

Abstract
To begin to unravel the complexity of HIV-associated changes in the brain, broader, multifaceted analyses of cerebrospinal fluid (CSF) are needed that examine a wide range of proteins reflecting different functions. CSF from HIV-infected patients with a range of cognitive deficits was compared to CSF from uninfected, cognitively normal patients to begin to identify protein changes associated with HIV infection and neurological disease progression. Uninfected patients showed relatively consistent patterns of protein expression. Highly expressed proteins in CSF included monocyte chemotactic protein-1, tissue inhibitors of metalloproteases, granulocyte colony-stimulating factor, adiponectin, soluble tumor necrosis factor receptor-1, urokinase-type plasminogen activator receptor, and insulin-like growth factor binding protein-2. Inflammatory and anti-inflammatory cytokines were expressed at low levels. HIV-infected patients showed increases in inflammatory proteins (interferon-gamma, tumor necrosis factor-alpha), anti-inflammatory proteins (IL-13), and chemokines but these correlated poorly with neurological status. The strongest correlation with increasing severity of neurological disease was a decline in growth factors, particularly, brain-derived neurotrophic factor and NT-3. These studies illustrate that HIV infection is associated with parallel changes in both inflammatory and neuroprotective proteins in the CSF. The inverse relationship between growth factors and neurological disease severity suggests that a loss of growth factor neuroprotection may contribute to the development of neural damage and may provide useful markers of disease progression.
Keywords: Neurotrophin, Cytokine, Chemokine, Dementia, Inflammation, Brain
Introduction
Early in the course of HIV-1 infection, monocytes are thought to carry the virus into the central nervous system thereby initiating infection, inflammation, and progressive neural dysfunction (Koenig et al. 1986; Kolson 2002; Bouwman et al. 1998; Towfighi et al. 2004; Hult et al. 2008). The neural dysfunction may be due, in part, to the accumulation of macrophage- and microglia-derived toxins (Epstein and Gendelman 1993; Zheng and Gendelman 1997; Giulian and Noonan 1998; Xiong et al. 2000). Many studies have attempted to define the toxic factors in the cerebrospinal fluid (CSF) that correlate with disease progression in an effort to identify individuals at highest risk for neurological disease and provide clues to the processes that lead to neuronal dysfunction. These studies have identified a number of known proteins thought to reflect inflammatory activity (Perrella et al. 1992; Elovaara and Muller 1993; Achim et al. 1993; Cinque et al. 1998, 2004; Conant et al. 1999; Sporer et al. 2000, 2005; Enting et al. 2000; Bragg et al. 2000; Liuzzi et al. 2000; Pemberton and Brew 2001; Sabri et al. 2001; Towfighi et al. 2004) as well as unidentified factors which may contribute to neuropathogenesis (Giulian et al. 1996; Meeker et al. 1999; Turchan et al. 2003). Factors in the CSF that have shown a significant correlation with dementia include monocyte chemotactic protein-1 (MCP-1) (Conant et al. 1998; Cinque et al. 1998), urokinase-type plasminogen activator receptor (uPAR) (Sporer et al. 2005), quinolinate (Heyes et al. 1991), and correlates of damage such as neurofilament light chain proteins (Hagberg et al. 2000; Gisslen et al. 2007), lipid metabolites (Bandaru et al. 2007), and beta amyloid/Tau (Brew et al. 2005). Although progress is being made in the identification of disease markers, we still know very little about the processes that give rise to neuropathogenesis.
In vitro studies have identified many factors that could contribute to HIV-associated CNS dysfunction. The many pathogenic pathways that have been proposed highlight the complexity of the disease processes and emphasize the need to examine a broad range of markers that reflect the dynamic nature of the disease process including both toxic and protective functions (Bazan et al. 2005; Streit 2005; Turrin and Rivest 2006). Since studies often focus on a few inflammatory cytokines, we currently have an incomplete picture of the interactions of HIV with the CNS that collectively determine the extent of the dysfunction. In order to better appreciate the nature of these interactions, we used antibody-based arrays to examine the profile of 120 cytokines, chemokines, and growth factors in human CSF. These initial studies provide the first comprehensive view of inflammatory, anti-inflammatory, and neuroprotective proteins in both normal and HIV-1-infected CNS in an effort to begin to understand the complex changes in HIV-infected brain.
Materials and methods
Human CSF
Twenty HIV+ CSF samples collected from ten clinically characterized individuals with composite neurological scores ranging from 2 to 176 (see below) were used to evaluate protein profiles across a range of CNS disease. The study was done with IRB approval from the University of North Carolina, and all subjects gave informed consent before participating. Patients were pre-screened for the presence of opportunistic infections and were excluded from the study if any were present. The CSF samples were chosen to represent a wide range of clinical profiles including three patients that showed large decreases in cognitive function (mean composite neurological scores increased from 41 to 139), two patients that improved substantially (mean composite neurological scores decreased from 168 to 50), and five patients with moderate or minimal changes. The intention was to provide the best opportunity to correlate protein values across a wide range of cognitive disease including changes in cognitive status in a relatively small group of samples. Sixteen samples of control CSF collected from HIV-negative individuals with no dementing disease or related inflammation were used to define the normal constituents of CSF. This control group had a mean age (±SD) of 44.9±15.6 years and was composed of nine females and seven males. Most patients were under evaluation for lower back pain, degenerative disks, and peripheral muscle weakness. Symptoms in these patients included paresthesias, migraine, hydrocephalus, occipital neuralgia, and gait instability. Patients with any cognitive dysfunction or fever were excluded. Because HIV+ CSF samples were chosen based on changes in their neurological presentation, they were not specifically matched to the control group. The mean age of these patients was 39.2 years with seven males and three females. The CSF used on the protein arrays was collected between October of 2000 and February of 2004, centrifuged to remove cells, and stored in 250–500-μl aliquots at −80°C until assayed.
Neurological score
Assessment of HIV-infected patients was accomplished using the AIDS Clinical Trials Group full neurological evaluation developed by Price and Sidtis (1990). This contains a global assessment of HIV-associated dementia (HAD, AIDS Dementia Complex) stage varying from equivocal (0.5) to severe (3.0). In addition, a quantitative scoring procedure for the neurological evaluation was implemented, increasing the sensitivity of the instrument and providing domains of functioning (Robertson et al. 2004). This procedure provided a weighted score approach to the items of the neurological exam and yielded an overall neurological total score as well as scores for the domains of cognitive, frontal, pyramidal, extrapyramidal, cranial nerves, cerebellar, spinal, autonomic, and sensory/peripheral. The total score for neurologically normal controls is typically under 20 with a mean of 5.85 (SD=7.95). Scores around 100 represent mild dementia. Using a cutoff of 59 for the total neurological score, significant cognitive–motor deficits were detected with a sensitivity of 95% and specificity of 95%, with false positives at 4.8%.
Measurement of CSF toxicity
The presence of putative neurotoxins in CSF was assessed by adding 12.5 μl CSF to cultured rat forebrain neurons in a 48-well plate to a final volume of 200 μl as previously described (Meeker et al. 1999). After 24 h, the number of dead cells was quantified by adding 1 μM ethidium homodimer to the live cultures for a period of 40 min. Dead cell nuclei show bright red fluorescence with a high signal to noise for semiautomated image analysis using digital microscopy and Metamorph® imaging software (Molecular Devices, Inc). Cell death was corrected for basal cell death in the cultures and then normalized to the cell death in cultures treated with an artificial CSF as the percent increase in cell death. This normalization corrected for any run to run differences in background cell death which typically ranged from 2% to 4% of the total number of neurons. Toxicity data were collected on CSF samples at or near the time of collection and the data archived for future use. Data presented here represent the net accumulation of these data.
Protein macroarrays
Antibody arrays were purchased from RayBiotech. The protein array protocol was the same as provided in the RayBiotech C Series 1000 kit User Manual with the exception of an additional wash step after HRP-streptavidin which was used to further reduce background. Membranes were blocked for 30 min in serum-based blocking buffer then incubated in 1.0 ml of human CSF overnight (18 h). They were then washed five times using two separate wash buffers. After washing, the membranes were incubated in a biotin-conjugated antibody solution for 2 h. Membranes were again washed, as described above, and incubated in a solution containing HRP-conjugated streptavidin for 2 h. After incubation, the membranes were washed once more and incubated in detection buffer for 2 min. The membranes were exposed to Kodak Bio-Max X-ray film for 2 to 5 s, and kept at −20°C for future reference. Membranes were developed, digitized, and then analyzed by measuring the optical density (OD) of each spot using Metamorph image analysis software (Molecular Devices, Inc). Protein abbreviations are the same as used by RayBiotech and are summarized in Table 1.
Table 1.
