

Abstract
Cardiac sympathetic neurons stimulate heart rate and the force of contraction through release of norepinephrine. Nerve growth factor modulates sympathetic transmission through activation of TrkA and p75NTR. Nerve growth factor plays an important role in post-infarct sympathetic remodeling. We used mice lacking p75NTR to examine the effect of altered nerve growth factor signaling on sympathetic neuropeptide expression, cardiac norepinephrine, and ventricular function after myocardial infarction. Infarct size was similar in wildtype and p75NTR−/− mice after ischemia-reperfusion surgery. Likewise, mRNAs encoding vasoactive intestinal peptide, galanin, and pituitary adenylate cyclase activating peptides were identical in wildtype and p75NTR−/− cardiac sympathetic neurons, as was expression of the TrkA neurotrophin receptor. Norepinephrine content was elevated in the base of the p75NTR−/− ventricle compared to wildtype, but levels were identical below the site of occlusion. Left ventricular pressure, dP/dtMAX, and dP/dtMIN were measured under isoflurane anesthesia 3 and 7 days after surgery. Ventricular pressure decreased significantly 3 days after infarction, and deficits in dP/dtMAX were revealed by stimulating beta receptors with dobutamine and release of endogenous norepinephrine with tyramine. dP/dtMIN was not altered by genotype or surgical group. Few differences were observed between genotypes 3 days after surgery, in contrast to low pressure and dP/dtMAX previously reported in control p75NTR−/− animals. Seven days after surgery ventricular pressure and dP/dtMAX were significantly lower in p75NTR−/− hearts compared to WT hearts. Thus, the lack of p75NTR did not enhance cardiac function after myocardial infarction.
Keywords: cardiac ischemia-reperfusion, norepinephrine, nerve growth factor
Introduction
The sympathetic nervous system stimulates heart rate, cardiac conduction, and the force of contraction through the release of norepinephrine (NE) and activation of β1adrenergic receptors (β1AR). Myocardial infarction (MI) causes sympathetic dysfunction (Abe et al., 1997; Barber et al., 1983; Zipes, 1990) in addition to its effects on the myocardium. Cardiac sympathetic drive is increased after MI (Graham et al., 2002; Karlsberg et al., 1979), but at the same time there is a loss of sympathetic fibers in the infarct and peri-infarct myocardium (Barber et al., 1983; Inoue and Zipes, 1988; Li et al., 2004; Minardo et al., 1988; Stanton et al., 1989). The resulting chronic alterations in noradrenergic transmission contribute to electrical remodeling and the development of ventricular arrhythmias (Cao et al., 2000a; Cao et al., 2000b; Dae et al., 1997; Rubart and Zipes, 2005), but their effects on cardiac physiology are less clear.
Nerve Growth Factor (NGF) supports sympathetic neuron survival, stimulates axon growth into the heart during development (Crowley et al., 1994; Glebova and Ginty, 2004), and plays an important role in sympathetic remodeling after myocardial infarction (Abe et al., 1997; Hasan et al., 2006; Hiltunen et al., 2001; Oh et al., 2006; Wernli et al., 2009; Zhou et al., 2004). In sympathetic neurons NGF acts through the TrkA and p75 neurotrophin receptors (p75NTR)(Bamji et al., 1998; Birren et al., 1993; Zampieri and Chao, 2006). NGF stimulates sympathetic axon outgrowth (Kohn et al., 1999), expression of tyrosine hydroxylase (TH), NE synthesis (Max et al., 1978; Thoenen, 1972), and synapse formation between pre- and post-ganglionic sympathetic neurons through trkA (Sharma et al., 2010). NGF can also regulate sympathetic expression of neuropeptides (Shadiack et al., 2001). p75NTR can functionally antagonize NGF-stimulated TrkA signaling in sympathetic neurons (Hannila et al., 2004). Thus, NGF stimulates greater axon outgrowth (Kohn et al., 1999), elevated TH expression and NE content (Habecker et al., 2008; Lorentz et al., 2010), and additional synaptic inputs (Sharma et al., 2010) in sympathetic neurons that lack p75NTR. NGF protects cardiac sympathetic nerves from acute dysfunction after ischemia-reperfusion (Abe et al., 1997), but the longer term effects of NGF signaling on sympathetic transmission and cardiac function are not known.
Mice lacking p75NTR (Lee et al., 1992; Lee et al., 1994) provide an interesting model to examine the effect of enhanced TrkA signaling on cardiac NE content and ventricular function after myocardial infarction (MI). Previous studies in these mice have identified sympathetic innervation in the right ventricle that appears normal (Jahed and Kawaja, 2005), but decreased innervation density in adult atria (Habecker et al., 2008) and left ventricular subendocardium (Lorentz et al., 2010). The functional effects of altered innervation are mixed, as heart rate is low in p75NTR−/− mice (Habecker et al., 2008), but stroke volume is normal despite low left ventricular peak systolic pressure (LVP) and dP/dtMAX (Lorentz et al., 2010). NGF is elevated in the heart after myocardial infarction (Abe et al., 1997; Hasan et al., 2006; Hiltunen et al., 2001; Zhou et al., 2004), and sympathetic drive to the heart is increased (Graham et al., 2002; Karlsberg et al., 1979). The effects of altered TrkA signaling may be enhanced after myocardial infarction and may blunt the loss of function in the left ventricle of p75NTR−/− hearts after myocardial infarction. Here we examine cardiac NE, left ventricular pressure, and dP/dt in WT and p75NTR−/− mice after ischemia-reperfusion.
