

Abstract
Glucocorticoid receptor (GR) exerts anti-inflammatory action in part by antagonizing proinflammatory transcription factors such as the nuclear factor kappa-b (NFKB). Here, we assess the crosstalk of activated GR and RELA (p65, major NFKB component) by global identification of their binding sites and target genes. We show that coactivation of GR and p65 alters the repertoire of regulated genes and results in their association with novel sites in a mutually dependent manner. These novel sites predominantly cluster with p65 target genes that are antagonized by activated GR and vice versa. Our data show that coactivation of GR and NFKB alters signaling pathways that are regulated by each factor separately and provide insight into the networks underlying the GR and NFKB crosstalk.
Glucocorticoids (GCs) are essential steroid hormones that regulate a variety of physiological processes including development, apoptosis, metabolism, and homeostasis. The biological actions of GCs are mediated through the ubiquitously expressed glucocorticoid receptor (GR), a ligand-activated transcription factor that belongs to the nuclear receptor superfamily. Unliganded GR resides in the cytoplasm as an inactive complex that dissociates upon hormone binding, and activated GR translocates to the nucleus to regulate transcription of its target genes (Schaaf and Cidlowski 2002; Pratt and Toft 2003; Nicolaides et al. 2010). GCs exert essential immunosuppressive and anti-inflammatory actions and have been widely used as drugs to treat immune and inflammatory disorders. Using single-gene approaches, several modes of action for GC's anti-inflammatory properties have been proposed (Necela and Cidlowski 2004; De Bosscher and Haegeman 2009; Coutinho and Chapman 2010). Direct binding of activated GR on GREs (glucocorticoid responsive elements; classical model) and interaction with NFKB and AP1 (nonclassical model) are the main mechanisms of regulation associated with glucocorticoid-mediated transactivation and transrepression (Yamamoto 1985; Konig et al. 1992; Beato et al. 1995; Gottlicher et al. 1998; De Bosscher et al. 2003; Necela and Cidlowski 2004).
NFKB is a family of constitutively expressed transcription factors that impact many biological processes such as cell growth, proliferation, development, and inflammatory and immune responses. Inactive NFKB, a dimer of the p50 and p65 (or other family members), remains in the cytosol due to its association with the inhibitory proteins of the NFKBI family (NFKBIA) (Vallabhapurapu and Karin 2009). In response to diverse internal and external inflammatory stimuli, such as the proinflammatory cytokine tumor necrosis factor alpha (TNF), NFKBIA is phosphorylated and rapidly degraded, releasing the NFKB dimer, which translocates to the nucleus. Activated NFKB binds to κB response elements (NFKB RE) and regulates expression of genes encoding various proteins such as proinflammatory cytokines, chemokines, receptors, and adhesion molecules (Barnes 1997; Barnes and Karin 1997; Baeuerle 1998; Hayden and Ghosh 2008).
Given that NFKB is a key mediator of immune and inflammatory responses and that GR exerts anti-inflammatory functions, the crosstalk between GR and NFKB signaling is of particular importance and has been the major focus of research for many years (Van Bogaert et al. 2010). The most extensively studied case has been the transrepression of NFKB and AP1 by GR. The inhibitory effect of GR is postulated to be largely due to recruitment of GR, via protein–protein interaction by DNA-bound NFKB or AP1 (tethering model) (Jonat et al. 1990; Cato and Wade 1996; McEwan et al. 1997; Karin 1998; De Bosscher et al. 2003). GR and p65 or JUN, an AP1 subunit, are suggested to physically interact and mutually antagonize each other's transcriptional activity (Konig et al. 1992; Ray and Prefontaine 1994; Gottlicher et al. 1998; Adcock et al. 1999). In addition, other mechanisms have been suggested for the anti-inflammatory effects of GR, such as modulation of chromatin environment (Ito et al. 2000; Tsaprouni et al. 2002; Beck et al. 2008) and competition for a limiting amount of cofactors, such as the acetyltransferases CREBBP and EP300 (Kamei et al. 1996). In principle, multiple layers of regulation seem to be involved in the crosstalk of GR and NFKB or AP1; however, the mechanism and extent of crosstalk has remained unresolved.
In the present study we set out to decipher the global GR and NFKB interaction on chromatin and to identify their targets genes. We mapped the GR- and p65-binding sites on a genome-wide scale upon activation of GR and NFKB separately or upon coactivation. In parallel, we determined RNA Pol II (RNAPII) occupancy as a direct measure of the transcriptional activity. We show that GC and NFKB signaling pathways are significantly rearranged following coactivation. By pairing distinct genomic binding patterns of GR and p65 with changes in transcriptional events, we provide an extensive insight into GR and NFKB crosstalk.
Results
Transcriptional read-out of GR and NFKB activation
We determined the direct global transcriptional changes induced upon activation of GR or NFKB pathways individually and upon coactivation by performing RNAPII ChIP-seq (see Methods). We used the well-studied HeLa B2 cells that express endogenous GR and p65 proteins and respond properly to glucocorticoids and proinflammatory cytokines (such as TNF). An overview of the number of tags that were sequenced and mapped in our experiments is presented in Supplemental Table 1. RNAPII occupancy throughout the gene body has been used as a direct read-out of transcriptional activity in a number of previous studies (Nielsen et al. 2008; Sultan et al. 2008; Welboren et al. 2009). We found 561 genes with significantly altered RNAPII occupancy upon treatment with triamcinolone acetonide (TA), a synthetic GR ligand (Fig. 1A). Furthermore, 1045 genes responded to treatment with the inflammatory cytokine TNF and 1003 genes showed altered RNAPII occupancy upon coactivation (TA + TNF). The RNAPII data were validated by assessing the mRNA levels of 44 randomly selected genes with altered RNAPII occupancy using RT–qPCR (Fig. 1B). The mRNA levels of the TA-responded genes strongly correlated with the RNAPII occupancy (top). However, we observed reduced correlation for genes responded to TA + TNF treatment (bottom) and poor correlation for TNF-responded genes (middle), most probably because genes involved in the inflammatory process respond rapidly to treatment and their mRNAs are short-lived (Hamilton et al. 2010), and thus not suitable for assessing transcriptional activity. In line, we found that the RNAPII occupancy correlated better with primary transcript (primRNA) levels than with the mRNA levels for 15 TNF-responded genes (Supplemental Fig. 1A). Therefore, the RNAPII occupancy is a more reliable read-out of transcription and is used as a measure for regulated genes in the present study. Collectively, 1932 genes showed significantly altered RNAPII occupancy in at least one condition tested. Comparison of the repertoires of regulated genes showed that 43% of the TA + TNF-regulated genes responded only when both ligands are added (Fig. 2A), indicating that GR and NFKB crosstalk alters signaling pathways that are regulated by each factor separately.
Figure 1.

