977 nM), while the addition of a large phenyl group led to complete loss of activity (9b). Moving from the furyl compound to the saturated tetrahydrofuran (9c) also led to an inactive compound. Substituting the furan for sulfur containing heterocycles, such as thiophene and thiazole, resulted in compounds that were equipotent (9d and 9f) or more potent (9e, 2-thiazole, 627 nM). Next, 6-membered heterocycles (9g–i) were evaluated with the 2-pyridyl compound, 9h, exhibiting the best potency (99.5 nM). Modulation of the basicity of the pyridine nitrogen led to a 10-fold loss in potency (pyrazine, 9g, 1780 nM; 4-pyrimidine, 9i, 1330 nM). Replacement of the heterocyclic amides with carbocyclic amides also led to much reduced potency (cyclohexyl, 9k, 1120 nM).
Having discovered a more potent replacement for the initial 5-bromo-2-furyl amide, we next turned our attention to the central core as this contains a 3-methoxy moiety which, when contained on the 2,4-dianiline species, could lead to bis-quinone formation. To this end, a number of substituted phenyl analogs were evaluated (Table 2). The 3-methoxy was replaced by both halogens (3-Cl, 10a, 517 nM; 3-F, 10b, 556 nM), trifluoromethyl (10c, 3750 nM) and ultimately removed (10d, 1650 nM). All of theese substitutions led to a significant decrease in activity; however, we were encouraged to see that halogen substitution was tolerated and generated compounds with submicrolar potency.
Table 2.
Human and Rat mGlu4 potency and % GluMax response (as normalized to standard 1) for selected central phenyl modification analogs.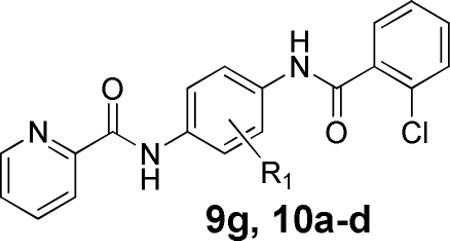
| Compd | R1 | hEC50 (nM)a | GluMax (%Cmpd 1)b | rEC50 (nM)a | GluMax (%Cmpd 1)b |
|---|---|---|---|---|---|
| 9g | 3-OMe | 99.5 ± 9 | 79.4 ± 1.7 | 106 ± 28 | 147.0 ± 4.3 |
| 10a | 3-Cl | 517 ± 57 | 77.9 ± 1.6 | 570 ± 131 | 128.2 ± 5.6 |
| 10b | 3-CF3 | 2090 ± 190 | 69.5 ± 1.5 | 2360 ± 230 | 101.9 ± 7.4 |
| 10c | H | 1605 ± 200 | 33.6 ± 0.5 | 2260 ± 260 | 49.6 ± 5.4 |
| 10d | 3-F | 556 ± 76 | 59.7 ± 1.1 | 739 ± 92 | 91.4 ± 2.9 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table).
Cmpd 1 is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative 1 response obtained in each day's run.
Next, our attention turned to the amide portion that originally was the 2-chlorophenyl amide (Table 3). Noting that the fluoro- and chloro-phenyl compounds were equipotent, we utilized both of these cores when assessing the right-hand amide SAR and a series of aryl, cycloalkyl, alkyl and heteroaromatic amides were evaluated. A number of interesting observations regarding the SAR were gleaned from this study. The 2-chlorophenyl (10a, 517 nM) was superior to other 2-subsituted compounds (2-fluorophenyl, 12f, 2380 nM; 2-methylphenyl, 12a, 958 nM; 2-methoxyphenyl, 12i, inactive). Moving the halogen around the ring or adding a second halogen generally led to compounds with much reduced potency (12g and 12h, inactive); however, the 2-chloro-4-fluoro compound (12d, 114 nM) was more active than the 2-chloro analog (10a, 517 nM). However, when the 2-chloro-4-fluoro was replaced with 2,4-difluoro (12g, inactive) there was a significant loss of potency (similar to the loss seen in 10a vs. 12f). Carbocycles were of equal potency (cyclohexyl, 12b, 426 nM vs. phenyl, 12c, 361 nM); however, the smaller isopropyl group lost significant activity (12e, 4270 nM). All of the heterocyclic substituents were much less active with the exception of 2-thiazole (12k, 403 nM).
