

Abstract
An efficient and simple source of nitroso reagents and their oxidation reactions are described. The combination of a Lewis acid and a metal nitrite is applied to the oxidation of silyl enol ethers. Amino acid and peptide derivatives were easily accessed through in situ C-C bond cleavage of fully substituted silyl enol ethers upon oxidation.
During the last three decades, new classes of nitroso compounds have emerged and have found numerous applications in organic synthesis.1-4 Since most of these compounds are toxic, explosive, and require several steps to prepare, their scopes of application are limited. The development of new practical and safe sources of nitroso compounds for oxidation reactions remains a challenge.
The combination of metal nitrite and Lewis acid could be a convenient route: triisopropylsilylchloride (TIPSCl) was exposed to silver nitrite in acetonitrile to generate silyl nitrite (eq 1).5 Immediately, a pale green color appeared with the precipitation of silver chloride, which clearly indicates the generation of the nitroso compound.6a Indeed, addition of silyl enol ether 1a into the reaction mixture exclusively resulted in the keto oxime 2a in 71% isolated yield.
 |
(1) |
The reaction seemed to be general and several Lewis acids could be used with silver nitrite: TiCl4 (40%), InCl3 (41%), FeCl3 (47%), and AlCl3 (45%). Further, other commercially available nitrite salts NaNO2/15-crown-5 (74%) and nBu4NNO2 (60%) could also be employed. Thus, this route appears to be a simple, easily accessible, and relatively safe source of nitroso reagents.6b
The oxygen bridge between the Lewis acid (LA) and nitroso moiety (LA-O-NO) plays a crucial role. The resonance stabilization of the nitroso moiety is overcompensated by the activation of oxophilic Lewis acid.
Experiments that probed the scope of this process under optimized conditions are summarized in Chart 1. A broad spectrum of silyl enol ethers, cyclic and acyclic, was employed to afford the desired products in good to excellent yields. Generally, E-oximes were observed except for the seven- and eight-membered rings (2d,e). The scale-up was also compatible: the reaction of silyl enol ether derived from α-tetralone was performed on gram scale and the desired product was isolated in 90% yield (Chart 1, 2j). Oxidation proceeded smoothly with silyl dienol ether to generate the 1,4-adduct (2k) exclusively. When triisopropyl silyl enol ethers were used, the desired oximes were isolated as TIPS-protected (2l–o). Acid sensitive heterocyclic furan substitution was also tolerated (2n).
Chart 1.

Oxidation of silyl enol ethers with metal nitritesa–d
aSi = TMS for 2b–k and 2u,v, TIPS for 2l-o, TBS for 2t, Si(TMS)3 for 2p–s (Z-silyl enol ethers were used). b Instead of silyl enol ether, sodium-enolates were used for 2w,x. c % isolated yields. d DMF was used as solvent for 2l–o.
While the reactions of silyl nitrite with aldehyde derived silyl enol ethers were sluggish under the optimized conditions, oxidation with aluminum nitrite proceeded cleanly and the α-hydroxyimino aldehydes 2p–s could be obtained in high yields. In contrast, conventional procedures to prepare these compounds require multistep transformations. Exposure of silyl ketene acetals and the sodium-enolates of active methylene derivatives furnished desired products in good yields (2t–x).
When excess aluminum nitrite derived from AlCl3 and nBu4NNO2 was used, the silyl enol ethers 1 produced the doubly oxidized products 3 as the major products (Chart 2). On the other hand, mixtures of 2 and 3 were isolated for 1e and 1g (entries 4,6). The ratio of 2/3 did not change even after the reaction times were prolonged. Surprisingly, no double oxidation product was observed for twelve-membered 1f (entry 5).
Chart 2.
