

Abstract
Like human immunodeficiency virus (HIV), feline immunodeficiency virus (FIV) invades and infects the central nervous system (CNS) soon after peripheral infection. The appearance of viral RNA is particularly prominent in the cerebrospinal fluid (CSF), suggesting an efficient route of virus transfer across the blood-CSF barrier. This raises the concern whether this route can establish a stable viral reservoir and also be a source of virus capable of reseeding peripheral systems. To examine this possibility, 200 μl of cell-free NCSU1 FIV or FIV-infected choroid plexus macrophages (ChP-Mac) was directly injected into the right lateral ventricle of the brain. Negative controls were sham inoculated with uninfected ChP-Mac or virus-free culture supernatant and positive controls were infected systemically by intraperitoneal (i.p.) injection. Intracerebroventricular (i.c.v.) inoculation with cell-free FIV resulted in high levels of plasma FIV RNA detected as early as 1 to 2 weeks post inoculation in all cats. In each case, the plasma viremia preceded the detection of CSF viral RNA. Compared to i.p. cats, i.c.v. cats had 32-fold higher CSF viral loads, 8-fold higher ratios of CSF to plasma viral load, and a 23-fold greater content of FIV proviral DNA in the brain. No FIV RNA was detected in plasma or CSF from the cats inoculated with FIV-infected ChP-Mac but an acute inflammatory response and a slight suppression of the CD4+:CD8+ ratio were observed. These results indicate that free FIV circulating in the CSF promotes infection of the CNS and provides a highly efficient pathway for the transfer of infectious virus to the periphery.
Keywords: dementia, HIV, macrophages, T cells, viremia
Introduction
Feline immunodeficiency virus (FIV) infection results in immunological and neurological changes similar to those seen in humans induced by human immunodeficiency virus type-1 (HIV-1) (Dow et al, 1990; Hurtrel et al, 1992; Phillips et al, 1994; Henriksen et al, 1995; Boche et al, 1996; Power et al, 1997, 1998; Meeker et al, 1997). Like HIV, FIV enters and infects the central nervous system (CNS) soon after peripheral infection (Dow et al, 1990; Hurtrel et al, 1992; Boche et al, 1996; Ryan et al, 2003, 2005). However, the temporal and mechanistic details of HIV-1 and FIV entry into the CNS and subsequent spread are still poorly understood. Studies with the animal lentiviruses, simian immunodeficiency virus (SIV) and FIV, in which the earliest penetration of virus could be carefully measured, have indicated that the virus appears in the cerebrospinal fluid (CSF) at approximately the same time as the peak plasma viremia (Dow et al, 1990; Hurtrel et al, 1991; Budka 1991b; Gray et al, 1996; Boche et al, 1996; Zink et al, 1999). The close temporal relationship between plasma and CSF viremia suggests that virus exchange from plasma to CSF, across the blood-brain barrier (BBB) and/or the blood-CSF barrier, is very efficient. Considerable evidence supports the idea that virus carried across the BBB by trafficking monocytes and possibly lymphocytes contributes significantly to infection of the brain (Hurtrel et al, 1993; Boche et al, 1996; Martin et al, 1998; Williams et al, 2001). The contribution of virus trafficking into the CSF through the blood-CSF barrier has been less well studied.
The choroid plexus (ChP), located in the cranial ventricles, is responsible for the production of CSF, and separates the blood from the CSF via the blood-CSF barrier (Segal, 2000; Redzic and Segal, 2004). This barrier is due to the presence of tight junctions between the cubical epithelium in the ChP (Redzic and Segal, 2004). Unlike the parenchymal capillaries with tight junctions between adjacent endothelial cells, ChP capillaries are fenestrated (Segal, 2000; Lossinsky and Shivers, 2004). The ChP stroma, lying between the capillaries and the epithelium, contains a high concentration of differentiated macrophages (Bragg et al, 2002b; Redzic and Segal, 2004). This structure provides an excellent opportunity for lentiviral interactions with macrophages or other cells trafficking through the ChP. Previous studies have shown that the choroid plexus is a prominent site for infection by HIV-1 (Falangola et al, 1995; Petito et al, 1999; Chen et al, 2000; Liu et al, 2000) or FIV (Bragg et al, 2002a, 2002b) and contains unique viral variants (Chen et al, 2000; Liu et al, 2000). These data suggest that this pathway may not only be an efficient route for infection of the brain but also a source for the development of local viral quasispecies. Although the early invasion of the CNS by lentiviruses does not cause significant brain disease, it is thought to seed the brain with virus and set the stage for the gradual spread of infection and a diffuse inflammatory activation (Hurtrel et al, 1992; Boche et al, 1996; Gray et al, 1996, 2001; Williams and Hickey, 2002). Subsequent to infection, the CNS may act as a persistent anatomical reservoir for lentiviruses. The nature of this potential reservoir is still under investigation. Although active lentivirus replication is difficult to detect in the brain of the asymptomatic host, a non-productive infection may be maintained (Boche et al, 1995; Blankson et al, 2002; Clements et al, 2002). Virus sequestered in the CNS remains a serious obstacle to the therapeutic eradication of HIV from infected individuals. Virus within the CNS may also give rise to the development of or sequestration of unique quasispecies. Compartmentalized viral variants have been identified in brain parenchyma and the choroid plexus including strains resistant to antiretroviral compounds (Shapshak et al, 1999; Chen et al, 2000; Cunningham et al, 2000; Antinori et al, 2003; Strain et al, 2005). All of these observations increase concerns whether the CNS can act as a stable reservoir for HIV with the potential to evolve independently from systemic virus and reseed the peripheral system with infectious virus if antiretroviral therapy is terminated. The ability of infectious virus to transfer from the brain to the systemic circulation via the CSF is largely unknown as few studies have directly assessed the potential of this pathway. In the following studies, we determined if virus penetrating into the CSF could establish a clinically significant primary infection of the brain and also evaluated the ability of infectious virus to penetrate from the CSF to the systemic circulation. Results from our in vivo studies demonstrated that the CSF serves as an important route both for infection of the brain and for the transfer of infectious virions into the periphery.
Results
Viral load in blood following i.p. and i.c.v. inoculation of FIV
The plasma viral RNA of infected cats was measured by real-time reverse transcriptase–polymerase chain reaction (RT-PCR). Treatment groups are summarized in Table 1. All infected cats receiving cell-free FIV showed similar kinetics for the plasma viremia regardless of the route of infection. A comparison of the mean viral load in plasma following intraperitoneal (i.p.) versus intracerebroventricular (i.c.v.) inoculation is shown in Figure 1. The plasma viremia appeared soon after infection and rapidly reached high levels (104 to 108 FIV copies/ml) 1 to 2 weeks after inoculation in all the cats receiving cell-free FIV. After the initial peak, plasma viral RNA dropped steadily and stabilized in the range of 103 to 106 FIV copies/ml until the end of the study (32 to 56 weeks post inoculation). However, the average peak viremia was higher and the plasma viremia was maintained at higher levels in the i.c.v. inoculated cats. Between 16 and 32 weeks post inoculation, the plasma FIV RNA levels in i.c.v. cats were relatively stable at an average value 1.3 log10 higher than that of cats receiving an i.p. inoculation (t = 5.14, df = 6, p = 0.002).
Table 1.