Materials and Methods
Animals and Experimental Group
Wildtype C57BL/6J and p75NTR−/− mice (B6.129S4-Ngfrtm1Jae/J) (Lee et al., 1992) were obtained from Jackson Laboratories. Mice were kept on a 12h:12h- light dark cycle with ad libitum access to food and water. Age and gender-matched male and female mice between 12–18 weeks old were used for all experiments. All procedures were approved by the Institutional Animal Care and Use Committee and comply with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH publication No. 85-23, revised 1996). The experimental groups used were sham-operated animals and animals that underwent ischemia-reperfusion surgery. Six animals were assigned to each group for each experiment, but in some groups only 4 animals survived the surgery and hemodynamic measurements.
Ischemia-Reperfusion Surgery
Anesthesia was induced with 4% inhaled isoflurane and maintained with 2% isoflurane. Mice were then intubated and mechanically ventilated. Core body temperature was monitored with a rectal probe and maintained at 37° C and two-lead ECG was monitored throughout the surgery using a PowerLab data acquisition system (ADInstruments). A left thoracotomy was performed in the 4th intercostal space and the pericardium was opened. The left anterior decending coronary artery (LAD) was reversibly ligated with a 8-0 suture for 30 minutes and then reperfused by release of the ligature. Occlusion was confirmed with ST segment elevation, regional cyanosis, and wall motion abnormalities. Reperfusion was confirmed by return of color to the myocardium distal to the ligation and disappearance of ST elevation. The suture remained within the wound for identification of the ligature site, and the chest and skin were closed in layers. After surgery, animals were returned to individual cages and given regular food and water for 3 days or 7 days before euthanasia and tissue harvest. Buprenex (0.1 mg/kg) was administered as needed to ensure the animals were comfortable following surgery. All surgical procedures were performed under aseptic conditions. Sham animals underwent the procedure described above except for the LAD ligation.
Infarct/Area at Risk Analysis
24 hours after the onset of reperfusion, mice were anesthetized with 4% isoflurane, intubated, and mechanically ventilated, and anesthesia was maintained with 2% inhaled isoflurane. The chest cavity was re-opened and the LAD was re-occluded using the same suture from the ischemia-reperfusion procedure. Fluorescent particles (4 mg/ml in deionized water with 0.01% Tween 20; Duke Scientific no. 34 –1, 2- to 8-μm size) were infused through a polyethelyne tube (PE10) with a 30-gauge needle tip in the left ventricle of the heart. Microspheres were infused at a rate of 400μL/min for 4 minutes, or until the heart stopped, in order to delineate the area at risk. The heart was then excised for infarct size analysis and cut into transverse sections 1mm thick using a cutting block. Both sides of each tissue section were photographed under ultraviolet light for measurement of area at risk. The slices were then placed in 2,3, 5-triphenyltetrazolium chloride solution (TTC, Sigma chemicals; 1% wt/vol in a sodium phosphate buffer at 37° C, pH 7.4) for 20 minutes. The staining procedure was carried out in the dark to prevent break down of the TTC by the light. The slices were then placed in 10% neutral buffered formalin overnight to increase the contrast of the stained and unstained tissue. Myocardium that did not stain red was presumed to be infarcted. Both sides of each 1mm section were photographed under white light and total area, area at risk and infarct areas for each slice were traced in Photoshop. The volume of myocardium at risk and infarcted myocardium were calculated from the measured areas and slice thickness. Infarct size was normalized as a fraction of the area at risk. All analyses were performed in a blinded fashion by two people. The data presented are the average of the two independent determinations of infarct/risk.
Real-time PCR
All RNA and PCR reagents were from Applied Biosystems. Stellate ganglia, which contain most of the sympathetic neurons that project to the heart, were removed and stored in RNAlater®. RNA was isolated using the RNAqueous micro kit, cDNA was generated, and real-time PCR was performed using ABI Taqman master mix and prevalidated Taqman gene expression assays for mouse vasoactive intestinal peptide (VIP), galanin, pituitary adenylate cyclase activating peptides (PACAP), TrkA and p75NTR. cDNA from a single ganglion was used to quantify all genes in duplicate. Left and right stellates were used interchangeably since previous studies indicated that similar changes occurred in both ganglia (Habecker et al., 2005). Peptide and TrkA mRNAs were normalized to GAPDH mRNA in the same sample, while p75NTR was normalized to actin in the same sample. Standard curves were generated for each gene tested. Differences between surgical groups within a genotype were determined by one-way ANOVA with Newman-Keuls post-test using Prism 5.0 software.
Hemodynamics
Mice were anesthetized with 4% isoflurane (Minrad Inc., Bethleham, PA) and maintained with 2–3% isoflurane. Mice were intubated and placed on a rodent ventilator. Body temperature was monitored and maintained at 37±0.2 ºC. A microtipped pressure transducer (1.0 French; Millar) was inserted into the right carotid artery and advanced into the left ventricle for measurement of left ventricular pressure using a PowerLab data acquisition system. A small polyvinyl catheter was placed in the left jugular vein for drug administration. When the animal was stable, it was given hexamethonium chloride (5 mg/kg; Sigma) to abolish ganglionic transmission. Previous experiments have confirmed that this dose is sufficient to fully inhibit ganglionic transmission in mice (Parrish et al., 2009a). Blockade of ganglionic transmission was required to assess basal function in the absence of neuronal input, and to abolish the baroreflex prior to administration of dobutamine and tyramine. After a new baseline was established, animals received a single bolus dose of the beta agonist dobutamine (32 μg/kg; Hospira Inc.) to assess beta receptor sensitivity. After the dobutamine washed out, another dose of hexamethonium was administered, followed by a bolus dose of tyramine hydrochloride (200 μg/kg; Sigma) to assess the cardiac response to release of endogenous NE. Left ventricular peak systolic pressure (LVP), dP/dtMAX, and dP/dtMIN were analyzed using ChartPro software (ADInstruments).