Table 3.
Human and Rat mGlu4 potency and % GluMax response (as normalized to standard 1) for selected acetamido amide analogs.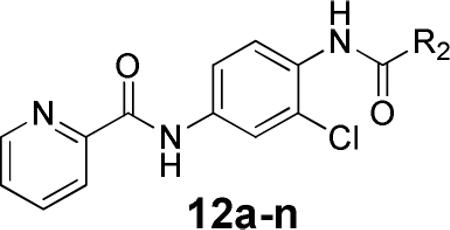
| Compd | R2 | hEC50 (nM)a | GluMax (%Cmpd 1)b | rEC50 (nM)a | GluMax (%Cmpd 1)b |
|---|---|---|---|---|---|
| 12a | 2-methylphenyl | 958 ± 48 | 72.5 ± 1.1 | 915 ± 161 | 123.1 ± 2.9 |
| 12b | cyclohexyl | 426 ± 58 | 59.2 ± 1.0 | 592 ± 70 | 106.0 ± 0.7 |
| 12c | phenyl | 361 ± 40 | 37.9 ± 1.3 | 518 ± 43 | 74.2 ± 8.7 |
| 12d | 2-chloro-4-fluorophenyl | 114 ± 26 | 61.1 ± 4.1 | 169 ± 28 | 118.8 ± 7.3 |
| 12e | isopropyl | 4270 ± 540 | 60.0 ± 1.1 | 4820 ± 1160 | 109.0 ± 3.4 |
| 12f | 2-fluorophenyl | 2380 ± 170 | 34.5 ± 1.0 | 4390 ± 1140 | 71.6 ± 10.0 |
| 12g | 2,4-difluorophenyl | inactivec | 24.6 ± 1.5 | 2100 ± 780 | 43.5 ± 12.0 |
| 12h | 3,5-dichlorophenyl | 2150 ± 448 | 37.4 ± 3.2 | 2340 ± 817 | 61.0 ± 9.0 |
| 12i | 2-methoxyphenyl | inactivec | 16.4 ± 1.3 | inactivec | 19.0 ± 2.1 |
| 12j | 5-thiazole | 2180 ± 310 | 86.7 ± 2.0 | 1730 ± 170 | 124.8 ± 8.7 |
| 12k | 2-thiazole | 403 ± 28 | 61.5 ± 2.0 | 533 ± 145 | 105.5 ± 2.4 |
| 12l | 2-thiophene | 1040 ± 100 | 57.8 ± 1.8 | 1130 ± 140 | 93.8 ± 3.8 |
| 12m | N-methyl-2-indole | 4720 ± 900 | 49.9 ± 4.1 | 2970 ± 290 | 87.1 ± 3.9 |
| 12n | 5-phenyl-2-furyl | inactivec | 24.9 ± 0.8 | 3350 ± 290 | 45.6 ± 10.8 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table).
Cmpd 1 is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative 1 response obtained in each day's run.
Inactive compounds are defined as %GluMax did not surpass 2× the EC20 value at the highest concentration (30 μM) for that day's run.
Lastly, as this structural class represents a 2,4-dianiline, we were interested to determine if we could replace the internal core structure with a non-aniline compound (see Table 1, Supporting Information). We utilized an indole core structure and when the NH of either amide was replaced with an alkyl substituent, a significant loss of potency was seen. Alkylation of either amide also led to inactive compounds (data not shown).
Table 1.
