

Abstract
Thiol alkylation is a powerful technique for the labeling of proteins. We report a new class of highly reactive, selective, and fluorogenic probes for thiols in aqueous solution at neutral pH, based on the 7-oxanorbornadiene (OND) framework. The maleate moiety in 7-oxabicyclo[2.2.1]hept-2,5-diene-2,3-dicarboxylic acid esters serves as both a tunable electrophile and an intramolecular quencher of an attached dansyl fluorophore. Thiols have been found to add with high rates (second-order rate constants of 40–200 M−1s−1) to give adducts that exhibit enhancements of fluorescence intensity up to 180-fold. The resulting adducts are also versatile with respect to cleavage (release) reactions by two different mechanisms. First, retro-Diels–Alder fragmentation occurs with half lives from days to weeks at room temperature, and an epoxide derivative is also reported that is incapable of cycloreversion cleavage. Second, monoamide OND derivatives undergo rapid closure to succinimide derivatives upon thiol addition, providing a thiol-triggered mechanism for immediate alcohol release. Peptides and proteins containing free thiol groups were labeled with OND electrophiles with high chemoselectivity. Since the system is so easily assembled from readily accessible modules, various functional groups can be added to OND linkers to allow the attachment of other molecules of interest.
Keywords: fluorogenic, Michael addition, oxanorbornadiene, thiol, peptide labeling, protein labeling, retro-Diels-Alder, cleavable linker
Introduction
The unique nucleophilicity of the thiol group makes cysteine side chains a popular site for selective bond formation to proteins. The alkylating agents normally used for such reactions are haloacetamides, maleimides, benzylic halides, β-chloroacrylamides, bromomethylketones, and epoxides, with the first two being the most popular.1 However, these agents are often not optimal in terms of selectivity, stability, or synthetic accessibility. Haloacetamides can react with other nucleophilic residues of proteins, such as N-terminal amine, lysine, methionine, histidine, and tyrosine, in addition to cysteine thiol.2 Maleimides are preferred in this regard, since they are more reactive with thiols3,4 and more selective with respect to other nucleophiles.4 It is commonly assumed that the relative reactivity of maleimides for thiol over amine is on the order of 1000:1 at pH 7, although we have observed less selective examples,5 and crosslinking with amines can occur.6 Maleimide synthesis can be challenging in some cases,7–10 and the deactivation of haloacetamides and maleimides by water (giving α-hydroxyamides and ring-opened amic acids, respectively) can compete significantly with thiol modification, particularly above pH 8.11–13
For these reasons, the development of selective thiol-reactive linkers is a subject of much current interest, especially systems that provide an optical signal upon conjugation. The fluorogenic bromobimanes14–17 are popular as analytical reagents for identifying cellular thiols and metabolites. Chromogenic vinylic sulfones,18 dye-maleimides,19 and cyanine-type linker electrophiles20 have been recently described, all of them highly reactive and selective. Michael acceptors generated in situ via Knoevenagel condensation have also been elegantly used to modify thiols selectively, serving as convenient photocleavable linkages or protecting groups.21 We describe here a system that is significantly easier to make than maleimides, more stable in aqueous solution, highly reactive with thiols, and which undergoes controllable fragmentation after thiol addition.
Results and Discussion
Synthesis and properties of thiol-reactive reagents
It has been previously reported that the attachment of a maleimide group to dye fragments such as dansylamide (5-dimethylamino-1-naphthalenesulfonamide) results in quenching of the chromophore, which is relieved when the π-system of the maleimide is disrupted.8,19,22 We applied the same strategy to electron-deficient oxanorbornadienes, which are readily obtained by [4+2] cycloaddition of electron-deficient alkynes with furans, and are known to react with thiolates23,24 and organic azides,25 much like the alkynes from which they are derived.
Reaction of furfurylamines 1 and 2 with dansyl chloride gave sulfonamides 3 and 4, which underwent smooth Diels–Alder cycloaddition with acetylenedicarboxylates to give adducts 5 and 6 (Figure 1). All of the components are inexpensive and the reactions can be performed on gram scale without difficulty. The compounds were characterized by standard methods, and the structure of 5c was confirmed by X-ray crystallographic analysis. The ester group distal to the sulfonamide was selectively hydrolyzed to monoacids 7,26 which proved resistant to coupling with several amines and alcohols under the influence of carbodiimide/base reagents. However, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinum chloride (DMT-MM),27 previously employed in a similar setting,28 provided acceptable yields of propargylamides 8a and 8b. Oxanorbornadienes 6 and 925 were synthesized in a similar manner. The propargyl substituents of 5c, 6, 8a, and 8b were chosen to facilitate further functionalization by azide-alkyne “click” cycloaddition,29–32 which is compatible with the electrophilic nature of the OND core.
Figure 1.

Preparation and reactions of electrophiles 5–9.
Reagents and conditions: (a) Et3N, CH2Cl2, 3h, room temp., 96–97%; (b) R2O2CC≡CCO2R2, neat, 60–80 °C, 2–4 h; (c) NaOH, THF/H2O, 0 °C, 1h, 80–85%; (d) DMT-MM, THF, room temp., 1h, 37–43%; (e) MeI, Cs2CO3, DMF, 20 min, 84%.
As expected, furans 3 and 4 were highly fluorescent and compounds 5, 6 and 9 were significantly quenched, presumably due to photoinduced electron transfer from the fluorophore excited state to the LUMO of the maleate moiety of the Diels–Alder adducts, analogous to the quenching mechanism of maleimide-dye pairs.22,33 Dansyl fluorescence was restored upon conjugate addition of thiol, resulting in high signal-to-noise ratios for the fluorogenic sensing of thiol nucleophiles (Figure 2 and Table 1).
Figure 2.

Excitation and emission spectra of 5a (100 µM) treated with glutathione at the indicated concentrations (0.1 M phosphate buffer, pH 7, containing 1% DMSO). Intensity (y-axis) is plotted in arbitrary units.
Table 1.
Reactivity, fluorogenicity, aqueous stability, and adduct stability of OND electrophiles.
| Cpd. | Structurea |
kobs (M−1s−1)b |
fluor. increase c |
OND half life (days)d |
Adduct half life (days)e |
|---|---|---|---|---|---|
| 5a | 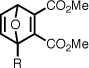 |
104 ± 3.2 | 178 | 9.3 ± 0.3 | 26.2 ± 0.5 |
| Me-5af | 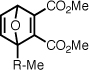 |
109 ± 6.1 | 36 | 5.6 ± 0.2 | 4.4 ± 0.2 |
| epoxy-5a | 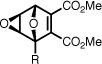 |
99.6 ± 2.9 | 100 | 3.2 ± 0.2 | no cleavage g |
| 5b | 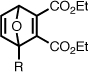 |
63.4 ± 2.4 | 108 | 20.6 ± 0.5 | 4.6 ± 0.1 |
| 5c | 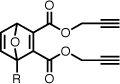 |
197 ± 11.3 | 30 | 1.8 ± 0.1 | 1.4 ± 0.1 |
| 6 | 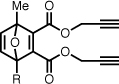 |
39.9 ± 1.9 | 12.4 | 3.2 ± 0.1 | 20.6 ± 0.1 |
| 7ch | 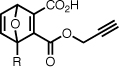 |
0.33 ± 0.01 | ca. 1 | 15.2 ± 0.3 | not determined |
| 8a | 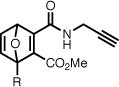 |
102 ± 3.8 | 60 | 9.0 ± 0.2 | 0.46 ± 0.02 i |
| 8b | 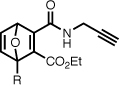 |
69.2 ± 3.3 | 60 | 17.1 ± 0.3 | 0.45 ± 0.01 i |
| 9 | 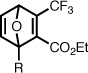 |
77.7 ± 5.6 | 25 | 1.1 ± 0.1 | 24.1 ± 0.4 |
R = CH2NHDansyl.
Second order rate constant for addition of compound to glutathione.
Ratio of emission intensity (550 nm, excitation 332 nm) observed 5 min after addition of 1 mM glutathione to 0.1 mM compound, vs. the emission intensity before addition, at room temperature.
Calculated from the first-order rate constant for the inactivation of the indicated electrophile by incubation of a 0.1 mM solution at room temperature.
Calculated from first-order rate constants for the retro Diels–Alder reaction of the β-mercaptoethanol adduct of the indicated compound,
R-Me = CH2NMeDansyl.
No cleavage is observed: the adduct maintains a covalent connection between thiol and electrophile fragments while decomposing by route(s) other than retro-Diels-Alder fragmentation, with a half life of 16.9 ± 0.5 days. The details are being explored and will be described elsewhere.
Rates of reaction determined by HPLC of aliquots; see text.