Genes with altered RNAPII occupancy upon TA and/or TNF treatment. (A) The number of genes with altered RNAPII occupancy (increased and decreased) in response to TA, TNF, and TA + TNF treatment, as indicated. (B) RNAPII occupancy vs mRNA levels. Scatter plots depicting the Pearson correlation (r) between changes in mRNA levels (y-axis) and in RNAPII occupancy (x-axis) for randomly selected regulated genes. Changes in mRNA and RNAPII occupancy were expressed as fold induction of the indicated treatment over DMSO (in log2 scale).
Figure 2.

Cluster analysis of genes regulated upon TA and/or TNF treatment. (A) Comparison of the genes regulated by TA, TNF, and TA + TNF. Venn diagram represents the overlap between the genes regulated upon each treatment. (B) Cluster analysis of the regulated genes. Using k-means clustering, the 1932 collectively regulated genes were clustered into six distinct clusters based on their changes in RNAPII occupancy upon treatment with TA, TNF, and TA + TNF, relative to DMSO, as depicted by the heatmap. The number of genes in each cluster is indicated. The cluster profiles are as follows: (Cluster 1) largely unaffected by TA and down-regulated by TNF and to a higher extent by TA + TNF; (Cluster 2) up-regulated by TA and TA + TNF, and down-regulated by TNF; (Cluster 3) up-regulated by TNF and down-regulated by TA and TA + TNF; (Cluster 4) largely unaffected by TA and strongly up-regulated in the presence of TNF (a subset of these genes is down-regulated by TA + TNF); (Cluster 5) mildly up-regulated by TA or TNF and strongly up-regulated by TA + TNF; (Cluster 6) up-regulated by TA and down-regulated by TNF and to a lesser extent by TA + TNF. The main characteristics of genes in clusters 2, 4, 5, and 6 are indicated at right.
Analysis of the profiles of regulated genes revealed six distinct clusters that were assigned to biological functions based on GO analysis (Fig. 2B; Table 1; Supplemental Fig. 1B; Supplemental Table 2). A detailed description of the six clusters is provided in the Supplementary Notes (description of Supplemental Fig. 1B). Concerning the effect of coactivation as compared with that of activation of GR and NFKB separately, four of the identified clusters are of particular interest: Genes in clusters 2 and 6 appear to comprise mainly genes up-regulated by TA that are either not affected (cluster 2) or suppressed by TNF (cluster 6) upon coactivation (TA + TNF). Cluster 4 includes genes up-regulated by TNF, 20% of which are significantly suppressed by TA upon TA + TNF treatment (proinflammatory genes), and cluster 5 contains genes synergistically up-regulated by TA and TNF. Intriguingly, both clusters 2 and 4 comprise genes involved in MAPK signaling pathways: Cluster 2 includes inhibitors of the MAPK signaling (such as DUSP1), which mediate at least part of the anti-inflammatory effects of Gcs, whereas cluster 4 contains genes that are involved in the activation of MAPK (such as MAP3K8, TNF, TGFB, TGFBR, and FAS) and downstream effectors (such as NFKB1 and NFKB2), indicating an active MAPK pathway involved in inflammatory crosstalk.
Table 1.
GO analysis for the genes of each of the six clusters (with the respective P-values) and examples of genes for the indicated GO terms

Taken together, our data indicate that coactivation of GR and NFKB alters expression of genes regulated by activation of each factor separately, thus leading to changes of signaling pathways regulated by TA or TNF
Genome-wide binding of activated GR and NFKB
We next assessed the chromatin-binding profile of GR and p65 on a genome-wide scale. We performed ChIP–seq to identify the GR-binding sites using chromatin from cells treated with TA or DMSO (see Methods). The number of mapped reads is presented in Supplemental Table 1. Analysis of our data revealed that the depth of sequencing was sufficient for the goals of our study (see legend to Supplemental Table 1) and further validated the quality of our data sets (Supplemental Fig. 2A,B). Peak calling identified 8306 GR-binding sites (P < 10−5, FDR < 0.05) upon treatment with TA, including sites in the promoters of known GR target genes such as PER1 (Fig. 3A). To confirm the specificity of our observations we developed HeLa B2 GR_KD cells that express very low levels of GR (described in Supplemental Fig. 2C–E). ChIP–qPCR experiments using independent biological replicates from WT and GR_KD cells validated our data (24/25 randomly selected sites) (Fig. 3B; Supplemental Fig. 3A). Similar results were obtained using either the monoclonal antibody used in ChIP–seq or a polyclonal antibody raised against a different epitope of human GR (data not shown). It should be noted that peaks were detected in the control (GR_DMSO); however, these sites could not be confirmed as hormone-independent GR-binding sites by ChIP–qPCR using the distinct polyclonal antibody (data not shown). Location analysis revealed that about half of the binding sites are intragenic (48%), a significant percentage are at >25 kb from transcription start sites (24%) and only a minor part in promoters (7%) (Fig. 3C), in agreement with recent studies for GR (Reddy et al. 2009), ER (Carroll et al. 2006; Lin et al. 2007; Welboren et al. 2009), and PPARγ:RXR (Nielsen et al. 2008). Our data set partially overlaps with a genome-wide GR profile in human A549 lung epithelial carcinoma cells (Fig. 3D; Reddy et al. 2009). The observed difference is likely to reflect the cell-type specificity of GR signaling with the overlapping binding sites being most probably common sites across different cell types, whereas nonoverlapping sites represent cell-type-specific regulatory elements. The ligand specificity (TA vs. Dex), may also play a significant role, given that the availability of intracellular steroids, and thus their potency, is altered by transporters that act in a ligand- and cell-type-specific manner in mammalian cells (Kralli and Yamamoto 1996). Antibody-dependent variation and sample handling could also account for the observed differences.
Figure 3.

Genome-wide GR- and p65-binding sites upon TA or TNF treatment. (A,E) RNAPII and GR (A) or p65 ChIP-seq data (E) illustrate RNAPII occupancy and GR and p65 binding in the promoter and/or within the gene body of target genes. Strong binding sites were detected upon TA (green) and TNF treatment (pink), whereas residual binding was observed in the absence of ligand (gray). Data were viewed in the University of California at Santa Cruz (UCSC) Genome Browser. The maximum number of overlapping tags, representing peak height, is indicated on the y-axis. (B,F) Validation of GR-binding sites (B) and p65-binding sites (F). Strip plots of GR ChIP–qPCR data for 25 randomly selected GR sites from WT and GR_KD cells (B) and p65 ChIP–qPCR data for 20 randomly selected p65 sites from WT and p65_KD cells (F) treated as indicated. Results are expressed as a percentage of input chromatin. Binding sites with fold induction (relative to DMSO) >2 are regarded as validated. Establishment of GR_KD and p65_KD cells are described in Supplemental Figure 2. (C,G) Genomic location analysis of GR-binding sites (C) and p65-binding sites (G). GR sites identified upon TA treatment (C) and p65 sites identified upon TNF treatment (G) were divided into the following categories based on their position relative to the nearest gene: distant (>25 Kb), within 5 Kb upstream, within 5 kb downstream, within 5–25 kb upstream, within 5–25 kb downstream and intragenic (exon, intron, and within 0–5 kb up- and downstream). The Pink Thing (http://pinkthing.cmbi.ru.nl) was used for the analysis. (D) Comparison of genome-wide GR profiling data. Venn diagram of the overlap of GR sites identified in the present study (8306 sites, ChIP-seq using HeLa B2 cells treated with TA for 4 h) with those identified in Reddy et al. (2009) (4392 sites, ChIP-seq using A549 cells treated with Dex for 1 h). (H) Comparison of genome-wide p65 profiling data. Venn diagram of the overlap of p65 sites identified in the present study (12,552 sites, ChIP-seq using HeLa B2 cells treated with TNF for 1 h) with those identified in Lim et al. (2007) (5858 sites, ChIP-PET using THP-1 cells treated with LPS for 1 h).