HPLC Analysis of Norepinephrine
NE levels were measured by HPLC with electrochemical detection as described previously (Parrish et al., 2009a). Hearts were excised and cut in 1–2 mm transverse cross sections, excluding the area spanning the LAD ligation. The base was processed as a single sample that included the top 2 mm of both ventricles. Below the site of LAD occlusion the left and right ventricles (LV, RV) were separated and processed individually for NE analysis. Heart tissue was homogenized in perchloric acid (0.1 M) containing 1.0 μM of the internal standard dihydroxybenzylamine (Sigma) to correct for sample recovery. Catecholamines were purified by alumina extraction before analysis by HPLC. Detection limits were ~0.05 pmol with recoveries from the alumina extraction >60%.
Results
Peptide mRNA expression
The ventricles are innervated by NGF-responsive sympathetic and sensory neurons whose production of neuropeptides can impact infarct size and might be altered by the lack of p75NTR (McMahon et al., 1995; Patel et al., 2000). Sensory neurons produce CGRP (Calcitonin Gene Related Peptide) and Substance P (Ieda et al., 2006), while cardiac sympathetic neurons express PACAP after ischemia-reperfusion (Alston et al., 2010). All of these peptides are cardio-protective and can decrease infarct size (Gasz et al., 2006; Huang et al., 2008; Li and Peng, 2002; Racz et al., 2008; Wang and Wang, 2005). Therefore, we examined sympathetic neuropeptide expression and infarct size in WT and p75NTR−/− mice. Sympathetic PACAP, galanin, and VIP mRNAs were essentially identical in sham animals from both genotypes, and did not change following MI (Table 1). Infarct size was measured in WT and p75NTR−/− mice and normalized to the area-at-risk within the same heart. The infarct/risk ratio was identical in both genotypes (Fig. 1).
Table 1.
Stellate ganglion peptide mRNAs normalized to GAPDH mRNA
| GAL/GAPDH | WT | P75−/− |
|---|---|---|
| Sham | 1.6 ± 0.3 | 1.9 ± 0.6 |
| 3d MI | 1.5 ± 0.2 | 2.0 ± 0.5 |
| 7d MI | 1.5 ± 0.1 | 1.8 ± 0.2 |
|
| ||
|
PACAP/GAPDH
| ||
| Sham (n=6) | 1.4 ± 0.2 | 1.5 ± 0.1 |
| 3d MI (n=6) | 2.0 ± 0.5 | 1.9 ± 0.4 |
| 7d MI | 1.6 ± 0.2 | 1.4 ± 0.2 |
|
| ||
|
VIP/GAPDH
| ||
| Sham | 0.9 ± 0.1 | 0.9 ± 0.1 |
| 3d MI | 1.0 ± 0.1 | 0.7 ± 0.1 |
| 7d MI | 1.2 ±0.03 | 0.6 ± 0.1 |
Values are mean±sem; n=4 except as noted.
Figure 1. Infarct size compared to area at risk.
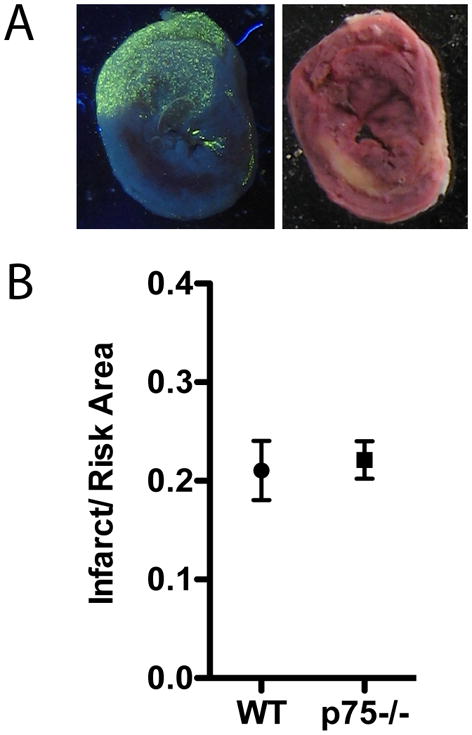
Area at risk (A) and Infarct (B) are shown from a representative section 24 hours after ischemia-reperfusion. The area at risk is devoid of fluorescent microspheres, while the infarct is yellow/white following TTC staining. C) Infarct and risk were quantified in 4 WT and 4 p75NTR−/− mice and the Mean ± SEM is shown.
Trk and p75NTR mRNA expression
To confirm that the absence of p75NTR did not alter expression of TrkA, we quantified TrkA mRNA in WT and p75NTR−/− stellate ganglia after sham or ischemia-reperfusion surgery. TrkA mRNA normalized to GAPDH was identical in both genotypes, and was not changed by myocardial infarction (new Fig 2A). Analysis of p75NTR mRNA in WT ganglia revealed that p75NTR expression was unaltered by ischemia-reperfusion (Fig. 2B).
Figure 2. TrkA and p75NTR mRNA expression.

A. TrkA mRNA normalized to GAPDH in WT and p75NTR−/− stellate ganglia. TrkA mRNA levels are identical in both genotypes and unchanged 3 days after myocardial infarction (3d MI). Data are mean±sem, n=4 for all groups. B. p75NTR mRNA normalized to actin in WT mice is similar after sham surgery (Con) or 24 hours (24h MI) or 3 days (3d MI) after myocardial infarction. Data are mean±sem, n=4 except 3d post-MI n=3.