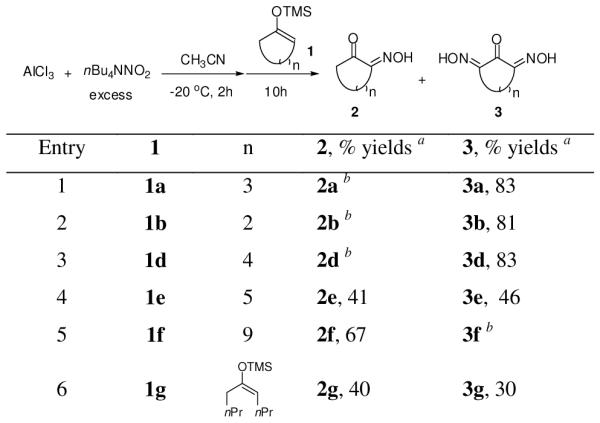
Double oxidation of silyl enol ethers 1
a Isolated yields. b Not observed.
Based on these observations, a possible path for the formation of 3 is depicted in Scheme 1. After the first mono-oxidation, oxime 2 complexes with the Lewis acid which induces further enolization of the ketone. The enol thus obtained further oxidizes to generate dioxime 3. We hypothesize that a syn-alignment of C=O and C=N bonds in 2 is required for double coordination with the aluminum ion. Such preferred alignment was observed from the crystal structure of 2b (five–membered ring, Scheme 1, A), which induced the enolization process required for further oxidation. In contrast, anti-alignment of C=O and C=N bonds of the twelve–membered ring 2f (Scheme 1, B) impedes the required coordination with aluminum, thus no further oxidation proceeds. A mixed situation can be considered for the formation of a mixture of 2 and 3 from eight–membered and acyclic silyl enol ethers (1e and 1g).
Scheme 1.
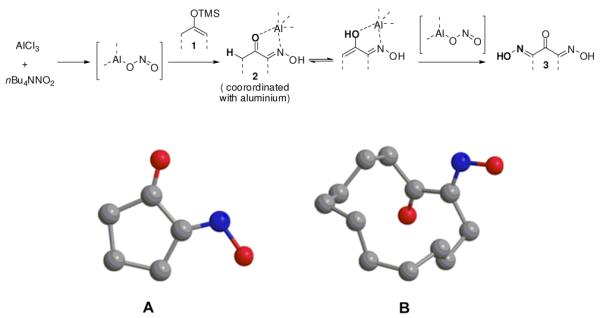
Plausible mechanism for the formation of 3
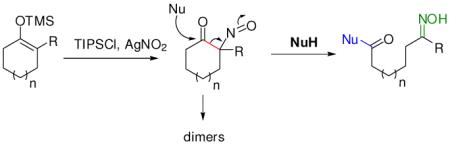
When fully substituted silyl enol ethers were used, the clean formation of the nitroso derivative was observed (eq 2). However, the nitroso compound easily dimerized (crystal structure in SI, p. S26) and precipitated from the reaction mixture.
 |
(2) |
Gratifyingly, switching the solvent from CH3CN to DMF retarded the dimerization process significantly and the successive treatment of external nucleophiles caused the carbon-carbon bond cleavage reaction (eq 2).7 Sodium methoxide produced the expected carbon-carbon cleavage products in high yields (Chart 3, 5a–c). Amazingly, both primary and secondary amines can also efficiently cleave the C-C bond to render amides (Chart 3, 6a–l). Importantly, free amino acid esters are also effective. L-valine, L-alanine, and L-proline methyl esters gave the expected peptides in good yields (6m–r). Various hetero atoms such as oxygen, sulfur, and nitrogen-containing derivatives were also effective for this cleavage process. It should be emphasized that secondary amines can also be reacted with high efficiency under very mild reaction conditions to generate tertiary amides, for which the amide synthesis methods are relatively rare.
Chart 3.

Oxidation of quaternary silyl enol ethers and one pot C-C bond cleavage reactionsa
a Isolated yields in %.
These substituted oximes 5,6 were chemoselectively reduced with indium metal to access protected amino acids and dipeptides 7 in excellent yields (Scheme 2).8
Scheme 2.
