

Abstract
Cellular stress, particularly in response to toxic and metabolic insults that perturb function of the endoplasmic reticulum (ER stress), is a powerful inducer of the transcription factor CHOP. The role of CHOP in the response of cells to injury associated with ER stress was examined in a murine deficiency model obtained by homologous recombination at the chop gene. Compared with the wild type, mouse embryonic fibroblasts (MEFs) derived from chop −/− animals exhibited significantly less programmed cell death when challenged with agents that perturb ER function. A similar deficit in programmed cells death in response to ER stress was also observed in MEFs that lack CHOP’s major dimerization partner, C/EBPβ, implicating the CHOP–C/EBP pathway in programmed cell death. An animal model for studying the effects of chop on the response to ER stress was developed. It entailed exposing mice with defined chop genotypes to a single sublethal intraperitoneal injection of tunicamycin and resulted in a severe illness characterized by transient renal insufficiency. In chop +/+ and chop +/− mice this was associated with the early expression of CHOP in the proximal tubules followed by the development of a histological picture similar to the human condition known as acute tubular necrosis, a process that resolved by cellular regeneration. In the chop −/− animals, in spite of the severe impairment in renal function, evidence of cellular death in the kidney was reduced compared with the wild type. The proximal tubule epithelium of chop −/− animals exhibited fourfold lower levels of TUNEL-positive cells (a marker for programmed cell death), and significantly less evidence for subsequent regeneration. CHOP therefore has a role in the induction of cell death under conditions associated with malfunction of the ER and may also have a role in cellular regeneration under such circumstances.
Keywords: Cell injury, gene targeting, animal model, nephrotoxicity, renal failure
The CHOP gene (encoding the C/EBP homologous protein-10, also known as GADD153) is regulated tightly by stress in a wide variety of cells. Initially isolated based on its inducibility by genotoxic stress (Fornace et al. 1988), subsequent studies revealed that the gene is most responsive to perturbations that culminate in the induction of stress in the endoplasmic reticulum (ER). CHOP expression is coordinately regulated with the ER chaperone BiP (Wang et al. 1996; Brewer et al. 1997; Halleck et al. 1997) and is inducible by agents that directly (Bartlett et al. 1992; Chen et al. 1992; Price and Calderwood 1992; Halleck et al. 1997) or indirectly (Carlson et al. 1993; Marten et al. 1994; Bruhat et al. 1997) lead to an impairment in the folding environment of the ER (ER stress). The mechanism by which ER stress leads to CHOP gene expression is not known, however, the signal for CHOP induction appears to emanate from the ER itself and is not simply a downstream consequence of impaired ER function (Wang et al. 1996).
ER stress regulates CHOP not only by inducing expression of the gene. The CHOP protein undergoes stress-inducible phosphorylation by stress-inducible members of the p38–MAP kinase family and phosphorylation is associated with enhanced transcriptional activation by CHOP (Wang and Ron 1996). This result, together with previous experiments that had shown that CHOP is a nuclear protein that forms stable heterodimers with C/EBP family members (Ron and Habener 1992) and that the dimers are capable of recognizing novel DNA target sequences (Ubeda et al. 1996), suggests that CHOP may have a role in transducing signals from the stressed ER to changes in gene expression. A role for CHOP in effecting significant alterations in cellular phenotypes is suggested by the observation that forced overexpression of the protein leads to induction of growth arrest (Barone et al. 1994; Zhan et al. 1994) and by the association between the expression of an altered form of CHOP—that encoded by the tumor-specific translocation-derived TLS–CHOP fusion oncogene—and the development of human liposarcoma (Crozat et al. 1993; Rabbitts et al. 1993).
ER stress is present in physiological and pathological conditions. Examples include tissue ischemia and excitotoxicity in neurons (Lowenstein et al. 1994; Kuznetov et al. 1996). These insults are associated with striking alterations in cellular phenotypes that include changes in gene expression, cell death, and in some cases, tissue regeneration. In naturally occurring cellular injuries, ER stress is but one component of a general perturbation in homeostasis and it is very difficult to determine which, if any, of the phenotypic changes observed are a response to stimuli arising specifically from that cellular compartment. Analysis of more defined cellular systems in which ER function is perturbed, suggest a link between ER stress and the induction of programmed cell death. This is the case both in cells cultured in the presence of tunicamycin, an inhibitor of protein glycosylation (Larsson et al. 1993; Pérez-Sala and Mollinedo 1995; Carlberg et al. 1996; Chang and Korolev 1996; Dricu et al. 1996) and in cells that harbor temperature-sensitive mutations in essential components of the ER glycosylation apparatus (Nakashima et al. 1993; Silberstein et al. 1995). The aforementioned experimental systems do not distinguish between a possible role for the ER stress signal in inducing cell death and the possibility that cell death is a downstream consequence of impaired ER function; as might occur, for example, if essential secreted or membrane-bound proteins fail to fold properly. Recent studies, however, suggest that the ER stress-signal may have a direct role in promoting cell death. BiP overexpression in Chinese hamster ovary (CHO) cells attenuates both the ER stress-signal and the cell death that is observed in response to calcium ionophore (Morris et al. 1997) and blocking the expression of BiP by means of antisense constructs increase the lethality of agents that promote ER stress (Little and Lee 1995; Liu et al. 1997). The mediators of a possible link between ER stress and cell death, however, remain unknown.
The tight linkage between ER stress and CHOP expression and activation suggests that CHOP may have a role in eliciting cellular responses to perturbations associated with ER stress. To explore this possibility, we have created mice that are nullizygous for the chop gene and subjected the mice and cells derived from them to insults that result in ER stress. Here we report that these mice and cells derived from them are defective in the development of programmed cell death in response to ER stress. We discuss the significance of this defect in the context of the adaptation of the organism to situations that promote ER stress.
Results
Cells that lack CHOP have a normal ER stress response
By homologous recombination in murine ES cells, the coding region of chop was replaced by a neo resistance gene. Aggregation chimeras prepared with these ES cells transmitted the ES clone through the germ line, and animals heterozygous for the disrupted chop allele were mated to produce the nullizygous state (Fig. 1A). chop −/− mice were born at the expected frequency, appeared phenotypically normal and had normal fertility and reproductive behavior. The mutant allele was maintained in two different genetic backgrounds with similar results (F2 crosses of 129SVJ;CD1 and 129SVJ;129SVEV).
Figure 1.
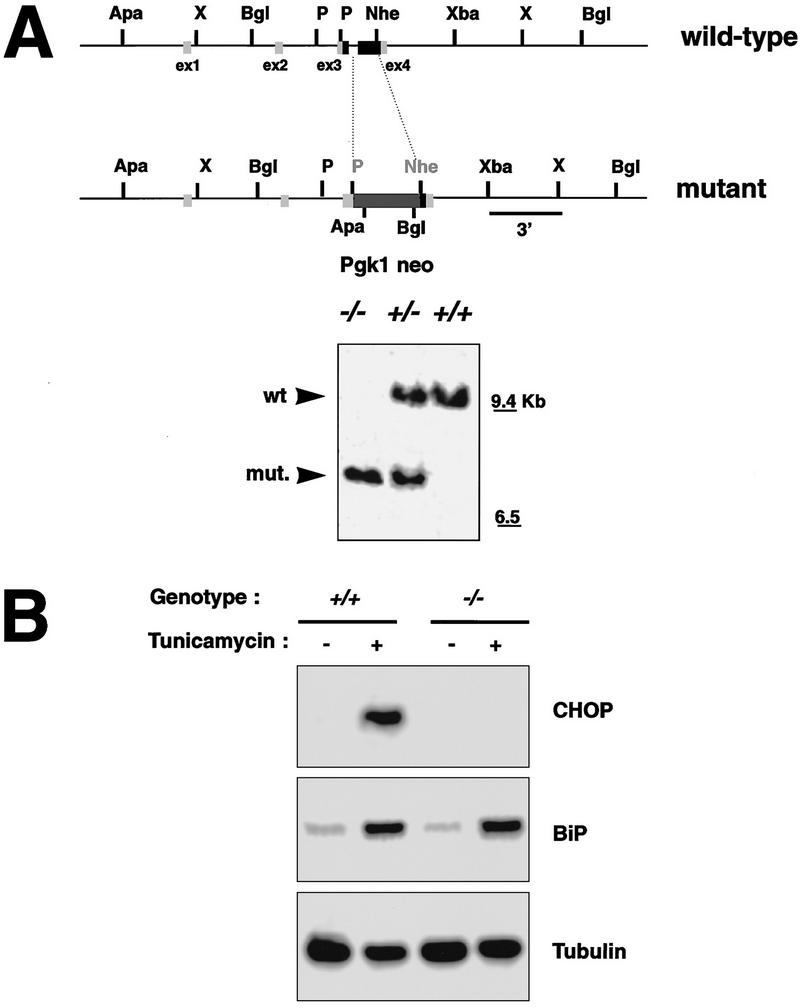
Deletion of the chop gene. (A) Structure of wild-type and mutant chop alleles. Exons are boxed, the coding region is shaded. In the targeted allele the chop-coding region, between the PmlI and the NheI sites is replaced by a PGK.neo gene (Apa–ApaI, X-XhoI, Bgl–BglII, P–PmlI, Nhe–NheI, Xba–XbaI). (Inset) An autoradiograph of a Southern blot of BglII-digested genomic DNA hybridized with a 3′ probe that is external to the targeting construct and detects both the wild-type and mutant chop alleles. (B) Northern blot analysis of CHOP, BiP, and tubulin in tunicamycin-treated (1 μg/ml for 6 hr) MEFs with the indicated chop genotypes. Note the absence of CHOP mRNA and the normal BiP induction in the chop −/− cells.