Treatment groups and weeks post inoculation at euthanasia
| Cat | Treatments | Weeks at euthanasia |
|---|---|---|
| 1 | Cell-free FIV i.c.v. inoculated | 56 |
| 2 | Cell-free FIV i.c.v. inoculated | 56 |
| 3 | Cell-free FIV i.c.v. inoculated | 56 |
| 4 | Cell-free FIV i.c.v. inoculated | 56 |
| 5 | Cell-free FIV i.c.v. inoculated | 12b |
| 6 | Cell-free FIV i.c.v. inoculated | 32 |
| 7 | FIV-free supernatant i.c.v. inoculated | 56 |
| 8 | FIV-free supernatant i.c.v. inoculated | 56 |
| 9 | FIV-free supernatant i.c.v. inoculated | 56 |
| 10 | Cell-free FIV i.p. inoculated | 32 |
| 11 | Cell-free FIV i.p. inoculated | 32 |
| 12 | Cell-free FIV i.p. inoculated | 32 |
| 13 | FIV infected ChP-Mac i.c.v. inoculated | 32 |
| 14 | FIV infected ChP-Mac i.c.v. inoculated | 32 |
| 15 | FIV infected ChP-Mac i.c.v. inoculated | 32 |
| 16 | FIV infected ChP-Mac i.c.v. inoculated | 32 |
| 17 | FIV infected ChP-Mac i.c.v. inoculatedc | — |
| 18 | Uninfected ChP-Mac i.c.v. inoculated | 32 |
i.c.v., intracerebroventricular; ChP-Mac, feline fetal choroid plexus macrophages; i.p., intraperitoneal.
Euthanized at 12 weeks post inoculation because of significant neurological signs.
inoculated with 3 × 105 FIV infected ChP-mac and euthanized at 16 h post inoculation.
Figure 1.

Comparison of the average plasma viral loads of cats infected i.c.v. (n = 6) or i.p. (n = 3) with the same dose (2 × 105 TCID50) of cell-free NCSU1 FIV at 0 week. RNA levels in plasma were determined by real time RT-PCR at the indicated time points. The plasma viremia appeared within the first week after infection and rapidly reached high levels (104 to 108 FIV copies/ml) 1 to 2 weeks after inoculation. After the initial viremia peak, plasma viral RNA dropped steadily. By 16 to 32 weeks the viremia in the i.c.v. cats stabilized at an average of 1.3 log10 higher than that of i.p. cats (P = .002). Values are mean ± SEM (* P < .05).
FIV provirus was detected in the peripheral blood mononuclear cells (PBMCs) of all i.c.v. and i.p. infected cats by 6 weeks post inoculation and remained positive for the remainder of the study. Changes in the PBMC proviral burdens over time are summarized in Figure 2. At 6 weeks post inoculation, the PBMC proviral DNA was approximately seven-fold greater in the i.c.v. cats (24,675 copies/106 cells). This difference increased to 163-fold by 16 weeks (103,344 copies/106 cells in the i.c.v. cats). With the exception of the 24-week time point, the PBMC proviral loads remained 7- to 36-fold greater in the i.c.v. cats (9001 to 11,445 copies/106 cells in the i.c.v. cats).
Figure 2.

Changes in the PBMCs proviral burden over time. With the exception of week 24, the average proviral burden (log copies FIV DNA/106 cells) of the i.c.v. cats ranged from 7- to 163-fold greater than the i.p. cats. The average proviral burden in the i.c.v. cats across all time points was 27, 594 ± 9954 versus 2086 ± 730 for the i.p. cats. Values are mean ± SEM (* P < .05).
Cats inoculated with FIV-infected ChP-Mac by i.c.v. failed to establish a detectable plasma viremia in each case (four of four cats). FIV viral DNA was detected in the macrophage inoculum by FIV env nested PCR, indicating that these cells had been infected in vitro. Control cats inoculated with uninfected CD4E cell supernatant or uninfected ChP-Mac were negative for viral RNA and DNA throughout the study period.
Lymphocyte subset changes following i.p. and i.c.v. inoculation with FIV
The lymphocyte subset changes in FIV infected cats or control cats were analyzed by flow cytometry. Cell-free FIV infection resulted in a rapid and progressive drop of the CD4+:CD8+ T-cell ratio in all the cats, which was slightly faster in the i.c.v. cats (Figure 3). Cats inoculated with FIV infected ChP-Mac showed a modest drop in the CD4+:CD8+ T-cell ratio to 67%–78% of the preinoculation values, which was significant by 4 weeks postinoculation (t = 19.36, df = 3, p = .0003). Control cats maintained CD4+:CD8+ T cell ratios at or slightly above the pre-inoculation values. Although CD4+ T cells dropped slightly at the first weeks, the rapid decrease in the CD4+:CD8+ T-cell ratio was largely due to a marked (~8-fold) and prolonged expansion of CD8+ T cells.
Figure 3.

Mean CD4+:CD8+ T-cell ratio of cats following i.c.v. inoculation with cell-free FIV (FIV-I.C.V., n = 6), i.p. inoculation with cell-free FIV (FIV-I.P., n = 3) or i.c.v. inoculation with FIV infected ChP macrophages (ChP-Mac, n = 4). The ratio after inoculation was normalized to the preinoculation CD4+:CD8+ ratio. Cell-free FIV infection resulted in a progressive drop of the CD4+:CD8+ ratio in all cats regardless of the inoculation route (Ps < .001). Cats inoculated with FIV infected ChP-Mac showed a mild but significant drop in the CD4+:CD8+ ratio to 67% to 78% of the preinoculation values (P = .0003). Values are mean ± SEM.
CSF viral loads following i.p. and i.c.v. inoculation with FIV
Viral RNA in the CSF, measured longitudinally by real time RT-PCR, was detected in all cats receiving i.p. or i.c.v. cell-free FIV inoculations, but not in the cats inoculated with FIV-infected ChP-Mac. Individual comparisons of the viral RNA kinetics within CSF and plasma of the cats inoculated i.c.v (cats 1 to 6) or i.p. (cats 10 to 12) with cell-free FIV is provided in Figure 4. Although direct inoculation into the lateral ventricle resulted in high CSF viral titers in all six i.c.v. cats, it failed to produce a rapid productive infection confined to the CSF compartment. Instead, the CSF viral RNA always appeared after the plasma viremia, typically by 1 to 3 weeks. Of note, five of the six i.c.v. cats showed a large second rise in the CSF viral peak (mean = 2.4 × 105 copies/ml), which was not correlated with a rise in plasma virus. CSF virus was also detected in the i.p. cats soon after systemic inoculation, similar to previous published results (Ryan et al, 2003). However, the level of CSF virus in i.p. cats was much lower than the i.c.v. cats (Table 2) and a significant secondary CSF viral peak never appeared.
Figure 4.

Virus kinetics in CSF and plasma in individual FIV infected cats after i.c.v. inoculation (cats 1 to 6) or i.p. inoculation (cats 10 to 12). The upper three graphs illustrate the three cats with highest CSF viral loads. The middle three figures illustrate three i.c.v. cats with moderate CSF viral loads and the bottom three figures illustrate the three i.p. cats. Five of six i.c.v. cats showed a large secondary viral peak in the CSF, which was not seen in the i.p. cats. Three cats had inversions of the CSF: plasma ratio (>1; ratio shown above each peak). FIV RNA levels in CSF and plasma were determined by real time RT-PCR at the indicated time points and the viral load presented as log10 FIV copies/ml. The detection limit of 10 copies is indicated as the baseline.
Table 2.
Comparison of mean CSF FIV RNA and CNS FIV DNA following i.c.v. versus i.p. inoculation.
| i.c.v. route | i.p. route | |
| Peak CSF viremia | 259,040 ± 124,195a | 7985 ± 5141a |
| Mean CSF: plasma ratiob | 1.34 ± 0.69 | 0.16 ± 0.16 |
| Mean CNS proviral burden | 83.6 ± 39.8c | 3.6 ± 8.7c |
the viral load is presented as FIV RNA copies/ml CSF.
calculated for paired samples at the CSF peak viremia showing the highest ratio.
the proviral burden is presented as FIV DNA copies/μg genomic DNA.
Table 2 compares the two groups (i.c.v. and i.p.) of cats with respect to the viral load in the CSF and CNS. The average CSF viral peak was 32-fold greater after direct inoculation into the ventricles (259, 040 ± 124, 195 FIV copies/ml) versus i.p. inoculation (7985 ± 5141 FIV copies/ml), and three of six i.c.v. cats showed an inversion in the CSF: plasma ratio (≥1). The average CSF: plasma ratios across all i.c.v. cats of 1.34 ± 0.69 was remarkably high relative to the i.p. cats (0.16 ± 0.16), suggesting a local CNS source of virus production. In addition, a significant proviral burden was found in CNS tissues of most i.c.v. cats and was greater and more extensive than the cats inoculated i.p. (Table 2 and below).