Norepinephrine content
Norepinephrine content in the base of the ventricles above the coronary artery ligation was significantly higher in p75NTR−/− hearts compared to WT in all surgical groups (Fig. 3A, B). In contrast, NE levels in the lower left ventricle (Fig. 3C, D) were significantly lower in p75NTR−/− sham hearts compared to WT 7 days after surgery (*p<0.05 WT sham vs. p75NTR−/− sham, two-way ANOVA with Bonferroni post-test). NE content decreased significantly in the lower left ventricle after MI compared to the same region in sham operated hearts, and post-infarct levels were similar in both genotypes.
Figure 3. Norepinephrine (NE) content in the ventricle.

NE was quantified in the base of the ventricles (A, B) or in the portion of left ventricle below the LAD ligation (C, D). NE from WT (open squares) and p75NTR−/− mice (filled squares) was assayed 3 days (A, C) and 7 days (B, D) after sham or ischemia- reperfusion surgery (MI). Differences between genotypes and surgical groups were analyzed by two-way ANOVA; Sham vs. MI differences are noted with a horizontal line, *p<0.05, **p<0.01, ***p<0.001, Mean ± SEM, n=6.
Hemodynamics
Basal left ventricular peak systolic pressure (LVP) and dP/dtMAX were similar in WT and p75NTR−/− hearts 3 days after MI (Fig. 4A, B), although dP/dtMAX was low in p75NTR−/− hearts after ganglionic inhibition. Myocardial infarction caused a decrease in LVP that was significant after blockade of ganglionic transmission (Fig. 4), but there were no significant decreases in dP/dtMAX in either genotype under basal conditions or after ganglionic block. dP/dtMIN values were altered little by genotype or surgical group (Table 2) at any of the time points examined. Heart rates were similar in WT and P75NTR−/− mice under basal conditions and after ganglionic block.
Figure 4. LVP and dP/dtMAX 3 days after ischemia-reperfusion.

LVP (A), dP/dtMAX (B), and heart rate (C) were measured in WT (open squares) and p75NTR−/− (filled squares) mice 3 days after sham (Sh) or ischemia-reperfusion (MI) surgery. Far left shows baseline values. LVP and dP/dtMAX decreased after ganglionic block (5 mg/kg hexamethonium), with a smaller effect on heart rate. The β1 agonist dobutamine (32 μg/kg) increased LVP and dP/dtMAX in both genotypes. After return to “blocked” baseline, tyramine-induced (200 μg/kg) release of endogenous NE stimulated LVP and dP/dtMAX. Dobutamine and tyramine stimulated increases in heart rate as well, but heart rate in p75NTR−/− mice was lower than wildtype. Differences between groups were analyzed by two-way ANOVA, and horizontal bars denote sham vs. MI differences, *p<0.05, **p<0.01; Mean ± SEM, n=6.
Table 2.
dP/dtMIN (mmhg/s), mean±sem
| 3 Day | WT Sham (n=6) | WT MI (n=4) | p75−/− Sham (n=6) | p75−/− MI (n=6) |
|---|---|---|---|---|
| Basal | −10458 ± 802 | −7803 ± 1256 | −8955 ± 1192 | −7381 ± 621 |
| Gang. Block | −6394 ± 721 | −4949 ± 502 | −4965 ± 856 | −4184 ± 381 |
| Dobutamine | −7723 ± 341 | −7449 ± 704 | −6792 ± 903 | −6624 ± 313 |
| Tyramine | −8811 ± 1055 | −7938 ± 914 | 8309 ± 757 | 5982 ± 475 |
|
| ||||
| 7 Day | WT Sham (n=6) | WT MI (n=6) | p75−/− Sham (n=7) | p75−/− MI (n=6) |
|
| ||||
| Basal | −10587 ± 1400 | −8666 ± 379 | −9165 ± 1122 | −9197 ± 843 |
| Gang. Block | −7790 ± 923 | −6237 ± 484 | −5620 ± 94 | −5124 ± 397 |
| Dobutamine | −9831 ± 1016 | −8746 ± 608 | −7333 ± 868 | −7968 ± 665 |
| Tyramine | −11335 ± 1321 | −8268 ± 197 | −8807 ± 753 | −7667 ± 594 |
Beta 1 adrenergic receptor (β1AR) expression is low in p75NTR−/− ventricles compared to WT (Lorentz et al., 2010), while NE content in the base of the p75NTR−/− ventricle is elevated (Fig. 3). We used the β1agonist dobutamine to directly assess cardiac responses, and we used tyramine to provoke release of endogenous NE. Dobutamine stimulation of β1AR resulted in similar LVP and dP/dtMAX in both genotypes three days after surgery (Fig. 4), despite decreased β1AR in p75NTR−/− hearts. LVP was not significantly lower after infarction, but dP/dtMAX was decreased by MI. Stimulating release of endogenous NE with tyramine led to similar LVP and dP/dtMAX in both genotypes, with both parameters significantly lower in the post-MI hearts (Fig. 4). Heart rate was lower in p75NTR−/− mice compared to WT following stimulation with dobutamine or tyramine, but myocardial infarction did not alter dobutamine or tyramine-stimulated heart rate (Fig. 4).
A week after sham or ischemia-reperfusion surgery several significant differences were observed between the genotypes, which was a striking change from the earlier time point. Basal LVP and dP/dtMAX were significantly lower in p75NTR−/− hearts compared to WT (Fig. 5). Myocardial infarction further depressed LVP in both genotypes, but did not alter dP/dtMAX (Fig. 5). Differences between the genotypes persisted after ganglionic blockade, with p75NTR−/− parameters low compared to WT. Dobutamine stimulation of β1AR resulted in similar dP/dtMAX in both genotypes, but dobutamine-stimulated LVP was significantly lower in p75NTR−/− hearts compared to WT and lower in the post-MI hearts compared to shams. Given the low basal dP/dtMAX in p75NTR−/− sham hearts, we calculated the difference after dobutamine and found that dobutamine stimulated a significantly greater increase in dP/dtMAX in p75NTR−/− shams (5978±950 mmHg/s) than in WT shams (2390±979 mmHg/s) (*p<0.05, two-way ANOVA with Bonferroni post-test). Stimulating release of endogenous NE with tyramine resulted in LVP and dP/dtMAX that were significantly lower in the p75NTR−/− genotype. Myocardial infarction significantly decreased the pressure generated by the left ventricle after tyramine administration, but dP/dtMAX was not altered significantly by MI. Heart rate was not significantly different between the genotypes or surgical groups a week after surgery (data not shown).