8a and 8b give the same imide thiol adduct, and so show identical rates of retro-Diels–Alder cleavage. All addition and decomposition measurements were performed in 0.1 M potassium phosphate buffer, pH 7, containing 10% DMSO.
OND reagents were found to be highly selective for thiols. Compound 5a was used as a representative electrophile (0.1 mM) in reactions with 1 mM of a selection of potentially nucleophilic amino acids at pH 7 (10% DMSO in 0.1M phosphate buffer). Lysine and histidine gave a very small (< 2%) increase in fluorescence after 60 minutes relative to the signal generated by thiol within seconds after mixing. The other amino acids, as well as the disulfide cystine, induced even less (Figure 3). The subsequent addition of excess thiol to these samples gave rise to an immediate and strong increase in fluorescence, verifying that no reaction had occurred in the presence of non-thiol nucleophiles.
Figure 3.

Fluorogenic activity of 5a with various amino acids. Relative fluorescence intensity of 0.1 mM 5a (1% DMSO in 0.1 M phosphate buffer, pH 7) at 550 nm (λex = 332 nm) after incubation with 1 mM analyte (1 min for cysteine and glutathione; 60 minutes for all others).
The rates of glutathione (0.11 mM) conjugate addition to OND probes (0.1 mM) were conveniently followed by the increase in fluorescence at 550 nm (Figure 2). Second-order kinetic behavior was observed with rate constants in the range of 40–200 M−1s−1 at pH 7 (Table 1), comparable to the reported values for the most reactive maleimides.12 (In a competition for limiting glutathione, 5a and N-ethylmaleimide were found to be equally reactive; see Supporting Information.) Methyl, ethyl, and propargyl ester substituents, having similar steric and electronic properties, did not change thiol reactivities very much (propargyl 5c > methyl 5a > ethyl 5b, but within a factor of 3 for the series), and the installation of a bridgehead methyl group was inhibitory (6 vs. 5c). A single amide group had little influence, with compounds 8a and 8b being as reactive toward thiol as their ester analogues 5a and 5b. Thus, additional functional groups such as alkyne can be easily introduced via an amide linkage without sacrificing the desired electrophilic reactivity of the OND core.
OND adducts may lose activity by conjugate addition of water or by hydrolysis of a conjugated ester group to the less electron-deficient carboxylate anion. (Carboxylic acid derivative 7c was by far the most sluggish electrophile toward glutathione in the series, being nearly 600 times less reactive with glutathione than its propargyl ester precursor 5c.) To gauge OND stability toward such deactivation, each compound was incubated in aqueous buffer and the amount remaining in solution was determined by HPLC as a function of time (Table 1). The observed trends were also reproduced by a different method, in which aliquots were removed and fluorogenic signals measured after addition of excess glutathione, compared to the same treatment of freshly-prepared solutions (data not shown). Methyl and ethyl diesters 5a and 5b were found to be significantly more stable (half lives in excess of 9 days at room temperature and pH 7) than propargyl esters 5c and 6 (half lives of 2–3 days), which are somewhat more electron deficient. Both methylation of the sulfonamide nitrogen (Me-5a) and epoxidation of the electron-rich double bond (epoxy-5a) made the electrophile moderately more sensitive to decomposition (half lives of 5.6 and 3.2 days vs. 9.3 days for 5a), without appreciably changing reactivity with thiol. As would be expected from the diminished electron-withdrawing power of carboxylate anions and amides relative to esters, 7c, 8a, and 8b were also very stable toward aqueous deactivation at pH 7. The stable members of the OND family are therefore longer lived in water than most maleimides;11,12 an example (5a vs. N-ethylmaleimide) is shown in Supporting Information. Trifluoromethyl derivative 9, previously used for reactions with organic azides,25,34 was the least stable electrophile tested.35
The rates of thiol addition to the OND electrophiles and their aqueous decomposition were therefore not directly proportional to each other (Table 1). With the exception of 7c, the OND compounds all reacted with glutathione at similar rates, but differed widely in their aqueous phase stabilities. Those reagents that are highly reactive toward thiols while being most stable toward decomposition in aqueous buffers are the most useful; 5b/8b and 5a/8a fit these criteria best. The difference in reactivity between 5c and 6 is also instructive. The introduction of a methyl group at the furan bridgehead (adjacent to the site of thiol attack, see Figure 4) strongly inhibited reaction with glutathione but did not change the stability of the electrophile very much. This suggests that decomposition may not occur by a similar conjugate addition of water; the mechanism of this process is under investigation. For practical purposes, all of the OND electrophiles may be used in water or buffer if the aqueous solutions are freshly prepared, and some of these solutions are stable for many days or weeks (Table 1).
Figure 4.

Structure and retro-Diels–Alder decomposition of thiol adducts.
(a) 200 mM 5a, 180 mM nucleophile in CH3CN, iPr2NEt (cat.), 5 min, RT, >80% isolated yield; (b) 10% DMSO in aqueous buffer, 20 °C
Linear pH-log(rate) profiles in aqueous buffers and a strong correlation of rate with solvent polarity in nonaqueous solvents (Supporting Information, Figures S4 and S5) support the assumption that thiolate anions are far more reactive with OND electrophiles than neutral thiols.36,37 For this reason, reactions in anhydrous acetonitrile were sluggish even at high millimolar concentrations, but the addition of trace amounts of triethylamine or diisopropylethylamine resulted in very fast reactions signaled by the immediate appearance of strong yellow-green fluorescence. Compound 7c, bearing a deprotonated carboxylate at pH 7, presented a special case. Its relatively electron-rich core does not engage in photoinduced electron transfer quenching of the fluorophore, so the molecule was highly fluorescent throughout, requiring HPLC of aliquots to determine rate constants for both thiol addition and aqueous decomposition. Its pH-reactivity profile was distinguished by compensating trade-offs between accelerating increases in thiolate concentration at higher pH and deactivating increases in the percentage of OND electrophile in the anionic carboxylate form (see Supporting Information).
Adducts 10a and 10b, resulting from syn-addition of thiol to the less-hindered site of the electrophile,23,24 were isolated as single diastereomers in high yield (Figure 4). Under the same conditions, monoamide derivative 8a underwent additional ring closure upon thiol addition to give succinimide 13, without observation of adduct 12. This process is likely triggered by the conversion of the electrophilic sp2 centers to sp3, making ring closure less strained. It has the additional and very useful feature of inducing the release of an alcohol fragment in response to thiol attack (see below). The identity of 13 was confirmed by standard mass and NMR spectra as well as the presence of cross-peaks between propargyl protons and both carbonyl groups in a 1H-13C HMBC spectrum. Each of these adducts underwent retro-Diels–Alder fragmentation to furan 3 and either thiomaleate 11 or thiomaleimide 14 (Figure 4).24,38
Thiol addition and retro-Diels–Alder fragmentation of electron-deficient oxanorbornadiene adducts has been used as a cleavable linkage strategy for solid-phase synthesis and polymer derivatization.23,39–42 We measured the cycloreversion rates of the β-mercaptoethanol addition products of most of the OND electrophiles described here (Table 1). Four categories of stability were identified: (a) cleavable but highly stable (thiol adducts of 5a, 6, and 9, half lives greater than 20 days at room temperature), (b) intermediate in stability (thioethers from 5b and Me-5a, half lives approximately 4.5 days), (c) rapidly cleaved (adducts from 5c and 8a/8b, half lives 12–36 hours), and (d) non-cleavable (thioether from epoxy-5a, which underwent one or more reactions with half life of approximately 17 days at room temperature, but without cleaving into two fragments43). The thiol adduct 10a and its dansylsulfonamide N-methylated analogue Me-10a exhibited a six-fold difference in retro-Diels–Alder decomposition rate in water (Table 1), with the latter structure being the less stable. This suggests that an intramolecular hydrogen bond between the sulfonamide NH and the adjacent ester group may help to stabilize compounds such as 10a. The four categories of thioethers each include members that exhibited high rates of thiol addition, so OND systems can be efficiently assembled while being tuned to provide variable rates of release.
Peptide and protein labeling
To test the bioconjugation reactivity of representative OND labels, a 1 mM solution of the nonapeptide 15 (KCIRGDTFG*, where G* represents a C-terminal glycine N-propargylamide) containing amine, carboxylate, guanidine, and alkyne functional groups in addition to thiol, was mixed with an equimolar amount of 5a, 8a, or 8b in pH 7 buffer containing 10% DMSO (Figure 5). A strong fluorescent signal was generated immediately in each case, reaching completion within one minute. Analysis by HPLC and MALDI-TOF mass spectrometry of the crude reaction mixtures showed no unreacted starting material (Supporting Information, Figures S6 and S7) and a major product corresponding to the addition of one electrophile in each case, along with some retro-Diels–Alder fragmentation induced in the MALDI analysis.44 When the peptide was pretreated with N-ethylmaleimide (1 mM) no reaction was observed with the OND electrophiles, confirming the thiol as the site of reaction.
