

Abstract
Invasion of human immunodeficiency virus (HIV) into the central and peripheral nervous system produces a wide range of neurological symptoms, which continue to persist even with adequate therapeutic suppression of the systemic viremia. The development of therapies designed to prevent the neurological complications of HIV require a detailed understanding of the mechanisms of virus penetration into the nervous system, infection, and subsequent neuropathogenesis. These processes, however, are difficult to study in humans. The identification of animal lentiviruses similar to HIV has provided useful models of HIV infection that have greatly facilitated these efforts. This review summarizes contributions made from in vitro and in vivo studies on the infectious and pathological interactions of feline immunodeficiency virus (FIV) with the nervous system. In vivo studies on FIV have provided insights into the natural progression of CNS disease as well as the contribution of various risk factors. In vitro studies have contributed to our understanding of immune cell trafficking, CNS infection and neuropathogenesis. Together, these studies have made unique contributions to our understanding of (1) lentiviral interactions at the blood–cerebrospinal fluid (CSF) barrier within the choroid plexus, (2) early FIV invasion and pathogenesis in the brain, and (3) lentiviral effects on intracellular calcium deregulation and neuronal dysfunction. The ability to combine in vitro and in vivo studies on FIV offers enormous potential to explore neuropathogenic mechanisms and generate information necessary for the development of effective therapeutic interventions.
Keywords: AIDS, dementia, human immunodeficiency virus, neurons, microglia, astrocytes
Introduction
Rapid penetration of human immunodeficiency virus (HIV) into the central nervous system (CNS), viral persistence in the brain, and neurological disease progression continue to be significant problems associated with HIV infection (McArthur et al. 2005). Even with current antiretroviral regimens, the prevalence of cognitive–motor dysfunction continues to increase in HIV-infected individuals. Therapeutic interventions are badly needed but the development of effective treatments has proven to be difficult because of the unique characteristics of CNS infection. Within the CNS, lentiviruses such as HIV are thought to replicate in cells of monocyte lineage including perivascular macrophages and microglia (Brinkmann et al. 1993; Dow et al. 1999; Fischer-Smith et al. 2001; Gonzalez-Scarano and Martin-Garcia 2005; Gorry et al. 2001; Hein et al. 2000; Koenig et al. 1986; Lane et al. 1996; Petito 2004; Williams et al. 2001; Zenger et al. 1997). Control of virus replication in the brain has been difficult as a result of the poor penetration of most antiretroviral drugs across the blood–brain and blood–cerebrospinal fluid (CSF) barriers as well as the presence of mechanisms for active transport of these compounds from the brain. Thus, CNS disease continues (Anthony et al. 2005; Neuenburg et al. 2002; Sacktor et al. 2002) even with suppression of both the systemic and CSF viral load (Cysique et al. 2005). To develop effective therapeutic interventions we need to better understand (1) how the virus gains access to the brain, (2) when and how CNS pathogenesis begins, (3) the unique properties of the brain viral reservoir, and (4) the mechanisms that underlie neuronal damage. Because of the difficulty of exploring these processes in humans, animal models have played an essential role in this endeavor.
Within 2 years of the identification of the HIV, two similar, naturally occurring animal lentiviruses were isolated. Simian immunodefciency virus (SIV), isolated from macaques (Kanki et al. 1985), and feline immunodeficiency virus (FIV) (Pedersen et al. 1987), isolated from domestic cats, are genetically and functionally similar to HIV including similar cell tropism and the ability to produce a severe acquired immune deficiency syndrome (AIDS). The study of infection in these animals has provided significant insights into the mechanisms of pathogenesis applicable to our understanding and treatment of HIV (for reviews, see Gendelman et al. 2005). In this review, we summarize in vitro and in vivo studies on FIV infection that have helped to shed light on early immune cell trafficking, CNS infection, the progression of CNS disease, and the mechanisms of neuronal dysfunction. Representative examples are used throughout to illustrate many of the key findings from these studies.
FIV: A lentivirus similar to HIV
FIV infection is seen in domestic and feral cat species worldwide (Carpenter and O’Brien 1995) with estimates of seroprevalence rates in the domestic cat ranging from approximately 1% to 14%. The genomic organization of FIV is similar to, although less complex than HIV-1 (Olmsted et al. 1989; Talbott et al. 1989); it has also been suggested, based on phylogenetic analysis, that FIV may represent a more primitive lentivirus that has evolved independently of HIV and SIV (Elder and Phillips 1993; Olmsted et al. 1989). FIV, like HIV-1(Livingstone et al. 1996; Potter et al. 2003), infects CD4+ and CD8+T lymphocytes (Brown et al. 1991; English et al. 1993) as well as monocyte-derived cells (Beebe et al. 1994; Brunner and Pedersen 1989; Dow et al. 1992). Natural transmission of the virus is thought to occur predominantly via bites received during fighting or sexual activity, although efficient transmission can also occur after vaginal inoculation (Burkhard et al. 2002; Burkhard and Dean 2003; Jordan et al. 1998; Obert and Hoover 2002). FIV is also transmitted vertically to offspring with a rate of 22–50% (Allison and Hoover 2003b; O’Neil et al. 1996) with transmission occurring both in utero (~20% infection rate) and during nursing (~13% infection rate) (O’Neil et al. 1996). However, offspring infected by this route often failed to show a sustained infection with FIV (Allison and Hoover 2003a) or clinical disease (Wasmoen et al. 1992).
Infection results in a clinical syndrome initially characterized by an inversion of the CD4/CD8 ratio, followed by a progressive decline in circulating CD4+T cells (Ackley et al. 1990; Tompkins et al. 1991; Torten et al. 1991) and a sustained increase in activated CD8+T cells (Bucci et al. 1998; Gebhard et al. 1999; Hoffmann-Fezer et al. 1992; Novotney et al. 1990; Willett et al. 1993). These CD8+T cells are thought to play an important role in the control of systemic and brain infection (Bucci et al. 1998; Gebhard et al. 1999; Hohdatsu et al. 2000; Hohdatsu et al. 2002). Eventually, a severe immunodeficiency develops with accompanying opportunistic infections (English et al. 1994). In addition, FIV also infects B lymphocytes (English et al. 1993) in vivo, thereby exhibiting a slightly broader host cell range than HIV-1(Levy 1993). Although most primary isolates of FIV will infect both T lymphocytes and monocytes, productive replication in vivo is largely restricted to FIV-infected T lymphocytes (Beebe et al. 1994; Brunner and Pedersen 1989; Dow et al. 1992; Dow et al. 1999). In contrast to HIV-1, FIV uses CD134 as a primary receptor instead of CD4 (de Parseval et al. 2004; Shimojima et al. 2004). Like HIV-1, FIV uses the alpha chemokine receptor, CXCR4, as a coreceptor (Egberink et al. 1999; Hosie et al. 1998; Richardson et al. 1999; Willett et al. 1997). Human and feline CXCR4 share significant homology and the receptor combination of CD134 and CXCR4 for FIV would be expected to confer similar tropism and mechanisms of infection as HIV. Although feline peripheral blood mononuclear cells (PBMCs) have been shown to express a CCR5 receptor mRNA with 68% homology to human CCR5 (Kovacs et al. 1999), the utilization of this receptor for infection is controversial (Johnston and Power 2002; Lerner and Elder 2000; Willett et al. 2002).
FIV infection results in the synthesis of neutralizing antibodies that are targeted to various envelope domains with variable loop 3 and the carboxy terminus of envelope consisting of major antigenic sites (de Ronde et al. 1994; Lombardi et al. 1995; Richardson et al. 1996). As with HIV-1, the cross-reactivity of sera for various FIV strains is highly variable (de Ronde et al. 1994; Del Mauro et al. 1998; Hohdatsu et al. 1997; Osborne et al. 1994; Richardson et al. 1996) and specific mutations can confer resistance (Siebelink et al. 1995). The ability of antibodies to target the FIV virion depends heavily on the exposure of epitopes on the envelope surface (Richardson et al. 1996), indicating that neutralizing antibodies face similar challenges for FIV as for HIV. Neutralizing antibodies are also found in the CSF of infected cats (Dow et al. 1990; Phillips et al. 1994). These antibodies differed from the profile in serum and showed a tendency to preferentially recognize envelope versus other FIV proteins (e.g., gag proteins). These observations suggested a local synthesis of the CSF antibodies. However, the presence of antibodies did not correlate well with the level of detectable virus or CNS disease. The extent to which neutralizing antibodies control virus in the CSF is not known, but studies showing the development of profiles of FIV envelope expression in the CSF that are unique from plasma are consistent with a role of local antibodies in variant selection (Liu et al. 2006b). In this case, a subset of FIV variants present in plasma appears within the CSF with the infrequent emergence of a variant that is not present in plasma. However, the restriction of FIV variants within the CSF does not appear to involve a specific selection because no consistent pattern of variants appears in the CSF.
Although there are some notable differences between HIV-1 and FIV, the pathogenesis of FIV infection in the brain and systemic tissues is remarkably similar to HIV-1. Studies on FIV pathogenesis have contributed to our understanding of the nature of lentiviral infections on many fronts (Burkhard and Dean 2003). These contributions are highlighted by the recent approval of the first animal lentivirus vaccine (Huang et al. 2004; Pu et al. 2001; Uhl et al. 2002). In addition, studies on FIV have contributed to our understanding of the mechanisms of immune suppression (Vahlenkamp et al. 2005) and have been used for the evaluation of antiretroviral therapeutics (Arai et al. 2002; Bisset et al. 2002; de Rozieres et al. 2004; Egberink et al. 1990; Hartmann et al. 1992; Hayes et al. 1995), the testing of virus penetration of mucosal barriers (Burkhard et al. 2002; Burkhard and Dean 2003; D’Cruz et al. 2004; Obert and Hoover 2002), the efficacy of topical antiviral treatments (D’Cruz et al. 2004), and the analysis of the efficacy of entry inhibitors (Garg et al. 2004). These studies highlight the diverse contributions of FIV to lentiviral pathogenesis, which cannot all be adequately covered in this review. In the following sections, contributions that the FIV model has made to our understanding of CNS pathogenesis are summarized.
FIV penetration into the brain and CSF
FIV trafficking across the blood–brain barrier
Like the primate lentiviruses, neurotropism of FIV is thought to be largely attributable to the penetration of infected monocytes/macrophages into the brain. Because of the tissue barriers and the relative lack of systemic immune cells, the penetration and dissemination of virus into the brain and CSF is unique from other tissues. There are two primary cell-associated entry pathways for FIV: (1) monocyte trafficking through the blood–brain barrier and (2) monocyte/macrophage trafficking across the blood–CSF barrier. The potential contribution of T cells is less well understood. In vitro models of the blood–brain barrier have been used to explore the mechanisms that control immune cell trafficking across the brain endothelium. These studies have used fetal feline microvascular endothelial cells grown on transwell insert membranes containing 3- to 5-μm pores. By combining these cultures with astrocytes and/or microglia, the role of parenchymal cells in the trafficking of PBMCs across the endothelium has been evaluated (Hudson et al. 2005). Monocytes, CD4+T cells, CD8+T cells, and B cells were all observed to cross the endothelium although the ability to cross was highly dependent on the presence of astrocytes. This positive role of astrocytes was unexpected inasmuch as they are instrumental in the formation of the blood–brain barrier. Exposure of the cultures to FIVNCSU1 did not induce any preferential trafficking of monocytes, in agreement with previous trafficking studies on human monocytes infected with HIV-1 (Persidsky et al. 1997). Of all the major PBMC subsets, only CD8+T cells were subject to positive regulation when the feline cultures were exposed to FIV. The addition of microglia to the cultures (feline endothelium + astrocytes + microglia) suppressed the trafficking of T cells, B cells, and monocytes (Hudson et al. 2005). The suppression of monocyte trafficking by the microglia was partially reversed when the cultures were inoculated with FIV. These results indicated that astrocytes facilitate trafficking whereas microglia exercise inhibitory control over the astrocytes. The broad range of trafficking cells seen in vitro is consistent with in vivo trafficking studies that have shown early invasion of T cells and B cells within the brain of cats 8–10 weeks after infection with FIVGL8 (Ryan et al. 2005). Provirus has been detected in the brain as early as 2–4 weeks postinoculation, suggesting that the early immune cell trafficking carries virus into the CNS during the initial systemic viremia (Poli et al. 1999; Ryan et al. 2003). FIV RNA was consistently recovered in brain tissue at 10 weeks when the infiltration of mononuclear cells was at a peak, suggesting that the trafficking cells fuel the virus production (Ryan et al. 2003). However, both the tissue pathology and the viral loads decreased substantially over time, indicating a significant recovery from this acute phase of infection. Tissue proviral burden was reduced to zero by 23 weeks, further suggesting that the acute phase of infection with FIVGL8 did not establish a persistent viral reservoir. Still, FIV-infected cats eventually develop a relatively high, sustained proviral burden in the brain (Macchi et al. 1998; Pistello et al. 1994). These and other studies also showed that a high proviral burden did not predict a high viral burden because it has often been difficult to demonstrate active virus replication in brain tissue carrying a significant proviral burden. The control of virus production in brain is not well understood and remains an important area of research.