Figure 5. LVP and dP/dtMAX 7 days after ischemia-reperfusion.

LVP (A) and dP/dtMAX (B) were measured in WT (open squares) and p75NTR−/− (filled squares) mice 7 days after sham (Sh) or ischemia-reperfusion (MI) surgery. Far left shows baseline LVP and dP/dtMAX, followed by ganglionic block (5 mg/kg hexamethonium). Both parameters were lower in p75NTR−/− hearts than WT. The β1 agonist dobutamine (32 μg/kg) increased LVP and dP/dtMAX in both genotypes, but LVP remained lower in p75NTR−/−. After return to “blocked” baseline, tyramine (200 μg/kg) was infused to induce release of endogenous NE, stimulating LVP and dP/dtMAX in both genotypes. Both parameters were lower in p75NTR−/− hearts than WT after tyramine. Differences between groups were analyzed by two-way ANOVA; horizontal bars in (A) denote sham vs. MI differences. *p<0.05, **p<0.01, ***p<0.001; Mean ± SEM, n=6, except WT MI n=4. In addition to the group differences that are shown, several subgroup differences were identified by a Bonferroni post-test. p75NTR−/− shams were significantly lower than WT shams in all conditions except LVP after ganglionic block, and dP/dtMAX after dobutamine. In contrast, dP/dtMAX after tyramine was the only condition where p75NTR−/− MI was significantly lower than WT MI.
Discussion
Several studies have examined sympathetic transmission and function in p75NTR−/− mice, and have identified effects on development, axon growth, and NE synthesis that might impact cardiac function. The lack of p75NTR alters synapse formation in sympathetic ganglia (Sharma et al., 2010), the excitability of post-ganglionic neurons (Luther and Birren, 2006; Luther and Birren, 2009), and increases cardiac NE content (Fig. 3)(Habecker et al., 2008; Lorentz et al., 2010). The lack of p75NTR also delays sympathetic innervation of the heart (Kuruvilla et al., 2004), which may alter myocyte development. In addition, the p75NTR−/− left ventricular subendocardium has few sympathetic fibers while the subepicardium has normal innervation density (Lorentz et al., 2010). Finally, arterial pressure is low in p75NTR−/− mice (Lorentz et al., 2010), possibly due to decreased sympathetic innervation of vascular smooth muscle (Long et al., 2009). Low arterial pressure is consistent with the low LVP and dP/dtMAX observed in the p75NTR−/− left ventricle. However, despite decreases in several hemodynamic parameters, stroke volume is normal in the p75NTR−/− heart (Lorentz et al., 2010). Thus, p75NTR impacts many different aspects of sympathetic cardiovascular control.
Sympathetic transmission to the heart is selectively stimulated following myocardial infarction (Ramchandra et al., 2009; Rundqvist et al., 1997), and this adaptation may be exacerbated in p75NTR−/− mice due to increased NGF signaling. First, transmission may be enhanced due to increased synapse formation (Sharma et al., 2010), neuron excitability (Luther and Birren, 2009), and NE content (Fig. 3)(Habecker et al., 2008; Lorentz et al., 2010). Second, sympathetic transmission may be enhanced due to elevated NGF in the heart following ischemia-reperfusion (Abe et al., 1997; Hiltunen et al., 2001), and increased Trk signaling in p75NTR−/− sympathetic neurons (Hannila et al., 2004). Our data show that TrkA expression is normal in p75NTR−/− cardiac sympathetic neurons, and that TrkA and p75NTR expression are not altered by ischemia-reperfusion. One limitation of this study is that we have not measured NGF levels in the p75NTR−/− ventricle after ischemia-reperfusion. NGF stimulates cardiac myocyte survival (Meloni et al., 2010), however, so the identical infarct/risk ratios in P75NTR−/− and WT hearts suggest that cardiac NGF levels are similar in both genotypes.
Enhanced sympathetic transmission in the heart might result in greater contractility and pressure generation, bringing p75NTR−/− left ventricular parameters up to the level seen in wildtype hearts. Three days after surgery LVP and dP/dtMAX were similar in both genotypes, consistent with enhanced sympathetic transmission, but that effect was transient so that seven days after surgery the p75NTR−/− hearts exhibited decreased LVP and dP/dtMAX compared to WT hearts. Comparison of the two time points revealed that WT hearts exhibited greater functional recovery at seven days compared to the p75NTR−/− hearts. For example, stimulation with dobutamine a week after MI generated an average LVP that was 15 mmHg higher in WT mice than seen 3 days after surgery, compared to a 6 mmHg increase in p75NTR−/−. A similar trend was observed in the sham operated animals, so that there were significant differences between genotypes in almost all ventricular parameters a week after surgery – similar to what was previously observed in unoperated control animals (Lorentz et al., 2010). The increased dobutamine responsiveness in WT hearts seven days after MI is consistent with decreased expression of β1AR in p75NTR−/− hearts (Lorentz et al., 2010). An important caveat is that LVP and dP/dtMAX are significantly affected by pre-load and after-load, which are decreased in p75NTR−/− mice (Lorentz et al., 2010). A load-independent assessment of LV function in WT vs. p75NTR−/− mice awaits further characterization, but measuring cardiac function by echocardiography revealed normal stroke volume in control p75NTR−/− hearts (Lorentz et al., 2010). This suggests that the decreased LVP and dP/dtMAX observed in p75NTR−/− hearts seven days after MI may not result in significantly altered cardiac output.