Figure 5.

Peptide labeling with OND electrophiles.
Treatment of bovine serum albumin (BSA, 1 mg/mL, 15 µM) with 50 µM of reagents 5b, 8b, or 16 (derived from 8b) under mild conditions provided efficient labeling of the single free cysteine residue at position 34 in two hours as revealed by the appearance of strong dansyl fluorescence in the protein band following denaturing gel electrophoresis (Figure 6). The denaturing protocol had some effect on the intensity of the fluorescent signal, consistent with the expected lability of the adduct. Thus, treatment of the BSA-5b conjugate with dithiothreitol (DTT) and urea at 95°C gave a significantly weaker fluorescent band than a room temperature protocol using the same denaturing reagents (lane 7 vs. 3). As observed with their β-mercaptoethanol conjugates (Table 1), the adduct of BSA with 8b was much less stable than with 5b, completely losing its fluorescence upon heating due to its faster retro-Diels–Alder fragmentation (lane 6 vs. 7). If one places a dye on the electrophilic fragment, however, the label is permanently attached. Thus, when treated with bis(dansyl) OND probe 16, BSA remained labeled even after denaturation at high temperature. The fluorescence intensity of the protein diminished by approximately one half (lane 5 vs. 1) because cycloreversion left one of the two dansyl units attached to the protein (17) and allowed the other to diffuse away, as shown.38 A non-cleavable linkage was also made with the epoxide derivative epoxy-5a. When analyzed after room temperature treatment, both 5a and epoxy-5a showed complete labeling (Figure 6, lanes 9 and 10); in contrast, only epoxy-5a provided a heat-stable linkage (lane 12 vs. 11).
Figure 6.

(Top) SDS-PAGE of BSA (1 mg/mL) labeled with the indicated OND reagents (50 µM, pH 7.0, 2 h, room temperature), followed by denaturation with DTT and urea at room temperature for 1 hour or at 95°C for 10 min, as indicated. The upper images show the gels visualized under ultraviolet irradiation before staining; the lower images show staining with Coumassie blue. (Bottom) Sequence of bond forming and breaking events with electrophile 16. The thiomaleimide adduct 17 may take on an additional nucleophile such as DTT or protein thiol.38
Conclusions
Oxanorbornadienedicarboxylates (OND), electrophilic adducts of furans and electron-deficient alkynes, have long been known as synthetic intermediates in Diels–Alder/retro-Diels–Alder sequences45,46 and more recently as reactive compounds toward cycloaddition47–49 and ene reactions.28,50,51 Their polar reactivity as Michael acceptors has, with one exception,23 received little attention. We show here that these readily available compounds provide excellent water stability while retaining rapid, selective, and fluorogenic reactivity toward thiols in small molecules, peptides, and proteins. Among fluoro- and chromogenic thiophiles recently described,18–20,52 the OND molecules are notable for their ease of synthesis and tunability in both addition and fragmentation processes. It is envisioned that other dyes with appropriate redox potentials can be used to provide better fluorogenic properties. The utility of these compounds for protein modification in vitro and in vivo, drug release, and polymer crosslinking is the subject of ongoing research in our laboratory.
Experimental Section
General
Reagents and solvents were purchased from commercial sources and used without further purification. THF, acetonitrile, diethyl ether, dichloromethane, and toluene were dried by passage through activated alumina columns.53,54 Routine mass spectra were obtained on an Agilent 1100 (G1946D) ESI-MSD instrument, using a standard C18 reversed-phase column eluted with 9:1 CH3CN:H2O containing 0.1% CF3CO2H. Elemental analyses were performed by Midwest MicroLab, LLC. Flash chromatography was performed on 230–400 mesh silica or SINGLE StEP pre-packed MPLC columns (Thomson Instrument Company, Oceanside, CA). Dye-containing materials were protected from light by wrapping the reaction and storage glassware in aluminum foil. Reactions of OND reagents with thiols were conducted in air rather than under inert atmosphere; for long-term storage the exposure of thiol reagents to air was minimized. Disulfide bond formation was not found to be significant under our reaction conditions.
Synthesis
Dipropargyl acetylenedicarboxylate
A slight modification of the published procedure55 was used. A suspension of acetylenedicarboxylic acid (5.15 g, 45.1 mmol), propargyl alcohol (5.20 g, 92.7 mmol) and p-toluenesulfonic acid monohydrate (0.5 g) in benzene (50 mL) was refluxed with a Dean-Stark trap for 24 h, collecting approximately 1.8 mL of water. The resulting clear brown mixture was cooled, diluted with diethyl ether (50 mL), and washed with saturated aqueous sodium bicarbonate (50 mL) and water (2×50 mL). The organic phase was dried over magnesium sulfate, concentrated and purified by flash chromatography (silica, 10–20% v/v ethyl acetate/hexanes) to provide the pure compound as a pale-yellow oil that solidified on storage at +4 °C to colorless crystals (4.78 g, 56%, mp 26–29 °C). Rf 0.56 (25% EtOAc/hexanes). Spectral data was in accordance with the literature.56 1H NMR (CDCl3, 400 MHz) δ 4.82 (d, 4H, J = 2.5), 2.57 (t, 2H, J = 2.5). 13C NMR (CDCl3, 100 MHz) δ 150.9, 76.7, 75.8, 74.9, 54.2.
N-Dansylfurfurylamine 3
To a stirred solution of 5-dimethylaminonaphthalenesulfonyl chloride (1.00 g, 3.70 mmol) and triethylamine (1.0 mL, 7.2 mmol) in CH2Cl2 (15 mL) was added a solution of furfurylamine (378 mg, 3.89 mmol) in CH2Cl2 (5 mL) via syringe under nitrogen. The resulting solution was stirred for 3 hours at room temperature and poured into 1.0 M pH 7 phosphate buffer (30 mL). The organic phase was separated, dried over magnesium sulfate, concentrated, and purified by flash chromatography (silica, 0–25% EtOAc/hexanes) to provide the title compound as a yellow-green oil which solidified on storage (1.18 g, 96%). Rf 0.35 (25% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.51 (app d, 1H, J = 8.5), 8.22 (m, 2H), 7.57–7.52 (m, 1H), 7.52–7.47 (m, 1H), 7.17 (app d, 1H, J = 7.6), 7.03 (dd, 1H, J = 1.8, 0.9), 6.04 (dd, 1H, J = 3.2, 1.8), 5.89 (dd, 1H, J = 3.2, 0.9), 4.98 (t, 1H, J = 6.0), 4.13 (d, 2H, J = 6.0), 2.88 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ 152.1, 149.5, 142.4, 134.8, 130.7, 130.0, 129.78, 129.7, 128.6, 123.3, 118.7, 115.3, 110.3, 108.1, 45.6, 40.4. ESI-MS (C17H18N2O3S) 331.0, [M+H]+. Note: the crude material may be precipitated by cooling a solution in 1:5 EtOAc:hexanes to obtain crystalline material of satisfactory purity.
N-dansyl(5-methylfurfuryl)amine (4)
To a stirred solution of 5-dimethylaminonaphthalene-sulfonyl chloride (472 mg, 1.75 mmol) and triethylamine (0.39 mL, 2.81 mmol) in CH2Cl2 (5 mL) was added a solution of furfurylamine (211 mg, 1.90 mmol) in CH2Cl2 (1 mL) via syringe under nitrogen. The resulting solution was stirred for 3 hours at room temperature and poured into 1.0 M pH 7 phosphate buffer (15 mL). The organic phase was washed with water (15 mL), dried over magnesium sulfate, concentrated, and purified by flash chromatography (silica, 0–25% EtOAc/hexanes) to provide the title compound as a yellow-green oil which solidified on storage (576 mg, 97%). Rf 0.40 (25% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.50 (app d, 1H, J = 8.5), 8.25–8.19 (m, 2H), 7.58–7.52 (m, 1H), 7.51–7.45 (m, 1H), 7.17 (app d, 1H, J = 7.6), 5.71 (d, 1H, J = 3.0), 5.55 (dq, 1H, J = 3.0, 1.0), 4.97 (t, 1H, J = 5.9), 4.08 (d, 2H, J = 6.0), 2.88 (s, 6H), 1.91 (app s, 3H). 13C NMR (CDCl3, 100 MHz) δ 152.2, 147.5, 135.0, 130.6 130.1, 129.9, 129.8, 128.5, 123.3, 118.9, 115.3, 109.1, 106.1, 45.6, 40.6, 13.3. ESI-MS (C18H20N2O3S) 345.1, [M+H]+.