Next, we extended our analysis to identify the global p65-binding profile upon treatment with TNF. ChIP-seq data revealed the presence of p65-binding sites in the promoter regions of known p65 target genes such as CCL2 (Fig. 3E). Peak calling revealed 12,552 TNF-induced p65-binding sites (P < 10−5, FDR < 0.05) that were validated by ChIP–qPCR using WT and p65_KD cells (20/20 randomly selected sites) (Fig. 3F; Supplemental Fig. 3B; HeLa B2 p65_KD cells expressing very low levels of p65 are described in Supplemental Fig. 2F,G). Location analysis revealed that the p65-binding sites are mainly intragenic (46%) and only 7% are located in promoters (Fig. 3G), in agreement with previous studies (Lim et al. 2007). Our p65 data set showed limited overlap with a genome-wide p65 profile identified by ChIP–PET in THP-1 human monocytic cells (Fig. 3H; Lim et al. 2007). The high accuracy, sensitivity, and sequence depth of ChIP–seq probably allowed for the identification of transient or weak binding sites as well as high-affinity sites in our data set. The observed difference is also likely due to the cell lines and ligands (TNF vs. LPS), as well as to sample handling.
To gain insight into the molecular crosstalk between GR and NFKB at the level of binding to chromatin, we mapped both GR- and p65-binding sites upon coactivation (TA + TNF). Peak calling revealed 8696 GR- and 12,713 p65-binding sites (P < 10−5, FDR < 0.05) (Fig. 4A) that were validated by ChIP–qPCR (Supplemental Fig. 3A,B; 25/25 and 20/20 randomly selected GR and p65 sites, respectively). Figure 4A summarizes the GR- and p65-binding sites detected upon TA, TNF, and TA + TNF treatment using the respective DSMO ChIP–seq data as control. To link the identified binding sites with the regulated genes, we calculated the median distance between the transcription start sites (TSS) of the regulated genes and the closest binding site, thus assigning one nearest peak to each regulated gene. Interestingly, promoter proximal binding of GR or p65 correlates well with up-regulation of their targets genes, whereas down-regulated genes are often associated with distal GR and p65 binding (Fig. 4B; Supplemental Fig. 3C), in agreement with similar findings for GR sites in human A549 cells (Reddy et al. 2009).
Figure 4.

Analysis of genome-wide GR- and p65-binding sites upon TA and/or TNF treatment. (A) The number of GR- and p65-binding sites identified upon TA, TNF, and TA + TNF treatment using the respective DMSO ChIP-seq data as control. (B) Median distance (in Kb) of GR- and p65-binding sites to the TSS of the nearest gene regulated by TA, TNF, and TA + TNF, as indicated. (C) Response elements for GR, NFKB, and AP1 identified as the predominant motifs in the GR and p65 binding sites. Motifs were identified by a de novo motif search and visualized using WebLogo. (D) Motif occurrence within the GR and p65 binding sites. The bar graph shows the percentage of GR and p65 sites containing the indicated motifs.
The above data provide the global profiles of GR and NFKB chromatin binding sites and show that coactivation does not strongly affect the number of binding sites identified upon single activation.
Motif analysis
De novo motif search in each set of GR- and p65-binding sites from different treatments identified significantly enriched DNA motifs (Fig. 4C; Supplemental Fig. 3D), which were used to scan each peak set. Analysis of scan results showed that GR and NFKB response elements (GRE and NFKB RE) are the most prevalent motifs identified in the 25%–30% or GR- and p65-binding sites, respectively (Fig. 4D). Interestingly, the AP1 response element (AP1 RE) is prominently present in both GR and p65 data sets under all conditions, thus corroborating and extending the important role of AP1 in glucocorticoid and TNF signaling pathways. We also found statistically significant enrichment of response elements for AP2, SP1, CEBPA, RORA1, TEAD1, and RREB1, in agreement with published data (Phuc Le et al. 2005; So et al. 2007); however, these motifs do not show differential occurrence between different categories of binding sites (Fig. 4D).
Our data show the presence of GRE and NFKB RE only to a fraction of GR- and p65-binding sites and suggest that other factors/mechanisms can facilitate their association with chromatin at the sites that do not contain the respective motifs.
Coactivation alters the GR- and NFKB-binding site repertoires and gene-expression profiles
Studies at the single gene level have provided evidence for a direct physical interaction between GR and inflammatory regulators such as NFKB and AP1 (Konig et al. 1992; Gottlicher et al. 1998); however, genome-wide assessment of the overlap and crosstalk between GR and NFKB has not been reported. Our analysis revealed that the majority of GR-binding sites occupied upon induction with TA + TNF were also occupied upon treatment with TA alone (Fig. 5A, maintained sites). Furthermore, 643 GR sites were lost upon coactivation with TNF. However, most of them were weak sites and ChIP–qPCR experiments for their validation were inconclusive (data not shown). Loss of these sites may also be the result of inefficient GR ChIP due to interaction of GR with other factors or modifications induced by coactivation, thus masking the epitope recognized by the antibody used in this study.
Figure 5.

Gained GR-binding sites and their correlation to gene expression profile. (A) Profile of GR-binding sites. Venn diagram of the overlap of GR sites detected upon treatment with TA or TA + TNF. (B) Tag density maps depicting the pattern of p65 occupancy around (peak mode ±2.5 kb) gained GR sites. Color density indicates the level of p65 occupancy (square root of tag density; see scale below) in a 250-bp window. The position of the example presented in E (ZBTB20) is indicated. (C) Motif occurrence within gained GR sites co-occupied by p65. The bar graph shows the percentage of sites containing the indicated motifs. (D) Boxplots of GR and p65 tag counts distributed under peak locations (log2 scale) of gained GR sites co-occupied by p65, upon TA + TNF treatment, in WT cells and the respective tag distributions under the same locations in p65_KD cells. (E) GR and p65 ChIP-seq data illustrate binding of GR and p65 at gained GR sites co-occupied by p65, detected upon the indicated treatments, in WT and p65_KD cells. Data were viewed in the UCSC Genome Browser. The maximum number of overlapping tags, representing peak height, is indicated on the y-axis. (F) Model of GR and p65 interaction at gained GR sites co-occupied by p65. (G) Enrichment of gained GR sites co-occupied by p65 that are assigned to the genes of each cluster. Peak enrichment fold ratio represents the ratio of the nonrandom enrichment (ratio of assigned binding sites to the total number of genes in each cluster) to the random enrichment (parallel enrichment calculation for 100 random sets of nonregulated genes, performed to assess the statistical significance of the peak enrichment analysis).