Cardiac output is affected by heart rate in addition to stroke volume, and previous studies indicate that heart rate is significantly lower in mice lacking p75NTR (Habecker et al., 2008; Lorentz et al., 2010). The significant decrease in heart rate previously identified in conscious p75NTR−/− animals was not apparent in this study, which showed only a trend toward lower heart rate. The blunted change in heart rate was likely due to the use of isoflurane anesthesia (Zuurbier et al., 2002). Heart rate is controlled by both sympathetic and parasympathetic transmission, and a recent study showed that mouse cardiac parasympathetic neurons express p75NTR (Hoard et al., 2008). However, analysis of the p75NTR−/− atrial innervation suggests that parasympathetic innervation density is normal, and blockade of cholinergic transmission with atropine raises heart rate to a similar extent in p75NTR−/− and WT mice (Habecker et al., 2008). Thus, any changes in heart rate observed in p75NTR null mice are likely due to altered sympathetic innervation.
Cardiac NE levels were also different between the genotypes, but there was regional variation in NE, consistent with previous studies (Li et al., 2004; Parrish et al., 2008; Parrish et al., 2009b). We expected that NE would remain similar after MI in the base of the ventricles but decreased in the middle/lower ventricle below the ligation due to loss of nerve fibers and depletion of TH. We hypothesized that increased TrkA signaling in the absence of p75NTR would blunt the loss of NE. NE content in the p75NTR−/− atria and right ventricle are higher than WT (Habecker et al., 2008; Lorentz et al., 2010), and NE content across the entire LV is identical p75NTR−/− and WT mice, despite fewer nerve fibers in the p75NTR−/− hearts (Lorentz et al., 2010). All of those data are consistent with enhanced NE production in p75NTR−/− sympathetic neurons. However, NE content was similar to WT or even decreased in the middle/lower p75NTR−/− ventricle after sham surgery and MI. Interpretation of NE data in the left ventricle is complicated by the fact that the subendocardial layer of muscle is essentially devoid of sympathetic nerve fibers (Lorentz et al., 2010). Tissue from the middle and lower portion of the ventricle likely contains a larger fraction of subendocardium than the samples collected from the base of the ventricle, and that may account for the dramatic difference in NE content in the p75NTR−/− base compared to lower in the heart. However, we cannot exclude the possibility that NE content per nerve fiber is significantly different in distinct areas of the left ventricle.
We examined GAL, PACAP, and VIP mRNA because previous studies identified increased expression of these peptides in cardiac sympathetic neurons (Alston et al., 2010; Habecker et al., 2005), and because altered neurotrophin signaling in mice lacking p75NTR leads to changes in peptide expression in sensory neurons (McMahon et al., 1995; Patel et al., 2000). Peptide mRNA levels were identical in cardiac sympathetic neurons from sham animals of both genotypes, suggesting that neurotrophin signaling is not a major regulator of peptide expression in sympathetic neurons from the stellate ganglia. No peptide mRNAs were increased significantly after MI in either genotype, which was surprising given the earlier data. GAL mRNA is increased in rat sympathetic neurons after MI (Habecker et al., 2005) but not in mouse (Alston et al., 2010), consistent with the lack of increased GAL mRNA in the current study. However, we expected PACAP and VIP mRNA to increase in both genotypes after MI since mouse studies revealed cytokine-induced increases in both peptides (Alston et al., 2010). The current study used a 30 minute LAD ligation, while the earlier work used a 45 min LAD ligation. The longer ligation time results in a significantly larger infarct (Parrish et al., 2009b), and presumably a larger inflammatory response which is a major stimulus for peptide expression. In any case, the lack of p75NTR did not affect sympathetic peptide expression after sham or myocardial infarction surgery
In summary, we used mice lacking p75NTR to determine if enhanced sympathetic transmission would blunt the loss of ventricular function after myocardial infarction. The lack of p75NTR did result in a transient “rescue” of ventricular parameters up to wildtype levels three days after sham or ischemia-reperfusion surgery, in contrast to low LVP and dP/dtMAX previously reported in p75NTR−/− unoperated control animals (Lorentz et al., 2010). However, week after surgery LVP and dP/dtMAX were once again low in p75NTR−/− compared to WT mice. The greatest differences were observed in sham animals when ganglionic transmission was intact, suggesting that increased sympathetic transmission after MI may continue to have a small effect. Nevertheless, high cardiac NE and enhanced transmission in p75NTR−/− sympathetic neurons are not sufficient to compensate in a sustained manner to restore cardiac function up to the levels seen in wildtype hearts.