OND structures 5
A mixture of N-dansylfurfurylamine (331 mg, 1.00 mmol) and the appropriate acetylenedicarboxylate (1.5–1.8 equiv) was dissolved in CH2Cl2 (0.5 mL). The solvent was removed under a stream of nitrogen at 50 °C and the reaction mixture was heated with stirring at 60–65 °C until the dye was consumed (ca. 3 hours). The reaction mixture was dissolved in a minimal amount of hot toluene and loaded directly on a pre-packed silica gel chromatography column and eluted with the appropriate solvent gradient to provide the pure product.
Dimethyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-7-oxanorborna-2,5-diene-2,3-dicarboxylate (5a)
Yellow foam, 81% yield. Rf 0.23 (25% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.28–8.22 (m, 2H), 7.59–7.51 (m, 2H), 7.19 (d, 1H, J = 7.6), 7.13 (dd, 1H, J = 5.2, 1.8), 6.89 (d, 1H, J = 5.3), 5.57 (d, 1H, J = 1.9), 5.17 (app t, 1H, J = 6.3), 3.78 (s, 3H), 3.71 (s, 3H), 3.71 (dd, 1H, J = 13.5, 7.0), 3.60 (dd, 1H, J = 13.5, 5.7), 2.88 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ 163.9, 162.8, 153.7, 152.2, 152.1, 145.0, 142.8, 134.4, 130.8, 130.1, 129.9, 129.7, 128.7, 123.32, 118.8, 115.5, 95.9, 84.1, 52.6, 52.6, 45.6, 42.1. ESI-MS 473.1, [M+H]+. Anal. calcd for C23H24N2O7S: C, 58.46; H, 5.12; N, 5.93. Found: C, 58.26; H, 5.16; N, 5.77.
Dimethyl 1-((5-(dimethylamino)-N-methylnaphthalene-1-sulfonamido)methyl)-7-oxanorborna-2,5-diene-2,3-dicarboxylate (Me-5a)
To a suspension of 5a (135 mg, 0.286 mmol) and cesium carbonate (186 mg, 0.573 mmol, 2.0 equiv) in dimethylformamide (0.6 mL) was added methyl iodide (57 mg, 0.40 mmol, 1.4 equiv) and the mixture was stirred for 20 minutes at room temperature. Aqueous potassium dihydrogen phosphate (5%, 1.5 mL) was added and the mixture was extracted with EtOAc (4×2 mL). The combined organic solutions were dried over magnesium sulfate, concentrated, and purified by flash chromatography (silica 30% EtOAc/hexanes) to provide the desired product as an orange foam (117 mg, 84%). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.42 (app d, 1H, J = 8.7), 8.15 (app d, 1H, J = 7.3), 7.58–7.51 (m, 2H), 7.19 (d, 1H, J = 7.6), 7.17–7.14 (m, 2H), 5.64 (d, 1H, J = 2.2), 4.19 (d, 1H, J = 15.1), 3.91 (d, 1H, J = 15.1), 3.87 (s, 3H), 3.79 (s, 3H) 2.88 (s, 6H), 2.86 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ 164.2, 162.9, 154.0, 152.5, 151.9, 144.7, 143.8, 133.8, 130.6, 130.5, 130.3, 130.0, 128.3, 123.3, 119.8, 115.4, 98.0, 84.1, 52.8, 52.5, 48.3, 45.6, 36.6. ESI-MS 487.1, [M+H]+. Anal. calcd for C24H26N2O7S: C, 59.25; H, 5.39; N, 5.76. Found: C, 59.26; H, 5.45; N, 5.63%.
The synthesis of 1-((5-(dimethylamino)-naphthalene-1-sulfonamido)methyl)-5,6-epoxy-7-oxanorborna-2-ene-2,3-dicarboxylate (epoxy-5a) is not optimized and will be described separately.43 Rf 0.10 (40% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.27–8.21 (m, 2H), 7.59–7.50 (m, 2H), 7.18 (app d, 1H, J = 7.6), 5.14 (dd, 1H, J = 8.2, 4.7), 5.03 (s, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.74 (dd, 1H, J = 13.7, 8.2), 3.74 (d, 1H, J = 3.5), 3.59 (d, 1H, J = 3.5), 3.41 (dd, 1H, J = 13.7, 4.7), 2.88 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ 163.1, 162.0, 152.2, 148.5, 147.4, 134.2, 130.9, 130.1, 129.9, 129.7, 128.8, 123.3, 118.6, 115.5, 90.6, 78.7, 57.4, 56.1, 53.0, 52.8, 45.6, 41.4. ESI-MS (C23H24N2O8S) 489.1, [M+H]+.
Diethyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-7-oxa oxanorborna-2,5-diene-2,3-dicarboxylate (5b)
Yellow foam, 80% yield. Rf 0.26 (25% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.27 (d, 1H, J = 8.5), ), 8.28–8.22 (m, 2H), 7.59–7.51 (m, 2H), 7.18 (d, 1H, J = 7.5), 7.12 (dd, 1H, J = 5.3, 1.9), 6.89 (d, 1H, J = 5.3), 5.55 (d, 1H, J = 1.9), 5.21 (t, 1H, J = 6.3), 4.22 (2×dq, 2H, J = 7.2, 2.0), 4.18 (q, 2H, J = 7.1), 3.72 (dd, 1H, J = 13.5, 6.9), 3.60 (dd, 1H, J = 13.5, 5.8), 2.88 (s, 6H), 1.30 (t, 3H, J = 7.1), 1.28 (t, 3H, J = 7.2). 13C NMR (CDCl3, 100 MHz) δ 163.6, 162.7, 153.5, 152.2, 151.6, 145.0, 142.8, 134.5, 130.8, 130.1, 129.9, 129.7, 128.7, 123.3, 118.8, 115.4, 95.9, 84.1, 61.9, 61.7, 45.6, 42.1, 14.2, 14.1. ESI-MS 501.1, [M+H]+. Anal. calcd for C25H28N2O7S: C, 59.99; H, 5.64; N, 5.60. Found: C, 59.24; H, 5.62; N, 5.31.
Dipropargyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-7-oxanorborna-2,5-diene-2,3-dicarboxylate (5c)
A mixture of N-dansylfurfurylamine (331 mg, 1.00 mmol), dipropargyl acetylenedicarboxylate (350 mg, 1.84 mmol), and toluene (0.5 mL) was heated at 60–65 °C with stirring for ca. 3 hours. After the reaction was complete, the resulting brown oil was loaded directly on a silica gel column and purified by flash chromatography (20–40% EtOAc/hexanes) to provide the title compound as an oil that solidified to yellow crystalline powder on trituration with Et2O and hexanes (428 mg, 82%). Rf 0.13 (25% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.56 (d, 1H, J = 8.5), 8.28–8.23 (m, 2H), 7.60–7.51 (m, 2H), 7.19 (d, 1H, J = 7.6), 7.14 (dd, 1H, J = 5.2, 1.9), 6.90 (d, 1H, J = 5.2), 5.60 (d, 1H, J = 1.9), 5.15 (t, 1H, J = 6.4), 4.77 (d, 2H, J = 2.5), 4.67 (2×dd, 2H, J = 15.5, 2.5), 3.73 (dd, 1H, J = 13.7, 7.1), 3.63 (dd, 1H, J = 13.7, 5.8), 2.89 (s, 6H), 2.50 (m, 2H). 13C NMR (CDCl3, 100 MHz) δ 162.6, 161.6, 153.9, 152.3, 152.2, 145.1, 142.9, 134.4, 130.9, 130.1, 130.1, 129.8, 128.8, 123.4, 118.8, 115.6, 96.2, 84.2, 76.0, 75.9, 53.3, 53.1, 45.7, 42.0. ESI-MS 520.1, [M+H]+. Anal. calcd for C27H24N2O7S: C, 62.30; H, 4.65; N, 5.38. Found: C, 62.37; H, 4.71; N, 5.28.
Dipropargyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-4-methyl-7-oxanorborna-2,5-diene-2,3-dicarboxylate (6)
Prepared according to the general procedure above; the reactants were heated for 5 hours at 80 °C. Yellow foam, 40% yield. Rf 0.27 (40% acetone/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.28–8.23 (m, 2H), 7.59–7.50 (m, 2H), 7.19 (app d, 1H, J = 7.1), 6.88 (app s, 2H), 5.28 (t, 1H, J = 6.4), 4.79 (d, 2H, J = 2.4), 4.62 (2×dd, 2H, J = 15.6, 2.4), 3.72 (dd, 1H, J = 13.8, 6.4), 3.67 (dd, 1H, J = 13.8, 6.6), 2.89 (s, 6H), 2.50 (m, 2H), 1.69 (s, 3H). 13C NMR (CDCl3, 100 MHz) δ 163.2, 162.1, 158.0, 152.2, 150.0, 147.6, 144.3, 134.6, 130.8, 130.1, 130.0, 129.8, 128.7, 123.4, 119.0, 115.5, 94.3, 93.2, 77.0, 75.9, 75.8, 53.1, 53.0, 45.7, 42.2, 15.1. ESI-MS 535.2, [M+H]+. Anal. calcd for C28H26N2O7S: C, 62.91; H, 4.90; N, 5.24. Found: C, 62.65; H, 4.99; N, 5.10%.
Ethyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-3-trifluoromethyl-7-oxanorborna-2,5-diene-2-carboxylate (9)
A mixture of N-dansylfurfurylamine (370 mg, 1.12 mmol), ethyl 4,4,4-trifluoro-2-butynoate (250 mg, 1.50 mmol), toluene (2 mL) was heated at 80 °C with stirring for 18 hours. The mixture was concentrated to half its original volume, loaded directly on a column of silica gel, and purified by flash chromatography (0–3% isopropanol/toluene) to give an oil which contained a minor fluorescent impurity, inseparable by chromatography. Two recrystallizations from Et2O/hexanes (2:3 v/v, 50 mL) afforded the pure compound as a yellow microcrystalline powder (350 mg, 63%). 1H NMR (CDCl3, 400 MHz) δ 8.56 (app d, 1H, J = 8.5), 8.28–8.23 (m, 2H), 7.59–7.51 (m, 2H), 7.21–7.15 (m, 2H), 7.10 (dd, 1H, J = 5.2, 2.0), 6.95 (d, 1H, J = 5.2), 5.51 (d, 1H, J = 2.0), 5.16 (t, 1H, J = 6.4), 4.17 (q, 1H, J = 7.0), 4.11 (q, 1H, J = 7.0), 3.76 (dd, 1H, J = 13.7, 7.1), 3.62 (dd, 1H, J = 13.7, 5.8), 2.89 (s, 6H), 1.25 (t, 3H, J = 7.1). 13C NMR (CDCl3, 100 MHz) δ 162.1, 152.0, 151.5, [150.21, 150.17, 150.12, 150.07] (q), 144.2, 143.1, 134.1, 130.7, 129.8, 129.8, 129.5, 128.5, [125.26, 122.54, 119.86, 117.18] (q, CF3), 123.1, 118.5, 115.2, 96.0, 82.7, 62.0, 45.4, 41.7, 13.7. ESI-MS 497.1, [M+H]+. Anal. calcd for C23H23F3N2O5S: C, 55.64; H, 4.67; N, 5.64. Found: C, 55.71; H, 4.71; N, 5.54%.
Mono-saponification of dialkyl oxanorbornadienedicarboxylates 5 (general)
To a stirred solution of oxanorbornadiene 5 (1 mmol) in THF (8 mL) was added freshly prepared aqueous NaOH (0.5 M, 4 mL) dropwise over 10 minutes at 0 °C. The reaction was monitored by TLC until the starting material was consumed (ca. 1 hour), at which point the solution was acidified with 1 M HCl to pH 3–4 and extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with brine, dried over magnesium sulfate, and concentrated to obtain the desired pure product.
1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-2-methoxycarbonyl-7-oxanorborna-2,5-diene-3-carboxylic acid (7a)
Yellow solid, 85%. 1H NMR (CDCl3, 400 MHz) δ 8.55 (d, 1H, J = 8.5), 8.27 (d, 1H, J = 8.5), 8.22 (d, 1H, J = 8.6), 7.59–7.50 (m, 2H), 7.22 (d, 1H, J = 7.6), 7.21–7.17 (m, 1H), 6.77 (d, 1H, J = 5.6), 5.64 (d, 1H, J = 1.8), 5.14 (t, 1H, J = 4.8), 3.99 (dd, 1H, J = 13.2, 8.4), 3.78 (s, 3H), 3.50 (dd, 1H, J = 13.2, 4.0), 2.88 (s, 6H). 13C NMR (CD3OD, 100 MHz) δ 165.9, 165.8, 157.8, 153.0, 151.4, 145.9, 143.9, 136.8, 131.3, 131.1, 131.0, 130.2, 129.2, 124.3, 120.6, 116.5, 97.4, 85.3, 52.9, 45.9, 42.9. ESI-MS (C22H22N2O7S) 459.1, [M+H]+.
1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-2-ethoxycarbonyl-7-oxanorborna-2,5-diene-3-carboxylic acid (7b)
Yellow solid, 80%. 1H NMR (CDCl3, 400 MHz) δ 8.55 (d, 1H, J = 8.5), 8.27 (d, 1H, J = 8.5), 8.22 (d, 1H, J = 8.6), 7.60–7.53 (m, 2H), 7.22 (d, 1H, J = 7.6), 7.18–7.21 (m, 1H), 6.82 (d, 1H, J = 5.3), 5.66 (d, 1H, J = 2.0), 4.96 (t, 1H, J = 4.8), 4.32 (dd, 1H, J = 10.9, 7.2), 4.24 (dd, 1H, J = 10.8, 7.2), 3.95 (dd, 1H, J = 13.2, 7.9), 3.56 (dd, 1H, J = 13.2, 4.7), 2.91 (s, 6H), 1.36 (t, 1H, J = 7.2). 13C NMR (MeOD, 100 MHz) δ 165.5, 165.3, 156.8, 152.8, 152.75, 145.9, 143.9, 136.8, 131.2, 131.0, 130.9, 130.2, 129.2, 124.4, 120.7, 116.5, 97.45, 85.2, 12.9, 45.9, 42.8, 14.2. ESI-MS (C23H24N2O7S) 473.1, [M+H]+.
1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-2-propargyloxycarbonyl-7-oxanorborna-2,5-diene-3-carboxylic acid (7c)
Yellow solid, 85%. 1H NMR (CDCl3, 400 MHz) δ 8.55 (d, 1H, J = 8.5), 8.35 (d, 1H, J = 8.7), 8.32 (d, 1H, J = 7.3), 7.64–7.59 (m, 2H), 7.34 (d, 1H, J = 7.6), 7.18–7.21 (m, 1H), 6.82 (d, 1H, J = 5.3), 5.61 (d, 1H, J = 2.0), 5.07 (t, 1H, J = 4.8), 4.83 (dd, 1H, J = 15.5, 2.5), 4.69 (dd, 1H, J = 15.5, 2.5), 3.98 (dd, 1H, J = 13.8, 8.0), 3.62 (dd, 1H, J = 13.8, 4.9), 3.03 (s, 6H), 2.63 (t, 1H, J = 2.4). 13C NMR (CD3OD, 100 MHz) 165.2, 164.2, 151.6, 145.9, 143.9, 137.2, 131.0, 130.5, 130.4, 129.1, 124.9, 122.0, 117.1, 97.7, 85.3, 78.0, 77.0, 53.8, 46.1, 42.7. ESI-MS (C24H22N2O7S) 483.1, [M+H]+.
Methyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-3-propargylcarbamoyl-7-oxanorborna-2,5-diene-2-carboxylate (8a)
To a suspension of acid 7a (130 mg, 0.283 mmol) and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride hydrate (87 mg, 0.314 mmol) in THF (1.5 mL) was added propargylamine (16 mg, 0.29 mmol). The reaction mixture was stirred for 4 hours at room temperature, concentrated to dryness, taken up in CH2Cl2, loaded on a column of silica, and purified by flash chromatography (20–40% acetone/hexanes) to yield the desired product as a yellow solid (52 mg, 37%). Rf 0.40 (40% acetone/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.75 (br t, 1H, J = 4.8), 8.57 (app d, 1H, J = 8.5), 8.29–8.21 (m, 2H), 7.59–7.51 (m, 2H), 7.22–7.17 (m, 2H), 6.75 (d, 1H, J = 5.2), 5.65 (d, 1H, J = 2.0), 5.00 (dd, 1H, J = 8.3, 4.1), 4.09 (ddd, 2H, J = 6.2, 4.8, 2.5), 3.96 (dd, 1H, J = 13.0, 8.3), 3.72 (s, 3H), 3.49 (dd, 1H, J = 13.0, 4.1), 2.89 (s, 6H), 2.25 (t, 1H, J = 2.6). 13C NMR (CDCl3, 100 MHz) δ 164.7, 161.8, 161.4, 152.2, 146.3, 145.8, 141.5, 134.4, 130.9, 130.1, 129.8, 129.8, 128.9, 123.2, 118.6, 115.6, 95.9, 84.6, 79.0, 71.8, 53.0, 45.6, 42.9, 29.4. ESI-MS 496.1, [M+H]+. Anal. calcd for C25H25N3O6S: C, 60.59; H, 5.08; N, 8.48. Found: C, 60.52; H, 5.18; N, 8.33%.