FIV trafficking across the blood–CSF barrier
In addition to penetration across the blood–brain barrier, large amounts of virus may also penetrate into the brain via the blood–CSF barrier. This barrier differs from the blood–brain barrier in several respects. First, there are no tight junctions at the vascular endothelium. This allows easier penetration of virus and immune cells. Second, the stromal region between the vascular endothelium and the choroidal epithelium contains a large population of macrophages as well as dendritic cells. Third, the blood–CSF barrier is formed by tight junctions between the cuboidal epithelial cells of the choroid plexus. In studies of mouse choroid plexus, the epithelium has been shown to constitutively express the adhesion molecules ICAM-1 and VCAM-1 (Steffen et al. 1996). In addition, ICAM-1, VCAM-1, and MAdCAM-1 were increased in response to inflammatory stimuli (Steffen et al. 1996). The expression of these adhesion molecules suggests that the epithelium exerts substantial control over trafficking of cells into the ventricular system and CSF, particularly under inflammatory conditions. Adhesion to the epithelium may facilitate penetration of cells through the tight junctions in much the same fashion as penetration of the blood–brain barrier. Thus, the choroid plexus is poised to be an important interface between the systemic immune system and the brain. However, the functional role of the choroid plexus in immune cell trafficking across the blood–CSF barrier is still poorly understood.
In addition to circulating immune cells, the trafficking of cells across the epithelium most likely includes resident choroid plexus macrophages. In the in vivo study on FIV infection of the brain by Ryan et al. (2005), a relatively robust infiltration of mononuclear cells was seen in the choroid plexus, meninges, and subarachnoid space as early as 4 weeks postinfection. This was accompanied by the appearance of low copy numbers of FIV RNA in the CSF, reinforcing the view that the blood–CSF barrier in the choroid plexus may be a significant site of the earliest immune cell and virus invasion. However, the relationship between the systemic viremia and virus in the CSF is complex. Detailed studies of plasma and CSF viral kinetics indicate that the transfer of virus is not a highly predictable process. As an example, Fig. 1 illustrates the time course of viremia in two cats intraperitoneally inoculated with FIVNCSU1. In most cases, the appearance of virus in CSF immediately follows the appearance in plasma (e.g., Cat #1). However, in Cat #2, virus appearance in the CSF is delayed several weeks in spite of the fact that it received the same inoculation and developed a rapid plasma viremia almost identical to Cat #1. The reason for the variation between plasma and CSF viral kinetics is still not well understood, but these analyses have indicated that virus penetration into the CSF can vary dramatically between animals. This variable delay may, in part, account for the low correlations typically seen when temporally matched plasma and CSF samples are compared. Although such correlations (e.g., r =0.310 for FIV RNA in CSF versus plasma) can reach statistical significance, they account for only about 10% of the variability in FIV-infected cats. CSF virus is certainly linked to plasma virus, but more work is needed to fully understand the constraints on virus exchange from the blood to the CSF compartment.
Fig. 1.
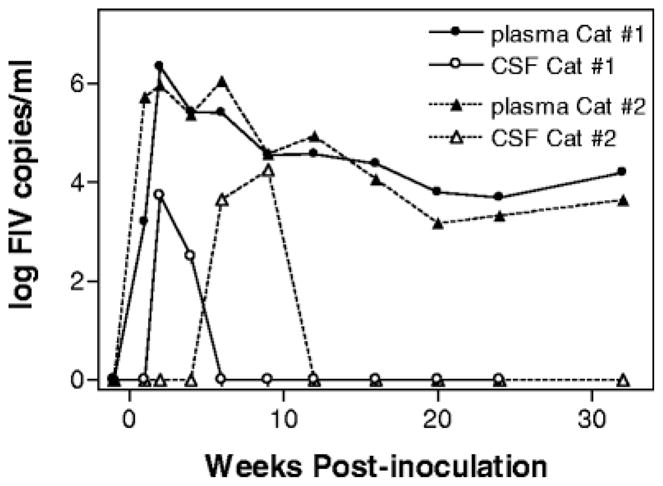
Example of the variable relationship between plasma and CSF viral kinetics. The time course for appearance of FIV RNA in the plasma and CSF is illustrated for two cats inoculated intraperitoneally with 2×105 median tissue culture infectious doses of FIVNCSU1. Although both cats developed almost identical plasma viremias, FIV appeared in the CSF of Cat #1 at 2 weeks but was delayed until 6 weeks postinoculation in Cat #2.
It has also been suggested based on studies of HIV that virus in the choroid plexus and CSF is likely to be a mix of virus from peripheral and CNS origin (Chen et al. 2000; Ellis et al. 2000; Strain et al. 2005). Studies on FIV have provided support for this view. High CSF/plasma ratios are relatively common in cats, suggesting a robust local virus production within the CSF and adjacent tissues (Liu et al. 2006a). Inversions of the FIV CSF/plasma ratio (CSF > plasma) often occur at times corresponding to the acute productive infection seen in brain parenchyma 4–10 weeks after infection (Ryan et al. 2003). Studies on FIV envelope diversity during the early stages of infection have suggested that the virus that first appears in the CSF is similar to plasma (Liu et al. 2006b). Over time, there can be significant divergence in the pattern of variants in the CSF versus plasma including the rare appearance of unique variants. These observations suggest that once established in the brain, local FIV synthesis and evolution can contribute to the pattern of variants seen in the CSF. Further evidence for the evolution of virus within the brain/CSF comes from the identification of neurovirulent brain-specific FIV sequences from the CSF (Power et al. 1997; Power et al. 1998). The evolution of sequences specific to the CNS also reinforces the view that FIV adapts to the neural environment much the same as HIV-1. As with HIV-1, many questions remain regarding compartmentalization, viral evolution, and local control of viral replication in the CNS. The FIV model provides a versatile system for the investigation of these processes through all stages of pathogenesis.
FIV neurotropism and neuropathogenesis
FIV neurotropism
FIV penetration into the brain initiates a long and slow decline in CNS function with few overt symptoms until the cats develop AIDS. As the cats become immune deficient, approximately 20% will develop significant neurological deficits. Early studies on FIV tropism demonstrated that virus could be recovered from brain and CSF of infected cats (Dow et al. 1990; Macchi et al. 1998; Power et al. 1997). In vitro studies of CNS cell tropism indicate that FIV can infect feline astrocytes (Billaud et al. 2000; Danave et al. 1994; Dow et al. 1990; Kawaguchi et al. 1992; Yu et al. 1998), microglia (Dow et al. 1990; Hein et al. 2000; Kawaguchi et al. 1992), choroid plexus macrophages (Bragg et al. 2002a), and perhaps brain microvascular endothelial cells (Steffan et al. 1994). In vivo studies indicate that most virus production in the CNS is maintained by macrophages and microglia (Dow et al. 1999; Hein et al. 2000).
Although microglia and macrophages are primary targets of FIV in the brain and choroid plexus, they support a relatively low-grade productive infection(Bragg et al. 2002a; Hein et al. 2000, 2001). However, the addition of PBMCs to infected microglia or macrophages can lead to a robust productive infection (Bragg et al. 2002a; Hein et al. 2000, 2001), indicating efficient transfer of virus to T cells. Interactions between macrophages and T cells could be particularly prominent within perivascular regions and the choroid plexus. Perivascular localization of infected macrophages is a common finding for HIV, SIV, and FIV infections. In addition, many studies have documented lentiviral transcripts within the choroid plexus of infected hosts (Beebe et al. 1994; Bragg et al. 2002a; Chen et al. 2000; Falangola et al. 1995; Petito et al. 1999). These studies suggested that the infected perivascular and choroid plexus macrophages are poised to interact with trafficking T cells in a fashion that could facilitate virus production. Such interactions might also promote the evolution of viral strains adapted for growth in both cell lineages, as suggested by the admixture of systemic and CNS viral genotypes seen in HIV-1-infected choroid plexus (Chen et al. 2000). The trafficking of virus and/or infected macrophages into the CSF might also contribute directly to pathogenesis, but the functional implications of macrophage and virus trafficking through the blood–CSF barrier are just beginning to be explored.
Although astrocytes can be infected with FIV in vitro, the role of astrocyte infection in vivo has been less clear because primary isolates of FIV are much less infectious than cell adapted strains and little evidence is available to support a widespread infection of astrocytes in the brain. However, the potential significance of astrocyte infection in vivo is underscored by studies of Gavrilin et al. (2002), showing that astrocytes could be infected in vitro when exposed to PBMC previously infected with FIVMD. In contrast, direct inoculation with cell-free wild-type FIVMD failed to infect the astrocytes. The subsequent passage of the virus from the PBMC–astrocyte cultures to fresh astrocyte cultures produced a new CXCR4-dependent, astrocyte-tropic strain of FIV (FIVMD-A). This evolution of the virus in response to cell–cell interactions has important implications for the development of reservoirs of viral quasispieces in the brain as well as their potential contribution to pathogenesis.
FIV neuropathogenesis
One of the early questions regarding lentiviral invasion of the CNS was whether CNS pathogenesis was progressive in nature. Because infection and pathogenesis could be closely followed in cats over time, a number of studies were performed to evaluate the evolution of CNS disease. These studies, using a variety of endpoints, showed that damage begins during the asymptomatic stages of disease but, as in humans, does not become severe until the development of AIDS. Neurological symptoms have been documented in cats experimentally inoculated with the primary isolates, FIVMD (Phillips et al. 1994; Podell et al. 1993, 1997, 1999; Prospero-Garcia et al. 1994), the infectious molecular clone, FIVPPR (Phillips et al. 1996, 2000), FIVNCSU1 (unpublished data), and the neurovirulent CSF-derived isolate, FIVV1CSF (Power et al. 1998). Symptoms observed in these cats included abnormal, stereotypic motor behaviors, anisocoria, increased aggression, increased cortical slow wave activity in quantitative electroencephalograms, prolonged latencies in brainstem evoked potentials, delayed righting and pupillary reflexes, decreased nerve conduction velocities, marked changes in sleep architecture, and deficits in cognitive–motor functions (Phillips et al. 1994, 1996; Podell et al. 1993, 1997; Prospero-Garcia et al. 1994; Steigerwald et al. 1999). Using proton magnetic resonance spectroscopy (MRS), Power et al. (1998) and Podell et al. (1999) demonstrated reductions in the concentrations of the neuronal marker, N-acetyl-aspartate (NAA) and the NAA/choline or NAA/creatine ratio within the brains of FIV-infected cats. Measurement of visual and auditory evoked potentials has also provided a sensitive index of progressive neurological dysfunction (Phillips et al. 1996; Podell et al. 1997). In each case, neurological deficits could be detected at relatively early stages of disease with progression over time. An understanding of the nature of the gradual CNS disease progression has become increasingly important as it most likely reflects the type of disease currently seen in patients on antiretroviral therapy.