Acknowledgments
This work was supported by AHA 0715669Z & 09PRE2110052 (C.U.L), AHA 0555553Z (B.A.H) and NIH HL093056 (B.A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abe T, Morgan DA, Gutterman DD. Protective role of nerve growth factor against postischemic dysfunction of sympathetic coronary innervation. Circulation. 1997;95:213–220. doi: 10.1161/01.cir.95.1.213. [DOI] [PubMed] [Google Scholar]
- Alston EN, Parrish DC, Hasan W, Tharp K, Pahlmeyer L, Habecker BA. Cardiac ischemia-reperfusion regulates sympathetic neuropeptide expression through gp130-dependent and independent mechanisms. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MJ, Mueller TM, Henry DP, Felten SY, Zipes DP. Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation. 1983;67:787–796. doi: 10.1161/01.cir.67.4.787. [DOI] [PubMed] [Google Scholar]
- Birren SJ, Lo L, Anderson DJ. Sympathetic neuroblasts undergo a developmental switch in trophic dependence. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, Lai WW, Karagueuzian HS, Wolf PL, Fishbein MC, Chen PS. Nerve sprouting and sudden cardiac death. Circ Res. 2000a;86:816–821. doi: 10.1161/01.res.86.7.816. [DOI] [PubMed] [Google Scholar]
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000b;101:1960–1969. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armaninl MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Dae MW, Lee RJ, Ursell PC, Chin MC, Stillson CA, Moise NS. Heterogeneous sympathetic innervation in German shepherd dogs with inherited ventricular arrhythmia and sudden cardiac death. Circulation. 1997;96:1337–1342. doi: 10.1161/01.cir.96.4.1337. [DOI] [PubMed] [Google Scholar]
- Gasz B, Racz B, Roth E, Borsiczky B, Ferencz A, Tamas A, Cserepes B, Lubics A, Gallyas F, Jr, Toth G, Lengvari I, Reglodi D. Pituitary adenylate cyclase activating polypeptide protects cardiomyocytes against oxidative stress-induced apoptosis. Peptides. 2006;27:87–94. doi: 10.1016/j.peptides.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Heterogeneous Requirement of NGF for Sympathetic Target Innervation In Vivo. J Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LN, Smith PA, Stoker JB, Mackintosh AF, Mary DA. Time course of sympathetic neural hyperactivity after uncomplicated acute myocardial infarction. Circulation. 2002;106:793–797. doi: 10.1161/01.cir.0000025610.14665.21. [DOI] [PubMed] [Google Scholar]
- Habecker BA, Bilimoria P, Linick C, Gritman K, Lorentz CU, Woodward W, Birren SJ. Regulation of cardiac innervation and function via the p75 neurotrophin receptor. Auton Neurosci. 2008;140:40–48. doi: 10.1016/j.autneu.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habecker BA, Gritman KR, Willison BD, Van Winkle DM. Myocardial Infarction Stimulates Galanin Expression in Cardiac Sympathetic Neurons. Neuropeptides. 2005;39:89–95. doi: 10.1016/j.npep.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Hannila SS, Lawrance GM, Ross GM, Kawaja MD. TrkA and mitogen-activated protein kinase phosphorylation are enhanced in sympathetic neurons lacking functional p75 neurotrophin receptor expression. Eur J Neurosci. 2004;19:2903–2908. doi: 10.1111/j.0953-816X.2004.03381.x. [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006;1124:142–154. doi: 10.1016/j.brainres.2006.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Hoard JL, Hoover DB, Mabe AM, Blakely RD, Feng N, Paolocci N. Cholinergic neurons of mouse intrinsic cardiac ganglia contain noradrenergic enzymes, norepinephrine transporters, and the neurotrophin receptors tropomyosin-related kinase A and p75. Neuroscience. 2008;156:129–142. doi: 10.1016/j.neuroscience.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Karve A, Shah I, Bowers MC, DiPette DJ, Supowit SC, Abela GS. Deletion of the mouse {alpha}-calcitonin gene-related peptide gene increases the vulnerability of the heart to ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2008;294:H1291–H1297. doi: 10.1152/ajpheart.00749.2007. [DOI] [PubMed] [Google Scholar]
- Ieda M, Kanazawa H, Ieda Y, Kimura K, Matsumura K, Tomita Y, Yagi T, Onizuka T, Shimoji K, Ogawa S, Makino S, Sano M, Fukuda K. Nerve growth factor is critical for cardiac sensory innervation and rescues neuropathy in diabetic hearts. Circulation. 2006;114:2351–2363. doi: 10.1161/CIRCULATIONAHA.106.627588. [DOI] [PubMed] [Google Scholar]
- Inoue H, Zipes DP. Time course of denervation of efferent sympathetic and vagal nerves after occlusion of the coronary artery in the canine heart. Circ Res. 1988;62:1111–1120. doi: 10.1161/01.res.62.6.1111. [DOI] [PubMed] [Google Scholar]
- Jahed A, Kawaja MD. The influences of p75 neurotrophin receptor and brain-derived neurotrophic factor in the sympathetic innervation of target tissues during murine postnatal development. Auton Neurosci. 2005;118:32–42. doi: 10.1016/j.autneu.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Karlsberg RP, Penkoske PA, Cryer PE, Corr PB, Roberts R. Rapid activation of the sympathetic nervous system following coronary artery occlusion: relationship to infarct size, site, and haemodynamic impact. Cardiovasc Res. 1979;13:523–531. doi: 10.1093/cvr/13.9.523. [DOI] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci. 1999;19:5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118:243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Lee KF, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–H2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Li YJ, Peng J. The cardioprotection of calcitonin gene-related peptide-mediated preconditioning. Eur J Pharmacol. 2002;442:173–177. doi: 10.1016/s0014-2999(02)01538-8. [DOI] [PubMed] [Google Scholar]