Relationship between CSF viral loads, plasma viral loads, and lymphocyte subsets
The above data suggested that the initial CSF viral peak was dependent upon the development of a systemic viremia. To determine if changes in lymphocytes and plasma viral loads were related to the appearance of CSF virus, the average kinetics of the CSF viral RNA were compared to the drop in the CD4+:CD8+ T-cell ratio and the viral kinetics in plasma (Figure 5). This analysis indicated that the first appearance of CSF virus followed the initial plasma viremia and was coincident with the drop in the CD4+:CD8+ T-cell ratio. In contrast, the plasma viremia appeared just before or during the initial drop in the CD4+:CD8+ T-cell ratio. Secondary CSF virus peaks occurred after the CD4+:CD8+ T-cell ratios had stabilized and were not correlated with any further lymphocyte changes or variation in plasma FIV.
Figure 5.
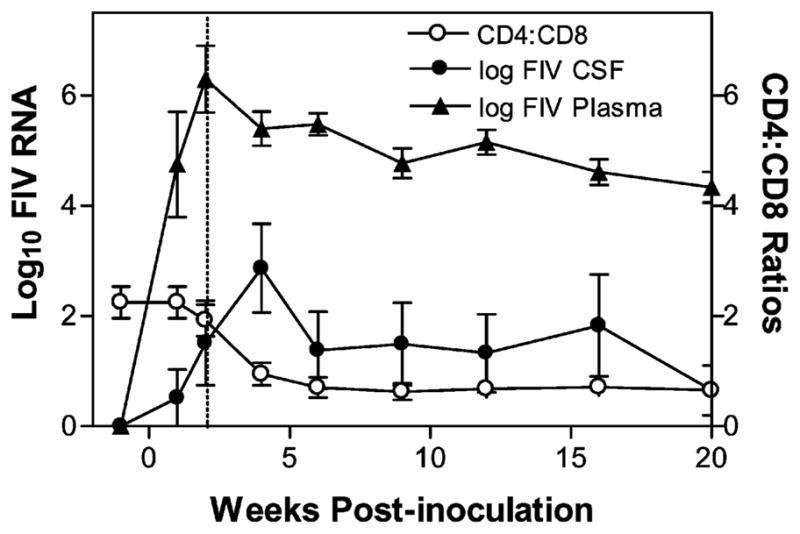
Comparison of the initial viral loads in CSF and plasma with the mean CD4+:CD8+ ratio in the six i.c.v. cats. The initial appearance of CSF virus followed the drop of plasma viremia and coincided with the decrease in the CD4+:CD8+ ratio. In contrast, the secondary virus peak that appeared in the CSF between 9 and 32 weeks p.i. occurred after the CD4+:CD8+ ratios had stabilized and were not correlated with any further changes in lymphocyte subsets. Values are mean ± SEM.
Although substantial published data suggested that parallel changes occur in plasma and CSF viral titers, correlations between CSF and plasma-associated lentiviral RNA in time-matched samples have generally been low (Brew et al, 1997; Cinque et al, 1998). We also observed a low correlation between temporally matched plasma and CSF FIV RNA levels (r2 = 0.096, P = 0.182). This poor correlation might be partially explained if the CSF FIV RNA levels lag behind the plasma levels. If the transfer of virus from plasma to CSF is slow, viral titers in CSF collected at later times might show improved correlations. When the plasma viremia was correlated with subsequent CSF viral loads (1 to 2 weeks’ delay), the r2 value improved to 0.3713 (P = 0.0159). The correlation was again lost when the CSF viral titers were offset by 3 weeks from the plasma values. The second CSF viral peak was not significantly correlated with plasma viral loads at any time.
FIV viral DNA in the brain
No viral RNA could be detected in the CNS of any infected cats by real time RT-PCR or by RT followed by nested PCR (data not shown), despite high levels of viral RNA (103 to 106 FIV copies/ml) in plasma at the time of euthanasia. Proviral (DNA) loads in the choroid plexus and four brain regions (frontal cortex gray matter [CTX gray], frontal cortex white matter [CTX white], caudate nucleus, and internal capsule–putamen [IC-Put]) were measured. The tissues were selected, in part, to contrast the FIV proviral DNA in regions adjacent to the ventricles (CTX white, caudate nucleus, ChP) versus deeper structures (IC-Put) or superficial regions adjacent to the subarachnoid space (CTX gray). A comparison of regional proviral loads in each cat is provided in Figure 6. Proviral DNA was detected in the brain or ChP from all infected cats. The average FIV DNA burden of the i.c.v. cats was 83.6 ± 39.8 FIV copies/μg DNA versus 3.6 ± 8.7 FIV copies/μg DNA for the i.p. cats, indicating a 23-fold greater proviral burden following inoculation into the lateral ventricle (P < 0.05, Table 2). This difference was not reflected in the average FIV DNA levels in lymph nodes (superficial cervical, retropharyngeal, popliteal), which were similar in both groups (735.3 ± 289.8 FIV copies/μg genomic DNA for i.c.v. cats versus 787.1 ± 196.4 FIV copies/μg genomic DNA for i.p. cats). However, the average FIV DNA in the brain of the i.c.v. cats did correlate with the PBMC FIV DNA isolated at euthanasia (r2 = 0.902, P < 0.01). On the average, the viral DNA burden in the brain was 0.90 ± 0.21% of the PBMC viral DNA. No significant relationship was found between plasma FIV RNA and either brain FIV DNA or PBMC FIV DNA.
Figure 6.

FIV proviral DNA in various brain regions of individual i.c.v. cats (cats 1 to 6) or i.p. cats (cats 10 to 12). Total genomic DNA was extracted from 30 mg tissue pieces of four brain regions, and the choroid plexus. FIV proviral DNA was quantified by real-time PCR and the proviral loads were expressed as FIV copies per microgram total genomic DNA. FIV DNA was detected in all the i.c.v. cats although the regional levels of proviral DNA were quite variable between individual cats. The number of FIV copies/μg genomic DNA detected is indicated above each bar. The limit of detection for the PCR was approximately 10 copy equivalents/μg genomic DNA and values at or below this level are show numerically with no associated bar. The overall proviral loads in the four brain regions are substantially higher and more widespread in i.c.v. cats than i.p. cats. Regions sampled: frontal cortical gray matter, CTX-gray; frontal cortex white matter immediately under the gray matter, CTX-white; caudate nucleus, internal capsule–putamen, IC-Put; and choroid plexus, ChP.
Although FIV DNA was detected in CNS regions sampled from all cats, no consistent pattern emerged in the regional distribution of provirus. Cat 5, which showed neurological signs, had the highest total proviral burden, which was concentrated in the white matter. Cat 6 had the next highest proviral burden with the viral DNA concentrated in the caudate nucleus. Cats 2 and 3 had intermediate proviral loads in the brain with highest concentrations in the cortical gray matter and choroid plexus, respectively. Finally, cat 4 had lower proviral loads that were near the detection limit. In addition, the location of virus was not restricted to structures adjacent to the ventricles.
Unlike the i.c.v. cats, in which FIV proviral DNA was detected in many brain regions, FIV proviral DNA was detected in one brain region in each of two i.p. cats. In each case, FIV DNA levels were just above the detection limit. Proviral loads in the choroid plexus from i.p. cats (2/3) were slightly higher than the brain.