chop +/− females were mated to chop +/− males and mouse embryonic fibroblasts (MEFs) were prepared from embryonic day 13.5 (E13.5) embryos. When treated with tunicamycin, the chop gene is induced to high levels in the chop +/+ cells but is undetectable in the chop −/− cells, consistent with lack of expression of the protein in the nullizygous cells. Induction of the ER chaperone BiP is indistinguishable in the two cell populations, indicating that CHOP is not required for the ER stress response and suggesting further that cells with both chop genotypes are indistinguishable from the point of view of the development of ER stress (Figs. 1B and 2A).
Figure 2.
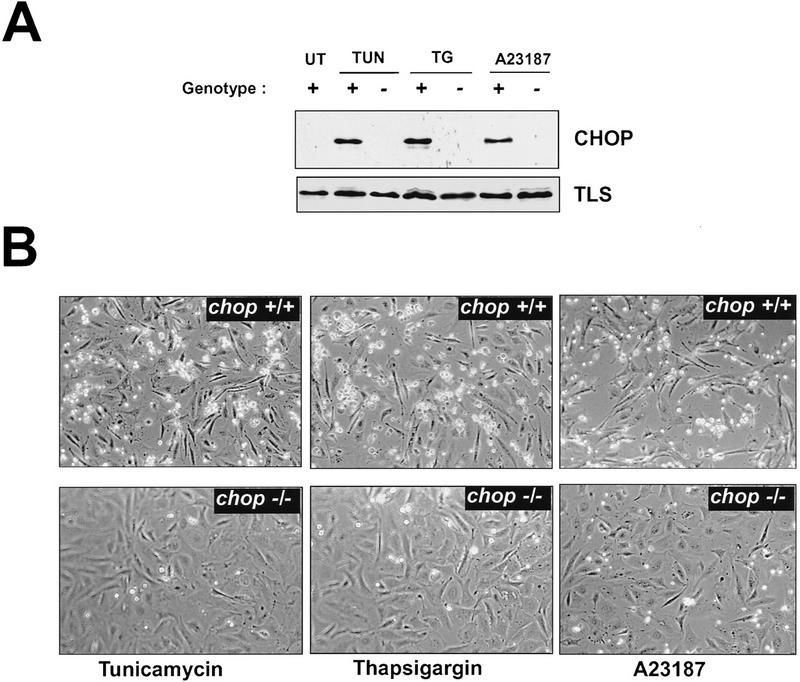

chop-deficient cells have enhanced survival when challenged with toxins that induce ER stress. (A) Western blots of CHOP and TLS (an internal control) proteins in lysates from MEFs with the indicated chop genotype following 6 hr of treatment with tunicamycin (TUN, 1 μg/ml), thapsigargin (TG, 2 μm), and calcium ionophore (A23187, 2 μm). (B) Phase-contrast photomicrograph of MEFs treated for 24 hr with the indicated compounds (320×). (C) Quantification of the fraction of dead cells as a function of time in tunicamycin treated MEFs with the indicated genotypes. (D) Survival of cells plated at low density and treated for 24 hr with the indicated dose of tunicamycin and then changed to normal growth media. At each dose, the number of colonies at 10 days in the treated plates is compared with the number of colonies in an untreated plate of the same density. Shown are mean and s.d. of a typical experiment performed in triplicate and repeated three times using independently prepared pools of MEFs.
Cells lacking chop have increased resistance to the death-promoting effects of agents that cause ER stress
CHOP is markedly induced by tunicamycin, thapsigargin (an ER-specific calcium ATPase inhibitor) and A23187 (a calcium ionophore)—all agents that cause ER stress (Price and Calderwood 1992; Fig. 2A). We treated pools of early-passage MEFs derived from sibling embryos with chop +/+ or chop −/− genotypes with agents that cause ER stress and observed the cells periodically by phase-contrast microscopy over a period of 48 hr. A reproducible pattern was noted. After a lag period (that ranged from ∼10 hr in the case of A23187 to ∼16 hr in the case of tunicamycin), the chop +/+ cells began to round up and became strikingly hyper-refringent, ultimately the cells detached from the substratum and floated in the media. The entire process was complete in 48 hr. The chop −/− cells on the other hand were strikingly resistant to these changes. At the time when the chop +/+ cells began to round up, the chop −/− cells were for the most part flat and adherent (Fig. 2B). Eventually the chop −/− cells did die in response to the toxins, but this was at a considerable delay with respect to the chop +/+ cells, and chop −/− cells remained attached to the substratum long after all the chop +/+ cells had detached. The temporal profile of the response of the cells with the two genotypes is depicted in graphic form in Figure 2C.
To determine if the differences in cell morphology observed between the two genotypes following tunicamycin treatment were also reflected in differences in cell viability, clonigenic experiment were performed. In these, cells with either genotype were plated at low density, treated with tunicamycin for 24 hr, and then allowed to recover and form colonies. A similar assay has been used by Lee and co-workers to reveal a role for BiP in promoting survival of cells in response to ER stress (Little and Lee 1995). For each dose of tunicamycin used, the chop −/− cells formed three- to fivefold more colonies than the chop +/+ ones (Fig. 2D). This indicates that a chop −/− genotype is associated with increased viability in cells challenged with the toxin.
Similar results to those shown above were observed with three different pools of MEFs procured on separate occasions, and were also found consistently when comparing individual clones with chop +/+ and chop −/− genotypes. The MEFs in Figure 2, B–D, were derived from embryos that had an equal contribution of genes from an outbred CD1 strain and the 129/SVJ strain (from which the targeted ES cells were derived). The experiment was also repeated, with similar results, in MEFs derived from more inbred animals with an equal contribution of 129/SVJ and 129/SVEV. To the extent, however, that 129/SVJ has recently been shown to be a “contaminated inbred strain” (Threadgill et al. 1997), none of these pools of MEFs can be considered truly isogenic. The use of sibling embryos and the pooling of MEFs from multiple embryos, however, minimizes the effects variation in the genetic composition of the cells may have on the phenotype in question. chop +/+ cells were also compared with chop −/+ cells in the aforementioned assays and no differences between these two genotypes were detected. We conclude that the absence of CHOP promotes increased survival of cells exposed to ER stress and that, within the resolution limits of our assay, a chop gene-dose effect is not discernible.
Programmed cell death in response to ER stress is attenuated in chop −/− cells
Tunicamycin has been reported previously to induce programmed cell death in a variety of cultured cells (Larsson et al. 1993; Carlberg et al. 1996; Dricu et al. 1996). To determine if the increased resistance of chop −/− cells to the death-promoting effects of tunicamycin is attributable to a decrease in programmed cell death, we performed FACS analysis of DNA content of tunicamycin-treated MEFs. After 24 hr of treatment with 1 μg/ml of tunicamycin, a distinct population of cells with sub-diploid DNA content, typical of cells undergoing programmed cell death, was evident in the chop +/+ cells but not in the chop −/− cells (Fig. 3A). To confirm these results, the free 3′ OH ends of the DNA from the tunicamycin-treated cells from both genotypes were labeled in situ by incorporating fluorescent-labeled nucleotides using terminal deoxynucleotide transferase (so-called TUNEL assay). The chop +/+ population contained many more labeled cells than the chop −/− population, indicating the presence of cells with degraded DNA, a feature of programmed cell death. Finally, the cells were counterstained with the fluorescent DNA-binding dye H33258, which revealed the presence of many more cells with condensed chromatin in the chop +/+ population than in the chop −/− one. Quantification of these results reveals a fourfold lower frequency of markers of programmed cell death in the chop −/− population when compared with the chop +/+ one (Fig. 3). Based on these results, we conclude that the decrease in cell death in the chop −/− population is attributable to a decrease in the proportion of cells with morphological and biochemical properties of programmed cell death.