Importantly, a fraction of GR sites (12%) is detected only upon coactivation (gained sites) (Fig. 5A), and could be validated by ChIP–qPCR experiments (18/19 randomly selected sites; Supplemental Fig. 4A). Tag density maps for lost, maintained, and gained GR sites clearly show that our ChIP–seq data analysis is comprehensive, and there is a clear distinction between the different categories of binding sites (Supplemental Fig. 4B). Given that the frequency of NFKB RE and AP1 RE is significantly increased in the gained sites (Supplemental Fig. 4C), we looked for GR and p65 co-occupancy at these sites. Interestingly, we identified that a large fraction of gained GR sites were co-occupied by p65, as illustrated by the tag density map showing the p65 occupancy around these gained GR sites (Fig. 5B, left) and the presented example (Fig. 5E). ChIP–reChIP–qPCR experiments validated their co-occupancy (15/19 randomly selected sites) (Supplemental Fig. 4D). The NFKB RE and AP1 RE occurred at the highest frequency in these gained GR sites co-occupied by p65 (Fig. 5C). It is therefore likely that TNF-induced binding of NFKB (or for that matter, AP1) facilitates GR entry at the gained sites. In line with this hypothesis, p65 KD abolished binding of GR (and p65) at the gained GR sites co-occupied by p65, as shown by the box plots from GR and p65 ChIP_seq in p65_KD cells (Fig. 5D) and illustrated for the ZBTB20 gene (Fig. 5E). These data support the recruitment of GR by activated chromatin-bound p65 at the gained GR sites (Fig. 5F). GR and p65 ChIP–qPCR experiments in WT and p65_KD cells validated the results (Supplemental Fig. 4E). A fraction of gained GR sites without p65 co-occupancy was also detected (the p65 occupancy pattern around these gained GR sites upon treatment with TA + TNF is similar to that of DMSO-treated cells) (Fig. 5B, right). These sites show increased occurrence of GRE and AP1 RE (Supplemental Fig. 5A). Strikingly, ChIP-seq using WT and p65_KD cells showed that binding of GR at these sites was still dependent on the presence of p65 (Supplemental Fig. 5B). Inefficient ChIP of p65 due to epitope masking could account for its absence from these gained sites.
A similar analysis was performed for the p65-binding sites. The majority of p65 sites are maintained upon single treatment or coactivation (Fig. 6A). Most of the 1075 p65 sites lost upon coactivation with TA and TNF were weak binding sites (data not shown), and at present we cannot exclude the possibility of inefficient ChIP of p65 at these sites upon cotreatment. Interestingly, 10% of the p65 sites were identified only upon coactivation (gained sites) (Fig. 6A) and validated by ChIP–qPCR (15/19 randomly selected sites, Supplemental Fig. 6A). Tag density maps depict the three categories of p65 sites (Supplemental Fig. 6B). Since these sites are highly enriched for GRE (Supplemental Fig. 6C), we searched for GR binding within the gained p65 sites and defined sites with, as well as without, GR co-occupancy (high and low GR occupancy around these gained p65 sites, respectively) (Fig. 6B,E). ChIP–reChIP–qPCR experiments confirmed the co-occupancy of both p65 and GR (11/12 randomly selected sites, Supplemental Fig. 6D). Motif analysis revealed the predominant occurrence of a GRE in the co-occupied gained sites but not in the sites without GR co-occupancy (Fig. 6C ; Supplemental Fig. 5D). Importantly, GR and p65 ChIP-seq in GR_KD cells showed reduced binding of GR as well as p65 in the co-occupied sites (Fig. 6D,E), thus confirming the recruitment of p65 by activated chromatin-bound GR at the gained p65 sites (Fig. 6F). These ChIP–seq data were validated by GR and p65 ChIP–qPCR experiments (Supplemental Fig. 6E). Binding of p65 at the gained sites without GR co-occupancy was also dependent on the presence of GR (Supplemental Fig. 5E). GR ChIP–qPCR with the monoclonal antibody used for ChIP–seq and a polyclonal antibody raised against a different epitope of GR did not reveal GR co-occupancy at these gained p65 sites (data not shown). However, at present we cannot exclude the possibility of inefficient GR ChIP at these sites due to epitope masking.
Figure 6.

Gained p65-binding sites and their correlation to gene expression profile. (A) Profile of p65 binding sites. Venn diagram of the overlap of p65 sites detected upon treatment with TNF or TA + TNF. (B) Tag density maps depicting the pattern of GR occupancy around (peak mode ±2.5 kb) gained p65 sites. Color density indicates the level of GR occupancy (square root of tag density; see scale below) in a 250-bp window. The position of the example presented in E (ZFAND5) is indicated. (C) Motif occurrence within gained p65 sites co-occupied by GR. The bar graph shows the percentage of sites containing the indicated motifs. (D) Boxplots of GR and p65 tag counts distributed under peak locations (log2 scale) of gained p65 sites co-occupied by GR, upon TA + TNF treatment, in WT cells, and the respective tag distributions under the same locations in GR_KD cells. (E) p65 and GR ChIP-seq data illustrate binding of p65 and GR at gained p65 sites co-occupied by GR, detected upon the indicated treatments, in WT and p65_KD cells. Data were viewed in the UCSC Genome Browser. The maximum number of overlapping tags, representing peak height, is indicated on the y-axis. (F) Model of GR and p65 interaction at gained p65 sites co-occupied by GR. (G) Enrichment of gained p65 sites co-occupied by GR that are assigned to the genes of each cluster. Peak enrichment fold ratio is as described in Figure 5G.
Comparative analysis of maintained GR and p65 sites showed that a substantial number of sites are common for GR and p65 (Supplemental Fig. 7A–C). Comparison of ChIP–seq data from WT, GR_KD, and p65_KD cells revealed that binding of GR or p65 on the maintained sites does not depend on the presence of the other factor (Supplemental Fig. 7D). These data show that GR and p65 binds to the maintained sites independently of each other, and therefore validate the effect of KDs on the gained sites as specific.