Effects of i.c.v. infusion of FIV-infected ChP-Mac
One cat inoculated with 3.6 × 105 macrophages showed a rapid post-surgical increase in body temperature to 41.4°C, restlessness, vocalizations, and aggressive behavior. Treatment with flunixin meglumine effectively reduced body temperature but the cat continued to be very depressed. The cat could be roused with strong noxious stimuli and could ambulate. Pupils were dilated and equal and menace response was present bilaterally. Due to the level of depression, a decision was made to euthanize the animal at 16 hours post inoculation. The cat was submitted to the resident pathologist for an unbiased assessment of the damage. At necropsy, a moderate meningeal hemorrage was seen within the longitudinal and transverse fissures with a mild herniation of the occipital lobes and moderate herniation of the cerebellum into the foramen magnum. The hemorrage was not considered to be sufficient to directly cause brain swelling and herniation. A soft friable brown focus approximately 4 mm wide was seen at the site of needle penetration extending into the cortex. No ventricular hemorrage was seen. In microscopic examination, a regionally and focally extensive neutrophilic and necrotizing encephalitis was seen with fibrinoid vascular necrosis. The mononuclear infiltration included the cortex, basal ganglia, brainstem, and cerebellum. Minimal reaction was seen within the ventricular system. The number of neutrophils within the parenchyma was above that expected for such a short clinical course, suggesting the involvement of proinflammatory mediators. In light of this response, additional precautions were therefore taken to minimize the inflammatory response in subsequent cats. The remaining four cats, dosed with fewer macrophages (105/200 μl), showed rapid increases in body temperature which were effectively prophylaxed with flunixin meglumine. These cats showed no overt signs of distress during and after recovery. Both infected and uninfected macrophages caused a temperature rise although the magnitude of the rise could not be compared due to the pre-treatment with flunixin meglumine. After 24 h, no effects of the infusion were noted. Of the four cats inoculated with FIV-infected macrophages for which FIV RNA was assessed, none showed signs of systemic or intracranial infection. Plasma and CSF FIV RNA were undetectable in each cat throughout the course of the experiment. Macrophages prepared for infusion were positive for FIV DNA by nested PCR for env, indicating that they had been infected. Tissue levels of FIV DNA also failed to exceed the detection limit, indicating a failure of the macrophages or virus to penetrate into the parenchyma. Overall, these findings indicated that macrophages cleared the ventricular compartment quickly and were not an effective vehicle for delivery of infectious virus to the CNS.
Discussion
In both human and animal lentiviral infections, viral RNA can be detected in the CSF at about the same time as the initial viremia peak in the blood (Dow et al, 1990; Hurtrel et al, 1991, 1992; Budka, 1991a; Davis et al, 1992; Boche et al, 1996; Clements et al, 2005). The close relationship between the appearance and disappearance of viral loads in plasma and CSF has suggested a mechanism for efficient transfer across the blood-CSF barrier. Further support for this possibility comes from previous demonstrations that the choroid plexus is a significant site of infection with HIV (Falangola et al, 1995; Petito et al, 1999; Petito, 2004) and FIV (Bragg et al, 2002a; 2002b), as well as a site for the development of unique quasispecies of HIV (Chen et al, 2000; Liu et al, 2000). Therefore, lentiviruses that traverse the choroid plexus-CSF pathway may contribute uniquely to CNS infection. To directly investigate the contribution of CSF virus to neuroinvasion and neuropathogenesis, we modeled virus trafficking across the blood-CSF barrier by infusing a bolus of FIV directly into the lateral ventricle and then followed the resulting infection in CSF, blood, and brain. This procedure allowed direct access of the virus to the brain compartment while keeping the blood-brain barrier intact. By restricting the initial access of the virus to the ventricles, we were able to address a number of questions regarding the impact of virus circulating in the CSF.
Is virus penetration into the CSF an effective means for establishing a persistent productive infection within the brain and could the CSF serve as an effective means by which infectious virus exits the CNS?
These two questions were answered simultaneously and unambiguously by the demonstration of the rapid appearance of a high plasma viral load after direct cell-free FIV injection into the lateral ventricle. Thus, the virus quickly exited the CSF and failed to produce an immediate, productive local infection. The plasma viremia preceded the appearance of virus in the CSF, suggesting that, at least initially, the appearance of virus within the CSF compartment was largely dependent upon the development of a systemic viremia. The appearance of the initial CSF viral peak coincided with the reduction in the CD4+:CD8+ T-cell ratio, suggesting that the immune response to FIV somehow facilitates entry of virus into the ventricles. This could be due to an increase in immune cell trafficking through the choroid plexus. The importance of virus trafficking into and through the choroid plexus is supported by a number of observations. The consistent infection of the choroid plexus in this (8/9 cats) and other studies with HIV, SIV, or FIV illustrates that this structure is a prominent target for lentivirus infection (Falangola et al, 1995; Petito et al, 1999; Chen et al, 2000; Liu et al, 2000; Bragg et al, 2002a; Ryan et al, 2005). The greatly improved correlation between plasma FIV and CSF FIV when a temporal lag is introduced suggests that virus is not rapidly transferred across this barrier but may be delayed by local immune and infectious interactions. If true, this lag would help to explain the relatively low correlations that have been observed in analyses of CSF and plasma HIV sampled at a single time point. The rapid clearance of virus from the CSF in most cats (8/9) is similar to SIV studies in which the postacute active viral replication was suppressed after an initial productive infection (Clements et al, 2002, 2005; Barber et al, 2004, 2005). Although the ventricular inoculation failed to induce a primary productive infection within the brain compartment, it was effective in generating levels of CSF virus well in excess of systemic infections (32-fold greater). Thus, the ultimate impact on the CSF viral load was substantial.
The subsequent appearance of a unique, large second viral peak in the CSF of the i.c.v. cats in spite of declining plasma FIV RNA values and a stable CD4+:CD8+ T-cell ratio also indicated that the ventricular virus somehow set the stage for the subsequent appearance of virus in the CSF, not normally seen with systemic infections. This result is similar to the rebound CSF viral peak seen in SIV infection protocols designed to produce rapid CNS disease (Clements et al, 2002, 2005; Barber et al, 2004. In addition, half of the i.c.v. cats achieved an inversion of the CSF:plasma ratio (≥1) again suggesting a local CNS source of virus production. Such high ratios are less common following systemic infection and are only occasionally seen in acquired immunodeficiency syndrome (AIDS) patients with HIV-1 encephalitis (Di Stefano et al, 1997). Overall, the result indicates that virus trafficking through the ventricular system can have a substantial impact on the intensity and kinetics of the viral load subsequently seen in the CSF.
Comparison of the plasma viremia kinetics in i.c.v cats with those of i.p. cats further indicated the presence of a highly efficient pathway for the transfer of infectious virions from the CSF to systemic compartments. All i.c.v. cats showed a pattern of plasma viremia similar to cats receiving an i.p. inoculation of FIV but there were also notable differences. Compared to i.p. cats, the initial mean viremia was slightly higher and the plasma viral RNA levels stabilized at much higher levels in the i.c.v. cats. Thus, the i.c.v. pathway produced a more robust infection than i.p. inoculation. Although the precise mechanism of virus transfer from the CSF is not known, it is likely that bulk flow of CSF rapidly removes the free virions. Studies have shown that clearance of macromolecules from the brain is rapid due to the high rate of CSF exchange. CSF volume turns over approximately 2 to 3 times in 24 h (Segal, 2000). Tracer studies with infusions of 125I- human serum albumin into the CSF recovered 17.7% ± 2.7% of the tracer in plasma 6 h after intracranial infusion into the CSF (Boulton et al, 1997). Although a major portion of CSF exits via the arachnoid villi in the sinuses, physiological studies have also suggested that a large proportion of CSF may flow into the extracranial lymphatics through the roots of cranial and spinal nerves (Boulton et al, 1997, 1998; Cserr and Knopf, 1997). Approximately 14% of radioiodinated protein was recovered in lymph 6 h after intracranial infusion into the CSF of cats (Cserr and Knopf, 1997). Injection of 50 μl of India ink into the cisterna magna of rats resulted in selectively blackening of deep cervical lymph nodes by 30 min (Kida et al, 1993). Although there has been considerable debate regarding the size of the pores at sites of CSF exit, studies have indicated that particles as large as 500 nm may rapidly exit the CSF (Mann et al, 1979). Collectively, these studies and our observations indicate that CSF bulk flow provides a rapid means for the clearance of macromolecules and perhaps virus particles into either the cervical lymph nodes or the cerebral sinuses.