Mean protein array optical density (OD) values for 16 HIV-negative control CSF samples
| Proteins expressed in control CSF
|
Positive signals that failed to reach the p<0.001 cutoff
|
No reliable detection
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | SEM | n | OD | SEM | n | OD | SEM | n | |||
| MCP-1 | 2.173 | 0.195 | 16 | FGF-6 | 0.183 | 0.025 | 16 | Leptin | 0.119 | 0.022 | 16 |
| TIMP-2 | 1.720 | 0.198 | 16 | Eotaxin-3 | 0.169 | 0.027 | 16 | GRO | 0.117 | 0.056 | 16 |
| GCSF | 1.213 | 0230 | 16 | M-CSF | 0.111 | 0.018 | 16 | Fas | 0.097 | 0.013 | 16 |
| Acrp30 | 1.009 | 0.137 | 16 | TNF-b | 0.098 | 0.026 | 16 | NAP-2 | 0.096 | 0.015 | 16 |
| IGFBP-2 | 0.894 | 0.178 | 16 | BDNF | 0.087 | 0013 | 16 | RANTES | 0.074 | 0.017 | 16 |
| sTNF RI | 0.641 | 0.083 | 16 | IL-6R | 0.079 | 0.016 | 16 | MCP-2 | 0.065 | 0.010 | 16 |
| TIMP-1 | 0.636 | 0.065 | 16 | IL-la | 0.077 | 0.013 | 16 | TNF-a | 0.060 | 0.012 | 16 |
| uPAR | 0.627 | 0.106 | 16 | SCF | 0.076 | 0.016 | 16 | MCP-3 | 0.053 | 0.010 | 16 |
| Angiogenin | 0.443 | 0.067 | 16 | 1GFBP-1 | 0.075 | 0.017 | 16 | FGF-4 | 0.048 | 0.017 | 16 |
| sgpl30 | 0.320 | 0.042 | 16 | TGF-b3 | 0.067 | 0.011 | 16 | IL-12 p40 | 0.048 | 0.009 | 16 |
| IL-6 | 0.303 | 0.149 | 16 | VEGF | 0.061 | 0.010 | 16 | TRAIL R4 | 0.046 | 0.007 | 16 |
| IGFBP-6 | 0.270 | 0.035 | 16 | MIG | 0.054 | 0.013 | 16 | MCP-4 | 0.046 | 0.012 | 16 |
| IL-1ra | 0.187 | 0.031 | 16 | IL-lb | 0.054 | 0.014 | 16 | FGF-9 | 0.045 | 0.008 | 16 |
| sTNF RII | 0.184 | 0.030 | 16 | IGFBP-3 | 0.053 | 0.010 | 16 | Fit-3 Ligand | 0.045 | 0.009 | 16 |
| Osteoprotegerin | 0.162 | 0.026 | 16 | CNTF | 0.051 | 0.019 | 16 | MIF | 0.039 | 0.007 | 16 |
| IL-3 | 0.160 | 0.020 | 16 | BMP-4 | 0.047 | 0.014 | 16 | Eotaxin | 0.034 | 0.023 | 16 |
| Fractalkine | 0.160 | 0.023 | 16 | SDF-1 | 0.046 | 0.009 | 16 | CKb 8-1 | 0.026 | 0.012 | 16 |
| IL-8 | 0.143 | 0.055 | 16 | IL-4 | 0.041 | 0.008 | 16 | GDNF | 0.025 | 0.008 | 16 |
| EGF | 0.138 | 0.104 | 16 | Thrombopoietin | 0.040 | 0.008 | 16 | GITR | 0.024 | 0.005 | 16 |
| MIP-lb | 0.124 | 0.050 | 16 | MSP-a | 0.039 | 0.005 | 16 | GRO-a | 0.021 | 0.005 | 16 |
| MIP-ld | 0.116 | 0.017 | 16 | LIGHT | 0.036 | 0.013 | 16 | IL-10 | 0.019 | 0.028 | 16 |
| IL-1 R4/ST2 | 0.115 | 0.013 | 16 | Angiopoietin | 0.036 | 0.009 | 16 | TARC | 0.018 | 0.004 | 16 |
| MDC | 0.111 | 0.018 | 16 | VEGF-D | 0.035 | 0.008 | 16 | Eotaxin-2 | 0.018 | 0.017 | 16 |
| MIP-la | 0.098 | 0.029 | 16 | PDGF-BB | 0.035 | 0.013 | 16 | AgRP | 0.018 | 0.005 | 16 |
| EGF-R | 0.098 | 0.012 | 16 | INF-g | 0.021 | 0.005 | 16 | bFGF | 0.014 | 0.005 | 16 |
| GTR-Ligand | 0.094 | 0.012 | 16 | ENA-78 | 0.020 | 0.005 | 16 | BMP-6 | 0.013 | 0.007 | 16 |
| PARC | 0.093 | 0.027 | 16 | PIGF | 0.019 | 0.005 | 16 | IL-17 | 0.011 | 0.004 | 16 |
| NT-3 | 0.079 | 0.023 | 16 | IL-5 | 0.017 | 0.006 | 16 | IGF-I | 0.010 | 0.014 | 16 |
| MIP-3a | 0.077 | 0.016 | 16 | CTACK | 0.014 | 0.006 | 16 | BTC | 0.009 | 0.004 | 16 |
| TRAIL R3 | 0.068 | 0.015 | 16 | IL-7 | 0.013 | 0.009 | 16 | HCC-4 | 0.008 | 0.003 | 16 |
| HGF | 0.063 | 0.013 | 16 | IL-2 Ra | 0.012 | 0.004 | 16 | IL-13 | 0.001 | 0.009 | 16 |
| Lymphotactin | 0.061 | 0.008 | 16 | IL-15 | 0.012 | 0.010 | 16 | CCL-28 | -0.001 | 0.002 | 16 |
| I-309 | 0.049 | 0.007 | 16 | Amphiregulin | 0.008 | 0.004 | 16 | FGF-7 | -0.001 | 0.011 | 16 |
| TGF-b1 | 0.047 | 0.010 | 16 | IL-16 | 0.007 | 0.010 | 16 | GCP-2 | -0.002 | 0.008 | 16 |
| MIP-3b | 0.044 | 0.007 | 16 | GM-CSF | 0.003 | 0.011 | 16 | ||||
| NT-4 | 0.044 | 0.007 | 16 | BLC | 0.000 | 0.011 | 16 | ||||
| IL-2 | 0.043 | 0.015 | 16 | ||||||||
| ICAM-1 | 0.042 | 0.016 | 16 | ||||||||
| Dtk | 0.039 | 0.009 | 16 | ||||||||
| TECK | 0.037 | 0.008 | 16 | ||||||||
| I-TAC | 0.035 | 0.006 | 16 | ||||||||
| IL-12 p70 | 0.034 | 0.004 | 16 | ||||||||
| Oncostatin M | 0.034 | 0.008 | 16 | ||||||||
| IL-1 RI | 0.031 | 0.006 | 16 | ||||||||
| b-NGF | 0.030 | 0.008 | 16 | ||||||||
| IGF-I SR | 0.030 | 0.006 | 16 | ||||||||
| IL-11 | 0.024 | 0.005 | 16 | ||||||||
| IGFBP-4 | 0.017 | 0.010 | 16 | ||||||||
| ICAM-3 | 0.014 | 0.005 | 16 | ||||||||
| AxI | 0.012 | 0.004 | 16 | ||||||||
The first column represents proteins with significant expression that reached the upper 99.9% confidence limit relative to the same array processed with a protein-free aCSF (array background). The middle column represents spots with intermediate signals which did not make the cutoff. The right column contains proteins on the array for which no detectable signal was obtained. Proteins are rank-ordered by abundance in each column Protein abbreviations and synonyms (http://raybiotech.com/cytokine_full_names.asp): Acrp30 adipocyte complement-related protein of 30 kDa; AgRP agouti-related protein; Amphiregulin; Angiogenin; Angiopoietin; Axl tyrosine-protein kinase receptor UFO; BDNF brain-derived neurotrophic factor; bFGF beta fibroblast growth factor; BLC (CXCL13) B cell lymphocyte chemoattractant; BMP-4 bone morphogenic protein-4; BMP-6 bone morphogenic protein-6; b-NGF beta nerve growth factor; BTC betacellulin; CCL-28 C-C motif chemokine 28; CNTF ciliary neurotrophic factor; CTACK (CCL27) cutaneous T cell attracting chemokine; Dtk developmental tyrosine kinase; EGF epidermal growth factor; EGF-R epidermal growth factor receptor; ENA-78 (CXCL5) epithelial neutrophil-activating peptide-78; Eotaxin; Eotaxin-2; Eotaxin-3; Fas tumor necrosis factor receptor superfamily member 6, cell death-inducing factor; FGF-4 fibroblast growth factor-4; FGF-6 fibroblast growth factor-6; FGF-7 fibroblast growth factor-7; FGF-9 fibroblast growth factor-9; Fit-3 Ligand Fms-like tyrosine kinase-3 Ligand; Fractalkine (CX3CL1); GCP-2 (CXCL6) granulocyte chemoattractant protein-2; GCSF granulocyte colony-stimulating factor; GDNF glial-derived neurotrophic factor GITR glucocorticoid-induced tumor necrosis factor receptor family-related gene; GITR-Ligand ligand for glucocorticoid-induced tumor necrosis factor receptor family-related gene; GM-CSF granulocyte-macrophage colony-stimulating factor; GRO growth-related oncogene; GRO-a growth-related oncogene-alpha; HCC-4 (CCL16, LEC, LMC, NCC-4) hemofiltrate CC chemokine 4; HGF hepatocyte growth factor; I-309 (CCL1) T lymphocyte-secreted protein I-309; ICAM-1 intercellular adhesion molecule-1; ICAM-3 intercellular adhesion molecule-3; IGFBP-1 insulin-like growth factor binding protein-1; IGFBP-2 insulin-like growth factor binding protein-2; IGFBP-3 insulin-like growth factor binding protein-3; IGFBP-4 insulin-like growth factor binding protein-4; IGFBP-6 insulin-like growth factor binding protein-6; IGF-I insulin-like growth factor-I; IGF-I SR insulin-like growth factor-I soluble receptor; IL-1 R4/ST2 interleukin-1, receptor 4/ST2; IL-1 RI interleukin-1 receptor 1; IL-10 interleukin-10; IL-11 interleukin-11; IL-12 p40 interleukin 12, 40 kDa subunit; IL-12 p70 interleukin 12, 70–75 kDa subunit; IL-13 interleukin-13; IL-15 interleukin-15; IL-16 interleukin-16; IL-17 interleukin-17; IL-1a interleukin-1 alpha; IL-1b interleukin-1 beta; IL-1ra interleukin-1 receptor antagonist; IL-2 interleukin-2; IL-2 Ra interleukin-2 receptor alpha chain; IL-3 interleukin-3; IL-4 interleukin-4; IL-5 interleukin-5; IL-6 interleukin-6; IL-6R interleukin 6 receptor; IL-7 interleukin-7; IL-8 interleukin-8 (CXCL8); INF-g interferon-gamma; I-TAC (CXCL11) interferon-inducible T cell alpha chemoattractant; Leptin; LIGHT (TNF soluble factor 14) tumor necrosis factor ligand superfamily member 14; Lymphotactin (XCL-1); MCP-1 monocyte chemoattractant-1; MCP-2 monocyte chemoattractant-2; MCP-3 monocyte chemoattractant-3; MCP-4 monocyte chemoattractant-4; M-CSF macrophage colony-stimulating factor; MDC macrophage-derived chemokine; MIF macrophage migration inhibitory factor; MIG (CXCL9) monocyte induced by interferon-gamma chemokine; MIP-1a macrophage inflammatory protein-1 alpha; MIP-1b macrophage inflammatory protein-1 beta; MIP-1d macrophage inflammatory protein-1 delta; MIP-3a macrophage inflammatory protein-3 alpha; MIP-3b macrophage inflammatory protein-3 beta; MSP-a macrophage-stimulating protein alpha; NAP-2 neutrophil-activating peptide; NT-3 neurotrophin-3; NT-4 neurotrophin-4; Oncostatin M; Osteoprotegerin; PARC (CCL-18) C-C motif chemokine 18; PDGF-BB platelet-derived growth factor-BB; PlGF placental growth factor; RANTES (CCL-5) C-C motif chemokine 5; SCF stem cell factor; SDF-1 (CXCL12) stromal cell derived factor-1; sgp130 soluble IL-6 signal-transducing glycoprotein 130; sTNF RI soluble tumor necrosis factor receptor I; sTNF RII soluble tumor necrosis factor receptor II; TARC (CCL17) thymus and activation-regulated chemokine; TECK (CCL25) thymus-expressed chemokine, C-C motif chemokine 25; TGF-b1 transforming growth factor beta 1; TGF-b3 transforming growth factor beta 3; Thrombopoietin; TIMP-1 tissue inhibitor of matrix metalloproteinases-1; TIMP-2 tissue inhibitor of matrix metalloproteinases-2; TNF-a tumor necrosis factor-alpha; TNF-b tumor necrosis factor beta; TRAIL R3 TNF receptor related apoptosis-inducing ligand receptor 3; TRAIL R4 TNF receptor related apoptosis-inducing ligand receptor 4; uPAR (CD87) urokinase-type plasminogen activator receptor; VEGF vascular endothelial growth factor; VEGF-D vascular endothelial growth factor-D