Figure 3.



The enhanced survival of chop −/− MEFs is attributable to less programmed cell death when challenged with tunicamycin. (A) Analysis of DNA content by FACS of fixed, propidium iodide-stained MEFs with the indicated chop genotype that had been treated for 24 hr with tunicamycin (1 μg/ml). Note the presence of a population of cells with hypo-diploid DNA content typical of programmed cell death (double arrowhead). (B) (Top panel) Fluorescent photomicrographs (200×, 400×, inset) of MEFs with the indicated chop genotype after 24 hr of tunicamycin treatment (1 μg/ml) whose DNA was 3′-end labeled with fluorescent nucleotides (TUNEL assay). (Bottom panel) Cells stained with the DNA-binding dye H33258 to reveal the presence of cells with condensed chromatin (arrows). The fraction of TUNEL-positive cells after 24 hr of tunicamycin (1 μg/ml) was compared in four individual clones of chop +/+ MEFs and chop −/− MEFs (28.2% ± 4.8% and 6.75% ± 3.6 mean and s.d. respectively, ρ = 0.02, two-tailed t-test). (C) MEFs with either chop genotype undergo cell cycle arrest when exposed to tunicamycin. Shown is a dual-channel FACS analysis of MEFs released from serum starvation in the presence of tunicamycin (1 μg/ml) and 12 hr later pulsed with BrdU for 2 hr before harvest, fixed and stained with anti-BrdU to reveal the fraction of cells in S-phase and propidium iodide to estimate DNA content. Note that the fraction of cells in S-phase and M2 is lower in the treated than in the untreated cells with an even greater reduction in the chop −/− compared with the chop +/+ cells.
Overexpression of CHOP protein is capable of inducing growth arrest in cycling cells (Barone et al. 1994; Zhan et al. 1994). Tunicamycin treatment has also been linked to the induction of growth arrest (Carlberg and Larsson 1993). To determine if CHOP has a role in the ability of tunicamycin to block cell-cycle progression, cycling MEFs with chop +/+ and chop −/− genotypes were synchronized by serum deprivation and then re-fed serum containing media in the presence or absence of tunicamycin (1 μg/ml). Twelve hours later, the cells were pulsed with BrdU for 2 hr to label newly synthesized DNA, fixed and stained with a fluorescent antibody to BrdU and the fluorescent DNA-binding dye propidium iodide. Dual channel FACS analysis showed that tunicamycin treatment resulted in a significant reduction in the fraction of cells incorporating BrdU (Fig. 3C)—consistent with the ability of tunicamycin to induce cell-cycle arrest (Carlberg and Larsson 1993). The reduction in BrdU labeling, however, was not significantly different in cells with different chop genotypes. If anything, a trend toward lower incorporation of BrdU in tunicamycin-treated cells was apparent in the chop −/− genotype when compared with the chop +/+ one, and we noted that the tunicamycin-treated chop −/− cells had a flatter morphology than the chop +/+ ones (Fig. 2B). Therefore, CHOP does not appear to have an important role in the cell-cycle arrest in response to this toxin.
Programmed cell death in response to ER stress is also attenuated in cells lacking CHOP’s major dimerization partner C/EBPβ
The predominant dimerization partner of CHOP in rodent fibroblasts is C/EBPβ (Barone et al. 1994; Zinszner et al. 1994). To the extent that CHOP does not form homodimers (Ubeda et al. 1996), its activity should be dependent on the presence of a dimerization partner and therefore one would expect that MEFs deficient in C/EBPβ would exhibit an attenuated programmed cell death response when challenged with tunicamycin. Pools of MEFs from embryos derived by mating c/ebpβ +/− females and c/ebpβ −/− males (a gift of Valeria Poli, University of Dundee, UK) were treated with tunicamycin and observed by phase microscopy. The c/ebpβ +/− MEFs exhibited a normal response to tunicamycin as observed for the wild-type MEFs from the chop stock. The c/ebpβ −/− MEFs, however, experienced significantly delayed onset of death and detachment from the plate, similar to the chop −/− cells (Fig. 4A,C). H33258 staining of the cells confirmed that the differences in survival were attributable to differences in the proportion of cells exhibiting morphological features of programmed cell death (Fig. 4B). It has been suggested previously that C/EBPβ may have a role in the induction of the chop gene in response to stress (Fawcett et al. 1996). We therefore examined the induction of CHOP protein in tunicamycin-treated MEFs with different c/ebpβ genotypes. A robust induction of CHOP was evident in both c/ebpβ +/− and c/ebpβ −/− MEFs, indicating that at least in this stress-inducible system C/EBPβ is not required for CHOP induction (Fig. 4D). This result also suggests that cells with both c/ebpβ genotypes respond to tunicamycin with an equally vigorous ER stress response. The latter conclusion is also supported by the observation that BiP mRNA induction is comparable in both c/ebpβ genotypes (data not shown). These results are consistent with a role for a CHOP–C/EBPβ heterodimer in the development of programmed cell death in response to ER stress. A role for C/EBPβ overexpression in promoting cell death has been uncovered in myeloid cells (Muller et al. 1995); however, it is not known whether that process proceeds by a CHOP-dependent pathway or independently of CHOP.
Figure 4.
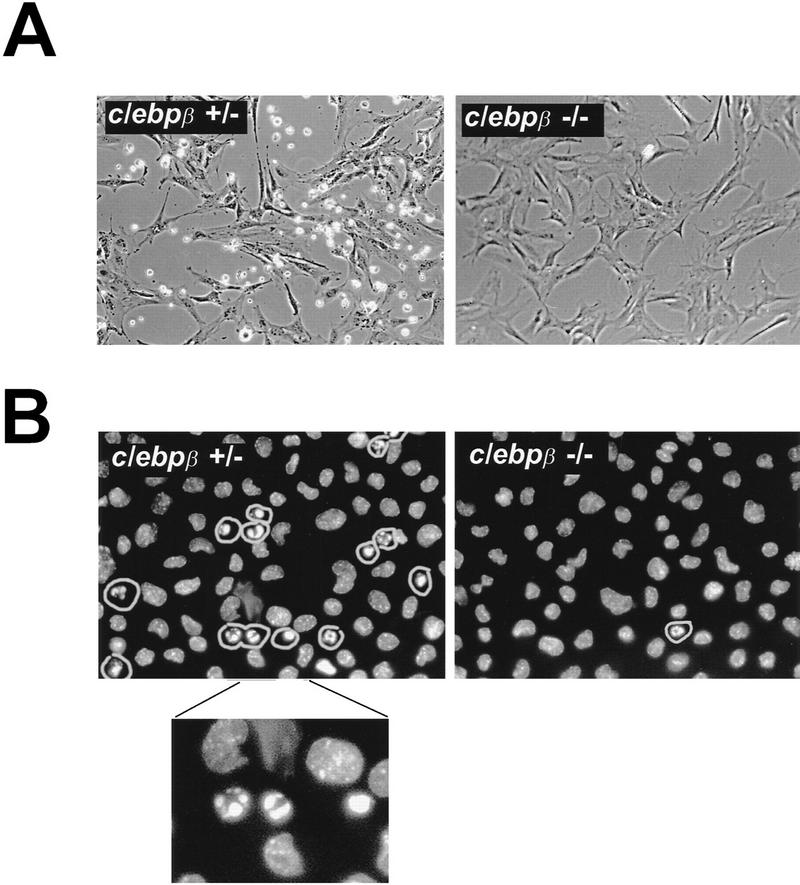


MEFs lacking CHOP’s major dimerization partner, C/EBPβ, are also resistant to the death-promoting effects of tunicamycin. (A) Photomicrograph (300×) of MEFs with the indicated c/ebpβ genotype 24 hr after treatment with tunicamycin (1 μg/ml). (B) Nuclear morphology of the same cells fixed and stained with the DNA-binding dye H33258. Note the increased fraction of cells with condensed chromatin in the c/ebpβ +/− population. (C) Quantification of the fraction of dead cells as a function of time in the two populations, analyzed as in Fig. 2C. (D) Intact CHOP response to tunicamycin in the c/ebpβ mutant cells. Shown is a Western blot of CHOP and TLS (the internal control) in untreated and tunicamycin-treated cells with the indicated genotypes.