Ethyl 1-((5-(dimethylamino)naphthalene-1-sulfonamido)methyl)-3-propargylcarbamoyl-7-oxanorborna-2,5-diene-2-carboxylate (8b)
The procedure is the same as for 8a, using 7b (87 mg, 0.184 mmol), propargylamine (11 mg, 0.200 mmol), and DMT-MM (56 mg, 0.202 mmol); yield 40 mg (43%). 1H NMR (CDCl3, 400 MHz) δ 8.76 (br t, 1H, J = 4.9), 8.56 (d, 1H, J = 8.5), 8.28–8.21 (m, 2H), 7.59–7.51 (m, 2H), 7.19 (d, 1H, J = 7.7), 7.17 (dd, 1H, J = 5.3, 2.0), 6.75 (d, 1H, J = 5.3), 5.65 (d, 1H, J = 2.0), 4.99 (dd, 1H, J = 7.9, 4.5), 4.17 (2×dq, 2H, J = 7.1, 3.6), 4.09 (ddd, 2H, J = 6.5, 5.4, 2.6), 3.91 (dd, 1H, J = 13.1, 7.9), 3.53 (dd, 1H, J = 13.1, 4.5), 2.89 (s, 6H), 2.24 (t, 1H, J = 2.6), 1.30 (t, 3H, J = 7.1). 13C NMR (CDCl3, 100 MHz) δ 164.1, 161.2, 161.1, 151.9, 146.4, 145.4, 141.4, 134.1, 130.7, 129.8, 129.6, 129.5, 128.6, 123.0, 118.5, 115.3, 95.7, 84.3, 78.8, 71.6, 62.5, 45.3, 42.6, 29.2, 13.7. ESI-MS (C26H27N3O6S) 510.1, [M+H]+.
Thiol adducts (general)
To a solution of oxanorbornadiene (0.100 mmol) in acetonitrile (0.5 mL) was added a thiol (0.090 mmol), followed by diisopropylethylamine (1 µL) to give an instant strong increase in fluorescence. On smaller scale, the amine was introduced as a vapor from a Pasteur pipette. After 5 minutes the reaction was quenched by the addition of acetic acid (5 µL), concentrated, and purified by flash chromatography or preparative TLC.
Compound 10a
Yellow foam, yield 95%, >90% purity. Rf 0.17 (1:1 EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.56 (d, 1H, J = 8.5), 8.28–8.21 (m, 2H) (m, 2H), 7.58–7.51 (m, 2H), 7.19 (d, 1H, J = 7.5), 6.42 (d, 1H, J = 5.7), 6.36 (dd, 1H, J = 5.7, 1.7), 5.04 (t, 1H, J = 6.4), 4.83 (d, 1H, J = 1.7), 3.72 (m, 2H), 3.63 (s, 3H), 3.61 (s, 3H), 3.48 (dd, 1H, J = 13.8, 6.0), 3.38 (dd, 1H, J = 13.8, 6.8), 3.18 (s, 1H), 2.94 (dt, 2H, J = 11.7, 5.5), 2.89 (s, 6H), 1.97 (m, 1H). 13C NMR (CDCl3, 100 MHz) δ 169.9, 169.7, 152.2, 137.7, 135.5, 134.4, 130.9, 130.1, 129.9, 129.7, 128.8, 129.7, 128.8, 123.3, 118.8, 115.5, 91.2, 84.7, 64.7, 60.9, 57.5, 52.8, 52.5, 45.6, 43.3, 34.7. ESI-MS (C25H30N2O8S2) 551.1, [M+H]+.
Compound 10b
Yellow solid, yield 80%, >90% purity. 1H NMR (CDCl3, 400 MHz) δ 8.56 (d, 1H, J = 8.5), 8.32–8.26 (m, 2H), 7.58–7.51 (m, 2H), 7.19 (d, 1H, J = 7.3), 6.41 (d, 1H, J = 5.7), 6.36 (dd, 1H, J = 5.6, 1.3), 4.99 (t, 1H, J = 6.4), 4.83 (d, 1H, J = 1.4), 4.15 (q, 2H, J = 7.2), 3.63 (s, 3H), 3.60 (s, 3H), 3.47 (dd, 1H, J = 13.8, 6.2), 3.34 (dd, 1H, J = 13.8, 6.6), 3.18 (s, 1H), 2.94 (dt, 2H, J = 11.7, 5.5), 2.89 (s, 6H), 2.55 (t, 2H, 7.3), 1.27(t, 3H, 7.2). 13C NMR (CDCl3, 100 MHz) δ 171.3, 169.8, 169.3, 152.0, 137.5, 135.3, 134.1, 130.7, 129.9, 129.8, 129.7, 128.6, 123.1, 118.6, 115.3, 90.9, 84.2, 64.7, 60.8, 57.3, 52.5, 52.2, 45.4, 43.1, 33.7, 26.3, 14.2. ESI-MS (C28H34N2O9S2) 607.1, [M+H]+.
Compound Me-10a
Yellow foam, yield 90%, >90% purity. Rf 0.30 (2:1 EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz) δ 8.55 (app d, 1H, J = 8.5), 8.32 (app d, 1H, J = 8.7), 8.12 (app d, 1H, J = 7.3), 7.59–7.49 (m, 2H), 7.19 (d, 1H, J = 7.5), 6.61 (d, 1H, J = 5.6), 6.42 (dd, 1H, J = 5.6, 1.2), 4.89 (d, 1H, J = 1.2), 3.90 (d, 1H, J = 5.6), 3.86 (d, 1H, J = 4.4), 3.79–3.74 (m, 2H), 3.71 (s, 3H), 3.66 (s, 3H), 3.41 (s, 1H), 3.08–3.03 (m, 2H), 2.92 (s, 3H), 2.88 (s, 6H). 13C NMR (CDCl3, 100 MHz) δ 170.1, 169.9, 151.9, 138.5, 135.1, 133.9, 130.7, 130.4, 130.3, 130.0, 128.4, 123.3, 119.7, 115.4, 93.0, 85.0, 64.5, 61.0, 57.9, 52.8, 52.5, 49.6, 45.6, 36.7, 34.8. ESI-MS (C26H32N2O8S2) 565.2, [M+H]+.
Compound 12
Yellow solid, yield 83%, >90% purity. 1H NMR (CDCl3, 400 MHz) δ 8.56 (app d, 1H, J = 8.5), 8.33–8.26 (m, 2H), 7.60–7.53 (m, 2H), 7.19 (app d, 1H, J = 7.5), 6.40 (dd, 1H, J = 5.7, 1.5), 6.19 (d, 1H, J = 5.8), 5.36 (t, 1H, J = 6.3), 5.09 (d, 1H, J = 1.5), 4.05 (d, 2H, J = 2.4), 3.84–3.73 (m, 2H), 3.66 (dd, 1H, J = 13.8, 6.1), 3.53 (dd, 1H, J = 13.8, 6.6), 3.06–3.01(m, 2H), 2.91 (s, 1H), 2.89 (s, 6H), 2.17 (t, 1H, J = 2.5). 13C NMR (CDCl3, 100 MHz) δ 173.5, 171.2, 152.2, 136.6, 135.9, 134.2, 131.0, 130.3, 130.1, 129.7, 128.8, 123.4, 118.7, 115.4 91.0, 84.3, 75.6, 72.0, 61.8, 59.8, 55.6, 45.6, 43.8, 33.7, 28.2. ESI-MS (C26H27N3O6S2) 542.2, [M+H]+.
Compound 16
To a solution of tris-(benzyltriazolylmethyl) amine (11 mg, 0.02 mmol), 5-Dimethylamino-naphthalene-1-sulfonic acid (3-azidopropyl) amide (23 mg, 0.069 mmol) and 8b (35 mg, 0.069 mmol) in DMSO (1.6 mL) was added CuSO4 (0.2 mL, 50 mM) and sodium ascorbate (0.2 mL, 100 mM). After 2 hours, water was added to the reaction to form a pale-yellow solid which was filtered, washed with water, and purified by preparative TLC (3% MeOH/CHCl3) to yield the desired product (44 mg, 77%). Rf 0.35 (3% MeOH/CDCl3). 1H NMR (CDCl3, 500 MHz) δ 7.63–7.53 (m, 4H), 7.44 (s, 1H, triazole), 7.38–7.35 (m, 1H, CONH), 7.28–7.22 (m, 2H), 7.12 (dd, J=5.2, 1.9, 1H, OND-H5), 6.82 (dd, J=5.2, 1H, OND-H6), 5.97–5.90 (m, 2H, SO2NH), 5.49 (d, J=1.9, 1H, OND-H4), 4.44 (dd, J = 15.9, 5.3, 1H, CH2NHCO), 4.41 (dd, J = 15.9, 5.3, 1H, CH2NHCO), 4.21 (t, J = 6.9, 2H), 4.00 (m, 2H), 3.72 (dd, J = 14.0, 7.0, 1H, OND-CH2NH), 3.72 (dd, J = 14.1, 5.6, 1H, OND-CH2NH), 2.85 (s, 6H), 2.84 (s, 6H), 2.79 (m, 2H), 1.88 (quint, J = 7.8, 2H) 1.08 (t, J = 7.1, 3H). 13C NMR (CDCl3, 125 MHz) δ 165.0, 162.7, 161.6, 153.1, 153.1, 146.8, 145.8, 145.0, 143.2, 136.1, 136.1, 131.3, 131.2, 130.7, 130.4, 130.3, 130.3, 129.2, 124.4, 124.3, 123.6, 119.8, 116.3, 100.6, 96.9, 85.3, 80.5, 63.0, 47.9, 45.7, 43.3, 40.9, 35.7, 30.8, 14.0. ESI-MS (C39H42N8O8S2) 843.2, [M+H]+.