Human and Rat mGlu4 potency and % GluMax response (as normalized to standard 1) for selected furyl amide analogs.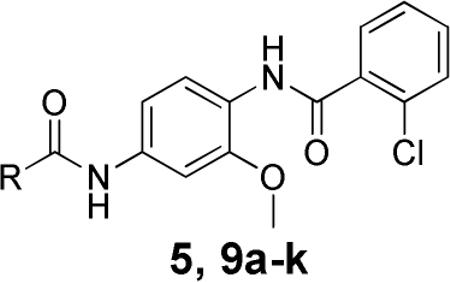
| Compd | R | hEC50 (nM)a | GluMax (%Cmpd 1)b | rEC50 (nM)a | GluMax (%Cmpd 1)b |
|---|---|---|---|---|---|
| 5 | 5-bromo-2-furan | 1440 ± 100 | 87.0 ± 1.2 | 1000 ± 90 | 125.4 ± 6.7 |
| 9a | 2-furan | 977 ± 99 | 62.7 ± 2.4 | 1010 ± 200 | 122.3 ± 2.9 |
| 9b | 5-phenyl-2-furan | inactivec | 16.3 ± 1.5 | inactivec | 16.3 ± 2.7 |
| 9c | 2-tetrahydrofuran | inactivec | 21.3 ± 2.9 | >10,000 | 27.1 ± 3.0 |
| 9d | 2-thiophene | 1170 ± 150 | 63.4 ± 1.5 | 860 ± 90 | 96.7 ± 8.5 |
| 9e | 2-thiazole | 627 ± 80 | 77.6 ± 1.7 | 377 ± 101 | 133.9 ± 6.6 |
| 9f | 4-thiazole | 845 ± 39 | 86.8 ± 2.3 | 753 ± 164 | 132.4 ± 6.6 |
| 9g | 2-pyridine | 99.5 ± 9 | 79.4 ± 1.7 | 106 ± 28 | 147.0 ± 4.3 |
| 9h | pyrazine | 1780 ± 140 | 74.3 ± 0.4 | 1120 ± 180 | 111.7 ± 6.6 |
| 9i | 4-pyrimidine | 1330 ± 90 | 77.9 ± 3.5 | 894 ± 203 | 133.1 ± 6.2 |
| 9j | phenyl | cannot fitd | cannot fitd | ||
| 9k | cyclohexyl | 1120 ± 270 | 26.9 ± 5.3 | 1520 ± 630 | 43.2 ± 13.2 |
| 1 | 4740 ± 51 | 2820 ± 44 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table).
1 is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative 1 response obtained in each day's run.
Inactive compounds are defined as %GluMax did not surpass 2× the EC20 value at the highest concentration (30 μM) for that day's run.
Curve could not be fit due to insolubility at higher concentrations.
With our best compounds evaluated for human and rat potency, we next turned our attention to evaluation of the compounds' ability to shift the glutamate response curve to the left, i.e. glutamate potentiation (fold shift, FS) as well as to assessment of the microsomal stability (intrinsic clearance) and the ability of the compounds to bind plasma proteins (see Table 4). For the most part, the GluMax (% 1) was mirrored in the fold shift data, although a few exceptions are worth noting. First, the 2-thiazole (9e) and 2-pyridyl (9g) both had high GluMax values (133.9% and 147.0%, respectively); however, this relatively small difference produced a significant difference in fold shift (32.2 vs. 72.0, respectively). Both of these values are the largest fold shifts reported to date for an mGlu4 PAM. As observed for other allosteric modulators, the fold shift data, representative of compound efficacy, did not always track with the potency values (10a, 570 nM, 18.3 FS vs. 12d, 169 nM, 6.7 FS).
Table 4.