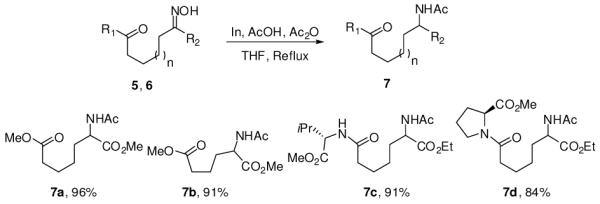
Synthesis of protected amino acids and dipeptides by indium mediated reduction of oximes
The synthetic utility of this new oxidation/cleavage strategy was further verified by short synthesis of the peptidoglycan mimetic peptide analogue.9 Scheme 3 demonstrates the power of this process by the remarkably short step synthesis of aminopimelyl-L-alanyl-L-alanine (9), a biologically important peptide analogue.
Scheme 3.
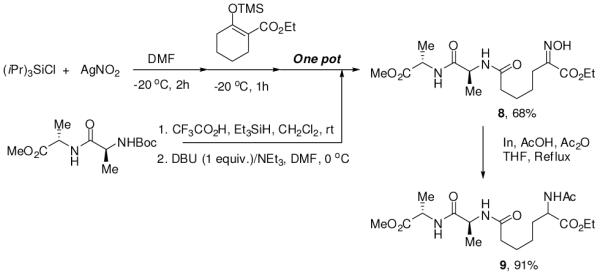
Synthesis of protected peptidoglycan mimetic peptide analogue
In conclusion, we have developed a new and flexible route to metal nitrite which is useful for α- and/or α,α’-oxidation of carbonyl compounds under very mild conditions. For α,α-disubstituted ketones, the resultant nitroso compounds smoothly cleaved under O- and N-nucleophiles to generate polyfunctional amides. This methodology can be used for the rapid synthesis of amino acid and peptide derivatives.
Supplementary Material
Acknowledgements
We gratefully acknowledge NIH for its grant (Asymmetric Oxidation: 2R01GM068433-05) supporting this research. We would also like to thank Dr. Antoni Jurkiewicz, Dr. Ian Steele, and Dr. Jin Qin for sharing their expertise in NMR, X-ray crystallography, and mass spectrometry, respectively.
Footnotes
Supporting Information. Complete experimental procedure and characterization data for prepared compounds. This material is available for free of charge via the internet at http://pubs.acs.org
REFERENCES
- (1).Reviews: Kirby GW. Chem. Soc. Rev. 1977;6:1. Iwasa S, Fakhruddin A, Nishiyama H. Mini-Reviews in Organic Chemistry. 2005;2:157. Bodnar BS, Miller MJ. Angew. Chem. Int. Ed. 2011;50:5630. doi: 10.1002/anie.201005764.
- (2).For cyanonitroso compound: Horsewood P, Kirby GW. Chem. Commun. 1971:1139. For α-halonitroso compounds: Wichterle O. Coll. Czech, Chem. Comm. 1947;12:292. Oppolzer W, Tamura O. Tetrahedron Lett. 1990;31:991. Oppolzer W, Tamura O, Sundarababu G, Signer M. J. Am. Chem. Soc. 1992;114:5900. Zhang D, Suling C, Miller MJ. J. Org. Chem. 1998;63:885. doi: 10.1021/jo971696q. For vinyl nitroso compounds Faragher R, Gilchrist TL. Chem. Commun. 1976:581. Gilchrist TL, Roberts TG. J. Chem. Soc., Perkin Trans. 1. 1983:1283. For nitrosoimines: Gilchrist TL, Peek ME, Rees CW. Chem. Commun. 1975:913. Gilchrist TL, Harris CJ, Hawkins DG, Moody CJ, Rees CW. J. Chem. Soc., Perkin Trans. 1. 1976:2166. For nitrosoamidines: Miller CA, Batey RA. Org. Lett. 2004;6:699. doi: 10.1021/ol0363117. For N-phosphinoylnitroso compounds: Ware RW, Jr., King SB. J. Am. Chem. Soc. 1999;121:6769. Ware RW, Jr., King SB. J. Org. Chem. 2000;65:8725. doi: 10.1021/jo001230z. Ware RW, Jr., Day CS, King SB. J. Org. Chem. 2002;67:6174. doi: 10.1021/jo0202839. For nitroso formate ester: Kirby GW, McGuigan H, Mackinnon JWM, Mclean D, Sharma RP. J. Chem. Soc., Perkin Trans 1. 1985:1437.