FIV neuropathology
Most FIV-infected cats show characteristic neuropathological changes similar to patients infected with HIV-1, although typically less severe than HIV-1 encephalitis. Diffuse astrogliosis and microgliosis, myelin pallor, microglial nodules, and rare, multinucleated giant cells have been observed in brains of FIV-infected cats (Abramo et al. 1995; Boche et al. 1996; Hurtrel et al. 1992; Poli et al. 1997; Silvotti et al. 1997). Astrogliosis, microgliosis, and a diffuse perivascular monocyte infiltration are most commonly seen (Fig. 2). In addition, a significant neuronal loss has been seen within the cortex and basal ganglia of FIVNCSU1-infected cats beginning as early as 2–3 years postinfection (Meeker et al. 1997; Power et al. 1997), whereas the cats were still asymptomatic. Large pyramidal cells within layers two, three, and five of the frontal/parietal cortex and large cells within the striatum appeared to be most vulnerable to early damage (Meeker et al. 1997). The loss of neurons in the parietal lobe and striatum correlated with decreases in the CD4/CD8 ratio, suggesting a close relationship to the systemic immune response (Meeker et al. 1997). An average 32% decrease in the number of large neurons was similar to numbers reported for HIV (Wiley et al. 1991). However, it is important to note that as other neurons were minimally affected (as with HIV), the decrease in total neuron density was a modest 2.3%. Although the extent of neuronal loss continued to increase with systemic disease severity, the general picture was one of a slow, progressive degeneration. Further support for FIV-associated neurodegeneration was provided by Jacobson et al. (1997), who found that large pyramidal neurons in cortical layers three and five show an increased immunoreactivity for neurofilament protein, and Koirala et al. (2001), who showed decreased expression of MAP-2 and GAD. These latter measures may reflect neuronal dysfunction that is likely to be a more sensitive index of CNS disease than cell death (Dou et al. 2003; Nagra et al. 1993; Toggas et al. 1996; Zheng et al. 2001). These observations also reinforce the idea that much of the pathology associated with FIV and HIV infection is potentially reversible. Reports of increased cortical synaptophysin immunoreactivity at early stages of disease progression (Meeker et al. 1997) and increased Timms staining (sprouting) in the hippocampus of FIV-infected cats (Mitchell et al. 1999) further suggest that compensatory processes may help to maintain neural function during the asymptomatic stages of disease. These observations emphasize the dynamic interplay between destructive and intrinsic neuroprotective responses during the course of disease.
Fig. 2.

Example of neuropathology seen in cats infected with FIV. Infiltration of monocytic cells positive for Mac387 has been seen in cats along the cortical surface (a), scattered through the parenchyma of the basal ganglia (b, arrows), particularly along blood vessels (c) and occasionally within and adjacent to the choroid plexus (d, arrows). In addition, diffuse activated microglia, positive for feline CD18, can be detected in white matter (e), and GFAP-immunoreactive astrocytes are prominent in the cortex (f) and other regions. These observations indicate the presence of a low-grade diffuse inflammatory process that becomes increasingly severe as the cats become immune deficient.
The appearance of neuronal loss and neuronal dysfunction in asymptomatic cats is consistent with the early appearance of altered sleep architecture at 10–12 months postinoculation (Prospero-Garcia et al. 1994), cortical atrophy by MRI at 12 months (Podell et al. 1993), decreased NAA and NAA/choline at 14 months (Podell et al. 1999), motor and spatial memory deficits at 12 months (Steigerwald et al. 1999), and the appearance of neurotoxic activity in the CSF of FIV-infected cats as early as 4 months (Bragg et al. 2002b). These findings clearly indicate the need for early therapeutic intervention to suppress neurodegeneration and highlight the potential utility of the feline model for the development and testing of such interventions.
Cellular mechanisms of FIV neurotoxicity
In vitro characterization of neurotoxicity
One of the advantages of the FIV model is the ability to perform both in vitro and in vivo experiments. This allows the efficient translation of in vitro information on cellular mechanisms to development of therapeutics in the naturally infected host. Most cell types within the feline CNS have been successfully cultured including neurons, astrocytes, microglia, macrophages, endothelial cells, and choroid plexus epithelium. Studies that have examined the interactions of FIV and macrophage-derived toxins with primary forebrain neural cultures have led to a number of important insights into the mechanisms of neuropathogenesis. Early studies using the primary isolate, FIVNCSU1, demonstrated relatively rapid effects on neuronal function and survival in culture (Meeker et al. 1996). Inoculation of primary feline neural cultures with FIV alone produced minimal cell death. However, the addition of a small, normally subtoxic concentration of glutamate (20 μM) resulted in swelling of neurons within 20 min followed by death of a subset of neurons (Meeker et al. 1996). The presence of FIV shifted the concentration–effect curve for glutamate toxicity approximately threefold to the left, indicating an increase in the neuronal sensitivity to the glutamate. The effects of FIV were blocked by an N-methyl-D-aspartate (NMDA) glutamate receptor antagonist. Toxicity increased progressively over the first week of exposure to FIV, suggesting the cumulative release of soluble toxic factors. Small amounts of FIV provirus were detected in some of these cultures at 17–24 days postinoculation, but no evidence of productive infection was observed during the period of toxin generation, which peaked at approximately 6 days. Studies using FIV envelope protein from the PPR strain reproduced most of the toxic features of live virus (Bragg et al. 1999; Gruol et al. 1998). The toxic effects were not induced by the envelope protein derived from FIV34TF10, a less neurovirulent strain (Bragg et al. 1999). As with the live virions, the purified FIVPPR envelope protein exhibited little intrinsic toxicity, but facilitated neuronal swelling, cell death (Meeker et al. 1996) and NMDA-evoked calcium entry into the cell (Gruol et al. 1998). Similar toxicity profiles were seen when cultured neurons were exposed to conditioned medium from FIVNCSU1-infected choroid plexus macrophages (Bragg et al. 2002b). Like HIV-1, infection of macrophages (Giulian et al. 1990; Pulliam et al. 1991; Xiong et al. 1999; Yeh et al. 2000; Zheng et al. 1999b) leads to the release of substances into the culture medium, which are toxic when applied to neuronal cultures. These experiments clearly demonstrated that the destructive inflammatory interactions of FIV with macrophages and microglial-enriched neural cultures recapitulated the effects of HIV and illustrated that toxin production did not depend on significant virus production.
FIV and intracellular calcium homeostasis
A diverse range of HIV-associated, macrophage-derived neurotoxins with various actions have been proposed (Gonzalez-Scarano and Martin-Garcia 2005; Jones and Power 2006). In general, the mechanisms of toxicity are thought to involve aberrant activation of various receptors and signal transduction pathways by viral proteins or macrophage-derived substances. The actions of these substances lead to a toxic accumulation of intracellular calcium, oxidative stress, and generation of proapoptotic signals. Many of these pathways have been the target of therapeutic strategies designed to protect neurons (Perry et al. 2005; Turchan et al. 2003). Control of intracellular calcium has been an important focus of many of these efforts.
Because excessive intracellular calcium accumulation is widely considered a final common pathway leading to neuronal dysfunction and death, Bragg et al. (2002c) used feline neural cultures to evaluate the relative contribution of various extra- and intracellular sources of calcium to neuronal death in the presence of macrophage- or microglial-derived toxins. In these experiments, cultured feline neurons were exposed to infectious virions (FIVNCSU1) or conditioned medium from choroid plexus macrophages previously inoculated with FIVNCSU1. Both challenges resulted in very small acute increases in intracellular calcium followed by a gradual rise to high levels. The conditioned medium typically provoked a greater toxic response than cell-free FIV virions, consistent with the idea that the macrophages are a primary source of the toxins. An example of the intracellular calcium response in neurons exposed to FIV virions isolated from the CSF of an infected cat is illustrated in Fig. 3a. A cluster of neurons and a large microglial (MG) cell adjacent to a neuron (arrow) is shown at times ranging from 0 to 40 min after exposure. A few neurons show a small acute calcium response (0.17 min), whereas most have a negligible response. After approximately 16 min, neuronal calcium begins to rise and by 40 min some cells show high calcium. Typical responses of individual neurons exposed to FIV in the presence or absence of the sodium channel antagonist, tetrodotoxin (TTX), are illustrated in Fig. 3b. Although a small acute calcium response is seen, no delayed accumulation of calcium is seen in the absence of synaptic transmission, emphasizing the importance of neuronal activity for the gradual deregulation of calcium. Microglia in the cultures also respond to the virus or macrophage-conditioned medium with a calcium increase although responses of individual microglia tend to be highly variable. Sustained exposure of cultured neurons to FIV results in dendritic beading, fragmentation, and the death of a subset of neurons. This neuronal pathology can be seen with MAP-2 staining, as illustrated in Fig. 4, and is similar to changes seen in the brain of patients with HIV encephalitis (Masliah et al. 1997).
Fig. 3.

Example of intracellular calcium changes in cultured feline neurons isolated from fetal cortex–hippocampus. (a) A typical cluster of neurons is shown with low resting calcium at 0 min. The intensity of the Fluo-3 fluorescence is proportional to the cytosolic calcium concentration and is color-coded from low to high as: purple>blue>green>yellow>red>white. Addition of FIV isolated from the CSF of an infected cat resulted in a small transient increase in a few neurons (0.17 min), followed by a gradual increase beginning as early as 11 min. Moderately high calcium levels developed in many neurons by 40 min. Individual neuronal responses can be quite variable, ranging from minimal responsiveness to robust increases as seen in an isolated neuron (arrow) adjacent to a microglial cell (MG). (b) Effect of synaptic activity on the calcium rise can be seen in a comparison between neurons exposed to FIV in the presence or absence of 1 μM tetrodotoxin (TTX). A small acute increase is seen in both preparations but the TTX-treated neurons failed to show the delayed calcium deregulation, indicating that synaptic activity is necessary for the delayed calcium rise.
Fig. 4.
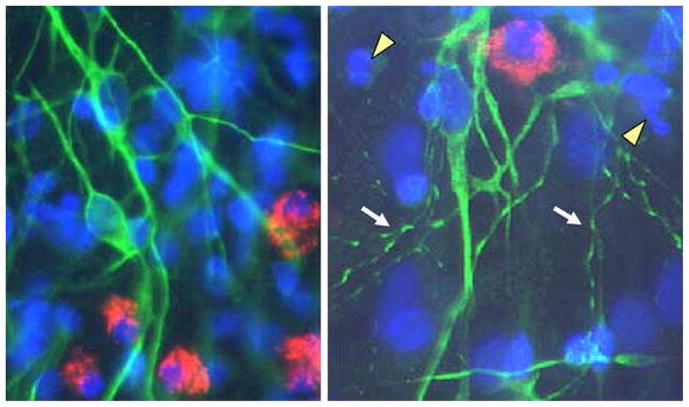
Dendritic damage seen in cultured neurons after 3 days of exposure to FIVNCSU1. (a) Mixed culture of normal feline neurons stained with MAP-2 (green) and microglia stained for feline CD18 (red). The nuclei are counterstained with bisbenzimide (blue). Astrocytes were not stained. (b) Beading and fragmentation of dendrities (arrows) is the most common pathological finding. Some increased cell death is apparent from condensed chromatin and apoptotic nuclei (arrowheads) but it is substantially less extensive than the dendritic changes.