Rapid entry of FIV into the lymph nodes, in particular, may provide the opportunity to contact and infect susceptible cells and result in high levels of plasma viremia. Harling-Berg et al (1999, 2002) have proposed a model for antigen transfer from the CNS whereby CSF drainage from the brain into cervical lymph nodes provides a relatively high concentration of antigen which stimulates a Th2-biased T-cell response and antibody production from B cells. The contribution of the cervical lymph nodes to antibody production was much greater than that of the spleen, indicating that antigen clearance and presentation via CSF drainage into the lymph nodes was immunologically more effective than clearance via the sinuses and blood (Harling-Berg et al, 1999, 2002). In addition, direct comparisons of a single dose of antigen inoculated into the CSF versus peripheral sites indicated that the induction of serum antibody was more effective following the CSF inoculation (Cserr et al, 1992; Harling-Berg et al, 2002). Trafficking of the FIV from the CSF into the lymphatics would be consistent with the rapid and robust plasma viremia which we observed following i.c.v. inoculation. In addition, the efficiency of this pathway would allow for a continuous cycling of virus between ventricular and systemic compartments.
Can infectious virions in the CSF establish a local CNS infection?
Given the rapid systemic infection and the lack of a primary CSF viral load, it is likely that a large proportion of virions injected into CSF were rapidly removed by bulk flow and spent little time in the brain compartment. In this regard, the CSF did not appear to provide a favorable environment for a sustained productive infection within the brain. Nevertheless, the high viral DNA burden in the brains of the i.c.v. cats relative to i.p. cats (in spite of similar peak plasma viremias) indicated that the inoculation into the ventricles facilitated infection of the brain. Although the mechanisms for such facilitation are not known, there are at least two possibilities. First, the infection of parenchymal cells or resident ventricular macrophages may be immediate but non-productive. Virus production may be dependent on interactions with other cells such as monocytes, T cells, or local macrophages, all of which would be present in low numbers at the time of inoculation. Increased trafficking of immune cells following systemic infection would then provide the cells necessary for virus production. It is also possible that free virus penetrated across the ependyma into the brain with the gradual development of a low grade infection. This would account for the higher proviral burden in the CNS of the i.c.v. cats. Few studies have directly addressed the ability of infectious free virions to move from the CSF into brain. Most studies with viral vectors have indicated that ependymal and meningeal tissues are effectively targeted but penetration of the viral particle into the parenchyma is highly restricted. Although the flow of CSF has been shown to efficiently clear macromolecules, several studies have indicated that small residual amounts may remain within the perivascular space surrounding the pial arteries and arterioles and may be associated with antigen presenting cells such as macrophages (Kida et al, 1993; Boulton et al, 1996; Harling-Berg et al, 1999). Small “pockets” of virus may exist that serve as a catalyst for local virus production and subsequent trafficking. Thus, although virus in the CSF did not produce an immediate productive infection, several observations suggest that conditions were established which supported subsequent local virus production.
Second, it is also possible that infection of the brain occurred exclusively after the systemic infection had been established. In this case, virus could be carried into the brain by infected mononuclear cells in the blood or by the trafficking of infected macrophages from the choroid plexus. These two processes differ in important ways. At the blood-brain barrier, most studies support the idea that infected monocytes traffic into the brain where they differentiate into macrophages. These macrophages then produce virus and transfer the infection to other cells. In the choroid plexus, a large population of macrophages normally exists between the blood and the epithelium. Virus penetrating through the fenestrated choroid plexus vasculature would come into contact with this local macrophage population. These differentiated macrophages are thought to traffic into the CSF in addition to modest numbers of monocytes and T cells. Because these macrophages can be infected by FIV (Bragg et al, 2002a), they constitute an important additional source of virus entry that is not well understood. The density of macrophages in the choroid plexus and level of infection could provide an efficient, sustained source of infected macrophages that has greater impact than the brief interactions between virus and resident macrophages in the CSF at the time of inoculation. However, the failure of infected ChP macrophages to produce a rapid, compartmentalized infection or to effectively transfer infection to the periphery suggested that CNS infection via local ChP macrophages is, at the very least, not an efficient process. Although we confirmed that the cells harbored FIV provirus, the infectious titer and the amount of virus production may have been too low to establish an infection. Even though the macrophages did not serve as an efficient delivery vehicle for infection, they induced an acute inflammatory response that was not seen with the virus infusions. In addition, they caused a small decrease in the CD4:CD8 ratio suggesting that they influenced the systemic immune response. It is possible that the inflammatory activation stimulated by these cells might prime the brain for subsequent trafficking and infection but more data are needed to fully appreciate their potential.
The above options of direct infection or facilitation of subsequent viral trafficking are not mutually exclusive and both could have contributed to infection of the brain. Regardless of the mechanism of entry, our studies clearly establish that virus trafficking through the CSF can have a substantial impact on infection of the brain. The persistence of viral DNA in the brain after elimination of virus from the CSF is consistent with other studies of HIV, SIV, or FIV, which have shown that the proviral load can persist in the brain throughout infection even when viral RNA or antigen is undetectable (Sinclair et al, 1994; Boche et al, 1996, 1999; Di Stefano et al, 1996; Chen et al, 2000; Hein et al, 2000; Clements et al, 2002).
It was expected that structures in close proximity to the lateral ventricles (e.g., caudate, cortical white matter) would have a greater proviral burden than structures remote from the ventricles (internal capsule/putamen) or structures more closely associated with subarachnoid CSF (superficial gray matter of the cortex). However, each i.c.v. cat gave a unique CNS profile. The region most consistently positive for FIV DNA was the choroid plexus with 6/6 i.c.v. cats and 2/3 i.p. cats showing infection. This is consistent with the results from many studies of HIV, SIV, and FIV infection (Falangola et al, 1995; Petito et al, 1999; Ryan et al, 2003). The highest level of proviral DNA was seen in the subcortical white matter of cat 5. Moderate levels of FIV DNA were seen in the caudate and gray matter of the cortex. This general pattern is consistent with a ventricular dissemination but the high variability between cats indicated that proximity to the ventricles was unlikely to be a major determinant of brain infection. A similar analysis of acute infection with FIVGL8 by Ryan et al (2003) indicated that FIV infection of brain tissue is apparent by 4 to 10 weeks after an intravenous inoculation. Both viral and proviral loads in the brain decreased greatly by 23 weeks post inoculation, indicating that much of the virus in the brain is rapidly cleared. As in our studies, no consistent pattern emerged to indicate a preferred route for CNS infection. It is notable that the appearance of the highest CNS virus at 10 weeks with a subsequent drop by 23 weeks is consistent with the large secondary CSF viral peak seen in our cats. This would suggest that the second rise in CSF FIV RNA may reflect substantial transient virus production in brain parenchyma.
Overall, our studies demonstrate that free NCSU1 FIV that enters the CSF compartment is rapidly cleared and does not establish a local productive infection. However, a significant latent infection may be established through this route, which is capable of reactivation by unknown stimuli, giving rise to a secondary CSF viral peak. Importantly, virus exiting the brain via the CSF is highly efficient at inducing a systemic infection. This reinforces the view that virus compartmentalized in the CNS has the potential to rekindle a systemic infection. A better understanding of these pathways is crucial to the development of therapies designed to limit brain infection, control CNS disease progression, and prevent the spread of infectious virus from the brain.