Our data suggest that TNF-induced binding of p65 facilitates GR recruitment at the gained GR sites (Fig. 5F). Similarly, TA-induced binding of GR facilitates binding of p65 at the gained p65 sites (Fig. 6F). To assess the involvement or consequence of GR and p65-binding sites that are gained upon coactivation in the regulation of clusters of genes defined by RNAPII occupancy (Fig. 2B), we assigned these gained sites to the genes of each cluster and then assessed the peak enrichment (see Methods). Interestingly, gained GR sites co-occupied by p65 are enriched in the cluster containing proinflammatory genes (up-regulated by TNF), some of which are suppressed by TA (Fig. 5G, cluster 4), thus suggesting that recruitment of GR by p65 mediates, a least in part, the anti-inflammatory function of GR. Assignment of the gained p65 sites co-occupied by GR uncovered enrichment to clusters containing GR targets that are partially suppressed by additional TNF treatment (Fig. 6G, cluster 6). These data indicate that recruited p65 antagonizes GR function on these genes. In contrast, the high enrichment of gained p65 sites to GR targets that are largely unaffected by additional TNF treatment (cluster 2) indicates that recruitment of p65 alone is insufficient and that other factors and/or conditions are required for cross-regulation to occur. The gained GR and p65 sites not co-occupied by p65 and GR, respectively, are enriched in all four clusters of regulated genes tested (2, 4, 5, and 6) but, in general, to a lesser extent as compared with the sites co-occupied by both factors (Supplemental Fig. 5C,F). The maintained sites co-occupied by GR and p65 were significantly enriched to clusters containing proinflammatory genes, some of which are down-regulated by TA (cluster 4) or genes strongly up-regulated by TA and TNF synergism (cluster 5) (Supplemental Fig. 7E).
Taken together, our data provide evidence that the genome-wide GR- and p65-binding repertoires are significantly affected by the combination of stimuli and support the notion that GR and p65 are recruited by each other (and probably other factors such as AP1) to new binding sites gained upon coactivation in a mutually dependent manner. Gained GR sites are enriched in the cluster containing proinflammatory genes, part of which are suppressed by TA, while gained p65 sites are enriched in the cluster of TA targets suppressed by TNF.
Discussion
To unravel the global crosstalk between GR and NFKB we applied ChIP-seq and mapped GR- and p65-binding sites and changes in RNAPII occupancy upon activation of GR and NFKB alone or in combination. We identified a large number of GR and p65-binding sites as well as differentially regulated genes. By pairing the binding-site data and the gene expression data, we provide a global overview of the GR and NFKB crosstalk.
Comparison of the regulated gene repertoires determined by RNAPII ChIP–seq revealed that coactivation of both GR and p65 strongly affects gene expression, as half of the TA + TNF-affected genes do not respond to either TA or TNF alone. This extensive and highly significant reprogramming of gene regulation appeared to be orchestrated to a large extent by the changes in GR- and p65-binding patterns. The striking discordance between the number of regulated genes and the GR- and p65-binding sites suggests that multiple sites are involved in the regulation of a single gene and/or that binding of a transcription factor is not sufficient to drive gene expression.
Comprehensive analysis of GR- and p65-binding sites revealed that GRE and NFKB RE are the predominantly occurring motifs, respectively. Nevertheless, only 25%–30% of the binding sites contain the respective motifs (our data, and Martone et al. 2003; Lim et al. 2007; Reddy et al. 2009), indicating that other (transcription) factors can facilitate the association of GR and p65 with chromatin. The high occurrence of GREs in gained p65-binding sites occupied only upon coactivation and, conversely, NFKB RE in gained GR sites, underscores the extent of GR and p65 mutual recruitment at these sites. In addition, the occurrence of the AP1 RE in the 20%–25% of GR- and p65-binding sites suggests that AP1 is likely to mediate part of the GR and p65 binding to chromatin. This is further supported by the binding of FOS in GR-binding sites containing AP1 REs (our unpublished observations).
Comparison of GR- and p65-binding site repertoires revealed significant changes upon coactivation of GR and p65. Although coactivation does not affect the majority of binding sites, it induces binding at a considerable number of new sites. The dependence of GR and p65 on each other for binding at these gained sites was confirmed and extended by ChIP–seq data from GR_KD and p65_KD cells and uncovered the functional and probably physical interaction of GR and p65 on chromatin on a genome-wide scale. The sites that are bound by GR only upon coactivation and that are co-occupied by p65 are enriched in the cluster containing proinflammatory genes, part of which are suppressed by liganded GR (cluster 4). A typical example is the inflammatory cytokine CCL2 gene that is induced by p65 and is effectively suppressed by GR upon coactivation. This p65-mediated recruitment of GR at inflammatory genes that are down-regulated upon coactivation corroborates the previously proposed mechanisms for the anti-inflammatory action of glucocorticoids, and extends it as a widely used mechanism of cross-regulation. Similarly, recruitment of p65 to sites occupied by GR in clusters 2 and 6 suggests that the GR and p65 crosstalk works in a bidirectional way, i.e., p65 is also targeted to GR on glucocorticoid activated genes and down-regulates them, as seen for genes of cluster 6. Our data also show binding of GR and p65 at common sites (maintained) that are not dependent or influenced by each other. These loci contain juxtaposed cis-acting elements involved in the regulation of shared target genes enriched in clusters 4 and 5 (as exemplified by trans-activation of the anti-inflammatory NFKBIA gene for cluster 4 and TNFAIP3 gene for cluster 5).
The recruitment of GR and p65 to gained sites that are not occupied by the other factor, but require its presence, was rather unexpected. Our data suggest that activated GR (or p65) may be required to induce chromatin modifications that will create a more accessible environment for binding of the other factor upon coactivation. In this case, GR (or p65) itself is likely to bind at a distant site and exerts its effect by chromatin looping. Alternatively, GR (or p65) could bind transiently on chromatin to potentiate binding of the other factor. In line with this, molecular chaperones (such as the p23, and to a lesser extent, the HSP90) have been shown to localize to genomic response elements and to promote disassembly of GR and NFKB transcriptional regulatory complexes, thus regulating responses to signaling changes (Freeman and Yamamoto 2002). Although the absence of GR and p65 co-occupancy at the gained p65 sites was confirmed by using two antibodies against GR, at present we cannot exclude the possibility of inefficient GR and p65 ChIP at gained p65 and GR sites, respectively, because of “epitope masking” due to protein–protein interactions/modifications induced by coactivation.
A major part of the GR- and p65-binding sites are located at long distances from the annotated genes. GR (or p65) bound at distal sites is likely to be involved in long-range chromatin interactions with GR (or p65) bound at other sites and probably other factors, thus forming loop structures. Such loop structures/hubs have been globally described for the ERα-binding sites that interact with each other and function as “anchor” regions; most genes close to these hubs are up-regulated in an ERα-dependent manner (Fullwood et al. 2009). In line with this observation, our data also show that binding sites occurring close to the TSS correlate well with up-regulated genes. At the single gene level, binding of NFKB at a distal site was shown to regulate CCL2 (also known as MCP-1) gene transcription by enabling binding of SP1 at a promoter-proximal site in an EP300-dependent manner (Teferedegne et al. 2006). Similarly, GR has been shown to activate the CIZ1-LCN2 locus by loop structure formation (Hakim et al. 2009). Chromatin looping may also enable the GR and p65 co-occupancy at the sites gained upon coactivation. Such chromatin loops/hubs have been proposed to serve as sites for recruitment of transcription factors that enable coordinated transcriptional regulation in a stimulus- and cell-specific manner.