As noted previously, exposure to virus, viral proteins, or macrophage-conditioned medium all lead to enhanced glutamate receptor activation and accumulation of intracellular calcium. Although there is strong evidence implicating glutamate receptors in the neuropathogenesis, the specific actions of putative toxins are still not well defined. On the one hand, studies have demonstrated that macrophages release glutamate and other substances that may directly stimulate glutamate receptors. On the other hand, numerous studies have indicated that glutamate receptors are indirectly modulated via undefined mechanisms. To better appreciate the role of glutamate receptor activation in the FIV model, we compared the response of neurons in macrophage-conditioned medium to the neuronal response to a moderate concentration of glutamate (100 μM). A comparison of these conditions would give an indication of the relative excitotoxic potency of the conditioned medium. Typical neuronal responses to macrophage-conditioned medium (macrophage–FIV) or 100 μM glutamate are illustrated in Fig. 5. Several important observations came from these experiments. First, the acute increase in the calcium signal was much smaller (5–35%) than that seen following application of a moderate concentration of glutamate (~100–200% increase), indicating that, under these conditions, the toxins did not have a pathologically significant direct excitatory effect on the neurons. The small acute response that was seen most likely reflected the presence of small concentrations of excitatory substances released from the macrophages. Second, although neurons exposed to the macrophage toxins showed a small acute response, a large delayed calcium rise is still seen (often larger than that seen following a glutamate challenge). This type of response is uniquely different from a classical excitotoxic response that is driven by the strong acute depolarization and calcium entry with subsequent development of a glutamate receptor-independent delayed calcium deregulation (Tymianski et al. 1993; Tymianski 1996). In contrast to glutamate excitotoxicity, virus and macrophage-derived toxins could induce a delayed calcium deregulation in the absence of a powerful acute stimulation. These observations suggested that there was an underlying dysfunction that was responsible for the development of pathological increases in intracellular calcium. This dysfunction would be expected to synergize with secretion of excitatory substances, inhibition of glutamate uptake, and processes that indirectly facilitate NMDA glutamate receptor function to encourage the accumulation of intracellular calcium. Thus, although the neuronal pathology may involve different pathways, they share a common endpoint in the generation of a delayed calcium deregulation. Finally, the delayed increase in intracellular calcium was not restricted to neurons. A subset of microglia also showed delayed increases in intracellular calcium (often very large) and astrocytes showed very small delayed increases (not shown) (Bragg et al. 2002c). Thus, although neurons are highly susceptible, all cells appear to be affected by the toxins. These observations further suggested the FIV-associated dysfunction may involve a general process common to many cell types.
Fig. 5.
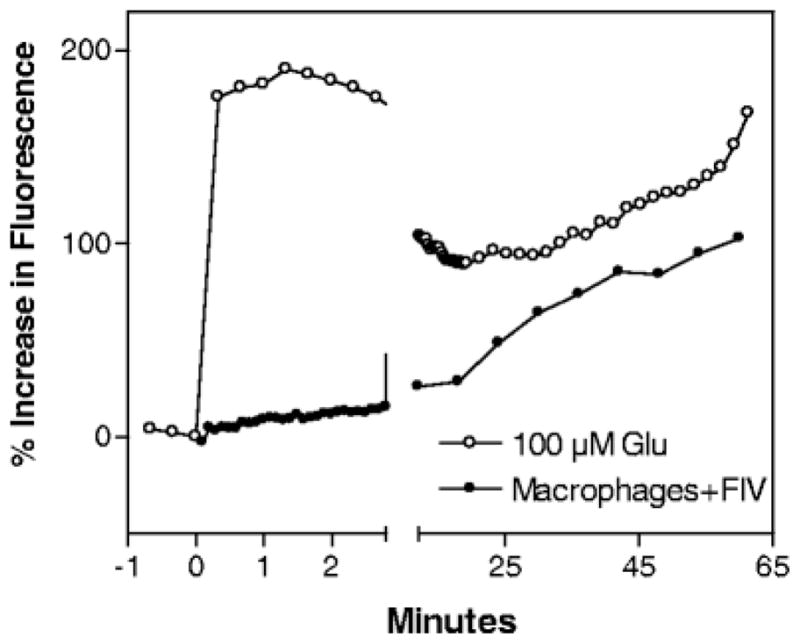
Comparison of the differential responses of cultured feline neurons after chronic application of 100 μM glutamate or conditioned medium from FIV-infected choroid plexus macrophages (macrophages+FIV). The medium was ultrafiltered (100 kDa) to remove free virions and large viral proteins. Glutamate evokes a large acute increase in intracellular calcium, which decreases and then begins to destabilize. The acute increase is not seen in most neurons exposed to macrophage-conditioned medium (although a small subset of neurons shows modest increases of approximately 10–40%). Even in the absence of a strong acute response, the neurons exposed to the macrophage-conditioned medium show a gradual delayed calcium deregulation.
If FIV exposure does not give rise to a strong excitotoxic glutamate response, what then is responsible for the observed increases in neuronal responsiveness to glutamate and the delayed calcium deregulation? Two observations in the feline neural cultures helped to provide a possible explanation (Bragg et al. 2002c). First, examination of the response to a brief pulse of glutamate in cultures inoculated with FIV, illustrated in Fig. 6a, not only showed a greater peak calcium response to the glutamate but also a delay in the recovery to baseline. The rate of recovery can be estimated from the slope of the plot of the log of the fluorescence at each time point (F) relative to the peak (F 0) versus time. A considerable slowing of the initial recovery rate is evident in the kinetics plot in Fig. 6b. The eventual recovery to baseline suggests that a single pulse of glutamate is not sufficient to establish a sustained rise in calcium. Similar effects have subsequently been reported after incubation of rat cortical neurons with CSF from HIV-infected patients (Meeker et al. 1999b, 2005) or CSF from FIV-infected cats (unpublished data), suggesting the presence of similar toxic processes in vivo. In companion experiments, antagonists that block ligand-gated calcium entry, voltage-gated calcium entry, intracellular calcium release, or synaptic transmission were all found to have neuroprotective efficacy during a 24-h exposure to medium collected from FIV-treated choroid plexus macrophages. Although the NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid (AP-5) showed the greatest neuroprotection (80%), antagonists of voltage-gated calcium entry (nimodipine, omega-conotoxin), internal IP3 receptor-mediated calcium release at the endoplasmic reticulum (xestospongin C), phospholipase C-linked G proteins (pertussis toxin), and synaptic activity (TTX), all showed partial neuroprotective efficacy ranging from 21% to 66% (Bragg et al. 2002c). These observations paralleled the findings from similar in vitro studies with HIV proteins and human macrophage conditioned medium (Haughey et al. 1999; Holden et al. 1999; Lo et al. 1992; Zheng et al. 1999a, b). The broad efficacy of such a diverse array of antagonists suggested that all major sources of calcium contributed to neuropathogenesis. Furthermore, partial protection afforded by TTX in this (Bragg et al. 2002c) and other paradigms (Hegg and Thayer 1999; Lo et al. 1992) indicated that much of the toxicity was dependent on synaptic activity. This observation indicated that a single “hit” with viral proteins or macrophage-derived excitotoxins, while perhaps contributing to the calcium accumulation, was not solely responsible for the long-term calcium deregulation. Together, these findings suggested a defect in a common downstream pathway that would facilitate calcium accumulation in response to all routes of calcium entry. The most general downstream pathways that might account for the above findings are those responsible for the recovery of basal intracellular calcium levels through the export of calcium from the cytosol. Such a deficit would be highly consistent with the decreased calcium recovery in Fig. 6 (see also Bragg et al. 2002c).
Fig. 6.

Characterization of the enhanced neuronal response to glutamate in mixed neural cultures exposed to FIV. (a) A brief (2 s) pulse of 100 μM glutamate applied to FIV-treated cultures (FIV+Glu) evokes a much larger neuronal calcium response than seen in neurons stimulated with glutamate in the absence of FIV (Glu). The FIV-treated neurons also develop a calcium plateau that delays recovery. (b) The rate of recovery of intracellular calcium can be calculated from a plot of the log of the ratio of calcium at each time point (F) to peak calcium (F 0) versus time. This analysis illustrates the dramatic decrease in the rate of recovery in neural cultures exposed to FIV (FIV+Glu).
Potential targets of macrophage-derived toxins that participate in the export of calcium from the cytosol include calcium transporters or exchangers at the plasma membrane, the endoplasmic reticulum calcium ATPase, or the mitochondrial uniporter (Bragg et al. 2002c). The characteristics of each recovery mechanism are summarized in Fig. 7. Recent studies with HIV-infected human CSF have suggested that the sodium–calcium exchanger (NCX) is the most likely cause of the calcium destabilization (Meeker et al. 2005). In these studies, the function of each calcium transporter was blocked and the deficit compared with the calcium deregulation seen in response to toxic HIV+ human CSF (Meeker et al. 2005). Only blockade of the NCX with benzamil or reversal of the sodium gradient (elimination of the driving force for exchange activity) were able to recapitulate the gradual rise in intracellular calcium seen with the HIV+ CSF. In addition, blockade of the NCX suppressed the rate of recovery of intracellular calcium after a brief pulse of glutamate. This deficit in the ability to export excess intracellular calcium would be expected to synergistically increase the magnitude of the intracellular calcium rise regardless of the source of calcium entry into the cytosol. Thus, it provides a common mechanism that would facilitate calcium accumulation in response to a variety of activation pathways. Even relatively modest increases in synaptic activity may lead to a gradual buildup of calcium if the calcium entry exceeds the limited capacity of the plasma membrane ATPase. Neuronal death would be expected only under conditions that provoke robust calcium entry into the cytosol. This scenario provides a consistent explanation for the neuroprotective effects of diverse receptor and channel antagonists (including TTX) and helps to explain why therapeutic agents targeted to any one source of calcium mobilization would have limited efficacy.
Fig. 7.

Model depicting the major processes that control intracellular calcium homeostasis. Cytosolic calcium can be taken up into endoplasmic reticulum by the sarcoplasmic–endoplasmic reticulum ATPase (SERCA) or into mitochondria by the uniporter. The endoplasmic reticulum normally maintains a high internal level of calcium and probably does not have a primary role in the clearance of cytosolic calcium. The mitochondrial uniporter exhibits an exponential increase in transport when the cytosolic calcium concentration exceeds ~1 μM. Most of the routine control of intracellular calcium is probably handled by the plasma membrane calcium ATPase (PMCA) and the sodium–calcium exchanger (NCX). PMCA is a high-affinity, low-capacity pump that uses ATP to drive the export of calcium from the cell. NCX is a low-affinity, high-capacity exchanger that uses the Na+ gradient across the plasma membrane to exchange 3 Na+ for 1 Ca2+. Recent studies designed to isolate the function of each process have suggested that loss of NCX function may be responsible for the gradual destabilization of intracellular calcium in neurons exposed to HIV, FIV, or associated toxins.