Materials and methods
Animals
Eighteen adult 10 to 18 month-old specific pathogen-free (SPF) cats were used in the study and divided into three groups (Table 1): In group I, six adult cats were intracerebroventricularly inoculated with a total of 200 μl (2 × 105 TCID50) cell-free NCSU1 FIV into the right lateral ventricle using a stereotaxic apparatus (see below), three adult cats were mock infected by injecting 200 μl of uninfected FCD4E cell (an interleukin [IL]-2 dependent feline CD4+ T-cell line used to prepare virus stocks) filtered cell-free supernatant. In group II, five cats were intracerebroventricularly inoculated with a total of 1–3.6 × 105 NCSU1FIV infected ChP-Mac in 200 μl HEPES-buffered artificial CSF (aCSF) composed of the following in mM concentrations: NaCl 137, KCl 5.0, CaCl2 2.3, MgCl2 1.3, glucose 20, HEPES 8, adjusted to pH 7.4 with NaOH, and one adult cat was mock inoculated by injecting 200 μl of aCSF containing 1 × 105 uninfected ChP-Mac. These cats were given an injection of flunixin meglumine (Banamine) at 1.1 mg/kg. Cats tolerated the procedure well and, with one notable exception (discussed in Results), no adverse effects were noted. To provide a comparison to systemic infection, in group III, three additional cats were inoculated intraperitoneally with an identical amount of cell free FIV (2 × 105 TCID50/200 μl). Blood and CSF samples were collected just prior to infection and at 1, 2, 4, 6, 9, 12, 16, 20, 24, and 32 weeks post inoculation. Some cats included samples at 40, 48, and 56 weeks post inoculation as well. The development of significant neurological disease or the complete clearance of viral RNA from the CSF in the absence of neurological disease was used as the endpoints for the studies. One cat (cat 5) manifested neurological symptoms at 12 weeks after infusion of cell-free virus. At necropsy, all animals were heparinized (2500 U via cardiac ventricular injection), and perfused with 0.9% saline using a peristaltic pump to remove virus-containing blood from the vasculature. All animal procedures were performed in accordance with National Institutes of Health guidelines and were reviewed and approved by the institutional animal care and use committee.
Preparation of virus and cellular inoculums
The NCSU1 strain of FIV, originally isolated from a naturally infected cat with lymphopenia and acute enteritis, has dual T-cell and macrophage tropism (Bragg et al, 2002a). Cell-free virus inoculum was prepared as described previously (Davidson et al, 1993). Briefly, pooled PBMCs from NCSU1 infected cats were cocultured with FCD4E cells. Cell supernatant containing high RT activity was collected, filtered (0.2 μm), and frozen as a source of virus stock. Virus titer was determined by the 50% tissue culture infectious dose (TCID50) method as described by Davidson et al (1993). For cell associated virus, feline fetal ChP macrophages were infected with NCSU1 FIV at a multiplicity of infection (MOI) of 1 as described in a previous publication (Bragg et al, 2002a). Twenty-four hours after inoculation, just prior to surgery, the cells were washed three times in sterile aCSF, incubated with 0.25% trypsin-EDTA (1.5 ml/35-mm dish) for 30 to 60 min, and harvested. The cells were then washed with 10-fold excess sterile aCSF and pelleted at 80 × g for 15 min. The supernatant was discarded and the pellet was washed and resuspended in aCSF at a concentration of 5 × 105 cells/ml. The cells were kept on ice until they were loaded into the injector.
Intracerebroventricular infusions
To test the infectious capabilities of virus in the CSF, the inoculum was infused directly into the right cerebral ventricle in a fashion that minimized any risk of blood contamination. SPF cats were anesthetized with isoflurane and the surface of the cranium exposed through a 3-cm skin incision. The bregma of the skull was identified and used as a landmark for the stereotaxic coordinates. A gas sterilized 23 gauge stainless steel injection needle was positioned at the intended coordinates: 2.5 mm anterior to bregma, and 3.0 mm lateral (right side). The injector was connected to approximately 60 cm of PE-50 tubing calibrated at 50-μl intervals. The virus stock was sterilized by passage through a 0.2-μm filter and then 250 μl of the inoculum was backloaded into the tubing. The injector was then lowered 5.0 mm below the surface of dura mater and the top of the tubing unclamped. A lack of flow under gravity feed verified that the injector was in tissue. The injector was then slowly lowered until it penetrated the ventricle as evidenced by an abrupt drop in the fluid level and the observation of pulsations. A volume of 200 μl of the inoculum was allowed to gradually flow into the ventricle under gravity feed. The injector was kept in place for an additional 2 min after delivery to ensure that the inoculum had fully penetrated. The needle was then withdrawn and any signs of blood or clear fluid noted. No blood was seen for any cat and a very slight reflux of clear fluid was seen for one cat (cat 5). The skull was sealed with bone wax and the fascia and skin sutured in place. Cats were carefully watched for the first 24 h after inoculation for any signs of toxicity or trauma. Seventeen of 18 cats recovered quickly with no adverse effects of the infusion.
Viral RNA in plasma and CSF
Viral RNA was extracted from CSF and plasma using QIAamp Viral RNA Mini Kit (Qiagen, CA). The viral RNA loads were determined by real time RT-PCR with the Taqman one-step RT-PCR master kit (Applied Biosystems, CA). The following specific primers and probe of NCSU1 FIV gag region were used: FIV NC-491F (5′-GATTAGGAGGTGAGGAAGT TCA GCT-3′), FIV NC-617R (5′-CTTTCA TCCAA TAT-TTCTTTATCTGCA-3′) and the labeled probe, FIV NC-555p (5′-FAM-CATGGCCACATTAATAAT GGC-GCA-TAMRA-3′) (Applied Biosystems) (Leutenegger et al, 1999). These reactions were performed with a Bio-Rad iCyclerTM iQ and analyzed using the manufacturer’s software. For each run, a standard curve was generated from the standard FIV-gag RNA dilutions with known copy numbers, and the RNA in the samples was quantified as FIV copy equivalents based on the standard curve. Values are expressed as FIV RNA copies/ml. The RNA gag standard was generated from The NCSU1 gag plasmid-pcDNA3.1gag kindly provided by Dr. Wayne Tompkins (North Carolina State University, Raleigh) by in vitro transcription with Ambion’s mMESSAGE mMACHINE kit (Austin, TX). The standard RNA concentration was determined by PhotoSpectroMeter and the copies/ml was calculated. The standards were run in serial 10-fold dilutions to generate a standard curve.
Proviral DNA in brain and PBMCs
Total genomic DNA was isolated from a 30-mg sample from each of four brain regions (frontal cortex gray matter, underlying cortical white matter, caudate nucleus, internal capsule-putamen), the choroid plexus and from 1 × 106 PBMCs using QIAamp DNA mini kit (Qiagen). The proviral DNA loads were determined by real-time PCR, with a Taqman Universal PCR master kit (Applied Biosystem) using the same primer set and probe as for real time RT-PCR. A standard curve was generated from the pcDNA3.1-gag plasmid sample.
Lymphocyte subset analysis
Whole blood from each cat was collected into EDTA anticoagulant tubes at various times post infection for a complete blood count with differential and lymphocyte phenotype analysis, as previously reported (English et al, 1993; Davidson et al, 1993). Percentages of CD4+ and CD8+T lymphocytes were determined by two- and three-color flow cytometry. The fluorochrome-conjugated monoclonal antibodies (mAbs) used in staining were PE-conjugated anti-CD3 (mAb 572), fluorescein isothiocyate (FITC)-conjugated anti-CD4 (mAb 30A), PE-conjugated anti-CD8 (mAb 3.357), anti-CD21 mAb (P. Moore, University of California, Davis, CA). Isotype-matched irrelevant antibodies were used as controls. The percent of positively stained lymphocytes was determined using a Becton Dickinson FACScan. Complete blood cell counts were determined by using a VetScan HMT hematology analyzer (Alaska Scientific), and the absolute lymphocyte number determined by differential counts. The number of cells within each lymphocyte subpopulation was then calculated based on the subset percentages and total lymphocytes.
Statistical analysis
A log10 transformation was performed on all the levels of FIV RNA or proviral DNA. The statistical significance between the mean values for the i.c.v. cats and i.p. cats was determined by a Student t test. Differences were considered significant if the P value was ≤ .05. To determine the correlation between levels of FIV RNA in CSF and plasma, and the proviral load in tissues and PBMCs, a linear regression analysis was performed and the results expressed as the square of the correlation coefficient (r2). All statistical calculations were done using Graphpad Prism software.