Analysis of proteins in CSF
Overall, there was moderate variation in the intensity of signal (OD) reflecting protein content between different CSF samples and across different runs. This variability was partially controlled by correcting for film background and normalizing expression to the total optical density of all spots on the array (relative OD). With this normalization, relatively consistent expression patterns were seen across patients with an average standard error of ±18.4% of the OD signal for any given protein. Thus, in most cases, approximately a 50% change in expression could reliably be detected in the HIV+ samples relative to normal CSF. By normalizing the data to a total OD value, the assay is not sensitive to uniform changes in protein expression such as global increases or decreases in most proteins. However, no such trends were seen in the raw data indicating that the normalization procedure should provide optimal control of inter-run variability. Negative controls consisting of a protein-free artificial CSF were also run to determine the non-specific background activity for each spot on the array. These negative controls were used to establish minimum baseline values for the array and were subtracted from the positive signals prior to normalization to provide an accurate estimate of protein expression.
Analysis of changes in CSF proteins
Changes in the expression of individual proteins were evaluated using a t test comparing the HIV-infected CSF to the uninfected controls. To provide sufficient stringency, proteins with a p value ≤0.01 were considered significant. Samples were also stratified into three groups reflecting minimal (neuro score=14.0±5.3, n=5), moderate (neuro score=45.4±2.9, n=10), and severe (neuro score=150.4± 12.0, n=5) neurological symptoms. A one-way ANOVA on the stratified data verified the changes seen with the t test but provided little additional information and less stringency. Thus, only the results from the t test are reported.
To provide greater sensitivity and disease specificity, correlations between the neurological scores and protein for each CSF sample were performed to determine if trends could be identified where changes in protein expression might track with cognitive status within the HIV-infected group. The GraphPad Prism software was used to calculate a correlation coefficient and level of significance for the regression of relative protein OD onto neuro score. Correlations with a p value less than 0.05 are reported as significant. Because the use of the arrays provides a highly detailed look at protein expression in a limited number of samples (sacrificing power for protein coverage), we also identified proteins which showed promising trends as having correlations with p values between 0.1 and 0.05.
Results
Pre- and post-highly active antiretroviral therapy toxicity of HIV+ CSF
In 1999, we reported the presence of neurotoxic activity in a subset of CSF samples collected from HIV-infected patients prior to the introduction of highly active antiretroviral therapy (HAART) (Meeker et al. 1999). In subsequent years following the introduction of HAART, the profile of toxicity in CSF began to change and we found it increasingly difficult to identify CSF samples with significant toxicity. Figure 1 illustrates changes in the toxicity profile of 40 HIV+ CSF samples collected from patients prior to the introduction of HAART and 136 HIV+ CSF samples collected after the introduction of HAART. Control CSF from HIV-negative individuals was used to establish the 95% confidence limit for uninfected individuals (dashed lines). After the introduction of HAART, there was a significant decrease in the relative toxicity of the CSF (p< 0.0001). The percentage of samples with toxic activity decreased from 47.5% to 8.1%. An unexpected finding was the extension of the distribution of values from the post-HAART samples into a range where there was less cell death (apparent neuroprotection). Two of the 40 pre-HAART CSF samples (5%) fell below the lower cutoff (the percentage expected by chance based on the 95% confidence limit), whereas 23.5% of the post-HAART CSF values fell below the cutoff. Although there are many factors that could contribute to these differences, we pursued the possibility that the post-HAART CSF may have neuroprotective as well as toxic activity with the net effect depending on the relative balance of each. To gain a better appreciation for the different types of proteins in HIV-infected CNS, we selected 20 HIV+ CSF samples representing a wide range of neurological disease for a broad analysis of 120 cytokines, chemokines, and growth factors. The limited availability of sufficient pre-HAART CSF required that samples come from patients after the introduction of HAART. Given the global nature of the screen, samples were selected to reflect a wide range of neurological disease in the hope of finding trends associated with disease progression with the understanding that this would reduce our ability to accurately identify changes in specific proteins.
Fig. 1.

Summary of toxic activity in human CSF collected prior to (pre) or after (post) the introduction of HAART. Toxic activity was assessed by measuring relative changes in cell death (percent increase in cell death) in rat neural cultures treated with a 1:10 dilution of the CSF in culture medium as compared to cultures treated with artificial CSF. All stages of disease were represented in the samples. HIV-negative control CSF was used to define the normal range of CSF for comparison which was defined as the mean toxicity ±1.96 SD units (p<0.05, dashed lines). Data points falling above this range were counted as toxic and represented 47.5% of all pre-HAART CSF samples (n=40). Two values in the pre-HAART group fell below this range which was expected by chance. Of the 136 CSF samples analyzed after the introduction of HAART, 8.1% were toxic and 23.5% were below the cutoff, suggesting protective activity
Normal cytokine, chemokine, and growth factor protein profiles in human CSF
To establish basal parameters for expression of each protein, 16 CSF samples from HIV-negative individuals were run on the RayBiotech human cytokine protein array VI and VII. An example of signals seen on the array is illustrated in Fig. 2. Mean OD values for HIV-negative control CSF for all proteins is summarized in Table 1. The heterogeneity of protein expression in the control samples was assessed to evaluate normal variation in the CSF composition. Standard error values ranged from 9.3% to 29.4% of the mean OD for 93% of the proteins indicating a relatively consistent level of expression in neurologically normal patients. The remaining 7% of the protein ODs had standard error values ranging from 30.4% to 71.6% indicative of more variation for these eight proteins. A test for normality indicated that the distribution of OD values for 118 proteins was consistent with a single normally distributed population (exceptions, fibroblast growth factor-4 and growth-related oncogene) verifying that the protein expression in the control samples was relatively homogeneous.
Fig. 2.

Example of the signals obtained on the RayBiotech membrane protein arrays after incubation in control CSF. The optical density of each spot was measured. Prominent signals and some proteins of interest are identified on the array
Proteins with significant concentrations in CSF are illustrated in Fig. 3. After correction for background and non-specific signals from array blanks, 50 proteins were found to be reliably expressed in the CSF (significantly above zero) using a 99.9% confidence limit (p<0.001). The mean OD and standard error for each protein is shown for 8 proteins highly expressed (Fig. 3a), 15 proteins expressed at moderate levels (Fig. 3b), and 27 proteins with lower expression (Fig. 3c). Variation was low and sensitivity was high with signals representing as little as 0.012 OD units reliably detected on the array. Negative controls included with the array had an average optical density of 0.00086±0.00191 giving a 14-fold signal to noise ratio on the membrane for the lowest signal considered to be statistically reliable (p<0.001). Notable among the eight proteins with the strongest signals were MCP-1 and tissue inhibitor of metalloproteinase (TIMP) 1 and 2. Other highly expressed proteins included granulocyte colony-stimulating factor (GCSF), adiponectin (Acrp30), soluble tumor necrosis factor (TNF) receptor-1 (sTNF RI), uPAR, and insulin-like growth factor binding protein-2 (IGFBP-2). Moderately high levels (OD=0.270–0.443, Fig. 3b) of angiogenin, sgp130, IL-6, and IGFBP-6 were seen. More modest levels (OD=0.111–0.187) of 11 additional proteins were seen (IL-1ra, sTNF RII, osteoprotegrin, IL-3, fractalkine, IL-8, EGF, monocyte inflammatory protein (MIP)-1b, MIP-1d, IL-1 R4/ST2, and macrophage-derived chemokine). Twenty-seven proteins had a low level of expression (Fig. 3c).