- (3).For nitroso benzene and pyridylnitroso compounds: Merino P, Tejero T. Angew. Chem. Int. ed. 2004;43:2995. doi: 10.1002/anie.200301760. Yamamoto H, Momiyama N. Chem. Commun. 2005:3514. doi: 10.1039/b503212c. Yamamoto H, Kawasaki M. Bull. Chem. Soc. Jpn. 2007;80:595. Labaziewicz H, Lindfors KR, Kejonen T,H. Heterocycles. 1989;29:2327.
- (4).Application in total synthesis: Aoyagi S, Tanaka R, Naruse M, Kibayashi C. J. Org. Chem. 1998;63:8397. Li J, Lang F, Ganem B. J. Org. Chem. 1998;63:3403. Faitg T, Soulie J, Lallemand J–Y, Richard L. Tetrahedron: asymmetry. 1999:2165. Blakemore PR, Kim S-K, White JD, Yokochi AFT. J. Chem. Soc., Perkin Trans. 1. 2001:1831. Ozawa T, Aoyagi S, Kibayashi C. J. Org. Chem. 2001;66:3338. doi: 10.1021/jo001589n. Li F, Brogan JB, Gage JL, Zhang D, Miller MJ. J. Org. Chem. 2004;69:4538. doi: 10.1021/jo0496796. Yamamoto Y, Yamamoto H. Eur. J. Org. Chem. 2006:2031. Microreview.
- (5).Weidenbruch M, Sabeit FZ. Naturforsch. 1976;31b:1212. [Google Scholar]
- (6).(a) More intense color was observed for the reaction with Ph3SiCl and (EtO)3SiCl. (b) (iPr)3SiONO is not volatile. Its boiling point is 40.5– 41.5 °C/1.9 mbar (from reference 5), which is much higher compared to commonly used isobutyl nitrite (b.p. = 66 °C at atmospheric pressure).
- (7).For C-C bond cleavage reactions using nitroso benzene and ketones see: Payetee JN, Yamamoto H. J. Am. Chem. Soc. 2008;130:12276. doi: 10.1021/ja804325f. Delgado A, Garcia JM, Mauleon D, Minguillon C, Subirats JR, Feliz M, Lopez F, Velasco D. Can. J. Chem. 1988;66:517. Rainier JD, Xu Q. Org. Lett. 1999;1:27. doi: 10.1021/ol990532o.
- (8).Harrison JR, Moody CJ, Pitts MR. Synlett. 2000:1601. Pitts MR, Harrison JR, Moody CJ, Pitts MR. J. Chem. Soc., Perkin Trans. 1. 2001:955.
- (9).Anderson JW, Adediran SA, Charlier P, Nguyen-Disteche M, Frere J-M, Nicholas RA, Pratt RF. Biochem. J. 2003;373:949. doi: 10.1042/BJ20030217. Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. FEMS Microbiol. Rev. 2006;30:673. doi: 10.1111/j.1574-6976.2006.00024.x. Sauvage E, Duez C, Herman R, Kerff F, Petrella S, Anderson JW, Adediran SA, Pratt RF, Frere J-M, Charlier P. J. Mol. Biol. 2007;371:528. doi: 10.1016/j.jmb.2007.05.071.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.