Fold shift, intrinsic clearance and plasma protein binding (PPB, rat) and CYP450 enzyme inhibition data for selected compounds.
| Compd | rEC50 (nM)a | GluMax (%Cmpd 1)b | rFS | Rat Clint (ClHEP), (mL/min/kg) | PPB (rat, %fu) | P450 (μM) |
|||
|---|---|---|---|---|---|---|---|---|---|
| 1A2 | 2C9 | 3A4 | 2D6 | ||||||
| 9e | 377 ± 101 | 133.9 ± 6.6 | 32.2 ± 2.4 | 185.3 (50.8) | 0.56 | ND | ND | ND | ND |
| 9g | 106 ± 28 | 147.0 ± 4.3 | 72.0 ± 4.5 | 42.6 (26.5) | 0.33 | >30 | 14.3 | >30 | 12.8 |
| 10a | 570 ± 131 | 128.2 ± 5.6 | 18.3 ± 1.3 | 44.9 (27.3) | 0.29 | >30 | 11.8 | >30 | 13.1 |
| 10d | 739 ± 92 | 91.4 ± 2.9 | 4.6 ± 0.1 | 31.2 (21.6) | 0.58 | ND | ND | ND | ND |
| 12a | 915 ± 161 | 123.1 ± 2.9 | 9.6 ± 0.6 | 46.2 (27.8) | 0.35 | >10 | 14.1 | >30 | 12.0 |
| 12d | 169 ± 28 | 118.8 ± 7.3 | 6.7 ± 2.0 | 54.0 (30.5) | ND | ND | ND | ND | ND |
| 12k | 533 ± 145 | 105.5 ± 2.4 | 6.0 ± 0.6 | ND | 0.65 | ND | ND | ND | ND |
| 15 | 1340 ± 200 | 107.1 ± 3.2 | 5.9 ± 1.1 | 605.2 (62.7) | 0.16 | ND | ND | ND | ND |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table).
Cmpd 1 is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative 1 response obtained in each day's run. ND = not determined.
Compounds were further evaluated by assessing their rate of intrinsic clearance in vitro as well as their ability to bind to plasma proteins (PPB) (see Table 2, Supporting Information). Two compounds possessed poor metabolic stability (9e and 15). These compounds possess a labile methoxy group which could be the contributing factor their instability. Interestingly, 9g also possesses a methoxy group but exhibits acceptable intrinsic clearance values (42.6 mL/min/kg). All of the other compounds tested exhibited acceptable intrinsic clearance (<50 mL/min/kg). All compounds evaluated were highly protein bound in rats (>99.5% bound). In addition to establishing in vitro pharmacology, metabolic stability and protein binding profiles, representative compounds (9g, 10a, and 12a) were tested for their inhibition of the major CYP450 enzymes (CYP1A2, 2C9, 3A4 and 2D6). These compounds showed no significant activity against CYP1A2 and CYP3A4 (>30 μM), but each of the compounds showed weak activity against CYP2C9 and CYP2D6 (>10 μM). Lastly, selectivity versus the other mGlu's was performed on 10a and this compound was selective versus the Group I and Group II mGlu's; but showed weak PAM activity (as assessed by the ability of 10a to left-shift the glutamate response curve at 30 μM) against mGlu7 (5.3 FS) and mGlu8 (3.8 FS) (see Table 2, Supporting Information). While compound is not completely selective for mGlu4, this compound seems to be group III mGlu preferring, which may allow for future rationally designed site directed mutagenesis studies to help confirm the binding location on the receptor.
Although there is a large range of fold shift values within compounds in this series, all other properties appear to be equivalent (intrinsic clearance, PPB, CYP inhibition profile); thus three representative compounds were chosen to further advance into in vivo PK studies (see Table 5). The hydrochloride salts of these compounds (9g, 10a, 12a) were synthesized and dosed subcutaneously (10 mg/kg) as an aqueous microsuspension containing 10% Tween-80, and the amount of compound present in brain and plasma was determined at 0.5, 1, 3, and 6 h after administration (Table 6). Uptake of 10a (VU0366037) was substantial in both plasma and brain with a brain:plasma ratio of 1.04. Plasma exposure of VU0366038, 12a and VU0415374, 9g were comparable to 10a; however, these compounds exhibited lower brain exposure with a brain:plasma ratio of 0.2 and 0.33, respectively.