Fig. 3.

Summary of the mean ± SEM (n=16) protein expression in CSF from HIV-negative controls that were significantly above membrane blanks (p<0.01). a Eight proteins were expressed at very high levels in CSF. b Fifteen proteins were expressed at moderate levels (note the change in OD scale). c Twenty-seven proteins were expressed at low levels. The standard error values were generally low indicating good consistency across patients for most proteins
Most classical inflammatory (INF-g, TNF-a, IL-1a, IL-1b, IL-12) and anti-inflammatory proteins (IL-10, IL-13, IL-4 and TGF-b1) were typically expressed at very low or negligible levels in the control CSF. However, given the high level of stringency, the lack of a significant signal should be interpreted cautiously. It does not mean that the protein is absent from CSF, merely that it was not detected with high reliability in this assay.
Effects of HIV infection on CSF proteins
Twenty CSF samples from HIV-infected patients representing different stages of neurological involvement were analyzed and compared to the profile of normal CSF described above. For the initial analysis, the overall impact of HIV infection was evaluated by pooling all HIV+ CSF samples and then comparing the mean level of expression of each protein to HIV-negative CSF. A significance level of 0.01 was used for each pairwise comparison to minimize false positives while allowing a relatively sensitive screen of protein changes associated with HIV infection. With this criterion, 26 proteins increased and 9 proteins decreased relative to the profile seen in HIV-negative control CSF samples. A summary of the proteins that increased is shown in Fig. 4. The proteins are rank-ordered based on the magnitude of the increase seen. Several pro-inflammatory proteins were increased including INF-g and TNF-a, as well as chemokines that might promote immune cell trafficking (eotaxin-2, C-C motif chemokine 28 (CCL-28), B cell lymphocyte chemoattractant, granulocyte chemoattractant protein-2, and cutaneous T cell attracting chemokine). While these changes are consistent with the presence of inflammatory activity, many increases were also seen in proteins that could be considered anti-inflammatory or neuroprotective. These proteins included glial-derived neurotrophic factor (GDNF), bone morphogenic proteins-4 and -6 (BMP-4, BMP-6), brain-derived neurotrophic factor (BDNF), basic fibroblast growth factor (bFGF), fibroblast growth factor-7 (FGF-7), granulocyte-macrophage colony-stimulating factor (GM-CSF), vascular endothelium growth factor (VEGF), and IL-13. Many other proteins were increased although the potential role of these in CSF is less clear.
Fig. 4.

Proteins increased in CSF from HIV-infected patients relative to HIV-negative controls. Bars represent the mean relative OD ± SEM for 20 CSF samples from patients with a wide range of neurological disease. Twenty-six proteins were significantly increased (t test, p<0.01) in the HIV+ CSF. Proteins are rank-ordered from greatest to least change
In addition to the significant increases summarized above, five additional proteins are worth noting due to the magnitude of the increases seen. In each case, greater than a 3-fold increase was seen relative to controls although the results were more variable. These proteins are insulin-like growth factor-I (IGF-I, 5.1-fold, p=0.0245), IGF binding protein-4 (IGFBP-4, 3.2-fold, p=0.0344) TNF-b (3.3-fold, p=0.0783), agouti-related protein (AgRP, 5.5-fold, p=0.1080), and IL-10 (4.5-fold, p=0.0453).
The nine proteins which decreased in HIV-infected individuals are summarized in Fig. 5. Notably, three of these proteins were among those with the highest expression in normal CSF: GCSF, uPAR, and sTNF RI. Smaller decreases were seen in FGF-6, eotaxin-3, IL-3, fractalkine, IL-1 R4/ST2, and MIP-3 alpha.
Fig. 5.
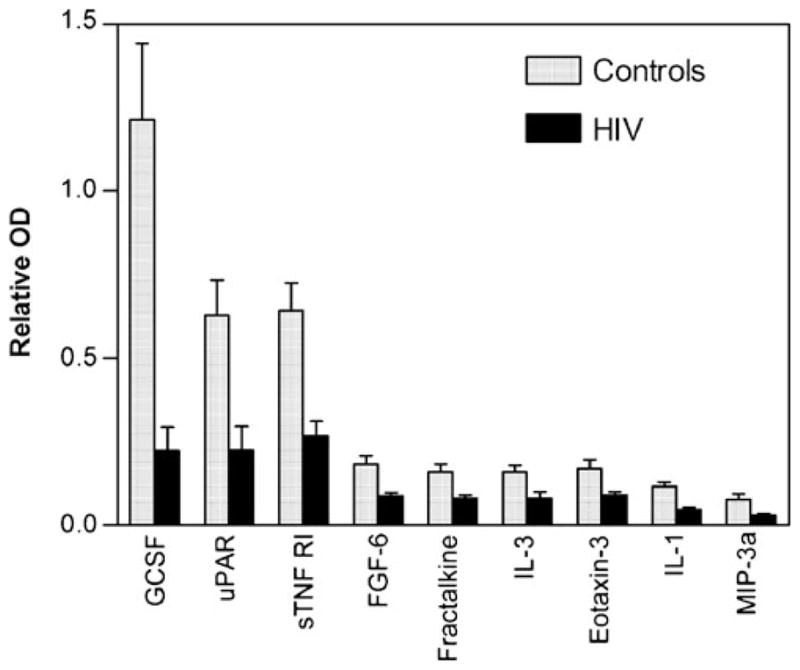
Proteins decreased in CSF from HIV-infected patients relative to HIV-negative controls. Bars represent the mean relative OD ± SEM for 20 CSF samples from patients with a wide range of neurological disease. Nine proteins were significantly decreased (t test, p<0.01) in the HIV+ CSF. Proteins are rank-ordered from greatest to least change
Relating changes in CSF protein content to HIV-associated neurological disease
To determine the extent to which changes in the protein profile in CSF were due to infection alone, we first asked if HIV-positive patients with minimal neurological disease differed from uninfected individuals. CSF samples from patients with neurological scores less than 30 (mean ± SEM neurological score=14.0±5.3; n=5) were compared to controls by t test. Using a cutoff of p≤0.01, a total of 11 proteins changed significantly in the HIV-infected individuals with little or no neurological symptoms indicating that HIV infection alone had a significant impact on the protein composition of the CSF. An additional 27 proteins were just above the cutoff (0.01<p<0.05), suggesting the potential for individual changes in a diverse array of proteins. These changes are summarized in Table 2 (increases) and Table 3 (decreases). Increases included the anti-inflammatory cytokine, IL-13, as well as the neuroprotective growth factors BMP-6 and GM-CSF. Decreases were seen in proteins with endogenous high content and included MCP-1, GCSF, Acrp30, sTNF RI, and uPAR.
Table 2.
Proteins increased in the CSF of HIV-positive individuals with no significant cognitive impairment (n=5) expressed as the OD change relative to uninfected controls (n=16)
| OD increase | SEM | Percent increase | Number | p | |
|---|---|---|---|---|---|
| IL-15 | 0.0601 | 0.0099 | 498.7 | 5 | 0.0004 |
| BMP-6 | 0.0592 | 0.0154 | 467.9 | 5 | 0.0085 |
| BLC | 0.0534 | 0.0101 | 15,7834.4 | 5 | 0.0015 |
| IL-13 | 0.0531 | 0.0093 | 8,188.3 | 5 | 0.0007 |
| GM-CSF | 0.0530 | 0.0114 | 1,588.2 | 5 | 0.0030 |
| HCC-4 | 0.0493 | 0.0080 | 637.2 | 5 | 0.0010 |
| IL-6R | 0.1349 | 0.0537 | 171.1 | 5 | 0.0455 |
| BDNF | 0.1124 | 0.0437 | 128.6 | 5 | 0.0423 |
| INF-g | 0.1084 | 0.0334 | 524.2 | 5 | 0.0214 |
| Dtk | 0.0961 | 0.0301 | 249.0 | 5 | 0.0200 |
| ICAM-1 | 0.0914 | 0.0258 | 215.0 | 5 | 0.0111 |
| BMP-4 | 0.0867 | 0.0279 | 184.3 | 5 | 0.0199 |
| bFGF | 0.0637 | 0.0234 | 456.2 | 5 | 0.0363 |
| CCL-28 | 0.0514 | 0.0187 | 4,320.1 | 5 | 0.0366 |
| SCF | 0.0509 | 0.0151 | 67.0 | 5 | 0.0269 |
| Axl | 0.0507 | 0.0142 | 411.8 | 5 | 0.0139 |
| IL-16 | 0.0506 | 0.0181 | 724.8 | 5 | 0.0309 |
| FGF-9 | 0.0498 | 0.0178 | 111.2 | 5 | 0.0304 |
| GDNF | 0.0487 | 0.0149 | 195.9 | 5 | 0.0164 |
| CTACK | 0.0464 | 0.0186 | 327.9 | 5 | 0.0462 |
| GITR | 0.0442 | 0.0137 | 181.5 | 5 | 0.0191 |
| FGF-7 | 0.0426 | 0.0118 | 3,126.7 | 5 | 0.0138 |
| IL-2 | 0.0412 | 0.0102 | 95.6 | 5 | 0.0246 |
| GCP-2 | 0.0395 | 0.0114 | 2,298.7 | 5 | 0.0129 |
| GRO-a | 0.0385 | 0.0130 | 181.2 | 5 | 0.0253 |
| Amphiregulin | 0.0366 | 0.0145 | 477.1 | 5 | 0.0450 |
| IL-11 | 0.0358 | 0.0114 | 152.2 | 5 | 0.0194 |
| IL-5 | 0.0323 | 0.0116 | 190.8 | 5 | 0.0313 |
| IL-2 Ra | 0.0314 | 0.0107 | 252.9 | 5 | 0.0261 |
| IL-1 RI | 0.0291 | 0.0085 | 95.2 | 5 | 0.0131 |
| TGF-b1 | 0.0267 | 0.0077 | 56.8 | 5 | 0.0410 |
The upper set of proteins represents changes that reached the significance cutoff of p<0.01 and the lower set of proteins represents changes with probabilities between 0.01 and 0.05. The percentage increase is also shown to indicate the relative increase. Values above 1,000 for percent increase are inflated due to very low control OD values
Table 3.