Tunicamycin-treated chop −/− mice have decreased programmed cell death in the renal proximal tubular epithelium
A major limitation to the study of cells procured from embryos with defined genotypes is the difficulty in discriminating between effects of the mutation on the process in question from the effects of the mutation on the adaptation of cells to culture conditions, with the latter influencing the process being studied. In vivo models circumvent this limitation. To develop an in vivo system for the study of the potential effects of chop on the phenotype of injured cells, mice were injected with various doses of tunicamycin and examined for the induction of CHOP mRNA and protein. Sublethal doses of the toxin (0.25–1 mg/kg) induced a profound increase in CHOP mRNA in the kidney. Levels of CHOP mRNA are seen to rise within a few hours of tunicamycin injection and elevated levels persist for 2-3 days (Fig. 6C, below). Tunicamycin injection also resulted in a reproducible clinical picture consisting of lassitude, lack of grooming, and weight loss that peaked between day 4 and 5 post-injection and was followed by recovery on days 7–8. The peak of weight loss also corresponded to an increase in blood urea nitrogen (BUN) and creatinine (a reflection of renal impairment), although these parameters proved more variable between animals than the weight loss and kidney histology (see below). These clinical phenomena are similar to those described in other species following tunicamycin injection (Koj et al. 1986; Finnie and O’Shea 1988, 1989). Frozen sections of tunicamycin-treated kidneys from wild-type mice were immunostained for CHOP protein. Bright staining was evident in the proximal tubular epithelium (Fig. 5A). We therefore focused our analysis on this CHOP-expressing cellular compartment.
Figure 6.




Regeneration is attenuated in the chop −/− mice. (A) In vivo BrdU labeling of proximal tubular epithelial cells 5 days after tunicamycin injection. The BrdU-positive nuclei stain darkly (top panels, 100×; bottom panel, 400×). (B) Vimentin staining of frozen kidney sections following tunicamycin treatment. Note the increase in the number of vimentin-positive cells that line the tubules of the treated chop +/− samples (arrowheads). The light asterisks indicate the position of glomeruli and the dark asterisks point to vascular structures, both of which normally contain vimentin-positive mesenchymal cells. (C) Analysis of LRF/ATF3 expression (a regeneration marker) in kidneys of animals with the indicated chop genotype following tunicamycin injection (1 mg/kg, IP). (D) Plot of the relative intensity of the LRF/ATF3 signal as a function of time in animals with either genotype. Note the lack of sustained expression of the marker in the chop −/− samples when compared with the chop +/+ samples. Shown is a typical experiment that was repeated three times with similar results.
Figure 5.

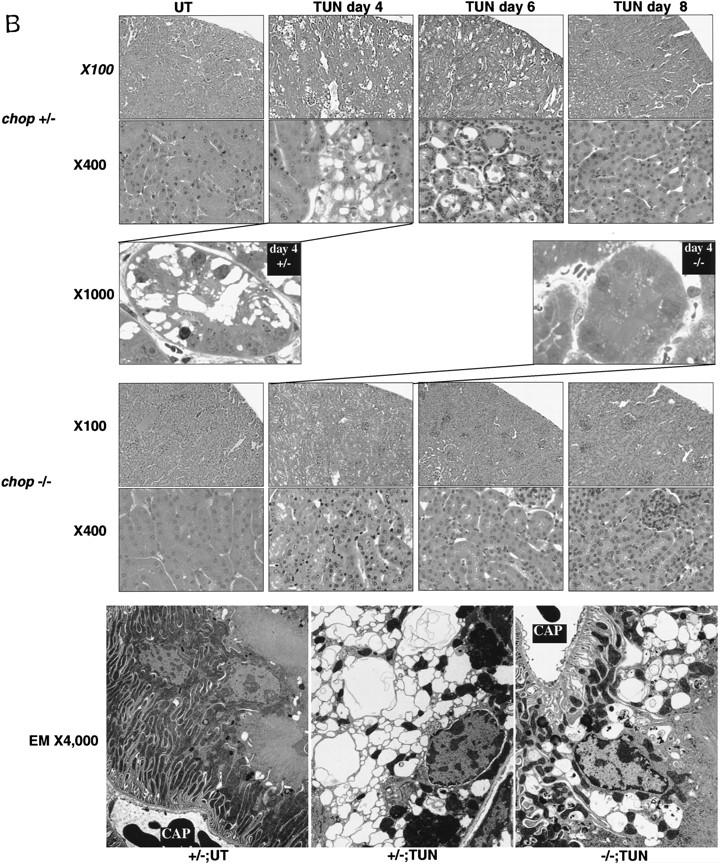


Attenuated tissue response to tunicamycin in chop −/− mice. (A) CHOP is induced in the proximal renal tubular epithelium in response to tunicamycin injection. Photomicrographs (100× and 400×, inset) of rabbit anti-CHOP immunofluorescence on frozen sections of kidneys from mice with the indicated genotypes 24 hr after tunicamycin injection (1 mg/kg, IP). Note the intense CHOP nuclear staining of the tubular cells (inset) in the cortical portions of the kidney of the treated chop +/+ sample. (B) Histological analysis of kidney sections from mice with indicated chop genotypes following tunicamycin injection (1 μg/kg). The hematoxylin and eosin stained samples are shown at magnifications of 100× and 400× and the ultrathin toludine-blue section is magnified 1000×. The electron micrographs (EM) are at a magnification of 4000×, “CAP” indicates a capillary. Note the presence of pyknotic nuclei and debris in the tubules of the +/− animals on days 4 and 6 and the absence of conspicuous light-microscopic changes in the −/− samples. At higher magnification (EM samples) dilated endosomes are visible in both genotypes. (C) TUNEL staining of day 4 kidney sections from untreated and treated mice of the indicated genotypes (100×). The insets (400×) are of identical fields stained for TUNEL and with the DNA-binding dye H33258 to reveal condensed chromatin (arrowheads). (D) The number of TUNEL-positive cells per high-powered field was quantified in two untreated wild-type (UT), nine tunicamycin-treated wild-type, and nine tunicamycin-treated −/− mice. Shown is the mean and s.d. of the number of TUNEL-positive cells per high-powered field in each sample. The means in each group were compared by a two-tailed t-test.
Tunicamycin treatment resulted in a highly reproducible histological picture in the kidneys of wild-type mice. By 72 hr, and peaking between 96 and 120 hr after treatment, the proximal tubular epithelial cells swell, followed by the appearance of cell ghosts with pyknotic nuclei and tubules with focal areas of denuded basal lamina. In the lumen of some of the tubules, cellular debris could be observed and this correlated with the appearance of cellular casts in the urine of the animals. The process was multifocal and temporally dispersed with affected tubules directly abutting normal appearing ones. Of note, the histological alterations were confined to the proximal tubular epithelium—the same compartment that expresses CHOP. The glomeruli, blood vessels, and distal tubular cells are spared (Fig. 5B). The appearance of pyknotic nuclei correlated with the presence of cells that stained positive for free 3′ OH DNA ends by the TUNEL assay (Fig. 5C). Evidence for regeneration was offered by the appearance of tubular cells that incorporated BrdU in animals injected with this S-phase marker (Fig. 6A) and by the emergence of many cells lining the tubule that stained positive for vimentin (Fig. 6B), a known regeneration marker in the proximal tubular epithelium (Wallin et al. 1992; Witzgall et al. 1994). By day 8 postinjection, all histological evidence of cellular damage had subsided and the kidneys appeared normal. This chain of events resembles that seen in other experimental toxic or metabolic insults to the kidney and is similar to the human disorder that is induced by ischemia, infection, and multiple toxins and is referred to as acute-tubular necrosis (Nonclercq et al. 1989; Wallin et al. 1992; Witzgall et al. 1994).