In conclusion, our results provide a novel genome-wide footprint of GR and NFKB crosstalk and substantially contribute to the understanding of the networks underlying the glucocorticoid and inflammatory signaling pathways.
Methods
Cell culture, Western blot, and luciferase assay
HeLa B2 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C in 5% CO2. HeLa B2 are HeLa cells stably transfected with the glucocorticoid-responsive mouse mammary tumor virus long terminal repeat–luciferase reporter vector (MMTV–Luc) (Hollenberg and Evans 1988), together with the neomycin resistance gene, using the calcium phosphate coprecipitation method. Eighteen hours after transfection, cells were washed with phosphate-buffered saline (PBS), fed with fresh medium, and 24 h later refed with medium containing geneticin (0.5 mg/mL of medium). Cells were fed with fresh geneticin-containing medium every 2–3 d, and colonies were isolated 3 wk later and tested for luciferase activity in the presence or the absence of 1 μM TA (T6501, Sigma-Aldrich). For GR analysis, cells were cultured in DMEM supplemented with 10% charcoal stripped FCS for 48–72 h before subsequent treatment and/or harvesting.
For Western blot analysis, whole-cell extracts (20–30 μg of protein) were analyzed by SDS-PAGE, followed by immunoblotting using antibodies against GR (Siriani et al. 2003) (MAb-NRhGR-050, Diagenode), p65 (sc-372, Santa Cruz Biotechnologies), and TBP (MAb-002-100, Diagenode). Proteins were visualized using ECL (GE healthcare).
For the luciferase assay, cells were treated with either DMSO or 1 μM TA for 16 h. Cells were harvested in Glo Lysis Buffer, and luciferase activity was measured using a Steady Glo Luciferase Assay system (Promega) in a Safire 2 plate reader. Luminescence readings were normalized with the protein content of the samples.
mRNA analysis
Total RNA was harvested using Qiagen RNeasy kit with on-column DNase treatment according to the manufacturer's protocol (Qiagen). A total of 1 μg of RNA was reverse transcribed to cDNA using iScript cDNA Synthesis kit (BIO-RAD) according to manufacturer's protocol. Intron-spanning primers were used to determine mature mRNA levels using RT–qPCR. Primers spanning intron/exon boundaries were used to quantify primary RNA transcripts. Expression levels were normalized to GAPDH or HPRT levels. The primer pairs used in this study are listed in Supplemental Table 3.
Lentiviral shRNA knockdown
shRNA lentiviral vector stock (pKLO.1-puro—MISSION shRNA, Sigma) containing shRNAi oligo DNA directed either against GR or p65 was produced in Human Embryonic Kidney 293T cells by transfection of the shRNA construct together with pHR8.2R and pCMV-VSV-G helper constructs using lipofectamine (Sigma). Next, HeLa B2 cells (1.5 × 106 in a 10-cm dish) were transduced with 3 mL of the virus containing supernatant supplemented with 8 μg/mL Polybrene (Sigma-Aldrich). Twenty-four hours after the first transduction, a second round of transduction was preformed similar to the first. The medium was refreshed 1 d post-infection and selection was performed using 6 μg/mL of puromycin (Sigma-Aldrich) for three passages. The mRNA and protein levels of GR and p65 were assessed in the KD (and WT) cells by RT–qPCR and Western blot, respectively.
ChIP, ChIP–reChIP (sequential ChIP), qPCR, and deep sequencing
HeLa B2 cells were cultured in DMEM supplemented with 10% charcoal-stripped FCS for 72 h and subsequently treated with either DMSO or 1 μM TA for 4 h with or without an additional treatment with 10 ng/mL TNF (T0157, Sigma-Aldrich) for the last 1 h. Chromatin was harvested and single ChIP followed by qPCR was preformed according to standard protocol (Denissov et al. 2007) with minor adjustments: Following a wash with buffer C (150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM HEPES at pH 7.6), the cells were resuspended in ChIP-incubation buffer at a concentration of 15 × 106 cells/mL and sheared using a Branson-250 sonicator (power 5.5, 12 beats, 10 sec per beat, 40-sec interval). Sonicated chromatin, equivalent to 3.5 × 106 cells, was incubated with the relevant antibody overnight at 4°C. Antibodies against human GR (MAb-NRhGR-050), RNAPII (AC-055-100, Diagenode), and p65 (sc-372) were used. ChIP–reChIP was preformed as described (Akkers et al. 2009) with minor adjustments: Chromatin was eluted from the beads and diluted in ChIP incubation buffer without SDS (5% Triton X-100, 0.75 M NaCl, 5 mM EDTA, 2.5 mM EGTA, 100 mM HEPES) to adjust the final SDS concentration to 0.15%, and then subjected to a second round of immunoprecipitation. ChIPed DNA was analyzed by real-time qPCR with specific primers (Biolegio, listed in Supplemental Table 3) using the 2x SYBR Geen mix (BIO-RAD) in a MyiQ thermocycler (BIO-RAD). Primers amplifying exon 2 of myoglobin (MB) were used as negative control. In addition, ChIPed DNA was prepared for sequencing and sequenced according to the manufacturer's instructions (Illumina) essentially as described (Nielsen et al. 2008; Welboren et al. 2009; Martens et al. 2010). Shortly, adaptor sequences were linked to generated ChIPed DNA, the library was sized selected (200–250 bp), and amplified by PCR. Subsequent sequencing was carried out on a Genome Analyzer (Illumina). Validation of GR- and p65-binding sites was performed with ChIP–qPCR experiments using the same antibodies as the ones used for ChIP–seq. In addition, a polyclonal antibody raised against amino acids 77–120 of hGR (DJ Mitsiou and MN Alexis, unpubl.) was used to validate GR sites and to confirm the absence of GR from the gained p65 sites without co-occupancy.
Read alignment and normalization
The image files generated by the Genome Analyzer were processed to extract DNA sequence data and the 32-bp tags were unambiguously mapped to the human genome (NCBI hg18) using the eland aligner, allowing, at most, 1 nt mismatch. The 32-bp sequence reads were directionally extended to 200 bp, corresponding to the length of the original fragments used for sequencing plus the ligated adapters. For each base pair in the genome the number of overlapping sequence reads was determined, averaged over a 10-bp window, and visualized in the University of California Santa Cruz Genome Browser (http://genome.ucsc.edu). All ChIP–seq raw data files have been submitted to the GEO database (accession no. GSE24518). As previously discussed (Zhang et al. 2008), large differences in sequencing depth may lead to an increase of FDR and result in false-positive peaks. In order to compensate for differences in sequencing depth and mapping efficiency among ChIP–seq samples at different conditions, the total number of tags of each sample was equalized by uniformly removing tags relatively to the sample with the lower number of tags (Nielsen et al. 2008). After equalization, the tracks contained the same number of sequence reads and can be compared quantitatively. The RNAPII, GR, and p65 data sets were normalized as independent groups.