Proteins decreased in the CSF of HIV-positive individuals with no cognitive impairment (n=5) expressed as the OD change relative to uninfected controls (n=16)
| OD decrease | SEM | Percent decrease | Number | p | |
|---|---|---|---|---|---|
| MCP-1 | −1.1265 | 0.1847 | −51.8 | 5 | 0.0005 |
| GCSF | −1.0517 | 0.0535 | −86.7 | 5 | 0.0003 |
| Acrp30 | −0.6638 | 0.0660 | −65.8 | 5 | 0.0002 |
| sTNF RI | −0.4480 | 0.0952 | −69.9 | 5 | 0.0025 |
| uPAR | −0.3938 | 0.0749 | −62.9 | 5 | 0.0048 |
| FGF-6 | −0.0788 | 0.0185 | −43.0 | 5 | 0.0157 |
| Leptin | −0.0620 | 0.0199 | −52.1 | 5 | 0.0409 |
| IL-1 R4/ST2 | −0.0490 | 0.0180 | −42.6 | 5 | 0.0381 |
| MIP-3a | −0.0412 | 0.0114 | −53.8 | 5 | 0.0420 |
The upper set of proteins represents changes that reached the significance cutoff of p<0.01 and the lower set of proteins represents changes with probabilities between 0.01 and 0.05. The percentage decrease is also shown to indicate the relative decrease
The above analyses provided a picture of the general changes in CSF associated with HIV infection but did not indicate how a given factor might be related to neuropathogenesis. We evaluated the strength of the relationship between each protein and disease severity by calculating the correlation between protein content and the matched neurological score. A significant relationship between neurological score and protein was found for six proteins illustrated in Figs. 6, 7, and 8.
Fig. 6.

Correlation between cytokines in CSF and neurological score (neuro score). A significant negative relationship was seen between the expression of the cytokines IL-5 and IL-6 and neurological disease severity. In each case, the open box on the Y axis shows the average content of each protein in CSF from the HIV-negative controls. Comparison to the control values illustrates that low neurological scores are associated with cytokine increases and higher scores with cytokine decreases
Fig. 7.

Correlation between chemokines in CSF and neurological score (neuro score). A significant positive relationship was seen between the expression of the chemokine MCP-1 and neurological disease severity. MCP-2 showed a significant negative correlation with neurological score. In each case, the open box on the Y axis shows the average content of each protein in CSF from the HIV-negative controls
Fig. 8.

Correlation between growth factors in CSF and neurological score (neuro score). A significant negative relationship was seen between the expression of the growth factors BDNF and NT-3 and neurological disease severity. The open box on the Y axis shows the average content of each protein in CSF from the HIV-negative controls. In most cases, there is an increase in growth factor expression in patients with low neurological scores and a decline as neurological disease severity increases
Two cytokines (IL-5 and IL-6), illustrated in Fig. 6, showed declines with disease severity with correlations of 0.444 (IL-5) and 0.443 (IL-6), each with a p value of 0.050. The decrease in IL-6 was paralleled by a decreasing trend in IL-6R (r=0.403, p=0.078, not shown). Similar trends were also seen for the cytokines IL-4, IL-13, and IL-17 that just fell short of significance (r=0.393, p=0.087; r=0.3942, p= 0.0855; and 0.426, p=0.061, respectively). The mean level of protein expression for uninfected control CSF is indicated on each figure by the open box on the Y axis. This value is included to illustrate that CSF levels of specific proteins may be increased or decreased relative to control levels depending on the neurological status. This explains why some proteins with positive increases in Fig. 4 show negative correlations with neurological score (increase in HIV-infected patients with low neuro scores and a decrease with high neuro scores). This also emphasizes the need to have CSF profiles for both uninfected patients as well as neurologically normal infected patients to discriminate between the response to the infection versus protein changes specifically associated with neurological decline.
One chemokine (MCP-2) decreased with disease severity (Fig. 7) (r=0.498, p=0.025) and one increased with disease severity (MCP-1, r=0.514, p=0.020). The box on the Y axis shows the average protein in control CSF. Two additional chemokines, CCL-28 and MIP-3a, showed decreasing trends with neurological impairment (not shown) that just failed to reach significance (p’s 0.063 and 0.066).
Two growth factors (BDNF and NT-3), illustrated in Fig. 8, also decreased with disease severity with r values of 0.482 (p=0.031) and 0.480 (p=0.032) for BDNF and NT-3, respectively. A similar trend was seen for EGF (r=0.431, p= 0.058), soluble EGF receptor (r=0.474, p=0.035), and CNTF (r=0.421, p=0.065).
A separate analysis examined which proteins in CSF best predicted subsequent neurological disease progression in the same patient. CSF protein at the first visit was correlated to the change in neuro score from the first to the second visit. Two patients with high neuro scores at entry were not included in the analysis. The average neuro score at the first visit was 34.8±7.1 and the average neuro score at the second visit was 70.4±23.0. The average interval from the first to the second visit was 7.1±0.9 months. Four proteins, illustrated in Fig. 9, showed a significant correlation to subsequent changes in neuro scores: BDNF (r=−0.7248, p=0.042), EGF (r=−0.7480, p=0.033), eotaxin-3 (r=−0.7075, p=0.050), and FGF-6 (r=−0.7707, p=0.025). Ten additional proteins showed promising trends: CNTF (p=0.0577), IL-5 (p=0.0586), HCC-4 (p=0.0750), IL-1RI (p=0.0762), eotaxin (p=0.0773), b-NGF (p=0.0845), Fit-3 ligand (p=0.0886), IL-16 (p=0.0937), IL-2 (p=0.0950), and GCSF (p=0.0953). Overall, growth factors were disproportionately represented in these results. Typical inflammatory cytokines were not associated with neurological disease progression. CSF HIV titers also showed a low correlation to the subsequent change in neuro score (r=0.106, NS).
Fig. 9.

Correlation between growth factor expression in the CSF (normalized OD) at the first visit and the change in the neurological score (neuro score) at the second visit. A significant negative relationship was seen for BDNF, EGF, eotaxin-3, and FGF-6, indicating that low levels of growth factors (BDNF, EGF, FGF-6) or chemokine (eotaxin-3) predicted a subsequent decline in neurological status
Discussion
HIV-associated neuronal damage is thought to be mediated in part by soluble factors secreted by macrophages and microglia. In vivo support for soluble toxins in the CNS comes from the demonstration that CSF from HIV-infected patients often contains substances that are toxic to neurons (Meeker et al. 1999, 2005). Our analysis of CSF samples collected after the introduction of HAART showed less neurotoxicity than pre-HAART samples. This is consistent with a general decrease in the severity of neurological disease with the introduction of HAART. However, these studies also identified potential protective activity in the post-HAART CSF raising the possibility that the CSF may contain a mix of proteins reflecting both destructive and repair processes. This is consistent with a growing awareness of the importance of the dual destructive/protective functions of microglia and macrophages in a variety of diseases (Streit 2005; Turrin and Rivest 2006).
The protein profiles presented in this paper represent an initial attempt to assess the overall balance of cytokines, chemokines, growth factors, and related substances in the CSF of normal and HIV-infected individuals. Because the focus was on establishing a broad profile of protein expression in a small number of samples, these studies have several limitations. First, it was not feasible to run large numbers of samples that would allow sensitive comparisons of individual proteins. Results were non-quantitative and required relatively large amounts of CSF (1 ml). The wide range of protein expression sometimes made it difficult to assess low and high abundance proteins on the same array, and careful background correction and blank arrays were necessary to reduce variability. These limitations, however, were offset by the ability to evaluate expression patterns of a large number of proteins with various functions. As such, our findings identified unexpected changes in protein expression including a potential role for changes in neuroprotective molecules in HIV pathogenesis.