In the chop −/− animals the histological picture was significantly milder than that observed in the chop +/− animals. Some swelling of the proximal tubular epithelial cells was apparent at higher magnification and electron micrographs revealed that in both genotypes, tunicamycin treatment resulted in typical changes consisting of severe dilation of the ER and the formation of pseudomyelin structures (Glassy and Ferrone 1981). Cell death leading to denuded basal lamina and nuclear pyknosis, however, were less frequent in the −/− kidneys (Fig. 5B). This correlated with significantly fewer cells that stained positive by the TUNEL assay in the chop −/− animals when compared with their chop +/+ littermates. Staining of the tunicamycin-treated chop +/+ kidneys with the DNA-binding dye H33258 revealed a chromatin condensation pattern typical of programmed cell death (Fig. 5C). The TUNEL staining in the tunicamycin-treated chop −/− animals was still eightfold higher than the “background” in untreated animals, indicating that in the absence of chop, tunicamycin is still capable of leading to programmed cell death (Fig. 5D). The reduced cell death also correlated with less evidence for cellular regeneration as reflected in fewer vimentin-positive tubules and BrdU-positive cells in the kidneys of tunicamycin-injected chop −/− mice compared with their chop+ littermates (Fig. 6A,B). The effect of the chop genotype on regeneration is also reflected in nonsustained expression of the regeneration marker LRF1/ATF3 in kidneys of chop −/− animals (Hsu et al. 1991)—a reproducible finding in all three experiments performed (Fig. 6C).
These genotype-related differences in response to tunicamycin were observed at doses of the toxin ranging from 0.25 mg/kg to 1 mg/kg (lower doses were without effect and higher doses were associated with early lethality by 36–48 hr). The uniformity of the clinical and histopathological response to tunicamycin injection in the chop +/− and chop +/+ animals, suggests that genetic variation in the outbred stock (in which both the wild-type and mutant chop alleles are maintained) has a minimal effect on the response to tunicamycin toxicity. Furthermore, to minimize any effect random variation in the gene pool may have on the parameters we studied, each animal was matched with a same sex sibling of a different chop genotype. Like the experiments on the MEFs described in the previous section, we used lines of mutant mice maintained in two different (albeit not completely inbred) genetic backgrounds—129SVJ;CD1 and 129SVJ;129SVEV. The effect of the chop genotype on the renal syndrome induced by tunicamycin was evident in both genetic backgrounds.
In spite of the attenuated histological response to tunicamycin injection in chop −/− kidneys, the clinical disorder induced by the toxin is no less severe in these animals than in their chop+ counterparts. Weight loss, lassitude, and impaired renal function, as reflected in elevated blood urea nitrogen and creatinine, were similar in the two groups of animals. These clinical observations correlate with the electron micrographs that shows severe ultra-structural changes in both genotypes (Fig. 5B). These results therefore support the conclusion that CHOP has a role in the induction of programmed cell death in response to ER stress not only in cultured cells but also in animals.
Discussion
The studies described herein, comparing cells and animals that do and do not express CHOP, point to a role for this transcription factor in controlling the development of programmed cell death in response to toxins that induce ER stress. This interpretation must be qualified by certain considerations. It is formally possible, that the targeted mutation that replaces the CHOP-coding region with a PGK–neo cassette is affecting the expression of a linked gene and it is the altered expression of the latter that gives rise to the observed phenotype. This is a generic concern in interpreting gene targeting experiments in mice and can only be definitively laid to rest by rescue of the phenotype through complementation in trans. The fact that c/ebpβ −/− MEFs that lack CHOP’s major dimerization partner, however, also exhibit a similar phenotype suggests that a defect in the chop–c/ebpβ pathway and not the inactivation of a chop-linked gene is responsible for the phenotype observed in the chop −/− cells. It is also formally possible that chop exerts its effect during ontogeny, by affecting developmental processes that have an impact on the manner in which cells are programmed to respond later to ER stress. We do not, however, favor this latter explanation. First, because CHOP is not normally expressed in cells and the chop −/− animals do not have other detectable developmental defects. Secondly, CHOP is strongly induced by ER stress and this induction precedes the development of the manifestations of programmed cell death by hours in the case of the cultured MEFs and by days in the case of the kidneys in vivo, making it likely that the phenotype observed in the chop −/− cells and animals is attributable to defective signaling downstream of chop. Finally, overexpression of CHOP has been linked to the induction of programmed cell death in growth-factor dependent 32D myeloid precursor cells, providing additional evidence for a causal link between CHOP expression and apoptosis (Friedman 1996).
We have attempted to rescue the CHOP-dependent death phenotype in chop −/− cell lines derived from MEFs by constitutive overexpression of CHOP and did not observe a reproducible effect of the transgene on cell death in response to ER stress. Interpretation of that experiment, however, is made difficult by the intrinsic variation in the death response exhibited by subclones of immortal rodent fibroblasts and by the fact that constitutive expression is also likely to be a poor substitute for the normally tightly regulated inducible expression of the endogenous gene. Constitutive expression of CHOP may be associated with down-regulation of components of the pathway needed for CHOP signaling. For example, we have noted that C/EBPβ protein levels are lowered by CHOP expression in NIH-3T3 cells (Zinszner et al. 1994). Furthermore, the very process of selection of CHOP-expressing cells for study may be biased against clones with an enhanced proclivity to undergo cell death. The ideal rescue experiment would be to introduce back into the cells or the animal a chop transgene that completely mimics the endogenous one. We are currently in the process of defining the sequences required for such a transgene.
Toxins that perturb ER function might be implicated in promoting cell death through multiple overlapping pathways. For example, failure to express essential cell surface receptors might result in triggering a death signal through growth factor deprivation (Dricu et al. 1996) and the changes that occur in intracellular Ca2+ dynamics during ER stress may also contribute to the process (Lam et al. 1994; Distelhorst et al. 1996; Liu et al. 1997). Therefore, in cells undergoing ER stress, it seems likely that chop carries out its role in promoting the manifestations of programmed cell death against a highly propitious background. Clearly signals other than CHOP expression are absolutely required, as overexpression of CHOP in MEFs does not induce programmed cell death (data not shown). This interdependence between multiple factors in promoting cell death seems to be the rule rather than the exception (White 1996). As a consequence perhaps, gain-of-function or loss-of-function mutations in single genes that have an impact on death pathways tend to have rather modest quantitative effects (for examples, see Bissonnette et al. 1992; Lowe et al. 1993; Graeber et al. 1996). When viewed against this background, the approximate fourfold difference in the manifestations of programmed cell death attributed to the effect of chop are entirely compatible with a significant biological role.
This study does not address the mechanism by which chop interacts with the death-promoting machinery. We note, however, that chop does not seem to have a role in the programmed cell death that occurs in response to DNA-damaging agents, such as the topoisomerase inhibitor etoposide (H. Zinszner, unpubl.), indicating that chop action may be specific to circumstances associated with ER stress. The parameters measured here—cell morphology, TUNEL staining, and cell survival, reflect the late manifestations of the process. Therefore, we do not know if chop regulates some aspect of cellular metabolism that modifies ER stress signals, if it participates in the decision to commit to the death pathway directly or if it has an impact on the manifestation of the death process once the pathway has been activated. BiP levels are similar in tunicamycin-treated MEFs with wild-type and mutant chop genotypes, suggesting that at least the upstream components of the ER stress pathway are intact in chop −/− cells. The fact that chop −/− cells are more sensitive to the growth-arresting properties of tunicamycin than chop+ cells (Fig. 3C) may also indirectly have an impact on their relative sensitivity to the toxin. These studies have not addressed possible chop action at the initiation versus execution of the death program, but we note with interest that one of the genes identified recently as being downstream of chop encodes a stress-inducible cytosolic protein with 49% sequence identity to gelsolin (X.Z. Wang et al., unpubl.), the latter has recently been implicated in the development of cytoskeletal changes in cells undergoing programmed cell death (Kothakota et al. 1997). Although we have not yet linked the product of this chop-inducible gene to the phenotype described here, it is tempting to speculate on the possibility that CHOP directs the expression of genes that participate in defining the manifestations of programmed cell death.