Acknowledgments
This study was supported by National Institutes of Health grant MH63646.
References
- Antinori A, Cingolani A, Giancola ML, Forbici F, De Luca A, Perno CF. Clinical implications of HIV-1 drug resistance in the neurological compartment. Scand J Infect Dis Suppl. 2003;35(Suppl 106):41–44. doi: 10.1080/03008870310009650. [DOI] [PubMed] [Google Scholar]
- Barber SA, Herbst DS, Bullock BT, Gama L, Clements JE. Innate immune responses and control of acute simian immunodeficiency virus replication in the central nervous system. J NeuroVirol. 2004;10(Suppl 1):15–20. doi: 10.1080/753312747. [DOI] [PubMed] [Google Scholar]
- Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- Boche D, Gray F, Chakrabarti L, Hurtrel M, Montagnier L, Hurtrel B. Low susceptibility of resident microglia to simian immunodeficiency virus replication during the early stages of infection. Neuropathol Appl Neurobiol. 1995;21:535–539. doi: 10.1111/j.1365-2990.1995.tb01100.x. [DOI] [PubMed] [Google Scholar]
- Boche D, Hurtrel M, Gray F, Claessens-Maire MA, Ganiere JP, Montagnier, Hurtrel B. Virus load and neuropathology in the FIV model. J NeuroVirol. 1996;2:377–387. doi: 10.3109/13550289609146903. [DOI] [PubMed] [Google Scholar]
- Boche D, Khatissian E, Gray F, Falanga P, Montagnier L, Hurtrel B. Viral load and neuropathology in the SIV model. J NeuroVirol. 1999;5:232–240. doi: 10.3109/13550289909015809. [DOI] [PubMed] [Google Scholar]
- Boulton M, Flessner M, Armstrong D, Hay J, Johnston M. Lymphatic drainage of the CNS: effects of lymphatic diversion/ligation on CSF protein transport to plasma. Am J Physiol. 1997;272:R1613–R1619. doi: 10.1152/ajpregu.1997.272.5.R1613. [DOI] [PubMed] [Google Scholar]
- Boulton M, Flessner M, Armstrong D, Hay J, Johnston M. Determination of volumetric cerebrospinal fluid absorption into extracranial lymphatics in sheep. Am J Physiol. 1998;274:R88–R96. doi: 10.1152/ajpregu.1998.274.1.R88. [DOI] [PubMed] [Google Scholar]
- Boulton M, Young A, Hay J, Armstrong D, Flessner M, Schwartz M, Johnston M. Drainage of CSF through lymphatic pathways and arachnoid villi in sheep: measurement of 125I-albumin clearance. Neuropathol Appl Neurobiol. 1996;22:325–333. doi: 10.1111/j.1365-2990.1996.tb01111.x. [DOI] [PubMed] [Google Scholar]
- Bragg D, Childers T, Tompkins M, Tompkins W, Meeker R. Infection of the choroid plexus by feline immunodeficiency virus. J NeuroVirol. 2002a;8:211–224. doi: 10.1080/13550280290049688. [DOI] [PubMed] [Google Scholar]
- Bragg D, Hudson L, Liang Y, Tompkins M, Fernandes A, Meeker R. Choroid plexus macrophages proliferate and release toxic factors in response to feline immunodeficiency virus. J NeuroVirol. 2002b;8:225–239. doi: 10.1080/13550280290049679. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Pemberton L, Cunningham P, Law MG. Levels of human immunodeficiency virus type 1 RNA in cerebrospinal fluid correlate with AIDS dementia stage. Infect Dis. 1997;175:963–966. doi: 10.1086/514001. [DOI] [PubMed] [Google Scholar]
- Budka H. Neuropathology of human immunodeficiency virus infection. Brain Pathol. 1991a;1:163–175. doi: 10.1111/j.1750-3639.1991.tb00656.x. [DOI] [PubMed] [Google Scholar]
- Budka H. The definition of HIV-specific neuropathology. Acta Pathol Jpn. 1991b;41:182–191. doi: 10.1111/j.1440-1827.1991.tb01645.x. [DOI] [PubMed] [Google Scholar]
- Chen H, Wood C, Petito CK. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: potential role of choroid plexus in the pathogenesis of HIV encephalitis. J NeuroVirol. 2000;6:498–506. doi: 10.3109/13550280009091950. [DOI] [PubMed] [Google Scholar]
- Cinque P, Vago L, Ceresa D, Mainini F, Terreni MR, Vagani A, Torri W, Bossolasco S, Lazzarin A. Cerebrospinal fluid HIV-1 RNA levels: correlation with HIV encephalitis. AIDS. 1998;12:389–394. doi: 10.1097/00002030-199804000-00007. [DOI] [PubMed] [Google Scholar]
- Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Jr, Tarwater PM, Lifson JD, Zink MC. The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002;186:905–913. doi: 10.1086/343768. [DOI] [PubMed] [Google Scholar]
- Clements JE, Ming Li, Gama L, Brandon Bullock, Carruth LM, Mankowski JL, Zink MC. The central nervous system is a viral reservoir in simian immunodeficiency virus-infected macaques on combined antiretroviral therapy: a model for human immunodeficiency virus patients on highly active antiretroviral therapy. J NeuroVirol. 2005;11(2):180–189. doi: 10.1080/13550280590922748-1. [DOI] [PubMed] [Google Scholar]
- Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992;2:269–276. doi: 10.1111/j.1750-3639.1992.tb00703.x. [DOI] [PubMed] [Google Scholar]
- Cserr HF, Knopf PM. Cervical lymphatics, the blood-brain barrier, and immunoreactivity of the brain. In: Keane RW, Hickey WF, editors. Immunology of the Nervous System. Oxford University Press; 1997. pp. 134–152. [Google Scholar]
- Cunningham PH, Smith DG, Satchell C, Cooper DA, Brew B. Evidence for independent development of resistance to HIV-1 reverse transcriptase inhibitors in the cerebrospinal fluid. AIDS. 2000;14:1949–1954. doi: 10.1097/00002030-200009080-00010. [DOI] [PubMed] [Google Scholar]
- Davidson MG, Rottman JB, English RV, Lappin MR, Tompkins MB. Feline immunodeficiency virus predisposes cats to acute generalized toxoplasmosis. Am J Pathol. 1993;143:1486–1497. [PMC free article] [PubMed] [Google Scholar]
- Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Monno L, Ramon FJ, Angarano G. In vivo evidence of HIV-1 productive infection in cerebrospinal fluid of patients with HIV-1 encephalopathy. AIDS. 1997;11:698–699. [PubMed] [Google Scholar]
- Di Stefano M, Wilt S, Gray F, Dubois-Dalcq M, Chiodi F. HIV type 1 V3 sequences and the development of dementia during AIDS. AIDS Res Hum Retroviruses. 1996;12:471–476. doi: 10.1089/aid.1996.12.471. [DOI] [PubMed] [Google Scholar]
- Dow S, Poss M, Hoover E. Feline immunodeficiency virus: a neurotropic lentivirus. J AIDS. 1990;3:658–668. [PubMed] [Google Scholar]
- English RV, Johnson CM, Gebhard DH, Tompkins MB. In vivo lymphocyte tropism of feline immunodeficiency virus. J Virol. 1993;67:5175–5186. doi: 10.1128/jvi.67.9.5175-5186.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falangola MF, Hanly A, Galvao-Castro B, Petito CK. HIV infection of human choroid plexus: a possible mechanism of viral entry into the CNS. J Neuropathol Exp Neurol. 1995;54:497–503. doi: 10.1097/00005072-199507000-00003. [DOI] [PubMed] [Google Scholar]
- Gray F, Adle-Biassette H, Chretien F, Lorin dlG, Force G, Keohane C. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol. 2001;20:146–155. [PubMed] [Google Scholar]
- Gray F, Hurtrel M, Hurtrel B. Early central nervous system changes in human immunodeficiency virus (HIV)-infection. Neuropathol Appl Neurobiol. 1996;19:3–9. doi: 10.1111/j.1365-2990.1993.tb00399.x. [DOI] [PubMed] [Google Scholar]