Normal CSF protein profiles
To understand disease-related changes in CSF proteins, we first needed a characterization of “normal” CSF to establish baseline levels of expression as well as the variation between individuals. A relatively consistent pattern of protein expression was seen in control CSF indicating that the intrinsic individual variation was not a limiting factor in the analysis. The normal profile of cytokines, chemokines, and growth factors in the CSF of uninfected individuals indicated a strong presence of proteins within specific functional categories. Among the most highly expressed were proteins that control monocyte entry and macrophage activity. The monocyte chemokine, MCP-1, was highly expressed as was the macrophage-stimulating factor, GCSF. The high level of MCP-1 suggested the presence of a robust endogenous signal for monocyte recruitment. TIMP-1 and TIMP-2 were also expressed at high levels, suggesting strong control of matrix metalloproteinases, which remodel the extracellular matrix and help to control cell trafficking. The TIMPs are thought to be neuroprotective against HIV-associated neural damage (Johnston et al. 2001; Suryadevara et al. 2003; Leveque et al. 2004) and may balance chemokine actions that favor recruitment of monocytes. Three additional substances expressed at high levels were sTNF RI, uPAR, and Acrp30 whose functions in the CNS are not well understood. High sTNF RI may reflect endogenous control of TNF activity and intrinsic protection from inflammatory damage (Nawashiro et al. 1997; Barone et al. 1997; Emsley et al. 2007). High sTNF RI levels were paralleled by moderately high sgp130 which may serve a similar function for IL-6 (Padberg et al. 1999; Jostock et al. 2001). Acrp30, also known as adiponectin, has been linked to the control of energy expenditure via lipid oxidation and glucose uptake, in part, through the translocation of glucose transporter 4 and sensitization of the insulin receptor (Berg et al. 2002; Qi et al. 2004). It has been correlated with mortality in chronic heart failure (Axelsson et al. 2005) and has been suggested as a systemic marker for wasting (Kistorp et al. 2005). Decreases resulting from HIV infection (Giralt et al. 2006) and an inverse correlation with body fat suggest a potential role in HIV-associated metabolic syndrome in HIV-infected adults and children (Tsiodras and Mantzoros 2006; Verkauskiene et al. 2006). In addition, decreased expression of the associated protein, leptin, in CSF of HIV-infected men has been linked to poor neuropsychological performance suggesting a link between proteins that control energy expenditure and neural disease (Huang et al. 2007). The role of uPAR in the CNS is not well understood, but studies have also linked changes in expression to HIV pathogenesis (Sidenius et al. 2004; Sporer et al. 2005). Pro-inflammatory and anti-inflammatory cytokines were generally low in uninfected CSF.
Changes in CSF proteins associated with HIV infection reflect both inflammatory and neuroprotective activity
Overall, the data illustrated that many changes in protein expression may be due simply to HIV infection, and these changes include alterations in anti-inflammatory and neuroprotective proteins as well as inflammatory proteins. The array profiles showed increases in the inflammatory cytokines, INF-g and TNF-a in response to HIV infection as previously observed. At the same time, the arrays clearly demonstrated that the response to infection includes increases in the cytokine IL-13 that has anti-inflammatory actions in the brain (Wong et al. 1997; Szczepanik et al. 2001) as well as a number of growth factors (GDNF, BMP-6, BDNF, VEGF, GM-CSF, and bFGF) with putative neuroprotective actions (Novikova et al. 2000; Stadelmann et al. 2002; Yabe et al. 2002; Storkebaum et al. 2004; Yasuhara et al. 2004; Husson et al. 2005; Kilic et al. 2005; Wu 2005; Gora-Kupilas and Josko 2005; Wang et al. 2005; Nakagawa et al. 2006; Kilic et al. 2006). The prominent increase in growth factors suggests the presence of restorative processes during HIV infection.
Fewer substances decreased in response to HIV infection but included potentially important proteins such as GCSF, uPAR, and sTNF RI. Increases in soluble uPAR in the CSF have previously been associated with neurological disease (Sidenius et al. 2004; Sporer et al. 2005). This is in contrast to our studies where an overall decrease in uPAR levels was seen with HIV infection. GCSF showed the greatest decrease in response to HIV infection, but little information is available on the potential role of this protein in HIV pathogenesis.
Is there a general pattern of protein changes associated with neurological decline in HIV infection?
Although there was a general increase in the pro-inflammatory cytokines, INF-g and TNF-a, there was little indication of a relationship to neurological disease. Two cytokines, IL-5 and IL-6, decreased in samples from patients with greater neurological disease. A similar decreasing trend was seen for the cytokines IL-4, IL-13, and IL-17 that just fell short of significance (r=0.393, p=0.087 and 0.426, p=0.061, respectively). While this might be indicative of a general loss of cytokine signaling, it is notable that INF-g and TNF-a remained high or increased with severity of neurological symptoms.
As previously reported, a significant increase in the chemokine, MCP-1, was seen with neurological decline (Conant et al. 1998; Kelder et al. 1998; Cinque et al. 1998). However, it is noteworthy that in the CSF samples analyzed in our studies, a decrease in MCP-1 was seen in infected patients with minimal neurological disease. Thus, the “increases” in MCP-1 with cognitive impairment may reflect a reversal of the pattern of MCP-1 secretion in neurologically asymptomatic versus symptomatic patients. This pattern may explain why some studies have observed decreases in MCP-1 with HIV infection (Enting et al. 2000; McCoig et al. 2004), while others show a positive correlation between MCP-1 CSF levels and neurological disease (Conant et al. 1998; Kelder et al. 1998; Cinque et al. 1998).
When proteins were directly correlated to neurological disease, an unexpected pattern emerged. Growth factors showed the strongest association with neurological decline. Two growth factors in particular (BDNF and NT-3) decreased in parallel with neurological decline with similar trends for two additional growth factors. Infected but unimpaired patients had higher than normal growth factor expression (hence the overall increase for the HIV group) which then decreased with more severe neurological disease. Further support for a role of growth factors comes from the correlation of growth factor expression to subsequent changes in neurological disease status. Only four proteins showed a significant relationship and three were growth factors (BDNF, EGF, FGF-6). Thus, a low concentration of these growth factors was predictive of subsequent neurological decline in the same patient. This would be consistent with the idea that neurological disease may progress, in part, due to a loss of endogenous neuroprotection. BDNF, in particular, has been shown to offer protection specifically against viral and gp120-associated neural damage (Mocchetti and Bachis 2004; Nosheny et al. 2005; Bachis and Mocchetti 2005; Meisner et al. 2008). Local administration of BDNF has been shown to reverse gp120-induced neurotoxicity both in vitro (Mocchetti and Bachis 2004; Bachis and Mocchetti 2005) and in vivo (Nosheny et al. 2007). The therapeutic effects of memantine have been attributed, in part, to an upregulation of BDNF in brain tissue of SIV-infected macaques (Meisner et al. 2008), and reversal of macrophage-associated damage in a SCID mouse model has been correlated with neurotrophin expression (Poluektova et al. 2004). Additional support for an interaction between BDNF expression and inflammation is found in various models. Decreases in BDNF expression have been associated with tissue damage in neurogenic inflammation of the retina (Bronzetti et al. 2007), atopic dermatitis lesions (Groneberg et al. 2007), brains of mice exposed to LPS (Schnydrig et al. 2007), and in the CSF of HIV-infected patients (Albrecht et al. 2006). In addition, in elderly patients, low plasma BDNF was found to be a robust biomarker of all cause mortality risk (Krabbe et al. 2009). The reason for the decline in BDNF and NT-3 is not known. It may reflect neural dysfunction or reduced production by other growth factor secreting cells such as microglia/macrophages (Elkabes et al. 1996). In post-mortem studies of AIDS patients, BDNF immunoreactivity was found to be co-localized to infiltrating, activated microglia/macrophages, suggesting that these cells may play a role in protection and repair (Soontornniyomkij et al. 1998). It is possible that altered functions of these cells may contribute to the loss of growth factor protection.
Overall, the above patterns of protein expression reinforce two important points. First, although inflammatory proteins were generally elevated, no clear relationship was seen with disease progression. Second, anti-inflammatory and neuroprotective activity in CSF must be factored into theories of HIV neuropathogenesis. The loss of growth factors, in particular, correlated with neurological disease and was best at predicting subsequent neurological decline. Although the number of samples was too small to draw definitive conclusions, the findings support a growing number of studies indicating that a loss of growth factor expression may not only be a useful marker of disease progression but also a potential target of therapeutic approaches designed to preserve the intrinsic neuroprotective activity in the CNS. The current observations have set the framework for more comprehensive studies of proteins released into the CNS in response to disease. The relative stability of the protein profiles across the control samples indicates that it is feasible to establish norms from which pathological samples can be judged. More work is needed to establish the functional significance of changes in neuroprotective and anti-inflammatory factors in HIV-infected individuals as well as the therapeutic potential of treatments designed to enhance these processes.
Acknowledgments
This work was supported by NIH Grants MH079726 and MH085606.
Contributor Information
Rick B. Meeker, Email: meekerr@neurology.unc.edu, Department of Neurology, University of North Carolina, CB #7025, 6113 Neuroscience Research Bldg, 115 Mason Farm Road, Chapel Hill, NC 27599, USA
Winona Poulton, Department of Neurology, University of North Carolina, CB #7025, 6113 Neuroscience Research Bldg, 115 Mason Farm Road, Chapel Hill, NC 27599, USA. Research Triangle Institute, Research Triangle Park, NC 27709, USA.
Silva Markovic-Plese, Department of Neurology, University of North Carolina, CB #7025, Physicians Office Bldg, Chapel Hill, NC 27599, USA.
Colin Hall, Department of Neurology, University of North Carolina, CB #7025, Physicians Office Bldg, Chapel Hill, NC 27599, USA.
Kevin Robertson, Department of Neurology, University of North Carolina, CB #7025, Physicians Office Bldg, Chapel Hill, NC 27599, USA.