Astrocytes and neuropathogenesis
As noted above, and illustrated in several in vitro studies (Bragg et al. 2002c; Gruol et al. 1998; Meeker et al. 1997), exposure of neural cultures to FIV enhances glutamate-mediated responses. We have hypothesized that this enhancement is attributable to a loss of calcium export by the NCX. However, the loss of NCX activity would not be apparent in the absence of receptor stimulation (i.e., no calcium signaling). Thus, the calcium overload in cells must be due to a combination of calcium mobilization and decreased calcium export. Indeed, numerous studies have implicated increased NMDA receptor activation in the pathogenesis (Epstein and Gelbard 1999; Gemignani et al. 2000; Gruol et al. 1998; Haughey et al. 2001; Kolson 2002; Lipton et al. 1991; Lipton 1992; Self et al. 2004; Toggas et al. 1996; Yeh et al. 2000). One prominent mechanism hypothesized to lead to enhancement of glutamatergic activity is the suppression of glutamate uptake by astrocytes (Fine et al. 1996). Glutamate transporters on astrocytes are responsible for the uptake of extracellular glutamate from the synaptic cleft. Failure of amino acid transporters results in prolonged exposure of neurons to glutamate and enhanced excitation. Support for this argument has been provided by reports of increased concentrations of glutamate in extracts prepared from brains of FIV-infected cats (Power et al. 1997) and the ability of FIV infection to inhibit uptake of glutamate by astrocytes in vitro (Yu et al. 1998). In these latter studies, feline astrocytes harboring a low-grade productive infection with the 34TF10 strain of FIV suffered a 50–60% reduction in the ability to transport glutamate (Yu et al. 1998). Given that astrocytes can be a target for infection by FIV (Danave et al. 1994; Dow et al. 1990; Zenger et al. 1995) and display a high density of FIV binding sites (Meeker et al. 1999a), including CXCR4 expression (Nakagaki et al. 2001), it is reasonable to postulate that virus in brain could induce a widespread inhibition of glutamate uptake that increases the levels of neuronal excitation. The extent of this effect in vivo is still unclear. The relatively poor infection of astrocytes in vivo by wild-type FIV has raised questions regarding the role of astrocytes in CNS pathogenesis. However, recent studies, summarized above, raise the possibility that the interaction of infected PBMC with astrocytes in vitro may be capable of generating astrocyte-tropic variants of FIV (Gavrilin et al. 2002). These observations will have important implications for CNS infection if similar interactions are shown to exist in vivo. It is also possible that deficits in sodium–calcium exchange in astrocytes and microglia may contribute to their dysfunction by altering calcium homeostasis. Calcium increases are observed in these cells under the same conditions that lead to neuronal calcium destabilization. The function of the NCX has not been explored in these cells and much remains to be done to determine the extent of the CNS dysfunction attributable to a loss of exchange activity in nonneuronal cells.
Interactions between drugs of abuse and lentiviral neuropathogenesis
The feline model of lentiviral pathogenesis is well suited for the study of drug interactions with viral pathogenesis as well as potential screening of therapeutic agents. It has been used to explore the interactions of both opiates and methamphetamine with infection. Multiple acute doses of morphine, designed to mimic human abuse patterns, were shown to delay the progression of FIV-induced disease (Barr et al. 2000, 2003). Methamphetamine, on the other hand, has been shown to increase FIV replication in cultured astrocytes when the infection is mediated by cell–cell interactions (Gavrilin et al. 2002). No effects were seen on direct infection of astrocytes or PBMC with cell-free virus. The result of these latter studies was an increased proviral burden in astrocytes, but not an increase in virus production. The impact of the interactions of methamphetamine with FIV in vivo are also beginning to be explored by taking advantage of the ability to image the feline brain with reasonable resolution. Proton magnetic resonance studies have shown decreased creatine and choline and elevations in GABA in methamphetamine-treated cats (Cloak et al. 2004). These early studies are beginning to generate valuable clinical information on the interactions between drugs of abuse and lentiviral infections.
Conclusion
FIV infection of the nervous system shares many common features with HIV including common coreceptor usage, similar cellular targets, rapid penetration of the CNS, evolution of neurovirulent FIV strains, and a diffuse inflammatory response with a progressive CNS pathogenesis. Differences between FIV and HIV include a more simplified virus structure, a different primary receptor, and a reduced severity of neuropathogenesis. In spite of the differences, the systemic and CNS endpoints of infection are remarkably similar. In vivo studies with FIV have provided insights into the natural progression of the disease in brain and a means to evaluate interactions between the virus and factors that increase the risk of infection and neurodegeneration. Results from in vitro studies of the effects of virus, viral proteins, and macrophage-derived toxins in primary feline neural cultures strongly parallel similar studies with HIV. These studies have (1) led to new insights into the role of astrocytes and microglia in immune cell trafficking, (2) provided valuable information on early infection, viral diversification, and disease progression in the CNS, (3) explored the impact of drugs of abuse on infection and pathogenesis, (4) fostered a greater understanding of the potential importance of the choroid plexus blood–CSF barrier as a site of virus entry, and (5) defined potential mechanisms that lead to the loss of neuronal calcium homeostasis. The ability to investigate the pathogenesis of FIV in specific-pathogen-free cats in parallel with in vitro studies of cultured feline cells provides the opportunity for translational studies that should facilitate the design and evaluation of new therapeutic strategies for the treatment of HIV-associated CNS disease.
Acknowledgments
This study was supported by NIMH Grant R01 MH063646.
References
- Abramo F, Bo S, Canese MG, Poli A. Regional distribution of lesions in the central nervous system of cats infected with feline immunodeficiency virus. AIDS Res Hum Retrovir. 1995;11:1247–1253. doi: 10.1089/aid.1995.11.1247. [DOI] [PubMed] [Google Scholar]
- Ackley CD, Yamamoto JK, Levy N, Pedersen NC, Cooper MD. Immunologic abnormalities in pathogen-free cats experimentally infected with feline immunodeficiency virus. J Virol. 1990;64:5652–5655. doi: 10.1128/jvi.64.11.5652-5655.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison RW, Hoover EA. Covert vertical transmission of feline immunodeficiency virus. AIDS Res Hum Retrovir. 2003a;19:421–434. doi: 10.1089/088922203765551764. [DOI] [PubMed] [Google Scholar]
- Allison RW, Hoover EA. Feline immunodeficiency virus is concentrated in milk early in lactation. AIDS Res Hum Retrovir. 2003b;19:245–253. doi: 10.1089/088922203763315759. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005;64:529–536. doi: 10.1093/jnen/64.6.529. [DOI] [PubMed] [Google Scholar]
- Arai M, Earl DD, Yamamoto JK. Is AZT/3TC therapy effective against FIV infection or immunopathogenesis? Vet Immunol Immunopathol. 2002;85:189–204. doi: 10.1016/s0165-2427(01)00426-3. [DOI] [PubMed] [Google Scholar]
- Barr MC, Billaud JN, Selway DR, Huitron-Resendiz S, Osborn KG, Henriksen SJ, Phillips TR. Effects of multiple acute morphine exposures on feline immunodeficiency virus disease progression. J Infect Dis. 2000;182:725–732. doi: 10.1086/315789. [DOI] [PubMed] [Google Scholar]
- Barr MC, Huitron-Resendiz S, Sanchez-Alavez M, Henriksen SJ, Phillips TR. Escalating morphine exposures followed by withdrawal in feline immunodeficiency virus-infected cats: a model for HIV infection in chronic opiate abusers. Drug Alcohol Depend. 2003;72:141–149. doi: 10.1016/s0376-8716(03)00195-9. [DOI] [PubMed] [Google Scholar]
- Beebe AM, Dua N, Faith TG, Moore PF, Pedersen NC, Dandekar S. Primary stage of feline immunodeficiency virus infection: viral dissemination and cellular targets. J Virol. 1994;68:3080–3091. doi: 10.1128/jvi.68.5.3080-3091.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billaud JN, Selway D, Yu N, Phillips TR. Replication rate of feline immunodeficiency virus in astrocytes is envelope dependent: implications for glutamate uptake. Virology. 2000;266:180–188. doi: 10.1006/viro.1999.0079. [DOI] [PubMed] [Google Scholar]
- Bisset LR, Lutz H, Boni J, Hofmann-Lehmann R, Luthy R, Schupbach J. Combined effect of zidovudine (ZDV), lamivudine (3TC) and abacavir (ABC) antiretroviral therapy in suppressing in vitro FIV replication. Antiviral Res. 2002;53:35–45. doi: 10.1016/s0166-3542(01)00190-5. [DOI] [PubMed] [Google Scholar]
- Boche D, Hurtrel M, Gray F, Claessens-Maire MA, Ganiere JP, Montagnier, Hurtrel B. Virus load and neuropathology in the FIV model. J Neurovirology. 1996;2:377–387. doi: 10.3109/13550289609146903. [DOI] [PubMed] [Google Scholar]
- Bragg D, Childers T, Tompkins M, Tompkins W, Meeker R. Infection of the choroid plexus by feline immunodeficiency virus. J Neurovirology. 2002a;8:211–224. doi: 10.1080/13550280290049688. [DOI] [PubMed] [Google Scholar]
- Bragg D, Hudson L, Liang Y, Tompkins M, Fernandes A, Meeker R. Choroid plexus macrophages proliferate and release toxic factors in response to feline immunodeficiency virus. J Neurovirology. 2002b;8:225–239. doi: 10.1080/13550280290049679. [DOI] [PubMed] [Google Scholar]
- Bragg DC, Boles JC, Meeker RB. Destabilization of neuronal calcium homeostasis by factors secreted from choroid plexus macrophage cultures in response to feline immunodeficiency virus. Neurobiol Dis. 2002c;9:173–186. doi: 10.1006/nbdi.2001.0459. [DOI] [PubMed] [Google Scholar]
- Bragg DC, Meeker RB, Duff BA, English RV, Tompkins MB. Neurotoxicity of FIV and FIV envelope protein in feline cortical cultures. Brain Res. 1999;816:431–437. doi: 10.1016/s0006-8993(98)01177-9. [DOI] [PubMed] [Google Scholar]
- Brinkmann R, Schwinn A, Muller J, Stahl-Hennig C, Coulibaly C, Hunsmann G, Czub S, Rethwilm A, Dorries R, Ter MV. In vitro and in vivo infection of rhesus monkey microglial cells by simian immunodeficiency virus. Virology. 1993;195:561–568. doi: 10.1006/viro.1993.1407. [DOI] [PubMed] [Google Scholar]
- Brown WC, Bissey L, Logan KS, Pedersen NC, Elder JH, Collisson EW. Feline immunodeficiency virus infects both CD4+ and CD8+ T lymphocytes. J Virol. 1991;65:3359–3364. doi: 10.1128/jvi.65.6.3359-3364.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D, Pedersen NC. Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J Virol. 1989;63:5483–5488. doi: 10.1128/jvi.63.12.5483-5488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci JG, English RV, Jordan HL, Childers TA, Tompkins MB, Tompkins WA. Mucosally transmitted feline immunodeficiency virus induces a CD8+ antiviral response that correlates with reduction of cell-associated virus. J Infect Dis. 1998;177:18–25. doi: 10.1086/513822. [DOI] [PubMed] [Google Scholar]
- Burkhard MJ, Dean GA. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr HIV Res. 2003;1:15–29. doi: 10.2174/1570162033352101. [DOI] [PubMed] [Google Scholar]