- Long JB, Jay SM, Segal SS, Madri JA. VEGF-A and Semaphorin3A: Modulators of vascular sympathetic innervation. Dev Biol. 2009;334:119–132. doi: 10.1016/j.ydbio.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentz CU, Alston EN, Belcik JT, Lindner JR, Giraud GD, Habecker BA. Heterogeneous ventricular sympathetic innervation, altered {beta} adrenergic receptor expression, and rhythm instability in mice lacking p75 neurotrophin receptor. Am J Physiol Heart Circ Physiol. 2010;298:H1652–H1660. doi: 10.1152/ajpheart.01128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol. 2006;96:946–958. doi: 10.1152/jn.01078.2005. [DOI] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. p75 and TrkA signaling regulates sympathetic neuronal firing patterns via differential modulation of voltage-gated currents. J Neurosci. 2009;29:5411–5424. doi: 10.1523/JNEUROSCI.3503-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Max SR, Rohrer H, Otten U, Thoenen H. Nerve growth factor-mediated induction of tyrosine hydroxylase in rat superior cervical ganglia in vitro. J Biol Chem. 1978;253:8013–8015. [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van LS, Spillmann F, Campesi I, Madeddu P, Quaini F, Emanueli C. Nerve Growth Factor Promotes Cardiac Repair following Myocardial Infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minardo JD, Tuli MM, Mock BH, Weiner RE, Pride HP, Wellman HN, Zipes DP. Scintigraphic and electrophysiological evidence of canine myocardial sympathetic denervation and reinnervation produced by myocardial infarction or phenol application. Circulation. 1988;78:1008–1019. doi: 10.1161/01.cir.78.4.1008. [DOI] [PubMed] [Google Scholar]
- Oh YS, Jong AY, Kim DT, Li H, Wang C, Zemljic-Harpf A, Ross RS, Fishbein MC, Chen PS, Chen LS. Spatial distribution of nerve sprouting after myocardial infarction in mice. Heart Rhythm. 2006;3:728–736. doi: 10.1016/j.hrthm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Hermes SM, Aicher SA, Nkadi P, Woodward WR, Stubbusch J, Gardner RT, Habecker BA. The absence of gp130 in dopamine {beta} hydroxylase-expressing neurons leads to autonomic imbalance and increased reperfusion arrhythmias. Am J Physiol Heart Circ Physiol. 2009a;297:H960–H967. doi: 10.1152/ajpheart.00409.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Nkadi P, Woodward WR, Schutz G, Habecker BA. Infarction-induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol. 2009b;95:304–314. doi: 10.1113/expphysiol.2009.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish DC, Gritman K, Van Winkle DM, Woodward WR, Bader M, Habecker BA. Postinfarct sympathetic hyperactivity differentially stimulates expression of tyrosine hydroxylase and norepinephrine transporter. Am J Physiol Heart Circ Physiol. 2008;294:H99–H106. doi: 10.1152/ajpheart.00533.2007. [DOI] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. doi: 10.1016/s0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Racz B, Gasz B, Gallyas F, Jr, Kiss P, Tamas A, Szanto Z, Lubics A, Lengvari I, Toth G, Hegyi O, Roth E, Reglodi D. PKA-Bad-14–3–3 and Akt-Bad-14–3–3 signaling pathways are involved in the protective effects of PACAP against ischemia/reperfusion-induced cardiomyocyte apoptosis. Regul Pept. 2008;145:105–115. doi: 10.1016/j.regpep.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Ramchandra R, Hood SG, Denton DA, Woods RL, McKinley MJ, McAllen RM, May CN. Basis for the preferential activation of cardiac sympathetic nerve activity in heart failure. Proc Natl Acad Sci U S A. 2009;106:924–928. doi: 10.1073/pnas.0811929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundqvist B, Elam M, Bergmann-Sverrisdottir Y, Eisenhofer G, Friberg P. Increased cardiac adrenergic drive precedes generalized sympathetic activation in human heart failure. Circulation. 1997;95:169–175. doi: 10.1161/01.cir.95.1.169. [DOI] [PubMed] [Google Scholar]
- Shadiack AM, Sun Y, Zigmond RE. Nerve Growth Factor Antiserum Induces Axotomy-Like Changes in Neuropeptide Expression in Intact Sympathetic and Sensory Neurons. J Neurosci. 2001;21:363–371. doi: 10.1523/JNEUROSCI.21-02-00363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Deppmann CD, Harrington AW, St HC, Chen ZY, Lee FS, Ginty DD. Long-Distance Control of Synapse Assembly by Target-Derived NGF. Neuron. 2010;67:422–434. doi: 10.1016/j.neuron.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton MS, Tuli MM, Radtke NL, Heger JJ, Miles WM, Mock BH, Burt RW, Wellman HN, Zipes DP. Regional sympathetic denervation after myocardial infarction in humans detected noninvasively using I-123-metaiodobenzylguanidine. J Am Coll Cardiol. 1989;14:1519–1526. doi: 10.1016/0735-1097(89)90391-4. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Comparison between the effect of neuronal activity and nerve growth factor on the enzymes involved in the synthesis of norepinephrine. Pharmacol Rev. 1972;24:255–267. [PubMed] [Google Scholar]
- Wang L, Wang DH. TRPV1 Gene Knockout Impairs Postischemic Recovery in Isolated Perfused Heart in Mice. Circulation. 2005;112:3617–3623. doi: 10.1161/CIRCULATIONAHA.105.556274. [DOI] [PubMed] [Google Scholar]
- Wernli G, Hasan W, Bhattacherjee A, van RN, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction. Basic Res Cardiol. 2009;104:681–693. doi: 10.1007/s00395-009-0033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri N, Chao MV. Mechanisms of neurotrophin receptor signalling. Biochem Soc Trans. 2006;34:607–611. doi: 10.1042/BST0340607. [DOI] [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95:76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
- Zipes DP. Influence of myocardial ischemia and infarction on autonomic innervation of heart. Circulation. 1990;82:1095–1105. doi: 10.1161/01.cir.82.4.1095. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Emons VM, Ince C. Hemodynamics of anesthetized ventilated mouse models: aspects of anesthetics, fluid support, and strain. Am J Physiol Heart Circ Physiol. 2002;282:H2099–H2105. doi: 10.1152/ajpheart.01002.2001. [DOI] [PubMed] [Google Scholar]