References
- Achim CL, Heyes MP, Wiley CA. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brains. J Clin Invest. 1993;91:2769–2775. doi: 10.1172/JCI116518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht D, Garcia L, Cartier L, Kettlun AM, Vergara C, Collados L, Valenzuela MA. Trophic factors in cerebrospinal fluid and spinal cord of patients with tropical spastic paraparesis, HIV, and Creutzfeldt–Jakob disease. AIDS Res Hum Retroviruses. 2006;22:248–254. doi: 10.1089/aid.2006.22.248. [DOI] [PubMed] [Google Scholar]
- Axelsson J, Heimburger O, Lindholm B, Stenvinkel P. Adipose tissue and its relation to inflammation: the role of adipokines. J Ren Nutr. 2005;15:131–136. doi: 10.1053/j.jrn.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Bachis A, Mocchetti I. Brain-derived neurotrophic factor is neuroprotective against human immunodeficiency virus-1 envelope proteins. Ann NY Acad Sci. 2005;1053:247–257. doi: 10.1196/annals.1344.022. [DOI] [PubMed] [Google Scholar]
- Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ. Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology. 2007;68:1481–1487. doi: 10.1212/01.wnl.0000260610.79853.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Marcheselli VL, Cole-Edwards K. Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann NY Acad Sci. 2005;1053:137–147. doi: 10.1196/annals.1344.011. [DOI] [PubMed] [Google Scholar]
- Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814–1820. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- Bragg DC, Robertson K, Hall CD, Meeker RB. Techniques to measure neurologic disease progression in HIV-1 patients. Science Online: NeuroAIDS. 2000;3:1–9. [Google Scholar]
- Brew BJ, Pemberton L, Blennow K, Wallin A, Hagberg L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology. 2005;65:1490–1492. doi: 10.1212/01.wnl.0000183293.95787.b7. [DOI] [PubMed] [Google Scholar]
- Bronzetti E, Artico M, Kovacs I, Felici LM, Magliulo G, Vignone D, D’Ambrosio A, Forte F, Di Liddo R, Feher J. Expression of neurotransmitters and neurotrophins in neurogenic inflammation of the rat retina. Eur J Histochem. 2007;51:251–260. [PubMed] [Google Scholar]
- Cinque P, Vago L, Mengozzi M, Torri V, Ceresa D, Vicenzi E, Transidico P, Vagani A, Sozzani S, Mantovani A, Lazzarin A, Poli G. Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. AIDS. 1998;12:1327–1332. doi: 10.1097/00002030-199811000-00014. [DOI] [PubMed] [Google Scholar]
- Cinque P, Nebuloni M, Santovito ML, Price RW, Gisslen M, Hagberg L, Bestetti A, Vago G, Lazzarin A, Blasi F, Sidenius N. The urokinase receptor is overexpressed in the AIDS dementia complex and other neurological manifestations. Ann Neurol. 2004;55:687–694. doi: 10.1002/ana.20076. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, McArthur JC, Griffin DE, Sjulson L, Wahl LM, Irani DN. Cerebrospinal fluid levels of MMP-2, 7, and 9 are elevated in association with human immunodeficiency virus dementia. Ann Neurol. 1999;46:391–398. doi: 10.1002/1531-8249(199909)46:3<391::aid-ana15>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Elkabes S, DiCicco-Bloom EM, Black IB. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elovaara I, Muller KM. Cytoimmunological abnormalities in cerebrospinal fluid in early stages of HIV-1 infection often precede changes in blood. J Neuroimmunol. 1993;44:199–204. doi: 10.1016/0165-5728(93)90043-x. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Illingworth K, Scarth S, Wickramasinghe V, Hoadley ME, Rothwell NJ, Tyrrell PJ, Hopkins SJ. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007;7:5. doi: 10.1186/1471-2377-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enting RH, Foudraine NA, Lange JM, Jurriaans S, van der PT, Weverling GJ, Portegies P. Cerebrospinal fluid beta2-microglobulin, monocyte chemotactic protein-1, and soluble tumour necrosis factor alpha receptors before and after treatment with lamivudine plus zidovudine or stavudine. J Neuroimmunol. 2000;102:216–221. doi: 10.1016/s0165-5728(99)00219-2. [DOI] [PubMed] [Google Scholar]
- Epstein L, Gendelman H. Human immunodeficiency virus type 1 infection of the nervous system: pathogenic mechanisms. Ann Neurol. 1993;33:429–436. doi: 10.1002/ana.410330502. [DOI] [PubMed] [Google Scholar]
- Giralt M, Domingo P, Guallar JP, Rodriguez de la Concepcion ML, Alegre M, Domingo JC, Villarroya F. HIV-1 infection alters gene expression in adipose tissue, which contributes to HIV-1/HAART-associated lipodystrophy. Antivir Ther. 2006;11:729–740. [PubMed] [Google Scholar]
- Gisslen M, Hagberg L, Brew BJ, Cinque P, Price RW, Rosengren L. Elevated cerebrospinal fluid neurofilament light protein concentrations predict the development of AIDS dementia complex. J Infect Dis. 2007;195:1774–1778. doi: 10.1086/518043. [DOI] [PubMed] [Google Scholar]
- Giulian D, Noonan CA. Neurotoxins from HIV-infected mononuclear phagocytes. In: Gendelman HE, Lipton SAEL, Swindells S, editors. The neurology of AIDS. International Thompson; New York: 1998. pp. 117–129. [Google Scholar]
- Giulian D, Yu J, Li X, Tom D, Li J, Wendt E, Lin S-N, Schwarcz R, Noonan C. Study of receptor-mediated neurotoxins released by HIV-1-infected mononuclear phagocytes found in human brain. J Neurosci. 1996;16:3139–3153. doi: 10.1523/JNEUROSCI.16-10-03139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gora-Kupilas K, Josko J. The neuroprotective function of vascular endothelial growth factor (VEGF) Folia Neuropathol. 2005;43:31–39. [PubMed] [Google Scholar]
- Groneberg DA, Fischer TC, Peckenschneider N, Noga O, Dinh QT, Welte T, Welker P. Cell type-specific regulation of brain-derived neurotrophic factor in states of allergic inflammation. Clin Exp Allergy. 2007;37:1386–1391. doi: 10.1111/j.1365-2222.2007.02790.x. [DOI] [PubMed] [Google Scholar]
- Hagberg L, Fuchs D, Rosengren L, Gisslen M. Intrathecal immune activation is associated with cerebrospinal fluid markers of neuronal destruction in AIDS patients. J Neuroimmunol. 2000;102:51–55. doi: 10.1016/s0165-5728(99)00150-2. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Brew B, Martin A, Markey SP, Price RW, Bhalla RB, Salazar A. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Adv Exp Med Biol. 1991;294:687–690. doi: 10.1007/978-1-4684-5952-4_94. [DOI] [PubMed] [Google Scholar]
- Huang JS, Letendre S, Marquie-Beck J, Cherner M, McCutchan JA, Grant I, Ellis R. Low CSF leptin levels are associated with worse learning and memory performance in HIV-infected men. J Neuroimmune Pharmacol. 2007;2:352–358. doi: 10.1007/s11481-007-9093-z. [DOI] [PubMed] [Google Scholar]
- Hult B, Chana G, Masliah E, Everall I. Neurobiology of HIV. Int Rev Psychiatry. 2008;20:3–13. doi: 10.1080/09540260701862086. [DOI] [PubMed] [Google Scholar]
- Husson I, Rangon CM, Lelievre V, Bemelmans AP, Sachs P, Mallet J, Kosofsky BE, Gressens P. BDNF-induced white matter neuroprotection and stage-dependent neuronal survival following a neonatal excitotoxic challenge. Cereb Cortex. 2005;15:250–261. doi: 10.1093/cercor/bhh127. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol. 2001;49:230–241. doi: 10.1002/1531-8249(20010201)49:2<230::aid-ana43>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268:160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol. 1998;44:831–835. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Hermann DM. TAT-GDNF in neurodegeneration and ischemic stroke. CNS Drug Rev. 2005;11:369–378. doi: 10.1111/j.1527-3458.2005.tb00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Wang Y, Bassetti CL, Marti HH, Hermann DM. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006;20:1185–1187. doi: 10.1096/fj.05-4829fje. [DOI] [PubMed] [Google Scholar]
- Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, Hildebrandt P. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation. 2005;112:1756–1762. doi: 10.1161/CIRCULATIONAHA.104.530972. [DOI] [PubMed] [Google Scholar]
- Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- Kolson DL. Neuropathogenesis of central nervous system HIV-1 infection. Clin Lab Med. 2002;22:703–717. doi: 10.1016/s0272-2712(02)00009-4. [DOI] [PubMed] [Google Scholar]
- Krabbe KS, Mortensen EL, Avlund K, Pedersen AN, Pedersen BK, Jorgensen T, Bruunsgaard H. Brain-derived neurotrophic factor predicts mortality risk in older women. J Am Geriatr Soc. 2009;57:1447–1452. doi: 10.1111/j.1532-5415.2009.02345.x. [DOI] [PubMed] [Google Scholar]
- Leveque T, Le Pavec G, Boutet A, Tardieu M, Dormont D, Gras G. Differential regulation of gelatinase A and B and TIMP-1 and -2 by TNF-alpha and HIV virions in astrocytes. Microbes Infect. 2004;6:157–163. doi: 10.1016/j.micinf.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Liuzzi GM, Mastroianni CM, Santacroce MP, Fanelli M, D’Agostino C, Vullo V, Riccio P. Increased activity of matrix metalloproteinases in the cerebrospinal fluid of patients with HIV-associated neurological diseases. J Neurovirol. 2000;6:156–163. doi: 10.3109/13550280009013159. [DOI] [PubMed] [Google Scholar]