- Burkhard MJ, Mathiason CK, O’Halloran K, Hoover EA. Kinetics of early FIV infection in cats exposed via the vaginal versus intravenous route. AIDS Res Hum Retrovir. 2002;18:217–226. doi: 10.1089/08892220252781284. [DOI] [PubMed] [Google Scholar]
- Carpenter MA, O’Brien SJ. Coadaptation and immunodeficiency virus: lessons from the Felidae. Curr Opin Genet Dev. 1995;5:739–745. doi: 10.1016/0959-437x(95)80006-q. [DOI] [PubMed] [Google Scholar]
- Chen H, Wood C, Petito CK. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: potential role of choroid plexus in the pathogenesis of HIV encephalitis. J Neurovirology. 2000;6:498–506. doi: 10.3109/13550280009091950. [DOI] [PubMed] [Google Scholar]
- Cloak CC, Chang L, Ernst T, Barr MC, Huitron-Resendiz S, Sanchez-Alavez M, Phillips TR, Henriksen S. Methamphetamine and AIDS: 1HMRS studies in a feline model of human disease. J Neuroimmunol. 2004;147:16–20. doi: 10.1016/j.jneuroim.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Cysique LA, Brew BJ, Halman M, Catalan J, Sacktor N, Price RW, Brown S, Atkinson JH, Clifford DB, Simpson D, Torres G, Hall C, Power C, Marder K, McArthur JC, Symonds W, Romero C. Undetectable cerebrospinal fluid HIV RNA and beta-2 microglobulin do not indicate inactive AIDS dementia complex in highly active antiretroviral therapy-treated patients. J Acquir Immune Defic Syndr. 2005;39:426–429. doi: 10.1097/01.qai.0000165799.59322.f5. [DOI] [PubMed] [Google Scholar]
- D’Cruz OJ, Waurzyniak B, Uckun FM. Antiretroviral spermicide WHI-07 prevents vaginal and rectal transmission of feline immunodeficiency virus in domestic cats. Antimicrob Agents Chemother. 2004;48:1082–1088. doi: 10.1128/AAC.48.4.1082-1088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danave IR, Tiffany Castiglioni E, Zenger E, Barhoumi R, Burghardt RC, Collisson EW. Feline immunodeficiency virus decreases cell–cell communication and mitochondrial membrane potential. J Virol. 1994;68:6745–6750. doi: 10.1128/jvi.68.10.6745-6750.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Parseval A, Chatterji U, Sun P, Elder JH. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc Natl Acad Sci USA. 2004;101:13044–13049. doi: 10.1073/pnas.0404006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ronde A, Stam JG, Boers P, Langedijk H, Meloen R, Hesselink W, Keldermans LC, van Vliet A, Verschoor EJ, Horzinek MC. Antibody response in cats to the envelope proteins of feline immunodeficiency virus: identification of an immunodominant neutralization domain. Virology. 1994;198:257–264. doi: 10.1006/viro.1994.1028. [DOI] [PubMed] [Google Scholar]
- de Rozieres S, Swan CH, Sheeter DA, Clingerman KJ, Lin YC, Huitron-Resendiz S, Henriksen S, Torbett BE, Elder JH. Assessment of FIV-C infection of cats as a function of treatment with the protease inhibitor, TL-3. Retrovirology. 2004;1:38. doi: 10.1186/1742-4690-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Mauro D, Matteucci D, Giannecchini S, Maggi F, Pistello M, Bendinelli M. Autologous and heterologous neutralization analyses of primary feline immunodeficiency virus isolates. J Virol. 1998;72:2199–2207. doi: 10.1128/jvi.72.3.2199-2207.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow S, Poss M, Hoover E. Feline immunodeficiency virus: a neurotropic lentivirus. J AIDS. 1990;3:658–668. [PubMed] [Google Scholar]
- Dow SW, Dreitz MJ, Hoover EA. Feline immunodeficiency virus neurotropism: evidence that astrocytes and microglia are the primary target cells. Vet Immunol Immunopathol. 1992;35:23–35. doi: 10.1016/0165-2427(92)90118-a. [DOI] [PubMed] [Google Scholar]
- Dow SW, Mathiason CK, Hoover EA. In vivo monocyte tropism of pathogenic feline immunodeficiency viruses. J Virol. 1999;73:6852–6861. doi: 10.1128/jvi.73.8.6852-6861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egberink H, Borst M, Niphuis H, Balzarini J, Neu H, Schellekens H, De Clercq E, Horzinek M, Koolen M. Suppression of feline immunodeficiency virus infection in vivo by 9-(2-phosphonomethoxyethyl)adenine. Proc Natl Acad Sci USA. 1990;87:3087–3091. doi: 10.1073/pnas.87.8.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egberink HF, De Clercq E, van Vliet AL, Balzarini J, Bridger GJ, Henson G, Horzinek MC, Schols D. Bicyclams, selective antagonists of the human chemokine receptor CXCR4, potently inhibit feline immunodeficiency virus replication. J Virol. 1999;73:6346–6352. doi: 10.1128/jvi.73.8.6346-6352.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder JH, Phillips TR. Molecular properties of feline immunodeficiency virus (FIV) Infect Agents Dis. 1993;2:361–374. [PubMed] [Google Scholar]
- Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, Abramson I, Grant I, McCutchan JA. Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology. 2000;54:927–936. doi: 10.1212/wnl.54.4.927. [DOI] [PubMed] [Google Scholar]
- English R, Johnson C, Gebhard DH, Tompkins MB. In vivo lymphocyte tropism of feline immunodeficiency virus. J Virol. 1993;67:5175–5186. doi: 10.1128/jvi.67.9.5175-5186.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English RV, Nelson P, Johnson CM, Nasisse M, Tompkins WA, Tompkins MB. Development of clinical disease in cats experimentally infected with feline immunodeficiency virus. J Infect Dis. 1994;170:543–552. doi: 10.1093/infdis/170.3.543. [DOI] [PubMed] [Google Scholar]
- Epstein LG, Gelbard HA. HIV-1-induced neuronal injury in the developing brain. J Leukoc Biol. 1999;65:453–457. doi: 10.1002/jlb.65.4.453. [DOI] [PubMed] [Google Scholar]
- Falangola MF, Hanly A, Galvao-Castro B, Petito CK. HIV infection of human choroid plexus: a possible mechanism of viral entry into the CNS. J Neuropathol Exp Sci. 1995;54:497–503. doi: 10.1097/00005072-199507000-00003. [DOI] [PubMed] [Google Scholar]
- Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271:15303–15306. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L’Heureux D, Regulier EG, Richardson MW, Amini S, Morgello S, Khalili K, Rappaport J. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirology. 2001;7:528–541. doi: 10.1080/135502801753248114. [DOI] [PubMed] [Google Scholar]
- Garg H, Fuller FJ, Tompkins WA. Mechanism of feline immunodeficiency virus envelope glycoprotein-mediated fusion. Virology. 2004;321:274–286. doi: 10.1016/j.virol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Gavrilin MA, Mathes LE, Podell M. Methamphetamine enhances cell-associated feline immunodeficiency virus replication in astrocytes. J Neurovirology. 2002;8:240–249. doi: 10.1080/13550280290049660. [DOI] [PubMed] [Google Scholar]
- Gebhard DH, Dow JL, Childers TA, Alvelo JI, Tompkins MB, Tompkins WA. Progressive expansion of an L-selectin-negative CD8 cell with anti-feline immunodeficiency virus (FIV) suppressor function in the circulation of FIV-infected cats. J Infect Dis. 1999;180:1503–1513. doi: 10.1086/315089. [DOI] [PubMed] [Google Scholar]
- Gemignani A, Paudice P, Pittaluga A, Raiteri M. The HIV-1 coat protein gp120 and some of its fragments potently activate native cerebral NMDA receptors mediating neuropeptide release. Eur J Neurosci. 2000;12:2839–2846. doi: 10.1046/j.1460-9568.2000.00172.x. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Lipton SA, Epstein LG, Swindells S. The Neurology of AIDS. 2. Oxford University Press; 2005. [Google Scholar]
- Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, Schols D, De Clercq E, Kunstman K, Wolinsky SM, Gabuzda D. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J Virol. 2001;75:10073–10089. doi: 10.1128/JVI.75.21.10073-10089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DL, Yu N, Parsons KL, Billaud JN, Elder JH, Phillips TR. Neurotoxic effects of feline immunodeficiency virus, FIV-PPR. J Neurovirology. 1998;4:415–425. doi: 10.3109/13550289809114540. [DOI] [PubMed] [Google Scholar]
- Hartmann K, Donath A, Beer B, Egberink HF, Horzinek MC, Lutz H, Hoffmann-Fezer G, Thum I, Thefeld S. Use of two virustatica (AZT, PMEA) in the treatment of FIV and of FeLV seropositive cats with clinical symptoms. Vet Immunol Immunopathol. 1992;35:167–175. doi: 10.1016/0165-2427(92)90129-e. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem. 1999;73:1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Hayes KA, Wilkinson JG, Frick R, Francke S, Mathes LE. Early suppression of viremia by ZDV does not alter the spread of feline immunodeficiency virus infection in cats. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:114–122. [PubMed] [Google Scholar]
- Hegg CC, Thayer SA. Monocytic cells secrete factors that evoke excitatory synaptic activity in rat hippocampal cultures. Eur J Pharmacol. 1999;385:231–237. doi: 10.1016/s0014-2999(99)00712-8. [DOI] [PubMed] [Google Scholar]
- Hein A, Martin JP, Dorries R. In vitro activation of feline immunodeficiency virus in ramified microglial cells from asymptomatically infected cats. J Virol. 2001;75:8090–8095. doi: 10.1128/JVI.75.17.8090-8095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein A, Martin JP, Koehren F, Bingen A, Dorries R. In vivo infection of ramified microglia from adult cat central nervous system by feline immunodeficiency virus. Virology. 2000;268:420–429. doi: 10.1006/viro.1999.0152. [DOI] [PubMed] [Google Scholar]
- Hoffmann-Fezer G, Thum J, Ackley C, Herbold M, Mysliwietz J, Thefeld S, Hartmann K, Kraft W. Decline in CD4+ cell numbers in cats with naturally acquired feline immunodeficiency virus infection. J Virol. 1992;66:1484–1488. doi: 10.1128/jvi.66.3.1484-1488.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohdatsu T, Fujimori S, Maeki M, Suma N, Motokawa K, Okada S, Koyama H. Virus neutralizing antibody titer to feline immunodeficiency virus isolates of subtypes A, B and D in experimentally or naturally infected cats. J Vet Med Sci. 1997;59:377–381. doi: 10.1292/jvms.59.377. [DOI] [PubMed] [Google Scholar]
- Hohdatsu T, Miyagawa N, Ohkubo M, Kida K, Koyama H. Studies on feline CD8+ T cell non-cytolytic anti-feline immunodeficiency virus (FIV) activity. Arch Virol. 2000;145:2525–2538. doi: 10.1007/s007050070006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohdatsu T, Sasagawa T, Yamazaki A, Motokawa K, Kusuhara H, Kaneshima T, Koyama H. CD8+ T cells from feline immunodeficiency virus (FIV) infected cats suppress exogenous FIV replication of their peripheral blood mononuclear cells in vitro. Arch Virol. 2002;147:1517–1529. doi: 10.1007/s00705-002-0827-1. [DOI] [PubMed] [Google Scholar]
- Holden CP, Haughey NJ, Nath A, Geiger JD. Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage-operated Ca2+ channels in human immunodeficiency virus type 1 gp120-mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience. 1999;91:1369–1378. doi: 10.1016/s0306-4522(98)00714-3. [DOI] [PubMed] [Google Scholar]
- Hosie MJ, Broere N, Hesselgesser J, Turner JD, Hoxie JA, Neil JC, Willett BJ. Modulation of feline immunodeficiency virus infection by stromal cell-derived factor. J Virol. 1998;72:2097–2104. doi: 10.1128/jvi.72.3.2097-2104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Conlee D, Loop J, Champ D, Gill M, Chu HJ. Efficacy and safety of a feline immunodeficiency virus vaccine. Anim Health Res Rev. 2004;5:295–300. doi: 10.1079/ahr200487. [DOI] [PubMed] [Google Scholar]
- Hudson LC, Bragg DC, Tompkins MB, Meeker RB. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005;1058:148–160. doi: 10.1016/j.brainres.2005.07.071. [DOI] [PubMed] [Google Scholar]