Peptide synthesis
The peptide 14 was prepared by standard Fmoc methods using Rink amide MBHA resin with an initial loading of 0.70 mmol/g. The resin was swollen in DMF for 45 min prior to synthesis. For sequence extension, the Fmoc-protected amino acid (4 equiv) was activated by treatment with HCTU (3.9 equiv) and N,N-diisopropylethylamine (500 µL per mmol of amino acid) in DMF (4 mL per mmol of amino acid) for 2 min. This solution was added to the free amine on resin, and the coupling reaction was allowed to proceed for 15 min (longer for difficult couplings) with intermittent stirring. After washing with DMF, Fmoc deprotection was achieved with 20% 4-methylpiperidine in DMF (2×7 min). The resin was washed again and the process was repeated for the next amino acid.
The symmetric anhydride of 2-bromoacetic acid (5 equiv of anhydride relative to free amine) was prepared by mixing the acid (10 equiv) with N,N'-diisopropylcarbodiimide (DIC, 5 equiv) in DMF at room temperature for 15 min. This mixture was then added to the resin and allowed to react for 1 h with intermittent stirring. The resin was washed and then treated with propargylamine (20 equiv) in DMF for 12–16 h. After washing, coupling of the next residue to the resultant secondary amine was achieved by preparing the symmetrical anhydride of that protected residue (5 equiv of anhydride) with DIC in DMF at 0°C for 15 min, and reacting this mixture with the amine for 1 h with occasional stirring. The following Fmoc-protected amino acids with side chain protecting groups were used as received from NovaBiochem: Fmoc-Ala-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Lys(Boc)-OH, Fmoc-Phe-OH, Fmoc-Thr(tBu)-OH.
The peptide was cleaved from the resin with 2% triisopropylsilane (TIS) in trifluoroacetic acid (TFA, approx. 1 mL TFA/TIS per g resin) for 2.5 h. The cleavage mixture (including resin) was mixed with cold ether to precipitate the peptide and then filtered. The filtrate was washed with cold ether, and 14 was extracted from the residue with H2O/MeCN mixtures (typically 5–20% MeCN in water) containing 0.1% TFA. The resulting solution was frozen and lyophilized to afford a white, solid product.
Reactivity
Stability study at pH 7 (Table 1)
1 mM stock solutions of each OND compound in DMSO (100 µL) were diluted with 0.1 M phosphate buffer (pH 7, 900 µL) at time 0 to initiate the experiment. At different time points, aliquots (50 µL) were injected on the HPLC, with peak detection at 332 nm.
Stability study of the β-mercaptoethanol adducts (Table 1)
A solution of each adduct was created by incubating 0.1 mM of the OND electrophile with an equimolar amount of β-mercaptoethanol in 100 mM phosphate buffer, pH 7, for one hour. HPLC analysis (peak detection at 332 nm) verified the complete formation of adduct in each case. These solutions were then allowed to stand at room temperature with periodic HPLC analysis, following the disappearance of the adduct peak with time. First-order rate constants were obtained by a standard linear plot of ln[adduct] vs. time.
Selectivity of 5b with amino acids (Figure 3)
1 mM stock solutions of 5b in DMSO (100 µL) were diluted with 100 mM phosphate buffer (pH 7, 800 µL). Each amino acid (0.1 mmol) was dissolved in 100 mM phosphate buffer (pH 7, 10 mL), and 10 µL of this solution was added to a solution of 5b (90 µL). The fluorescence intensity (λex 332 nm, λem 550 nm) was monitored every 30 seconds over 60 minutes.
Rate constant determination of 5, 6, 8 and 9 with glutathione at pH 7 (Table 1)
1 mM stock solutions of each OND compound in DMSO (100 µL) were diluted with 100 mM phosphate buffer (pH 7, 800 µL). Glutathione (1.1 mM, 10 µL) was added to these reagent solutions (111 µM, 90 µL) to final concentrations of 110 µM and 100 µM, respectively. The fluorescence intensity (λex 332 nm, λem 550 nm) was recorded every 30 seconds. Each reaction reached the same maximum emission intensity, which was the same as displayed by a freshly-prepared 100 µM solution of the authentic adduct of 5a under the same conditions, and was therefore assumed to represent 100% completion. Rate constants were determined by a standard fit of % completion (ratio of fluorescence intensity at time t to the maximum intensity) vs. time to the integrated second-order rate equation; error values represent the standard deviation of three independent runs.
Rate constant determination of 7c with glutathione, cysteine, and cysteine methyl ester
1 mM stock solutions of 7c in DMSO (100 µL) were diluted with buffer (800 µL) (100 mM phosphate buffer at pH 7, 25 mM citrate at pH 4, 5, or 6). Thiol (1.1 mM, 100 µL) was added to these solutions of 7c (111 µM, 900 µL) to final concentrations of 110 µM and 100 µM, respectively. Aliquots (150 µL) withdrawn at different time points were quenched with ethyl maleimide (1 µL, 100 mM), diluted with water containing 0.1% TFA (250 µL), and injected on the HPLC, with peak detection at 332 nm.
Peptide and BSA labeling (Figures 5, 6)
The OND electrophile (10 mM in DMSO, 10 µL and 5 µL, respectively) was added to peptide 14 (1 mM in 0.1 M phosphate buffer, pH 7, 90 µL) or BSA (1 mg/mL, 1 mL). Assuming a molar absorbitivy similar to that of the corresponding β-mercaptoethanol adduct, a labeling density of 1.0±0.2 attachments per BSA molecule was found in each case. Pre-treatment of the protein with N-ethylmaleimide eliminated the reaction with OND reagent.
Supplementary Material
Acknowledgments
We thank The Skaggs Institute for Chemical Biology and the National Institutes of Health (RR021886) for financial support, Dr. Reshma Jagasia for the synthesis of peptide 14, and Dr. Michal Sabat (University of Virginia) for the X-ray structural analysis of 5c.
Footnotes
Supporting Information Available. X-ray crystal structure of 5c, kinetic data for 7c, HPLC and MALDI data for the reactions with peptide 14, and 1H NMR spectra of 3, 5, and thiol adduct 10a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hermanson GT. Bioconjugate Techniques. Second Edition. San Diego: Academic Press; 2008. [Google Scholar]
- 2.Jullien M, Garel JR. Biochemistry. 1981;20:7021–7026. doi: 10.1021/bi00527a038. [DOI] [PubMed] [Google Scholar]
- 3.Schelté P, Boeckler C, Frisch B, Schuber F. Bioconjugate Chem. 2000;11:118–123. doi: 10.1021/bc990122k. [DOI] [PubMed] [Google Scholar]
- 4.Vanderhooft JL, Mann BK, Prestwich GD. Biomacromolecules. 2007;8:2883–2889. doi: 10.1021/bm0703564. [DOI] [PubMed] [Google Scholar]
- 5.When performing thiol alkylation reactions on cowpea mosaic virus capsids bearing surface cysteine residues, we have observed as much as 25% reactivity with lysines having diminished pKa values by virtue of their unique microenvironment. The assumption of high thiol selectivity for maleimides may therefore be invalid on occasion.
- 6.Ishii Y, Lehrer SS. Biophys. J. 1986;50:75–80. doi: 10.1016/S0006-3495(86)83440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller O, Rudinger J. Helv. Chim. Acta. 1975;58:531–541. doi: 10.1002/hlca.19750580224. [DOI] [PubMed] [Google Scholar]
- 8. Corrie JET. J. Chem. Soc., Perkin Trans. 1994:2975–2982. and references therein.