Peak detection and clustering
Detection of putative GR- and p65-binding sites was performed using MACS (Zhang et al. 2008) with P < 10−5 for GR and p65 normalized tracks at FDR level <0.05. DMSO tracks of GR and p65 ChIP–seq were used as controls for the detection of GR and p65 peaks, respectively. For each transcription factor, peak locations identified by MACS upon single activation (e.g., GR_TA) and coactivation (e.g., GR_TA + TNF) were combined in a common pool, and sequence tags were counted under each peak location of the pool independent of treatment. Next, the following Rc analysis was applied (peaks from GR and p65 were not combined):
where 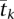 denotes the average number of tags under a specific genomic region (in this case, under each peak), averaged per k bp, and T1, T2 are the treatments for which samples are sequenced (for GR, T1 corresponds to TA + TNF and T2 to TA, while for p65, T1 corresponds to TA + TNF and T2 to TNF). Larger values of Rc indicate binding enrichment, while smaller values indicate binding depletion upon T1. Gained/lost peaks for each transcription factor were defined as peaks falling away from the median of Rc distribution ±2 × MAD. Final peaks for each treatment were defined by combining peaks called by MACS and peaks identified by Rc analysis, excluding redundancies, as follows:
denotes the average number of tags under a specific genomic region (in this case, under each peak), averaged per k bp, and T1, T2 are the treatments for which samples are sequenced (for GR, T1 corresponds to TA + TNF and T2 to TA, while for p65, T1 corresponds to TA + TNF and T2 to TNF). Larger values of Rc indicate binding enrichment, while smaller values indicate binding depletion upon T1. Gained/lost peaks for each transcription factor were defined as peaks falling away from the median of Rc distribution ±2 × MAD. Final peaks for each treatment were defined by combining peaks called by MACS and peaks identified by Rc analysis, excluding redundancies, as follows:
 |
where ∪ denotes the set union operator, ∩ denotes the set intersection operator, and \ denotes the set difference operator. In addition, MACS denotes peaks called by MACS and Rc peaks defined by Rc analysis.
Finally, PinkThing (http://pinkthing.cmbi.ru.nl/cgi-bin/index50.pl [F Nielsen, M Kooyman, HG Stunnenberg, and M Huynen, in prep.]) was used to determine the location distribution of identified peaks in relation to the distance to the closest genes.
RNAPII analysis (clustering, gene annotation, distance analysis)
To identify differentially expressed genes, the number of RNAPII sequence tags was counted for all NCBI hg18 (July 2007) annotated human genes (from +500 to end of gene) accessed from Ensemble (release 50; July 2008), excluding genes smaller than 750 bp. Next, sequence tags within gene bodies were retrieved and averaged per 750 bp, and the log2 fold ratio between average tags in treated and DMSO samples was calculated. Genes presenting a ratio away from the distribution median ± MAD for any of the TA, TNF, and TA + TNF conditions were classified as differentially expressed. An additional filter based on an exponential model, which empirically determines thresholds on the minimum sum of tags in DMSO and treatment samples required for a gene to be called expressed, in relation to the gene body length was applied. In order to identify groups of genes presenting similar expression patterns among the TA, TNF, and TA + TNF treatments, k-means clustering was performed using the Pearson correlation distance. The optimal number of clusters (six) was determined using the Gap statistic (Tibshirani et al. 2001) coupled with an iterative procedure implemented in Gene ARMADA software (Chatziioannou et al. 2009). Over-represented GO categories for each cluster of RNAPII-regulated genes were detected using WebGestalt (Zhang et al. 2005). The median distance from the summit of GR- or p65-binding sites to the closest regulated gene TSS was calculated using the web base tool “fetch closest feature” of Galaxy (Blankenberg et al. 2007).
Peak assignment and enrichment analysis
In order to associate the putative GR- and p65-binding sites with regulated genes for each condition, the distance of peaks falling within ±10 Kb relative to the TSS of each regulated gene was calculated so as to obtain a first distribution of nearby peaks to each gene (one peak is assumed to be assigned to multiple genes and vice versa). The final distribution of assigned peaks was obtained by applying the hypergeometric test to each peak located near regulated genes. This method detected peaks that are close to groups of regulated genes in an over-represented manner, relative to the whole genome that was used as background. To determine whether a peak set was enriched to certain clusters or not, the ratio of assigned peaks to each cluster, to the total number of genes in that cluster was used to determine a “peak per gene index” for each peak set and for each condition, according to the following formula:
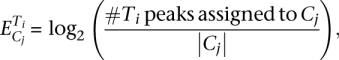 |
where Ti is the treatment (i = GR_TA, GR_TA + TNF, p65_TNF, p65_TA + TNF), Cj is the cluster of genes (j = 1…6) and | Cj | represents the cluster cardinality, or the number of genes in cluster j. In order to assess whether the enrichment scores occur by chance, the same peak sets were assigned to random sets of genes spanning the whole genome, devised to clusters of the same cardinality as the original ones. This procedure was repeated for n = 100 times, and statistical significance was assessed using as statistical measurement a modification of the estimation of the Achieved Significance Level for bootstrap (Efron and Tibshirani 1993):
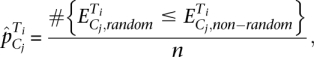 |
that is, the ratio of the number of times random enrichment is found greater than or equal to the nonrandom enrichment, to the number of repetitions. A cluster was enriched for a peak set if 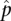 was <0.01. Supplemental Table 4 summarizes the assigned GR and p65 peaks.
was <0.01. Supplemental Table 4 summarizes the assigned GR and p65 peaks.
Motif search
The coordinates of peak maxima for each peak set were extended by 200 bp on each side to create 400-bp genomic regions. De novo motif search was performed using a combination of widely used software coupled with motif quality control, clustering, and statistical significance metrics (van Heeringen and Veenstra 2011) in each set of peaks for each treatment and for varying motif lengths (6–25 bp). The top scoring clustered motifs were curated and converted to Position Weight Matrices (PWMs), which were compared against the JASPAR v3 database using STAMP (Mahony and Benos 2007). The data-driven PWMs were subsequently used to scan all peak subsets for the occurrence of the respective motifs as previously described (Smeenk et al. 2008). A specific sequence background model was constructed for each scan using random nonbound sequences.
Data access
All ChIP-seq raw data files have been submitted to the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE24518.
Acknowledgments
We thank Simon van Heeringen and Arie B. Brinkman from the Department of Molecular Biology, Radboud University, Nijmegen, for generously providing some data analysis tools. This project is supported by the TI-Pharma (Netherlands, grant T2-101-1) and the European Union (MTKD-CT-2004-509836: “MACROGENEX”).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.118042.110.