- Hurtrel M, Ganiere J, Guelifi J, Chakrabarti L, Maire M, Gray F, Montagnier L, Hurtrel B. Comparison of early and late feline immunodeficiency virus encephalopathies. AIDS. 1992;6:399–406. doi: 10.1097/00002030-199204000-00007. [DOI] [PubMed] [Google Scholar]
- Jacobson S, Henricksen SJ, Prospero-Garcia O, Phillips TR, Elder JH, Young WG, Bloom FE, Fox HS. Cortical neuronal cytoskeletal changes associated with FIV infection. Journal Neurovirology. 1997;3:283–289. doi: 10.3109/13550289709029469. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Power C. Feline immunodeficiency virus xenoinfection: the role of chemokine receptors and envelope diversity. J Virol. 2002;76:3626–3636. doi: 10.1128/JVI.76.8.3626-3636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Power C. Regulation of neural cell survival by HIV-1 infection. Neurobiol Dis. 2006;21:1–17. doi: 10.1016/j.nbd.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Jordan HL, Howard JG, Bucci JG, Butterworth JL, English R, Kennedy-Stoskopf S, Tompkins MB, Tompkins WA. Horizontal transmission of feline immunodeficiency virus with semen from seropositive cats. J Reprod Immunol. 1998;41:341–357. doi: 10.1016/s0165-0378(98)00070-9. [DOI] [PubMed] [Google Scholar]
- Kanki PJ, McLane MF, King NW, Jr, Letvin NL, Hunt RD, Sehgal P, Daniel MD, Desrosiers RC, Essex M. Serologic identification and characterization of a macaque T-lymphotropic retrovirus closely related to HTLV-III. Science. 1985;228:1199–1201. doi: 10.1126/science.3873705. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Maeda K, Tohya Y, Furuya T, Miyazawa T, Horimoto T, Norimine J, Kai C, Mikami T. Replicative difference in early-passage feline brain cells among feline immunodeficiency virus isolates. Arch Virol. 1992;125:347–354. doi: 10.1007/BF01309653. [DOI] [PubMed] [Google Scholar]
- Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- Koirala TR, Nakagaki K, Ishida T, Nonaka S, Morikawa S, Tabira T. Decreased expression of MAP-2 and GAD in the brain of cats infected with feline immunodeficiency virus. Tohoku J Exp Med. 2001;195:141–151. doi: 10.1620/tjem.195.141. [DOI] [PubMed] [Google Scholar]
- Kolson DL. Neuropathogenesis of central nervous system HIV-1 infection. Clin Lab Med. 2002;22:703–717. doi: 10.1016/s0272-2712(02)00009-4. [DOI] [PubMed] [Google Scholar]
- Kovacs EM, Baxter GD, Robinson WF. Feline peripheral blood mononuclear cells express message for both CXC and CC type chemokine receptors. Arch Virol. 1999;144:273–285. doi: 10.1007/s007050050503. [DOI] [PubMed] [Google Scholar]
- Lane JH, Sasseville VG, Smith MO, Vogel P, Pauley DR, Heyes MP, Lackner AA. Neuroinvasion by simian immunodeficiency virus coincides with increased numbers of perivascular macrophages/microglia and intrathecal immune activation. J Neurovirology. 1996;2:423–432. doi: 10.3109/13550289609146909. [DOI] [PubMed] [Google Scholar]
- Lerner DL, Elder JH. Expanded host cell tropism and cytopathic properties of feline immunodeficiency virus strain PPR subsequent to passage through interleukin-2-independent T cells. J Virol. 2000;74:1854–1863. doi: 10.1128/jvi.74.4.1854-1863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JA. Pathogenesis of human immunodeficiency virus infection. Microbiol Rev. 1993;57:183–289. doi: 10.1128/mr.57.1.183-289.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton S. Models of neuronal injury in AIDS: another role for the NMDA receptor? TINS. 1992;15:75–80. doi: 10.1016/0166-2236(92)90013-x. [DOI] [PubMed] [Google Scholar]
- Lipton S, Sucher N, Kaiser P, Dreyer E. Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron. 1991;7:111–118. doi: 10.1016/0896-6273(91)90079-f. [DOI] [PubMed] [Google Scholar]
- Liu P, Hudson LC, Tompkins MB, Vahlenkamp TW, Colby B, Rundle C, Meeker RB. Cerebrospinal fluid is an efficient route for establishing brain infection with feline immunodeficiency virus and transferring infectious virus to the periphery. J Neurovirology. 2006a;12:294–306. doi: 10.1080/13550280600889567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Hudson LC, Tompkins MB, Vahlenkamp TW, Meeker RB. Compartmentalization and evolution of feline immunodeficiency virus between the central nervous system and periphery following intracerebroventricular or systemic inoculation. J Neurovirology. 2006b;12:307–321. doi: 10.1080/13550280600889575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone WJ, Moore M, Innes D, Bell JE, Simmonds P. Frequent infection of peripheral blood CD8-positive T-lymphocytes with HIV-1. Edinburgh Heterosexual Transmission Study Group. Lancet. 1996;348:649–654. doi: 10.1016/s0140-6736(96)02091-0. [DOI] [PubMed] [Google Scholar]
- Lo TK, Fallert CJ, Piser TM, Thayer SA. HIV-1 envelope protein evokes intracellular calcium oscillations in rat hippocampal neurons. Brain Res. 1992;594:189–196. doi: 10.1016/0006-8993(92)91125-x. [DOI] [PubMed] [Google Scholar]
- Lombardi S, Massi C, Tozzini F, Zaccaro L, Bazzichi A, Bandecchi P, La Rosa C, Bendinelli M, Garzelli C. Epitope mapping of the V3 domain of feline immunodeficiency virus envelope glycoprotein by monoclonal antibodies. J Gen Virol. 1995;76 (Pt 8):1893–1899. doi: 10.1099/0022-1317-76-8-1893. [DOI] [PubMed] [Google Scholar]
- Macchi S, Maggi F, Di Iorio C, Poli A, Bendinelli M, Pistello M. Detection of feline immunodeficiency proviral sequences in lymphoid tissues and the central nervous system by in situ gene amplification. J Virol Methods. 1998;73:109–119. doi: 10.1016/s0166-0934(98)00053-6. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- Meeker R, English R, Tompkins M. Enhanced excitotoxicity in primary feline neural cultures exposed to feline immunodeficiency virus (FIV) J Neuro-AIDS. 1996;1:1–27. doi: 10.1300/j128v01n03_01. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Azuma Y, Bragg DC, English RV, Tompkins M. Microglial proliferation in cortical neural cultures exposed to feline immunodeficiency virus. J Neuroimmunol. 1999a;101:15–26. doi: 10.1016/s0165-5728(99)00126-5. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Boles JC, Robertson KR, Hall CD. Cerebrospinal fluid from human immunodeficiency virus-infected individuals facilitates neurotoxicity by suppressing intracellular calcium recovery. J Neurovirology. 2005;11:144–156. doi: 10.1080/13550280590922757. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Robertson K, Barry T, Hall C. Neurotoxicity of CSF from HIV-infected humans. J Neurovirology. 1999b;5:507–518. doi: 10.3109/13550289909045380. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Thiede BA, Hall C, English R, Tompkins M. Cortical cell loss in asymptomatic cats experimentally infected with feline immunodeficiency virus. AIDS Res Hum Retrovir. 1997;13:1131–1140. doi: 10.1089/aid.1997.13.1131. [DOI] [PubMed] [Google Scholar]
- Mitchell TW, Buckmaster PS, Hoover EA, Whalen LR, Dudek FE. Neuron loss and axon reorganization in the dentate gyrus of cats infected with the feline immunodeficiency virus. J Comp Neurol. 1999;411:563–577. [PubMed] [Google Scholar]
- Nagra RM, Masliah E, Wiley CA. Synaptic and dendritic pathology in murine retroviral encephalitis. Exp Neurol. 1993;124:283–288. doi: 10.1006/exnr.1993.1198. [DOI] [PubMed] [Google Scholar]
- Nakagaki K, Nakagaki K, Takahashi K, Schols D, De Clercq E, Tabira T. CXCR4 is the primary receptor for feline immunodeficiency virus in astrocytes. J Neurovirology. 2001;7:487–492. doi: 10.1080/135502801753170354. [DOI] [PubMed] [Google Scholar]
- Neuenburg JK, Brodt HR, Herndier BG, Bickel M, Bacchetti P, Price RW, Grant RM, Schlote W. HIV-related neuropathology, 1985 to 1999: rising prevalence of HIV encephalopathy in the era of highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2002;31:171–177. doi: 10.1097/00126334-200210010-00007. [DOI] [PubMed] [Google Scholar]
- Novotney C, English R, Housman J, Davidson M, Nasisse M, Jeng CR, Davis W, Tompkins M. Lymphocyte population changes in cats naturally infected with feline immunodeficiency virus. AIDS. 1990;4:1213–1218. doi: 10.1097/00002030-199012000-00005. [DOI] [PubMed] [Google Scholar]
- O’Neil LL, Burkhard MJ, Hoover EA. Frequent perinatal transmission of feline immunodeficiency virus by chronically infected cats. J Virol. 1996;70:2894–2901. doi: 10.1128/jvi.70.5.2894-2901.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obert LA, Hoover EA. Early pathogenesis of transmucosal feline immunodeficiency virus infection. J Virol. 2002;76:6311–6322. doi: 10.1128/JVI.76.12.6311-6322.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted R, Hirsch V, Purcell R, Johnson P. Nucleotide sequence analysis of feline immunodeficiency virus:genome organization and relationship to other lentiviruses. Proc Natl Acad Sci. 1989;86:8088–8092. doi: 10.1073/pnas.86.20.8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne R, Rigby M, Siebelink K, Neil JC, Jarrett O. Virus neutralization reveals antigenic variation among feline immunodeficiency virus isolates. J Gen Virol. 1994;75 (Pt 12):3641–3645. doi: 10.1099/0022-1317-75-12-3641. [DOI] [PubMed] [Google Scholar]
- Pedersen NC, Ho EW, Brown ML, Yamamoto JK. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science. 1987;235:790–793. doi: 10.1126/science.3643650. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Gelbard HA. Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox Res. 2005;8:161–166. doi: 10.1007/BF03033827. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M. A model for monocyte migration through the blood–brain barrier during HIV-1 encephalitis. J Immunol. 1997;158:3499–3510. [PubMed] [Google Scholar]
- Petito CK. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J Neurovirology. 2004;10(Suppl 1):21–24. doi: 10.1080/753312748. [DOI] [PubMed] [Google Scholar]
- Petito CK, Chen H, Mastri AR, Torres-Munoz J, Roberts B, Wood C. HIV infection of choroid plexus in AIDS and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection. J Neurovirology. 1999;5:670–677. doi: 10.3109/13550289909021295. [DOI] [PubMed] [Google Scholar]
- Phillips T, Prospero-Garcia O, Puaoi D, Lerner D, Fox H, Olmsted R, Bloom F, Heriksen S, Elder J. Neurological abnormalities associated with feline immunodeficiency virus infection. J Gen Virol. 1994;75:979–987. doi: 10.1099/0022-1317-75-5-979. [DOI] [PubMed] [Google Scholar]
- Phillips TR, Prospero-Garcia O, Wheeler DW, Wagaman P, Lerner DL, Fox HS, Whalen LR, Bloom FE, Elder JH, Henricksen SJ. Neurologic dysfunctions caused by a molecular clone of feline immunodeficiency virus, FIV-PPR. J Neurovirology. 1996;2:388–396. doi: 10.3109/13550289609146904. [DOI] [PubMed] [Google Scholar]
- Phipps AJ, Hayes KA, Buck WR, Podell M, Mathes LE. Neurophysiologic and immunologic abnormalities associated with feline immunodeficiency virus molecular clone FIV-PPR DNA inoculation. J Acquir Immune Defic Syndr. 2000;23:8–16. doi: 10.1097/00126334-200001010-00002. [DOI] [PubMed] [Google Scholar]
- Pistello M, Menzo S, Giorgi M, Da Prato L, Cammarota G, Clementi M, Bendinelli M. Competitive polymerase chain reaction for quantitating feline immunodeficiency virus load in infected cat tissues. Mol Cell Probes. 1994;8:229–234. doi: 10.1006/mcpr.1994.1032. [DOI] [PubMed] [Google Scholar]
- Podell M, Hayes K, Oglesbee M, Mathes L. Progressive encephalopathy associated with CD4/CD8 inversion in adult FIV-infected cats. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15:332–340. doi: 10.1097/00042560-199708150-00002. [DOI] [PubMed] [Google Scholar]