What might be the significance of chop’s role in promoting the manifestations of programmed cell death? As pointed out in the introduction, ER stress and CHOP induction are features of tissue ischemia (Price and Calderwood 1992; H. Zinszner, unpubl.) and occur in response to important nephrotoxins such as cisplatin (Gately et al. 1996). These conditions are also associated with tissue damage and programmed cell death (Lieberthal and Levine 1996; Zager 1997). Therefore, a role for chop in the response of tissues to these insults could have broad clinical relevance. This study focuses on in vivo experiments using tunicamycin because it is a relatively pure system (the intracellular target of tunicamycin is defined) and because it is the least toxic and most powerful inducer of CHOP we know. Preliminary studies indicate that kidneys from chop −/− mice also exhibit less evidence of cell death when challenged with the nephrotoxin cisplatin or when subjected to temporary circulatory occlusion (H. Zinszner and D. Ron, unpubl.). The results reported here using tunicamycin appear, therefore, generalizable to other, more physiological forms of renal injury that are also associated with ER stress such as ischemia and chemotherapy-induced nephrotoxicity. The fact that CHOP induction by tunicamycin is also observed in the liver and in cultured primary cerebellar neurons (H. Zinszner and X.Z. Wang, unpubl.) suggests that the gene may be implicated in programmed cell death in response to perturbations that induce ER stress in those cell types as well. An important caveat is that we do not know the relative contribution of necrosis and apoptosis to the renal picture induced by tunicamycin—TUNEL positivity can be observed in both cellular processes. The presence of chromatin condensation in the nuclei of the proximal tubular cells (Fig. 5C) and the paucity of evidence for inflammation, however, suggest to us that programmed cell death is occurring and that the cellular changes that take place in the kidney are similar to those observed in the cultured embryonic fibroblasts. This interpretation does not conflict with the fact that the histological picture induced by tunicamycin in mice resembles the human disorder referred to historically as “acute tubular necrosis.” Recent work suggests the occurrence of programmed cell death in that disorder as well (Lieberthal and Levine 1996; Zager 1997).
The magnitude of the impairment in renal function and the clinical responses to tunicamycin are similar in animals with both chop genotypes; this is found to be the case in spite of the fact that the kidneys of tunicamycin-treated chop −/− mice are almost normal by light microscopy. Therefore, it appears that the injured cells in the chop −/− animals are incapable of performing their normal function—a finding that correlates with the presence of severe ultra-structural abnormalities in proximal tubular cells of the tunicamycin-treated mutant mice. At first glance, chop’s role in the response to ER stress appears counter-adaptive, leading to more, not less, cell death. The observation that with or without chop, renal cellular dysfunction is prominent in tunicamycin-treated mice, however, suggests an alternative explanation by which removal of the injured and dysfunctional cells by an active chop-dependent process of programmed cell death promotes regeneration and effects a more complete restoration of organ function. Regeneration presumably leads to the replacement of the damaged tubular cells with a new epithelium and is reflected in the increase in both vimentin positive and S-phase cells in the proximal tubules. The chop+ mice exhibit substantially more expression of these markers than their chop −/− siblings, although we do not know if the chop gene has an effect on regeneration that is independent of its effects on cell death.
In situations of cellular injury associated with DNA damage, failure to undergo programmed cell death has been linked to the subsequent development of cancer (Hartwell 1992). It is possible to imagine that even in the case of insults that do not primarily cause DNA damage, persistence of injured cells, or lack of vigorous regeneration signals may, in the long run, have adverse effects on tissue homeostasis. We also note that, whereas the C/EBP dimerization partners of CHOP have their homologs in flies and worms, the chop gene appears to be a relatively late addition in the genome of metazoans [the sequenced portion of the Caenorhabditis elegans genome has no obvious chop homolog and Drosophila cells do not have a detectable CHOP-like tunicamycin-inducible protein that dimerizes with C/EBPs (D. Ron, unpubl.)]. To the extent that tissue regeneration is part of the survival strategy of species with longer-lived and more complex individual organisms and is infrequently used by less complex metazoans, we propose that only the former had cause to evolve a gene such as chop. Admittedly, by day 8 post-injury, the kidneys of both chop −/− and chop +/− animals appear histologically indistinguishable and animals from both genotypes have recovered clinically. Therefore, at least in this acute model of toxicity and by the relatively crude histological parameters at our disposal for tracking the process, a regeneration defect in the chop −/− animals is not readily detectable. It remains possible, however, that longer follow-up and repeated exposure to toxin will reveal a lasting deficit in tissue integrity in chop −/− mice.
Materials and methods
Gene targeting
A targeted mutation was introduced into the murine chop gene by homologous recombination in embryonic stem cells, following an established protocol (Joyner 1993). The 5′ homology arm, in the targeting vector (pPNT; Tybulewicz et al. 1991), was the 3.5-kb XhoI–PmlI (partial) mouse genomic fragment, whose 3′ end is in chop exon 3 immediately upstream of the initiating methionine. The 3′ homology fragment was a 3-kb NheI–XbaI genomic fragment whose 5′ end is in chop exon 4. Homologous recombination results in the replacement of virtually all the chop-coding region with a PGK.neo cassette (Fig. 1). G418-resistant ES clones (3/150) in the R1 line (Nagy et al. 1993) were targeted successfully. Aggregation chimeras between targeted ES cells and CD1 morula were performed and founders bred to CD1 mice, transmission of targeted ES clone was ascertained by coat-color analysis and genotyping of the offspring. The targeted mutation was propagated in two different genetic backgrounds—intercrossing chop +/− F1s in a 129SVJ;CD1 outbred background and in a 129SVJ;129SVEV background. Some of the mice have been studied after >10 generations of chop +/− intercrossing with similar results. This level of intercrossing should permit all but the most tightly linked loci to segregate away from the mutant chop allele.
The c/ebpβ mutant mice from the colony at IRBM (Screpanti et al. 1995) were backcrossed for five generations into the inbred FVB/n strain and maintained by mating c/ebpβ −/− males to c/ebpβ −/+ females.
Cell culture and treatment
Mouse embryonic fibroblasts (MEFs) with defined genotypes were produced essentially as described (Wurst and Joyner 1993). Briefly, females were sacrificed at day 13.5 of gestation. Embryos were decapitated and eviscerated and carcasses were digested in trypsin for 30 min at 37°C. The digested and titurated cells were plated on gelatin-coated tissue-culture plates, two 100-mm plates per embryo. The cells were split 1:3 every 2–3 days and studied at passage 3–4. Cells from individual embryos were either pooled with sibling cells of identical genotype or analyzed as single embryo-derived pools. Comparisons were performed between sibling MEFs of identical passage.
MEFs at passage 3–4 were plated at 250,000 cells per 60-mm dish, in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum (Atlanta Biologicals) and studied 24 hr later at ∼75% confluence. Treatment of cells with tunicamycin, thapsigargin, and A23187 (all from Sigma) was performed essentially as described (Price and Calderwood 1992). Cells were photographed under phase-contrast with a Zeiss Axiovert 25 microscope. To quantify cell death, at the indicated time points, all the cells in the plate (adherent and floating) were combined, pelleted by slowspeed spin, resuspended in a small volume of phosphate-buffered saline (PBS, pH 7.4), and applied to an alcian blue (Sigma)-coated glass coverslip; virtually all the cells (alive and dead) adhere to the cover slip under these conditions and nuclear morphology is well preserved. The cells were fixed in PBS-buffered 4% formaldehyde and stained with the karyophilic dye H33258 and photographed through a fluorescent microscope. The live cells and cells undergoing programmed cell death were distinguished by the nuclear morphology and quantified. On parallel coverslips, the fixed cells were stained by the TUNEL procedure using a kit (Promega, apoptosis detection system) following the manufacturer’s instructions.
Analysis of clonal survival following exposure to different doses of tunicamycin was performed essentially as described (Little and Lee 1995). Briefly, early passage MEFs were plated at low dilution (∼2000 cells per 100-mm dish) in triplicate and treated 24 hr later for 24 hr with the indicated concentrations of tunicamycin. Ten days later the plates were fixed and stained with crystal violet and the number of colonies containing >50 cells were counted. Survival was calculated as the ratio between the number of colonies in the treated plates and the number of colonies in the untreated plate.
For cell cycle analysis, MEFs were plated at low density (∼25% confluence) and arrested in low serum (0.25%) for 48 hr. Serum-containing media with tunicamycin (1 μg/ml) was added for 12 hr. For the last 2 hr BrdU (Boehringer Mannheim) was added to the media at 10 μm final concentration. Cells were trypsinized and fixed in 70% EtOH for 30 min, resuspended in 2n HCl, 0.5% Triton X-100, for 30 min followed by neutralization in 0.1 m sodium borate. Cells were then staining with an anti-BrdU monoclonal antibody (Boehringer Mannheim) and an FITC-coupled secondary antibody. Before FACS analysis, cells were incubated for 30 min in a solution containing 50 μg/ml each of RNase A and propidium iodide at 37°C. Dual-channel flow cytometric analysis was performed on Becton Dickenson FACScan flow cytometer.