- McCoig C, Castrejon MM, Saavedra-Lozano J, Castano E, Baez C, Lanier ER, Saez-Llorens X, Ramilo O. Cerebrospinal fluid and plasma concentrations of proinflammatory mediators in human immunodeficiency virus-infected children. Pediatr Infect Dis J. 2004;23:114–118. doi: 10.1097/01.inf.0000109247.67480.7a. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Robertson K, Barry T, Hall C. Neurotoxicity of CSF from HIV-infected humans. J Neurovirol. 1999;5:507–518. doi: 10.3109/13550289909045380. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Boles JC, Robertson KR, Hall CD. Cerebrospinal fluid from human immunodeficiency virus-infected individuals facilitates neurotoxicity by suppressing intracellular calcium recovery. J Neurovirol. 2005;11:144–156. doi: 10.1080/13550280590922757. [DOI] [PubMed] [Google Scholar]
- Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, Meulen VT, Koutsilieri E. Memantine upregulates BDNF and prevents dopamine deficits in SIV-infected macaques: a novel pharmacological action of memantine. Neuropsychopharmacology. 2008;33:2228–2236. doi: 10.1038/sj.npp.1301615. [DOI] [PubMed] [Google Scholar]
- Mocchetti I, Bachis A. Brain-derived neurotrophic factor activation of TrkB protects neurons from HIV-1/gp120-induced cell death. Crit Rev Neurobiol. 2004;16:51–57. doi: 10.1615/critrevneurobiol.v16.i12.50. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res. 2006;1089:179–185. doi: 10.1016/j.brainres.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Martin D, Hallenbeck JM. Neuroprotective effects of TNF binding protein in focal cerebral ischemia. Brain Res. 1997;778:265–271. doi: 10.1016/s0006-8993(97)00981-5. [DOI] [PubMed] [Google Scholar]
- Nosheny RL, Mocchetti I, Bachis A. Brain-derived neurotrophic factor as a prototype neuroprotective factor against HIV-1-associated neuronal degeneration. Neurotox Res. 2005;8:187–198. doi: 10.1007/BF03033829. [DOI] [PubMed] [Google Scholar]
- Nosheny RL, Ahmed F, Yakovlev A, Meyer EM, Ren K, Tessarollo L, Mocchetti I. Brain-derived neurotrophic factor prevents the nigrostriatal degeneration induced by human immunodeficiency virus-1 glycoprotein 120 in vivo. Eur J Neurosci. 2007;25:2275–2284. doi: 10.1111/j.1460-9568.2007.05506.x. [DOI] [PubMed] [Google Scholar]
- Novikova LN, Novikov LN, Kellerth JO. Survival effects of BDNF and NT-3 on axotomized rubrospinal neurons depend on the temporal pattern of neurotrophin administration. Eur J Neurosci. 2000;12:776–780. doi: 10.1046/j.1460-9568.2000.00978.x. [DOI] [PubMed] [Google Scholar]
- Padberg F, Feneberg W, Schmidt S, Schwarz MJ, Korschenhausen D, Greenberg BD, Nolde T, Muller N, Trapmann H, Konig N, Moller HJ, Hampel H. CSF and serum levels of soluble interleukin-6 receptors (sIL-6R and sgp130), but not of interleukin-6 are altered in multiple sclerosis. J Neuroimmunol. 1999;99:218–223. doi: 10.1016/s0165-5728(99)00120-4. [DOI] [PubMed] [Google Scholar]
- Pemberton LA, Brew BJ. Cerebrospinal fluid S-100beta and its relationship with AIDS dementia complex. J Clin Virol. 2001;22:249–253. doi: 10.1016/s1386-6532(01)00196-2. [DOI] [PubMed] [Google Scholar]
- Perrella O, Guerriero M, Izzo E, Soscia M, Carrieri PB. Interleukin-6 and granulocyte macrophage-CSF in the cerebrospinal fluid from HIV infected subjects with involvement of the central nervous system. Arq Neuropsiquiatr. 1992;50:180–182. doi: 10.1590/s0004-282x1992000200008. [DOI] [PubMed] [Google Scholar]
- Poluektova L, Gorantla S, Faraci J, Birusingh K, Dou H, Gendelman HE. Neuroregulatory events follow adaptive immune-mediated elimination of HIV-1-infected macrophages: studies in a murine model of viral encephalitis. J Immunol. 2004;172:7610–7617. doi: 10.4049/jimmunol.172.12.7610. [DOI] [PubMed] [Google Scholar]
- Price RW, Sidtis JJ. Evaluation of the AIDS dementia complex in clinical trials. J Acquir Immune Defic Syndr. 1990;3(Suppl 2):S51–S60. [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10:524–529. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- Robertson KR, Kapoor C, Robertson WT, Fuscus S, Ford S, Hall C. No gender differences in the progression of nervous system disease of HIV infection. J Acquir Immune Defic Syndr. 2004;36:817–822. doi: 10.1097/00126334-200407010-00008. [DOI] [PubMed] [Google Scholar]
- Sabri F, de Milito A, Pirskanen R, Elovaara I, Hagberg L, Cinque P, Price R, Chiodi F. Elevated levels of soluble Fas and Fas ligand in cerebrospinal fluid of patients with AIDS dementia complex. J Neuroimmunol. 2001;114:197–206. doi: 10.1016/s0165-5728(00)00424-0. [DOI] [PubMed] [Google Scholar]
- Schnydrig S, Korner L, Landweer S, Ernst B, Walker G, Otten U, Kunz D. Peripheral lipopolysaccharide administration transiently affects expression of brain-derived neurotrophic factor, corticotropin and proopiomelanocortin in mouse brain. Neurosci Lett. 2007;429:69–73. doi: 10.1016/j.neulet.2007.09.067. [DOI] [PubMed] [Google Scholar]
- Sidenius N, Nebuloni M, Sala S, Zerbi P, Price RW, Gisslen M, Hagberg L, Vago L, Lazzarin A, Blasi F, Cinque P. Expression of the urokinase plasminogen activator and its receptor in HIV-1-associated central nervous system disease. J Neuroimmunol. 2004;157:133–139. doi: 10.1016/j.jneuroim.2004.08.038. [DOI] [PubMed] [Google Scholar]
- Soontornniyomkij V, Wang G, Pittman CA, Wiley CA, Achim CL. Expression of brain-derived neurotrophic factor protein in activated microglia of human immunodeficiency virus type 1 encephalitis. Neuropathol Appl Neurobiol. 1998;24:453–460. doi: 10.1046/j.1365-2990.1998.00134.x. [DOI] [PubMed] [Google Scholar]
- Sporer B, Koedel U, Goebel FD, Pfister HW. Increased levels of soluble Fas receptor and Fas ligand in the cerebrospinal fluid of HIV-infected patients. AIDS Res Hum Retroviruses. 2000;16:221–226. doi: 10.1089/088922200309313. [DOI] [PubMed] [Google Scholar]
- Sporer B, Koedel U, Popp B, Paul R, Pfister HW. Evaluation of cerebrospinal fluid uPA, PAI-1, and soluble uPAR levels in HIV-infected patients. J Neuroimmunol. 2005;163:190–194. doi: 10.1016/j.jneuroim.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26:943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Res Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Suryadevara R, Holter S, Borgmann K, Persidsky R, Labenz-Zink C, Persidsky Y, Gendelman HE, Wu L, Ghorpade A. Regulation of tissue inhibitor of metalloproteinase-1 by astrocytes: links to HIV-1 dementia. Glia. 2003;44:47–56. doi: 10.1002/glia.10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate A beta(1-42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol. 2001;113:49–62. doi: 10.1016/s0165-5728(00)00404-5. [DOI] [PubMed] [Google Scholar]
- Towfighi A, Skolasky RL, St Hillaire C, Conant K, McArthur JC. CSF soluble Fas correlates with the severity of HIV-associated dementia. Neurology. 2004;62:654–656. doi: 10.1212/01.wnl.0000110188.37546.51. [DOI] [PubMed] [Google Scholar]
- Tsiodras S, Mantzoros C. Leptin and adiponectin in the HIV associated metabolic syndrome: physiologic and therapeutic implications. Am J Infect Dis. 2006;2:141–152. doi: 10.3844/ajidsp.2006.141.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan J, Pocernich CB, Gairola C, Chauhan A, Schifitto G, Butterfield DA, Buch S, Narayan O, Sinai A, Geiger J, Berger JR, Elford H, Nath A. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology. 2003;60:307–314. doi: 10.1212/01.wnl.0000042048.85204.3d. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Molecular and cellular immune mediators of neuroprotection. Mol Neurobiol. 2006;34:221–242. doi: 10.1385/MN:34:3:221. [DOI] [PubMed] [Google Scholar]
- Verkauskiene R, Dollfus C, Levine M, Faye A, Deghmoun S, Houang M, Chevenne D, Bresson JL, Blanche S, Levy-Marchal C. Serum adiponectin and leptin concentrations in HIV-infected children with fat redistribution syndrome. Pediatr Res. 2006;60:225–230. doi: 10.1203/01.pdr.0000228335.64894.26. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kilic E, Kilic U, Weber B, Bassetti CL, Marti HH, Hermann DM. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain. 2005;128:52–63. doi: 10.1093/brain/awh325. [DOI] [PubMed] [Google Scholar]
- Wong ML, Bongiorno PB, Rettori V, McCann SM, Licinio J. Interleukin (IL) 1beta, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: pathophysiological implications. Proc Natl Acad Sci USA. 1997;94:227–232. doi: 10.1073/pnas.94.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D. Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRx. 2005;2:120–128. doi: 10.1602/neurorx.2.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Zeng YC, Lewis T, Zheng J, Persidsky Y, Gendelman HE. HIV-1 infected mononuclear phagocyte secretory products affect neuronal physiology leading to cellular demise: relevance for HIV-1-associated dementia. J Neurovirol. 2000;6(Suppl 1):S14–S23. [PubMed] [Google Scholar]
- Yabe T, Samuels I, Schwartz JP. Bone morphogenetic proteins BMP-6 and BMP-7 have differential effects on survival and neurite outgrowth of cerebellar granule cell neurons. J Neurosci Res. 2002;68:161–168. doi: 10.1002/jnr.10210. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Shingo T, Kobayashi K, Takeuchi A, Yano A, Muraoka K, Matsui T, Miyoshi Y, Hamada H, Date I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;19:1494–1504. doi: 10.1111/j.1460-9568.2004.03254.x. [DOI] [PubMed] [Google Scholar]
- Zheng J, Gendelman HE. The HIV-1 associated dementia complex: a metabolic encephalopathy fueled by viral replication in mononuclear phagocytes. Curr Opin Neurol. 1997;10:319–325. [PubMed] [Google Scholar]