Table 5.
Rat Pharmacokinetic Data for Compounds VU0415374 (9g), VU0366037 (10a), and VU0366038 (12a) following SC dosing (10 mg/kg; 10% tween 80 microsuspension). AUC=area under the curve as assessed from 0–6 hours.
| parameters | VU0415374, 9g | VU0366037, 10a | VU0366038, 12a |
|---|---|---|---|
| Plasma AUC (ng·hr/mL) | 938 | 657.8 | 766.1 |
| Plasma Cmax (ng/mL) | 265 | 198.1 | 219.2 |
| Brain AUC (ng·hr/g) | 312 | 681.0 | 155.4 |
| Brain Cmax (ng/g) | 69.1 | 163.2 | 59.4 |
| Tmax (h) | 1 | 0.5 | 0.5 |
| AUC0–6h, brain/AUC0–6h,plasma | 0.33 | 1.04 | 0.2 |
Conclusion
In summary, we have discovered and evaluated a novel series of N-(4-acetamido)-phenylpicolinamides as mGlu4 positive allosteric modulators suitable as in vivo tool compounds. Compounds 9g, 10a, and 12a exhibited submicromolar EC50's against both human and rat mGlu4, and all three robustly left-shifted the glutamate response curve with 9g and 10a possessing fold shifts >15. In addition, compounds 10a and 12a showed acceptable in vitro PK parameters (intrinsic clearance) and were further evaluated for CNS exposure. 10a showed good exposure in both plasma and brain, with a ratio of 1.04.
Experimental Section
General
The syntheses of selected compounds are described below. The general chemistry, experimental information, and syntheses of all other compounds are supplied in the Supporting Information. Purity of all final compounds was determined by HPLC analysis is >95%.
N-(4-(2-chlorobenzamido)-3-methoxyphenyl)picolinamide (9g)
To a solution of 8g (0.91 g, 2.98 mmol) in ethanol (EtOH):ethyl acetate (EtOAc) (200 mL, 1:1) was added Ra-Ni (~0.1 equivalents). An H2 atmosphere (1 atm) was applied to the rxn mixture. After 16 h, the rxn was filtered through Celite and concentrated. The crude reside was deemed >85% pure by LCMS and carried through without further purification. To a solution of the aforementioned aniline in dimethylformamide (DMF) (25 mL) and diisopropyl ethylamine (DIEA) (5 mL, 28.7 mmol) was added 2-pyridinecarbonyl chloride (0.53 g, 2.98 mmol). After 12 h at rt, the rxn was added to EtOAc:H2O (150 mL, 1:1). The organic layer was washed with H2O (2 × 50 mL), Brine (50 mL) and dried (MgSO4). After filtration and concentration, the residue was purified by mass-guided preparative HPLC to afford amide 9g (0.66 g, 58% yield over 2 steps). Analytical LCMS (Method 2): single peak (220 nm), RT = 1.167 min., m/z 382.0 [M + H]+; 1H NMR (400 MHz, DMSO-d6): δ 10.64 (s, 1H), 9.58 (s, 1H), 8.74 (d, J = 4.4 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 8.07 (ddd, J = 8.0, 8.0, 1.6 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 2.0 Hz, 1H), 7.68 (dd, J = 6.4, 4.8 Hz, 1H), 7.58–7.46 (m, 5H), 3.82 (s, 3H); HRMS, calc'd for C20H17N3O3Cl (M+H+), 382.0958; found 382.0958.