Western blot analysis of CHOP and TLS and Northern blot analysis of CHOP, BiP, Tubulin, and β-actin were all performed as described previously (Wang et al. 1996). The LRF/ATF3 Northern blot was hybridized to a murine cDNA fragment obtained by representational difference analysis of genes expressed in tunicamycin-treated chop +/− and chop −/− mouse kidneys (X.Z. Wang, unpubl.). This fragment lies between two DpnII sites in the 3′ UTR of the murine ATF3/LRF cDNA (nucleotides 1305–1718).
Animal experiments and tissue sample analysis
All animal experiments were pre-approved by NYU’s institutional animal care and utilization committee. Sibling mice (6- to 10-week-old), matched for sex and discordant at the chop locus, were given a single 1 μg/gram body weight intraperitoneal injection of a 0.05 mg/ml suspension of tunicamycin in 150 mm dextrose. At various times thereafter, the mice were killed by CO2 narcosis. Kidneys were removed and fixed in either Carnoy’s solution (6:3:1 vol/vol ratio of ethanol, chloroform and acetic acid, for hematoxylin and eosin staining), 4% PBS buffered formalin (for TUNEL assay and BrdU labeling), or snap-frozen in liquid nitrogen and stored at −80°C until analysis (for vimentin and CHOP immunohistochemistry). Fixed samples were paraffin embedded and 5 μm sections mounted on glass slides. Both histological analysis of stained slides and TUNEL staining were performed by a party unaware the animals genotype or treatment group (R.T. Lightfoot and J.L. Stevens).
CHOP immunostaining was performed on 5-μm cryosections fixed in 4% buffered formalin, after blocking with 1% donkey serum in PBS, the sections were incubated with rabbit anti-CHOP polyclonal serum (Ron and Habener 1992) at 1:1000 for 1 hr at room temperature and staining was revealed by a secondary FITC coupled donkey anti-rabbit serum at a dilution of 1:100 (Jackson ImmunoResearch labs).
BrdU immunostaining was performed on tissues from animals injected intraperitoneally with BrdU 100 μg/gram body weight (as a 5 mg/ml solution in 0.007 m NaOH) 2 hr before sacrifice. Paraffin sections of formalin-fixed kidneys from BrdU-injected animals were deparaffinized, treated with proteinase K (Boehringer Mannheim) in 0.1N HCl for 1 hr at 37°C, and bleached in a solution of 10% H2O2 in methanol for 10 min. After blocking with 1% normal rabbit serum, sections were incubated overnight with a rat anti-BrdU monoclonal antibody (Harlan Sera-lab Ltd.) and revealed by a rabbit anti-Rat IgG polyclonal antibodies coupled to horseradish peroxidase (Jackson ImmunoResearch labs). Enzyme activity was detected using a peroxidase substrate-DAB kit from Vector Lab Inc.
Vimentin immunostaining was performed on 5 μm cryosections of kidneys fixed in 100% EtOH for 15 min, air-dried, and rehydrated. After blocking, sections were incubated with a goat anti-vimentin antiserum (Medina et al. 1983) 1:100 overnight at 4°C. FITC-conjugated rabbit anti-goat immunoglobulin at 1:200 were used to reveal the staining (Jackson ImmunoResearch Labs). Electron micrographs and ultrathin (1 μm, toludine-blue-stained sections) were prepared from tissue fixed in 2.5% gluteraldehyde 0.1 m Na cacodylate (pH 7.4). For EM, the sample was embedded in epon resin EMbed-812 (EM Sciences, Fort Washington, Pennsylvania), polymerized, thin sectioned, and stained with uranyl acetate and lead citrate. The specimens were viewed with JEOL 100XII electron microscope.
Acknowledgments
We are grateful to Anna Auerbach from the NYU ES Cell and Transgenic (ESTG) facility for expert assistance in production of the chop− mice; to Valeria Poli for the gift of the c/ebpβ deficient mice; to Bonnie B. Asch for the gift of anti-vimentin goat serum; to Ed Skolnik, Lennart Philipson, and members of their labs and ours for useful discussions. This project was supported by U.S. Public Health Service awards ES08681 and DK47119 (to D.R.) and DK46267 and ES07847 (to J.L.S.). The core mouse transgenic facility at NYU is supported by National Cancer Institute. D.R. is a Stephen Birnbaum Scholar of the Leukemia Society of America. H.Z. was supported in part by a grant from Institute National de la Santé de la Recherche Médicale.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL ron@saturn.med.nyu.edu; FAX (212) 263-8591.
References
- Barone MV, Crozat AY, Tabaee A, Philipson L, Ron D. CHOP (GADD153) and its oncogenic variant, TLS-CHOP, differ in their ability to induce G1/S arrest. Genes & Dev. 1994;8:453–464. doi: 10.1101/gad.8.4.453. [DOI] [PubMed] [Google Scholar]
- Bartlett J, Luethy J, Carlson S, Sollott S, Holbrook N. Calcium ionophore A23187 induces expression of the growth arrest and DNA damage inducible CCAAT/enhancer-binding protein (C/EBP)-related gene, gadd153. J Biol Chem. 1992;267:20465–20470. [PubMed] [Google Scholar]
- Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:552–554. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- Brewer JW, Cleveland JL, Hendershot LM. A pathway distinct from the mammalian unfolded protein response regulates expression of endoplasmic reticulum chaperones in non-stressed cells. EMBO J. 1997;16:7207–7216. doi: 10.1093/emboj/16.23.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruhat A, Jousse C, Wang X-Z, Ron D, Ferrara M, Fafournoux F. Amino acid limitation induces expression of chop, a CCAAT/enhancer binding protein related gene at both transcriptional and post-transcriptional levels. J Biol Chem. 1997;272:17588–17593. doi: 10.1074/jbc.272.28.17588. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Larsson O. Role of N-linked glycosylation in cell-cycle progression and initiation of DNA synthesis in tumor-transformed human fibroblasts. Anticancer Res. 1993;13:167–171. [PubMed] [Google Scholar]
- Carlberg M, Dricu A, Blegen H, Kass G, Orrenius D, Larsson O. Short exposure to tunicamycin induce apoptosis in SV40 transformed cells but not in normal human fibroblasts. Carcinogenesis (Lond) 1996;17:2589–2596. doi: 10.1093/carcin/17.12.2589. [DOI] [PubMed] [Google Scholar]
- Carlson SG, Fawcett TW, Bartlett JD, Bernier M, Holbrook NJ. Regulation of the C/EBP-related gene, gadd153, by glucose deprivation. Mol Cell Biol. 1993;13:4736–4744. doi: 10.1128/mcb.13.8.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Korolev V. Specific toxicity in induction of programmed cell death of sympathetic neurons. Exp Neurol. 1996;137:201–211. doi: 10.1006/exnr.1996.0019. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yu K, Holbrook NJ, Stevens JL. Activation of the growth arrest and DNA damage-inducible gene gadd153 by nephrotoxic cysteine conjugates and dithiothreitol. J Biol Chem. 1992;267:8207–8212. [PubMed] [Google Scholar]
- Crozat AY, Åman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma with t(12;16)(q13;p11) Nature. 1993;363:640–644. doi: 10.1038/363640a0. [DOI] [PubMed] [Google Scholar]
- Distelhorst CW, Lam M, McCormick TS. Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene. 1996;12:2051–2055. [PubMed] [Google Scholar]
- Dricu A, Carlberg M, Wang M, Larsson O. Inhibition of N-linked glycosylation using Tunicamycin causes cell death in malignant cells: Role of down regulation of the insulin-like growth factor 1 receptor in induction of apoptosis. Cancer Res. 1996;57:543–548. [PubMed] [Google Scholar]
- Fawcett TW, Eastman HB, Martindale JL, Holbrook NJ. Physical and functional association between GADD153 and CCAAT/enhancer-binding protein beta during cellular stress. J Biol Chem. 1996;271:14285–14289. doi: 10.1074/jbc.271.24.14285. [DOI] [PubMed] [Google Scholar]
- Finnie J, O’Shea J. Pathological and pathogenetic changes in the central nervous system of guinea pigs given tunicamycin. Acta Neurologica (Berlin) 1988;75:411–421. doi: 10.1007/BF00687795. [DOI] [PubMed] [Google Scholar]
- ————— Acute hepatotoxicity with resultant pulmonary and cerebral embolism in guinea pigs given tunicamycin. Pathology. 1989;21:194–199. doi: 10.3109/00313028909061058. [DOI] [PubMed] [Google Scholar]
- Fornace A, Alamo I, Hollander M. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AD. GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res. 1996;56:3250–3256. [PubMed] [Google Scholar]