- Harling-Berg CJ, Hallett JJ, Park JT, Knopf PM. Hierarchy of immune responses to antigen in the normal brain. Curr Top Microbiol Immunol. 2002;265:1–22. doi: 10.1007/978-3-662-09525-6_1. [DOI] [PubMed] [Google Scholar]
- Harling-Berg CJ, Park TJ, Knopf PM. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J Neuroimmunol. 1999;101:111–127. doi: 10.1016/s0165-5728(99)00130-7. [DOI] [PubMed] [Google Scholar]
- Hein A, Martin JP, Koehren F, Bingen A, Dorries R. In vivo infection of ramified microglia from adult cat central nervous system by feline immunodeficiency virus. Virology. 2000;268:420–429. doi: 10.1006/viro.1999.0152. [DOI] [PubMed] [Google Scholar]
- Henriksen SJ, Prospero-Garcia O, Phillips TR, Fox HS, Bloom FE, Elder JH. Feline immunodeficiency virus as a model for study of lentivirus infection of the central nervous system. [review] Curr Top Microbio Immunol. 1995;202:167–186. doi: 10.1007/978-3-642-79657-9_12. [DOI] [PubMed] [Google Scholar]
- Hurtrel B, Chakrabarti L, Hurtrel M, Maire MA, Dormont D, Montagnier L. Early SIV encephalopathy. J Med Primatol. 1991;20:159–166. [PubMed] [Google Scholar]
- Hurtrel B, Chakrabarti L, Hurtrel M, Montagnier L. Target cells during early SIV encephalopathy. Res Virol. 1993;144:41–46. doi: 10.1016/s0923-2516(06)80010-5. [DOI] [PubMed] [Google Scholar]
- Hurtrel M, Ganiere J, Guelifi J, Chakrabarti L, Maire M, Gray F, Montagnier L, Hurtrel B. Comparison of early and late feline immunodeficiency virus encephalopathies. AIDS. 1992;6:399–406. doi: 10.1097/00002030-199204000-00007. [DOI] [PubMed] [Google Scholar]
- Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol. 1993;19:480–488. doi: 10.1111/j.1365-2990.1993.tb00476.x. [DOI] [PubMed] [Google Scholar]
- Leutenegger CM, Klein D, Hofmann-Lehmann R, Mislin C, Hummel U, Boni J, Boretti F, Guenzburg WH, Lutz H. Rapid feline immunodeficiency virus provirus quantitation by polymerase chain reaction using the TaqMan fluorogenic real-time detection system. J Virol Methods. 1999;78:105–116. doi: 10.1016/s0166-0934(98)00166-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tang XP, McArthur JC, Scott J, Gartner S. Analysis of human immunodeficiency virus type 1 gp160 sequences from a patient with HIV dementia: evidence for monocyte trafficking into brain. J Neurovirol. 2000;6(Suppl 1):S70–S81. [PubMed] [Google Scholar]
- Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Histol Histopathol. 2004;19:535–564. doi: 10.14670/HH-19.535. [DOI] [PubMed] [Google Scholar]
- Mann JD, Butler AB, Johnson RN, Bass NH. Clearance of macromolecular and particulate substances from the cerebrospinal fluid system of the rat. J Neurosurg. 1979;50:343–348. doi: 10.3171/jns.1979.50.3.0343. [DOI] [PubMed] [Google Scholar]
- Martin C, Albert J, Hansson P, Pehrsson P, Link H, Sonnerborg A. Cerebrospinal fluid mononuclear cell counts influence CSF HIV-1 RNA levels. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;17:214–219. doi: 10.1097/00042560-199803010-00005. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Thiede BA, Hall C, English R, Tompkins M. Cortical cell loss in asymptomatic cats experimentally infected with feline immunodeficiency virus. AIDS Res Hum Retroviruses. 1997;13:1131–1140. doi: 10.1089/aid.1997.13.1131. [DOI] [PubMed] [Google Scholar]
- Petito CK. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J NeuroVirol. 2004;10 (Suppl 1):21–24. doi: 10.1080/753312748. [DOI] [PubMed] [Google Scholar]
- Petito CK, Chen H, Mastri AR, Torres-Munoz J, Roberts B, Wood C. HIV infection of choroid plexus in AIDS and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection. J Neurovirol. 1999;5:670–677. doi: 10.3109/13550289909021295. [DOI] [PubMed] [Google Scholar]
- Phillips T, Prospero-Garcia O, Puaoi D, Lerner D, Fox H, Olmsted R, Bloom F, Heriksen S, Elder J. Neurological abnormalities associated with feline immunodeficiency virus infection. J Gen Virol. 1994;75:979–987. doi: 10.1099/0022-1317-75-5-979. [DOI] [PubMed] [Google Scholar]
- Power C, Buist R, Johnston JB, Del Bigio MR, Ni W, Dawood MR, Peeling J. Neurovirulence in feline immunodeficiency virus-infected neonatal cats is viral strain specific and dependent on systemic immune suppression. J Virol. 1998;72:9109–9115. doi: 10.1128/jvi.72.11.9109-9115.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, Moench T, Peeling J, Kong P-A, Langelier T. Feline immunodeficiency virus causes increased glutamate levels and neuronal loss in brain. Neuroscience. 1997;77:1175–1185. doi: 10.1016/s0306-4522(96)00531-3. [DOI] [PubMed] [Google Scholar]
- Redzic ZB, Segal MB. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Deliv Rev. 2004;56:1695–1716. doi: 10.1016/j.addr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Ryan G, Grimes T, Brankin B, Mabruk MJ, Hosie MJ, Jarrett O, Callanan JJ. Neuropathology associated with feline immunodeficiency virus infection highlights prominent lymphocyte trafficking through both the blood-brain and blood-choroid plexus barriers. J NeuroVirol. 2005;11:337–345. doi: 10.1080/13550280500186445. [DOI] [PubMed] [Google Scholar]
- Ryan G, Klein D, Knapp E, Hosie MJ, Grimes T, Mabruk MJ, Jarrett O, Callanan JJ. Dynamics of viral and proviral loads of feline immunodeficiency virus within the feline central nervous system during the acute phase following intravenous infection. J Virol. 2003;77:7477–7485. doi: 10.1128/JVI.77.13.7477-7485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal MB. The choroid plexuses and the barriers between the blood and the cerebrospinal fluid. Cell Mol Neurobiol. 2000;20:183–196. doi: 10.1023/a:1007045605751. [DOI] [PubMed] [Google Scholar]
- Shapshak P, Segal DM, Crandall KA, Fujimura RK, Zhang BT, Xin KQ, Okuda K, Petito CK, Eisdorfer C, Goodkin K. Independent evolution of HIV type 1 in different brain regions. AIDS Res Hum Retroviruses. 1999;15:811–820. doi: 10.1089/088922299310719. [DOI] [PubMed] [Google Scholar]
- Sinclair E, Gray F, Ciardi A, Scaravilli F. Immunohistochemical changes and PCR detection of HIV provirus DNA in brains of asymptomatic HIV-positive patients. J Neuropathol Exp Neurol. 1994;53:43–50. doi: 10.1097/00005072-199401000-00006. [DOI] [PubMed] [Google Scholar]
- Strain MC, Letendre S, Pillai SK, Russell T, Ignacio CC, Gunthard HF, Good B, Smith DM, Wolinsky SM, Furtado M, Marquie-Beck J, Durelle J, Grant I, Richman DD, Marcotte T, McCutchan JA, Ellis RJ, Wong JK. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J Virol. 2005;79:1772–1788. doi: 10.1128/JVI.79.3.1772-1788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–562. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M, Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE. High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol. 1999;73:10480–10488. doi: 10.1128/jvi.73.12.10480-10488.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]