N-(3-chloro-4-(2-chlorobenzamido)phenyl)picolinamide (10a)
Following the general procedure above for 9g, compound 10a (0.62 g, 81%) was obtained as a tan solid: Analytical LCMS (Method 2): single peak (214 nm); RT = 1.496 min.; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (s, 1H), 10.18 (s, 1H), 8.77 (d, J = 4.8 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.19 (d, J = 7.6 Hz, 1H), 8.10 (ddd, J = 8.0, 1.6, 1.6 Hz, 1H), 7.93 (dd, J = 8.8, 2.0 Hz, 1H), 7.73–7.70 (m, 1H), 7.63–7.46 (m, 5H); HRMS, calc'd for C19H14N2O2Cl2 [M + H]+, 386.0463; found 386.0467.
N-(3-chloro-4-(2-methylbenzamido)phenyl)picolinamide (12a)
To a solution of 6 (1.67 g, 9.69 mmol) and dimethylaminopyridine (DMAP) (0.12 g, 0.97 mmol) in tetrahydrofuran (THF) (50 mL) at rt was added (Boc)2O (6.33 g, 29.0 mmol). After 16 h, the rxn was added to EtOAc:water (200 mL, 1:1) and the organic layer was separated and washed with Brine (50 mL), dried (MgSO4), filtered and concentrated. To a solution of the crude residue in EtOAc (200 mL) was added Rainey-Ni (~0.1 equivalent) and an H2 atmosphere via balloon (1 atm) was applied. After 16 h, LCMS confirms loss of starting material, mixture was filtered through a Celite filter and concentrated and was carried on without purification. To a solution of the crude residue in dichloromethane (DCM) (20 mL) was added DIEA (3.4 mL, 24.2 mmol), followed by picolinoyl acid chloride hydrochloride (1.72 g, 9.69 mmol). After 16 h, the rxn was added to DCM:NaHCO3 (aq) (200 mL, 1:1) and the organic layer was separated. The organic layer was washed with Brine (50 mL), dried (MgSO4), filtered and concentrated to afford amide 11 which was carried on without purification. To a solution of amide 11 in DCM (100 mL) at 0 °C was added 4M HCl in 1,4-dioxane (20 mL). After 15 min, the ice bath was removed. The violet red solution stirred for 12 h at rt and after TLC confirmed loss of starting material, the solvent was removed under vacuo. To a mixture of the crude residue in DMF (20 mL) at 0 °C was added DIEA (4.78 mL, 34.0 mmol) followed by o-toluoyl chloride (1.26 mL, 9.69 mmol). After 15 min, the ice bath was removed. After an additional 12 h, the rxn was added to EtOAc:H2O (200 mL, 1:1). The organic layer was washed with H2O (2 × 50 mL), Brine (50 mL) and dried (MgSO4). After filtration and concentration, the residue was purified by mass-guided preparative HPLC to afford amide 12a (1.45 g, 41% overall yield). Analytical LCMS (Method 2): single peak (214 nm); RT = 1.482 min; 1H NMR (400 MHz, DMSO-d6): δ 10.90 (s, 1H), 9.94 (s, 1H), 8.77 (d, J = 4.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 8.10 (ddd, J = 8.0, 2.0, 2.0 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.71 (ddd, J = 7.6, 4.8, 1.6 Hz, 1H), 7.58 (d, J = 8.8 Hz, 5H), 7.54 (d, J = 7.6 Hz, 1H), 7.40 (dd, J = 7.2, 7.2 Hz, 1H), 7.33–7.30 (m, 2H), 2.46 (s, 3H); HRMS, calc'd for C20H17N3O2Cl [M + H]+, 366.1009; found 366.1009.
Supplementary Material
Acknowledgment
The authors thank Emily L. Days, Tasha Nalywajko, Cheryl A. Austin and Michael Baxter Williams for their critical contributions to the HTS portion of the project. In addition, we would like to thank Katrina Brewer, Ryan Morrison and Matt Mulder for technical assistance with the PK assays, and Chris Denicola, Nathan Kett, and Sichen Chang for the purification of compounds utilizing the mass-directed HPLC system. This work was supported by the National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, the Michael J. Fox Foundation, the Vanderbilt Department of Pharmacology and the Vanderbilt Institute of Chemical Biology.