- 9.Weh J, Duerkop A, Wolfbeis OS. ChemBioChem. 2007;8:122–128. doi: 10.1002/cbic.200600316. [DOI] [PubMed] [Google Scholar]
- 10.Lu ZK, Weber R, Twieg RJ. Tetrahedron Lett. 2006;47:7213–7217. doi: 10.1016/j.tetlet.2006.07.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machida M, Machida MI, Kanaoka Y. Chem. Pharm. Bull. 1977;25:2739–2743. [Google Scholar]
- 12.Tournier EJM, Wallach J, Blond P. Analytica Chim. Acta. 1998;36:33–44. [Google Scholar]
- 13.Kalia J, Raines RT. Bioorg. Med. Chem. Lett. 2007;17:6286–6289. doi: 10.1016/j.bmcl.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kosower EM, Pazhenchevsky B, Hershkowitz E. J. Am. Chem. Soc. 1978;100:6516–6518. [Google Scholar]
- 15.Hulbert PB, Yakubu SI. J. Pharm. Pharmacol. 1983;35:384–386. doi: 10.1111/j.2042-7158.1983.tb02962.x. [DOI] [PubMed] [Google Scholar]
- 16.Kosower EM, Kosower NS. Methods Enzymol. 1995;251:133–148. doi: 10.1016/0076-6879(95)51117-2. [DOI] [PubMed] [Google Scholar]
- 17. Sen CK, Roy S, Packer L. Methods Enzymol. 1999;299:247–258. doi: 10.1016/s0076-6879(99)99024-9. and references therein.
- 18.Holler M, Sin HS, James A, Burger A, Tritsch D, Biellmann J-F. Chem. Eur. J. 2000;6:2053–2062. doi: 10.1002/1521-3765(20000602)6:11<2053::aid-chem2053>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto T, Urano Y, Shoda T, Kojima H, Nagano T. Org. Lett. 2007;9:3375–3377. doi: 10.1021/ol071352e. [DOI] [PubMed] [Google Scholar]
- 20.Bouffard J, Kim Y, Swager TM, Weissleder R, Hilderbrand SA. Org. Lett. 2008;10:37–40. doi: 10.1021/ol702539v. [DOI] [PubMed] [Google Scholar]
- 21.Clarke KM, LaClair JJ, Burkart MD. J. Org. Chem. 2005;70:3709–3711. doi: 10.1021/jo0481396. [DOI] [PubMed] [Google Scholar]
- 22.Girouard S, Houle MH, Grandbois A, Keillor JW, Michnick SW. J. Am. Chem. Soc. 2005;127:559–566. doi: 10.1021/ja045742x. [DOI] [PubMed] [Google Scholar]
- 23.Albert S, Soret A, Blanco L, Deloisy S. Tetrahedron. 2007;63:2888–2900. [Google Scholar]
- 24.Mahajna M, Quistad GB, Casida JE. Chem. Res. Toxicol. 1996;9:241–246. doi: 10.1021/tx950127f. [DOI] [PubMed] [Google Scholar]
- 25.Van Berkel SS, Dirks AJ, Debets MF, van Delft FL, Cornelissen JJLM, Nolte RJM, Rutjes FPJT. ChemBioChem. 2007;8:1504–1508. doi: 10.1002/cbic.200700278. [DOI] [PubMed] [Google Scholar]
- 26.Niwayama S. J. Org. Chem. 2000;65:5834–5836. doi: 10.1021/jo0001986. [DOI] [PubMed] [Google Scholar]
- 27.Kunishima M, Kawachi C, Morita J, Terao K, Iwasaki F, Tani S. Tetrahedron. 1999;55:13159–13170. [Google Scholar]
- 28.Baran PS, Li K, O'Malley DR, Mitsos C. Angew. Chem.-Int. Edit. 2006;45:249–252. doi: 10.1002/anie.200503374. [DOI] [PubMed] [Google Scholar]
- 29.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 30.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3062. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 32.Rodionov VO, Presolski S, Gardinier S, Lim Y-H, Finn MG. J. Am. Chem. Soc. 2007;129:12696–12704. doi: 10.1021/ja072678l. [DOI] [PubMed] [Google Scholar]
- 33.Guy J, Caron K, Dufresne S, Michnick SW, Skene WG, Keillor JW. J. Am. Chem. Soc. 2007;129:11969–11977. doi: 10.1021/ja0738125. [DOI] [PubMed] [Google Scholar]
- 34.Van Berkel SS, Dirks AJ, Meeuwissen SA, Pingen DLL, Boerman OC, LaVerman P, van Delft FL, Cornelissen JJLM, Rutjes FPJT. ChemBioChem. 2008;9:1805–1815. doi: 10.1002/cbic.200800074. [DOI] [PubMed] [Google Scholar]
- 35.This elegant report by Cornelissen, Rutjes, and coworkers (ref. 25, see also ref. 34) describes the azide reactivity of compound 9 and related structures in the context of bioconjugation. The rate of this reaction is far slower than condensation with thiol. For example, we found 9 to engage glutathione with a rate constant of approximately 78 M−1s−1 (Table 1, room temperature) and benzyl azide at a rate of 2.3 × 10−3 M−1s−1 in methanol (37°C), the latter in excellent agreement with the value reported in ref. 25 for the analogous oxanorbornadiene lacking the dansylaminomethyl substituent.
- 36.Schmidt TJ, Ak M, Mrowietz U. Bioorg. Med. Chem. 2007;15:333–342. doi: 10.1016/j.bmc.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 37.Khatik GL, Kumar R, Chakraborti AK. Org. Lett. 2006;8:2432–2436. doi: 10.1021/ol060846t. [DOI] [PubMed] [Google Scholar]
- 38.The reactivity of a representative thiomaleate of structure 11 is discussed in Supporting Information. We are currently exploring the electrophilic and potential thiol crosslinking and exchange capabilities of thiomaleates and thiomaleimide adducts such as 14 and 17.
- 39.Takaguchi Y, Tajima T, Ohta K, Motoyoshiya J, Aoyama H, Wakahara T, Akasaka T, Fujitsuka M, Ito O. Angew. Chem., Int. Ed. 2002;41:817–819. doi: 10.1002/1521-3773(20020301)41:5<817::aid-anie817>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 40.Chen X, Dam MA, Ono K, Mal A, Shen H, Nutt SR, Sheran K, Wudl F. Science. 2002;295:1698–1702. doi: 10.1126/science.1065879. [DOI] [PubMed] [Google Scholar]
- 41.Gheneim R, Perez-Berumen C, Gandini A. Macromolecules. 2002;35:7246–7253. [Google Scholar]
- 42.Keller KA, Guo J, Punna S, Finn MG. Tetrahedron Lett. 2005;46:1181–1184. [Google Scholar]
- 43.The synthesis and reactivities of epoxy-5a and related compounds will be described in detail elsewhere.
- 44.Meng J-C, Averbuj C, Lewis WG, Siuzdak G, Finn MG. Angew. Chem. Int. Ed. 2004;43:1255–1260. doi: 10.1002/anie.200352803. [DOI] [PubMed] [Google Scholar]
- 45.Grigg R, Roffey P, Sargent MV. J. Chem. Soc. C. 1967:2327. [Google Scholar]
- 46.Mavoungou-Gomes L. Bull. Soc. Chim. Fr. 1967:1753–1758. [Google Scholar]
- 47.Allen A, Le Marquand P, Burton R, Villeneuve K, Tam W. J. Org. Chem. 2007;72:7849–7857. doi: 10.1021/jo7012884. [DOI] [PubMed] [Google Scholar]
- 48.Burton RR, Tam W. J. Org. Chem. 2007;72:7333–7336. doi: 10.1021/jo701383d. [DOI] [PubMed] [Google Scholar]
- 49.Burton RR, Tam W. Tetrahedron Lett. 2006;47:7185–7189. [Google Scholar]
- 50.Choony N, Kuhnert N, Sammes PG, Smith G, Ward RW. J. Chem. Soc., Perkin Trans. 2002;1:1999–2005. [Google Scholar]
- 51.Choony N, Sammes PG, Smith G, Ward RW. Chem. Commun. 2001:2062–2063. doi: 10.1039/b106669m. [DOI] [PubMed] [Google Scholar]
- 52.Pires MM, Chmielewski J. Org. Lett. 2008;10:837–840. doi: 10.1021/ol702769n. [DOI] [PubMed] [Google Scholar]
- 53.Alaimo PJ, Peters DW, Arnold J, Bergman RG. J. Chem. Educ. 2001;78:64. [Google Scholar]
- 54.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 55.Tom Dieck H, Munz C, Muller CJ. J. Organomet. Chem. 1990;384:243–255. [Google Scholar]
- 56.Hashmi ASK, Grundl MA, Nass AR, Naumann F, Bats JW, Bolte M. Eur. J. Org. Chem. 2001:4705–4732. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.