- Podell M, Maruyama K, Smith M, Hayes KA, Buck WR, Ruehlmann DS, Mathes LE. Frontal lobe neuronal injury correlates to altered function in FIV-infected cats. J Acquir Immune Defic Syndr. 1999;22:10–18. doi: 10.1097/00042560-199909010-00002. [DOI] [PubMed] [Google Scholar]
- Podell M, Oglesbee M, Mathes L, Krakowka S, Olmstead R, Lafrado L. AIDS-associated encephalopathy with experimental feline immunodeficiency virus Infection. J AIDS. 1993;6:758–771. [PubMed] [Google Scholar]
- Poli A, Abramo F, Di Iorio C, Cantile C, Carli MA, Pollera C, Vago L, Tosoni A, Costanzi G. Neuropathology in cats experimentally infected with feline immunodeficiency virus: a morphological, immunocytochemical and morphometric study. J Neurovirology. 1997;3:361–368. doi: 10.3109/13550289709030750. [DOI] [PubMed] [Google Scholar]
- Poli A, Pistello M, Carli MA, Abramo F, Mancuso G, Nicoletti E, Bendinelli M. Tumor necrosis factor-alpha and virus expression in the central nervous system of cats infected with feline immunodeficiency virus. J Neurovirology. 1999;5:465–473. doi: 10.3109/13550289909045375. [DOI] [PubMed] [Google Scholar]
- Potter SJ, Dwyer DE, Saksena NK. Differential cellular distribution of HIV-1 drug resistance in vivo: evidence for infection of CD8+ T cells during HAART. Virology. 2003;305:339–352. doi: 10.1006/viro.2002.1703. [DOI] [PubMed] [Google Scholar]
- Power C, Buist R, Johnston JB, Del Bigio MR, Ni W, Dawood MR, Peeling J. Neurovirulence in feline immunodeficiency virus-infected neonatal cats is viral strain specific and dependent on systemic immune suppression. J Virol. 1998;72:9109–9115. doi: 10.1128/jvi.72.11.9109-9115.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power C, Moench T, Peeling J, Kong PA, Langelier T. Feline immunodeficiency virus causes increased glutamate levels and neuronal loss in brain. Neuroscience. 1997;77:1175–1185. doi: 10.1016/s0306-4522(96)00531-3. [DOI] [PubMed] [Google Scholar]
- Prospero-Garcia O, Herold N, Phillips T, Elder J, Bloom F, Henriksen S. Sleep patterns are disturbed in cats infected with feline immunodeficiency virus. Proc Natl Acad Sci. 1994;91:12947–12951. doi: 10.1073/pnas.91.26.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu R, Coleman J, Omori M, Arai M, Hohdatsu T, Huang C, Tanabe T, Yamamoto JK. Dual-subtype FIV vaccine protects cats against in vivo swarms of both homologous and heterologous subtype FIV isolates. AIDS. 2001;15:1225–1237. doi: 10.1097/00002030-200107060-00004. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Herndier BG, Tang NM, McGrath MS. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest. 1991;87:503–512. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J, Fossati I, Moraillon A, Castelot S, Sonigo P, Pancino G. Neutralization sensitivity and accessibility of continuous B cell epitopes of the feline immunodeficiency virus envelope. J Gen Virol. 1996;77 (Pt 4):759–771. doi: 10.1099/0022-1317-77-4-759. [DOI] [PubMed] [Google Scholar]
- Richardson J, Pancino G, Merat R, Leste-Lasserre T, Moraillon A, Schneider-Mergener J, Alizon M, Sonigo P, Heveker N. Shared usage of the chemokine receptor CXCR4 by primary and laboratory-adapted strains of feline immunodeficiency virus. J Virol. 1999;73:3661–3671. doi: 10.1128/jvi.73.5.3661-3671.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan G, Grimes T, Brankin B, Mabruk MJ, Hosie MJ, Jarrett O, Callanan JJ. Neuropathology associated with feline immunodeficiency virus infection highlights prominent lymphocyte trafficking through both the blood–brain and blood–choroid plexus barriers. J Neurovirology. 2005;11:337–345. doi: 10.1080/13550280500186445. [DOI] [PubMed] [Google Scholar]
- Ryan G, Klein D, Knapp E, Hosie MJ, Grimes T, Mabruk MJ, Jarrett O, Callanan JJ. Dynamics of viral and proviral loads of feline immunodeficiency virus within the feline central nervous system during the acute phase following intravenous infection. J Virol. 2003;77:7477–7485. doi: 10.1128/JVI.77.13.7477-7485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirology. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- Self RL, Mulholland PJ, Nath A, Harris BR, Prendergast MA. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular Ca2+ that require function of an N-methyl-D-aspartate receptor polyamine-sensitive site. Brain Res. 2004;995:39–45. doi: 10.1016/j.brainres.2003.09.052. [DOI] [PubMed] [Google Scholar]
- Shimojima M, Miyazawa T, Ikeda Y, McMonagle EL, Haining H, Akashi H, Takeuchi Y, Hosie MJ, Willett BJ. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science. 2004;303:1192–1195. doi: 10.1126/science.1092124. [DOI] [PubMed] [Google Scholar]
- Siebelink KH, Bosch ML, Rimmelzwaan GF, Meloen RH, Osterhaus AD. Two different mutations in the envelope protein of feline immunodeficiency virus allow the virus to escape from neutralization by feline serum antibodies. Vet Immunol Immunopathol. 1995;46:51–59. doi: 10.1016/0165-2427(94)07005-r. [DOI] [PubMed] [Google Scholar]
- Silvotti L, Corradi A, Brandi G, Cabassi A, Bendinelli M, Magnan M, Piedimonte G. FIV induced encephalopathy: early brain lesions in the absence of viral replication in monocyte/macrophages. A pathogenetic model. Vet Immunol Immunopathol. 1997;55:263–271. doi: 10.1016/s0165-2427(96)05617-6. [DOI] [PubMed] [Google Scholar]
- Steffan AM, Lafon ME, Gendrault JL, Koehren F, De Monte M, Royer C, Kirn A, Gut JP. Feline immunodeficiency virus can productively infect cultured endothelial cells from cat brain microvessels. J Gen Virol. 1994;75:3647–3653. doi: 10.1099/0022-1317-75-12-3647. [DOI] [PubMed] [Google Scholar]
- Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am J Pathol. 1996;148:1819–1838. [PMC free article] [PubMed] [Google Scholar]
- Steigerwald ES, Sarter M, March P, Podell M. Effects of feline immunodeficiency virus on cognition and behavioral function in cats. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:411–419. doi: 10.1097/00042560-199904150-00001. [DOI] [PubMed] [Google Scholar]
- Strain MC, Letendre S, Pillai SK, Russell T, Ignacio CC, Gunthard HF, Good B, Smith DM, Wolinksy SM, Furtado M, Marquie-Beck J, Durelle J, Grant I, Richman DD, Marcotte T, McCutchan JA, Ellis RJ, Wong JK. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J Virol. 2005;79:1772–1788. doi: 10.1128/JVI.79.3.1772-1788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott RL, Sparger EE, Lovelace KM, Fitch WM, Pedersen NC, Luciw PA, Elder JH. Nucleotide sequence and genomic organization of feline immunodeficiency virus. Proc Natl Acad Sci USA. 1989;86:5743–5747. doi: 10.1073/pnas.86.15.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Mucke L. Prevention of HIV-1 gp120-induced neuronal damage in the central nervous system of transgenic mice by the NMDA receptor antagonist memantine. Brain Res. 1996;706:303–307. doi: 10.1016/0006-8993(95)01197-8. [DOI] [PubMed] [Google Scholar]
- Tompkins MB, Nelson PD, English RV, Novotney C. Early events in the immunopathogenesis of feline retrovirus infections. JAVMA. 1991;199:1311–1315. [PubMed] [Google Scholar]
- Torten M, Franchini M, Barlough JE, George JW, Mozes E, Lutz H, Pedersen NC. Progressive immune dysfunction in cats experimentally infected with feline immunodeficiency virus. J Virol. 1991;65:2225–2230. doi: 10.1128/jvi.65.5.2225-2230.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan J, Sacktor N, Wojna V, Conant K, Nath A. Neuroprotective therapy for HIV dementia. Curr HIV Res. 2003;1:373–383. doi: 10.2174/1570162033485113. [DOI] [PubMed] [Google Scholar]
- Tymianski M. Cytosolic calcium concentrations and cell death in vitro. Adv Neurol. 1996;71:85–105. [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl EW, Heaton-Jones TG, Pu R, Yamamoto JK. FIV vaccine development and its importance to veterinary and human medicine: a review FIV vaccine 2002 update and review. Vet Immunol Immunopathol. 2002;90:113–132. doi: 10.1016/S0165-2427(02)00227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahlenkamp TW, Tompkins MB, Tompkins WA. The role of CD4+CD25+ regulatory T cells in viral infections. Vet Immunol Immunopathol. 2005;108:219–225. doi: 10.1016/j.vetimm.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Wasmoen T, Armiger-Luhman S, Egan C, Hall V, Chu HJ, Chavez L, Acree W. Transmission of feline immunodeficiency virus from infected queens to kittens. Vet Immunol Immunopathol. 1992;35:83–93. doi: 10.1016/0165-2427(92)90123-8. [DOI] [PubMed] [Google Scholar]
- Wiley C, Masliah E, Morey M, Lemere C, DeTeresa R, Grafe M, Hansen L, Terry R. Neocortical damage during HIV infection. Ann Neurol. 1991;29:651–657. doi: 10.1002/ana.410290613. [DOI] [PubMed] [Google Scholar]
- Willett BJ, Cannon CA, Hosie MJ. Upregulation of surface feline CXCR4 expression following ectopic expression of CCR5: implications for studies of the cell tropism of feline immunodeficiency virus. J Virol. 2002;76:9242–9252. doi: 10.1128/JVI.76.18.9242-9252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett BJ, Hosie MJ, Callanan JJ, Neil JC, Jarrett O. Infection with feline immunodeficiency virus is followed by the rapid expansion of a CD8+ lymphocyte subset. Immunology. 1993;78:1–6. [PMC free article] [PubMed] [Google Scholar]
- Willett BJ, Picard L, Hosie MJ, Turner JD, Adema K, Clapham PR. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J Virol. 1997;71:6407–6415. doi: 10.1128/jvi.71.9.6407-6415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–915. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Zheng J, Thylin M, Gendelman HE. Unraveling the mechanisms of neurotoxicity in HIV type 1-associated dementia: inhibition of neuronal synaptic transmission by macrophage secretory products. AIDS Res Hum Retrovir. 1999;15:57–63. doi: 10.1089/088922299311718. [DOI] [PubMed] [Google Scholar]
- Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of L-cysteine. J Immunol. 2000;164:4265–4270. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]
- Yu N, Billaud JN, Phillips TR. Effects of feline immunodeficiency virus on astrocyte glutamate uptake: implications for lentivirus-induced central nervous system diseases. Proc Natl Acad Sci USA. 1998;95:2624–2629. doi: 10.1073/pnas.95.5.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenger E, Collisson EW, Barhoumi R, Burghardt RC, Danave IR, Tiffany-Castiglioni E. Laser cytometric analysis of FIV-induced injury in astroglia. Glia. 1995;13:92–100. doi: 10.1002/glia.440130203. [DOI] [PubMed] [Google Scholar]
- Zenger E, Tiffany-Castiglioni E, Collisson EW. Cellular mechanisms of feline immunodeficiency virus (FIV)-induced neuropathogenesis. Front Biosci. 1997;2:d527–d537. doi: 10.2741/a210. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ghorpade A, Niemann D, Cotter RL, Thylin MR, Epstein L, Swartz JM, Shepard RB, Liu X, Nukuna A, Gendelman HE. Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: implications for human immunodeficiency virus type 1-associated dementia. J Virol. 1999a;73:8256–8267. doi: 10.1128/jvi.73.10.8256-8267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Cotter RL, Lopez AL, Ghorpade A, Persidsky Y, Xiong H, Leisman GB, Che MH, Gendelman HE. HIV-1 infected and immune competent mononuclear phagocytes induce quantitative alterations in neuronal dendritic arbor: relevance for HIV-1-associated dementia. Neurotox Res. 2001;3:443–459. doi: 10.1007/BF03033203. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 1999b;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]