Footnotes
Supporting Information Available: Experimental procedures, spectroscopic data, and NMRdata for selected compounds and biological procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: SAR: structure-activity relationship; mGluR: metabotropic glutamate receptor; PAM: positive allosteric modulator; HTS: high-throughput screening; Clint: intrinsic clearance; PPB: plasma protein binding; AUC: area under the curve; GPCR: G-protein coupled receptor; PHCCC: N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide; CNS: central nervous system; PD: Parkinson's disease; GIRK: G protein-coupled inwardly-rectifying potassium channel.
References and Notes
- 1.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Ann. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 3.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev. Drug. Dis. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 5.Johnson MP, Nisenbaum ES, Large TH, Emkey R, Baez M, Kingston AE. Allosteric modulators of metabotropic glutamate receptors: lessons learnt from mGlu1, mGlu2 and mGlu5 potentiators and antagonists. Biochem. Soc. Trans. 2004;32:881–887. doi: 10.1042/BST0320881. [DOI] [PubMed] [Google Scholar]
- 6.Gasparini F, Kuhn R, Pin J-P. Allosteric modulation of group 1 metabotropic glutamate receptors: novel subtype-selective ligands and therapeutic perspectives. Exp. Opin. Pharmacol. 2002;2:43–49. doi: 10.1016/s1471-4892(01)00119-9. [DOI] [PubMed] [Google Scholar]
- 7.Knoflach F, Mutel V, Jolidon S, Kew JNC, Malherbe P, Vieira E, Wichman J, Kemp JA. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc. Nat. Acad. Sci. 2001;98:13402–13407. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlumberger C, Pietraszek M, Gravius A, Klein K-U, Greco S, Moré L, Danysz W. Comparison of the mGlu5 receptor positive allosteric modulator ADX47273 and the mGlu2/3 receptor agonist LY354740 in tests for antipyschotic-like activity. Eur. J. Pharmacol. 2009;623:73–83. doi: 10.1016/j.ejphar.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu5: Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg. Med. Chem. Lett. 2010;20:4390–4394. doi: 10.1016/j.bmcl.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ. (−)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 11.Marino MJ, Williams DL, Jr., O'Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Kinney GG, Conn PJ. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc. Nat. Acad. Sci. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs) J. Med. Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.East SP, Bamford S, Dietz MGA, Eickmeier C, Flegg A, Ferger B, Gemkow MJ, Heilker R, Hengerer B, Kotey A, Loke P, Schänzle G, Schubert H-D, Scott J, Whittaker M, Williams M, Zawadzki P, Gerlach K. An orally bioavailable positive allosteric modulator of the mGlu4 receptor with efficacy in an animal model of motor dysfunction. Bioorg. Med. Chem. Lett. 2010;20:4901–4905. doi: 10.1016/j.bmcl.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 14.Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C. Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. J. Comb. Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 15.All new compounds were characterized by LCMS and/or 1H NMR and found to be in agreement with their structures (>95% purity).
- 16.Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008;73:1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- 17.Williams R, Zhou Y, Niswender CM, Luo Q, Conn PJ, Lindsley CW, Hopkins CR. Re-exploration of the PHCCC scaffold: discovery of improved positive allosteric modulators of mGluR4. ACS Chem. Neurosci. 2010;1:411–419. doi: 10.1021/cn9000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engers DW, Gentry PR, Williams R, Bolinger JD, Weaver CD, Menon UN, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. Synthesis and SAR of novel, 4-(phenylsulfamoyl)phenylacetamide mGlu4 positive allosteric modulators (PAMs) identified by functional high-throughput screening (HTS) Bioorg. Med. Chem. Lett. 2010;20:5175–5178. doi: 10.1016/j.bmcl.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.