References
- Adcock IM, Nasuhara Y, Stevens DA, Barnes PJ 1999. Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: preferential targetting of NF-kappaB and lack of I-kappaB involvement. Br J Pharmacol 127: 1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Francoijs KJ, Stunnenberg HG, Veenstra GJ 2009. A hierarchy of H3K4me3 and H3K27me3 acquisition in spatial gene regulation in Xenopus embryos. Dev Cell 17: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA 1998. Pro-inflammatory signaling: last pieces in the NF-kappaB puzzle? Curr Biol 8: R19–R22 [DOI] [PubMed] [Google Scholar]
- Barnes PJ 1997. Nuclear factor-kappa B. Int J Biochem Cell Biol 29: 867–870 [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M 1997. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336: 1066–1071 [DOI] [PubMed] [Google Scholar]
- Beato M, Herrlich P, Schutz G 1995. Steroid hormone receptors: many actors in search of a plot. Cell 83: 851–857 [DOI] [PubMed] [Google Scholar]
- Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, De Bosscher K 2008. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J 27: 1682–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Taylor J, Schenck I, He J, Zhang Y, Ghent M, Veeraraghavan N, Albert I, Miller W, Makova KD, et al. 2007. A framework for collaborative analysis of ENCODE data: making large-scale analyses biologist-friendly. Genome Res 17: 960–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. 2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38: 1289–1297 [DOI] [PubMed] [Google Scholar]
- Cato AC, Wade E 1996. Molecular mechanisms of anti-inflammatory action of glucocorticoids. Bioessays 18: 371–378 [DOI] [PubMed] [Google Scholar]
- Chatziioannou A, Moulos P, Kolisis FN 2009. Gene ARMADA: an integrated multi-analysis platform for microarray data implemented in MATLAB. BMC Bioinformatics 10: 354 doi: 10.1186/1471-2105-10-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho AE, Chapman KE 2010. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 1: 2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Haegeman G 2009. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 23: 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G 2003. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24: 488–522 [DOI] [PubMed] [Google Scholar]
- Denissov S, van Driel M, Voit R, Hekkelman M, Hulsen T, Hernandez N, Grummt I, Wehrens R, Stunnenberg H 2007. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J 26: 944–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani R 1993. An introduction to the bootstrap. Chapman & Hall/CRC, Boca Raton, FL [Google Scholar]
- Freeman BC, Yamamoto KR 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296: 2232–2235 [DOI] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. 2009. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462: 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlicher M, Heck S, Herrlich P 1998. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med 76: 480–489 [DOI] [PubMed] [Google Scholar]
- Hakim O, John S, Ling JQ, Biddie SC, Hoffman AR, Hager GL 2009. Glucocorticoid receptor activation of the Ciz1-Lcn2 locus by long range interactions. J Biol Chem 284: 6048–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton T, Novotny M, Pavicic PJ Jr., Herjan T, Hartupee J, Sun D, Zhao C, Datta S 2010. Diversity in post-transcriptional control of neutrophil chemoattractant cytokine gene expression. Cytokine 52: 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S 2008. Shared principles in NF-kappaB signaling. Cell 132: 344–362 [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Evans RM 1988. Multiple and cooperative trans-activation domains of the human glucocorticoid receptor. Cell 55: 899–906 [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM 2000. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 20: 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonat C, Rahmsdorf HJ, Park KK, Cato AC, Gebel S, Ponta H, Herrlich P 1990. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62: 1189–1204 [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, et al. 1996. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85: 403–414 [DOI] [PubMed] [Google Scholar]
- Karin M 1998. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell 93: 487–490 [DOI] [PubMed] [Google Scholar]
- Konig H, Ponta H, Rahmsdorf HJ, Herrlich P 1992. Interference between pathway-specific transcription factors: glucocorticoids antagonize phorbol ester-induced AP-1 activity without altering AP-1 site occupation in vivo. EMBO J 11: 2241–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralli A, Yamamoto KR 1996. An FK506-sensitive transporter selectively decreases intracellular levels and potency of steroid hormones. J Biol Chem 271: 17152–17156 [DOI] [PubMed] [Google Scholar]
- Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, Sung WK, Lipovich L, Vega VB, Chen J, et al. 2007. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell 27: 622–635 [DOI] [PubMed] [Google Scholar]
- Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, et al. 2007. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet 3: e87 doi: 10.1371/journal.pgen.0030087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Benos PV 2007. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35: W253–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, Altucci L, Stunnenberg HG 2010. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 17: 173–185 [DOI] [PubMed] [Google Scholar]
- Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, et al. 2003. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci 100: 12247–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan IJ, Wright AP, Gustafsson JA 1997. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions. Bioessays 19: 153–160 [DOI] [PubMed] [Google Scholar]
- Necela BM, Cidlowski JA 2004. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc 1: 239–246 [DOI] [PubMed] [Google Scholar]
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E 2010. The human glucocorticoid receptor: molecular basis of biologic function. Steroids 75: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, et al. 2008. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev 22: 2953–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuc Le P, Friedman JR, Schug J, Brestelli JE, Parker JB, Bochkis IM, Kaestner KH 2005. Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet 1: e16 doi: 10.1371/journal.pgen.0010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO 2003. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med 228: 111–133 [DOI] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE 1994. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci 91: 752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf MJ, Cidlowski JA 2002. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol 83: 37–48 [DOI] [PubMed] [Google Scholar]
- Siriani D, Mitsiou DJ, Alexis MN 2003. Overexpressed glucocorticoid receptor negatively regulates gene expression under conditions that favour accumulation of non-hormone-binding forms of the receptor. J Steroid Biochem Mol Biol 84: 171–180 [DOI] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M 2008. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 36: 3639–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR 2007. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3: e94 doi: 10.1371/journal.pgen.0030094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. 2008. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960 [DOI] [PubMed] [Google Scholar]
- Teferedegne B, Green MR, Guo Z, Boss JM 2006. Mechanism of action of a distal NF-kappaB-dependent enhancer. Mol Cell Biol 26: 5759–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Walther G, Hastie T 2001. Estimating the number of clusters in a data set via the gap statistic. JR Stat Soc 63: 411–423 [Google Scholar]
- Tsaprouni LG, Ito K, Punchard N, Adcock IM 2002. Triamcinolone acetonide and dexamethasome suppress TNF-alpha-induced histone H4 acetylation on lysine residues 8 and 12 in mononuclear cells. Ann N Y Acad Sci 973: 481–483 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693–733 [DOI] [PubMed] [Google Scholar]
- Van Bogaert T, De Bosscher K, Libert C 2010. Crosstalk between TNF and glucocorticoid receptor signaling pathways. Cytokine Growth Factor Rev 21: 275–286 [DOI] [PubMed] [Google Scholar]
- van Heeringen SJ, Veenstra GJ 2011. GimmeMotifs: a de novo motif prediction pipeline for ChIP-sequencing experiments. Bioinformatics 27: 270–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG 2009. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J 28: 1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KR 1985. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet 19: 209–252 [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J 2005. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33: W741–W748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137 doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]