- Gately D, Sharma A, Christen R, Howell S. Cisplatin and taxol activate different signal pathways regulating cellular injury-induced expression of GADD153. Br J Cancer. 1996;73:18–23. doi: 10.1038/bjc.1996.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassy MC, Ferrone S. Ultrastructural alterations in human lymphoblastoid B cell lines treated with tunicamycin. Am J Pathol. 1981;103:1–9. [PMC free article] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours [see comments] Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Halleck M, Holbrook N, Skinner J, Liu H, Stevens J. The molecular response to reductive stress in LLC-PK1 renal epithelial cells: Coordinate transcriptional regulation of gadd153 and grp78 genes by thiols. Cell Stress Chaperones. 1997;2:31–40. doi: 10.1379/1466-1268(1997)002<0031:tmrtrs>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1992;71:543–546. doi: 10.1016/0092-8674(92)90586-2. [DOI] [PubMed] [Google Scholar]
- Hsu J-C, Laz T, Mohn K, Taub R. Identification of LRF-1, a leucine-zipper protein that is rapidly and highly induced in regenerating liver. Proc Natl Acad Sci. 1991;88:3511–3513. doi: 10.1073/pnas.88.9.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner A. Gene targeting—a practical approach. Oxford, UK: IRL Press; 1993. [Google Scholar]
- Koj A, Bereta J, Dubin A, Kurdowska A, Chindemi P, Regoeczi E. Alpha-1-acute phase globulin in the blood of tunicamycin-injected rats. Isolation of the non-glycosylated form, its inhibitory properties and synthesis in liver slices. Folia Histochem Cytobiol. 1986;24:7–14. [PubMed] [Google Scholar]
- Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT. Caspase-3-generated fragment of gelsolin: Effector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Kuznetov G, Bush K, Zhang P, Nigam S. Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc Natl Acad Sci. 1996;93:8584–8589. doi: 10.1073/pnas.93.16.8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Carlberg M, Zetterberg A. Selective killing induced by an inhibitor of N-linked glycosylation. J Cell Sci. 1993;106:299–307. doi: 10.1242/jcs.106.1.299. [DOI] [PubMed] [Google Scholar]
- Lieberthal W, Levine JS. Mechanisms of apoptosis and its potential role in renal tubular epithelial cell injury. Am J Physiol. 1996;271:F477–F488. doi: 10.1152/ajprenal.1996.271.3.F477. [DOI] [PubMed] [Google Scholar]
- Little E, Lee AS. Generation of a mammalian cell line deficient in glucose-regulated protein stress induction through targeted ribozyme driven by a stress-inducible promoter. J Biol Chem. 1995;270:9526–9534. [PubMed] [Google Scholar]
- Liu H, Bowes RCR, van de Water B, Sillence C, Nagelkerke JF, Stevens JL. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J Biol Chem. 1997;272:21751–21759. doi: 10.1074/jbc.272.35.21751. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Lowenstein D, Gwinn R, Seren M, Simon R, McIntosh T. Increased expression of mRNA encoding calbindin-D28K, the glucose regulated proteins, or the 72 kDA heat-shock protein in three models of acute CNS injury. Mol Brain Res. 1994;22:299–308. doi: 10.1016/0169-328x(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Marten NW, Burke EJ, Hayden JM, Straus DS. Effect of amino acid limitation on the expression of 19 genes in rat hepatoma cells. FASEB J. 1994;8:538–544. doi: 10.1096/fasebj.8.8.8181673. [DOI] [PubMed] [Google Scholar]
- Medina D, Miller F, Oborn CJ, Asch BB. Mitochondrial inclusions in selenium-treated mouse mammary epithelial cell lines. Cancer Res. 1983;43:2100–2105. [PubMed] [Google Scholar]
- Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ. Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem. 1997;272:4327–4334. doi: 10.1074/jbc.272.7.4327. [DOI] [PubMed] [Google Scholar]
- Muller C, Kowenz-Leutz E, Grieser-Ade S, Graf T, Leutz A. NF-M (chicken C/EBP beta) induces eosinophilic differentiation and apoptosis in a hematopoietic progenitor cell line. EMBO J. 1995;14:6127–6135. doi: 10.1002/j.1460-2075.1995.tb00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima T, Sekiguchi T, Kuraoka A, Fukushima K, Shibata Y, Komiyama S, Nishimoto T. Molecular cloning of a human cDNA encoding a novel protein, DAD1, whose defect causes apoptotic cell death in hamster BHK21 cells. Mol Cell Biol. 1993;13:6367–6374. doi: 10.1128/mcb.13.10.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonclercq D, Toubeau G, Laurent G, Tulkens PM, Heuson-Stiennon JA. Tissue injury and repair in the rat kidney after exposure to cisplatin or carboplatin. Exp Mol Pathol. 1989;51:123–140. doi: 10.1016/0014-4800(89)90013-0. [DOI] [PubMed] [Google Scholar]
- Pérez-Sala D, Mollinedo F. Inhibition of N-linked glycosylation induces early apoptosis in human promyelocytic HL-60 cells. J Cell Physiol. 1995;163:523–531. doi: 10.1002/jcp.1041630312. [DOI] [PubMed] [Google Scholar]
- Price B, Calderwood S. Gadd 45 and Gadd 153 messenger RNA levels are increased during hypoxia and after exposure of cells to agents which elevate the levels of glucose-regulated proteins. Cancer Res. 1992;52:3814–3817. [PubMed] [Google Scholar]
- Rabbitts TH, Forster A, Larson R, Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nature Genet. 1993;4:175–180. doi: 10.1038/ng0693-175. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant negative inhibitor of gene transcription. Genes & Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, Sellitto C, Scarpa S, Bellavia D, Lattanzio G, Bistoni F, Frati L, Cortese R, Gulino A, Ciliberto G, et al. Lymphoprolifrative disorder and imbalanced T-helper response in C/EBPβ-deficient mice. EMBO J. 1995;14:1932–1941. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein S, Collins P, Kelleher D, Gilmore R. The essential OST2 gene encodes the 16-kD subunit of the yeast oligosaccharyltransferase, a highly conserved protein expressed in diverse eukaryotic organisms. J Cell Biol. 1995;131:371–383. doi: 10.1083/jcb.131.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Yee D, Matin A, Nadeau JH, Magnuson T. Genealogy of the 129 inbred strains: 129/SvJ is a contaminated inbred strain. Mamm Genome. 1997;8:390–393. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V, Crawford C, Jackson P, Bronson R, Mulligan R. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Wang X-Z, Zinszner H, Wu I, Habener J, Ron D. Stress-induced binding of transcription factor CHOP to a novel DNA-control element. Mol Cell Biol. 1996;16:1479–1489. doi: 10.1128/mcb.16.4.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin A, Zhang G, Jones TW, Jaken S, Stevens JL. Mechanism of the nephrogenic repair response. Studies on proliferation and vimentin expression after 35S-1,2-dichlorovinyl-L-cysteine nephrotoxicity in vivo and in cultured proximal tubule epithelial cells. Lab Invest. 1992;66:474–484. [PubMed] [Google Scholar]
- Wang X-Z, Lawson B, Brewer J, Zinszner H, Sanjay A, Mi L, Boorstein R, Kreibich G, Hendershot L, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X-Z, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP-kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- White E. Life, death, and the pursuit of apoptosis. Genes & Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- Witzgall R, Brown D, Schwarz C, Bonventre J. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. J Clin Invest. 1994;93:2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurst W, Joyner A. Gene targeting—A practical approach. Oxford, UK: IRL Press; 1993. Production of targeted embryonic stem cell clones; pp. 36–41. [Google Scholar]
- Zager RA. Pathogenetic mechanisms in nephrotoxic acute renal failure. Semin Nephrol. 1997;17:3–14. [PubMed] [Google Scholar]
- Zhan Q, Liebermann DA, Alamo I, Hollander MC, Ron D, Kohn KW, Hoffman B, Fornace AJ. The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that cooperatively suppress cell growth. Mol Cell Biol. 1994;14:2361–2371. doi: 10.1128/mcb.14.4.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Albalat R, Ron D. A novel effector domain from the RNA-binding proteins TLS or EWS is required for oncogenic transformation by CHOP. Genes & Dev. 1994;8:2513–2526. doi: 10.1101/gad.8.21.2513. [DOI] [PubMed] [Google Scholar]