

Abstract
Budding yeast possesses a checkpoint-dependent mechanism of delaying G1 progression in response to UV and ionizing radiation DNA damage. We have shown that after a pulse of DNA damage in G1 with the alkylating agent MMS, there is also a MEC1-, RAD53-, and RAD9-dependent delay in G1. This delay occurs at or before Start, as the MMS-treated cells do not bud, remain sensitive to α-factor, and have low CLN1 and CLN2 transcript levels for a longer time than untreated cells. We further show that MMS directly and reversibly down-regulates CLN1 and CLN2 transcript levels. The initial drop in CLN transcript levels in MMS is not RAD53 dependent, but the kinetics of reaccumulation of CLN messages as cells recover from the damage is faster in rad53-11 cells than in wild type cells. This is not an indirect effect of faster progression through G1, because CLN transcripts reaccumulate faster in rad53-11 mutants arrested in G1 as well. In addition, the recovery of CLN mRNA levels can be also hastened by a SWI6 deletion or by overexpression of the truncated Swi4 (Swi4-t) that lacks the carboxy-terminal domain through which Swi4 associates with Swi6. This indicates that both Rad53 and Swi6 are negative regulators of CLN expression after DNA damage. Finally, Swi6 undergoes an MMS-inducible, RAD53-dependent phosphorylation in G1 cells, and Rad53, immunoprecipitated from MMS-treated cells, phosphorylates Swi6 in vitro. On the basis of these observations, we suggest that the Rad53-dependent phosphorylation of Swi6 may delay the transition to S phase by inhibiting CLN transcription.
Keywords: MMS, CLN1, CLN2, RAD53, SWI6, cell cycle, checkpoint
DNA damage checkpoints ensure that a cell with lesions in its DNA does not divide before the damage is eradicated. In the yeast Saccharomyces cerevisiae, three DNA damage-inducible checkpoints have been identified that operate in G1, S, and G2 phases of the cell cycle (Weinert and Hartwell 1989; Siede et al. 1993, 1994; Weinert et al. 1994; Paulovich and Hartwell 1995; Paulovich et al. 1997a). Two genes, MEC1/ESR1/SAD3 and RAD53/MEC2/SPK1/SAD1, appear important for the performance of all three checkpoints (Allen et al. 1994; Weinert et al. 1994; Paulovich and Hartwell 1995; Siede et al. 1996). In addition, RAD9, RAD17, RAD24, and MEC3 are involved in G1 and G2 checkpoints (Siede et al. 1993, 1994; Weinert et al. 1994). These checkpoint gene products delay the cell cycle when DNA damage is present, which allows time for repair and improves survival in the presence of damaging agents. MEC1 encodes a member of a PI kinase family and has homologs in fission yeast, Drosophila, mice, and humans (Al-Khodairy and Carr 1992; Hari et al. 1995; Morrow et al. 1995; Savitsky et al. 1995; Pecker et al. 1996). The kinase activity of Mec1 has not been demonstrated, although it is thought that Mec1, its close yeast homolog Tel1, and similar kinases, like ATM of humans, are protein, rather than lipid, kinases (Hunter 1995). RAD53 encodes a dual specificity kinase, whose catalytic activity has been demonstrated, but its substrate specificity remains unidentified (Zheng et al. 1993; Sun et al. 1996).
Rad9, Rad17, Rad24, and Mec3 are thought to be involved in recognition of damage and initiating a signal transduction cascade that activates Mec1 and Rad53 (Lydall and Weinert 1995; Navas et al. 1996; Sanchez et al. 1996; Sun et al. 1996). In turn, Mec1 and Rad53 transmit the signal to critical targets including cell cycle machinery and DNA repair enzymes. Two biochemical events have been detected upon DNA damage in cells. First, Rad53 undergoes phosphorylation that is dependent on Mec1, Rad9, Rad17, Rad24, and Mec3 (Navas et al. 1996; Sanchez et al. 1996; Sun et al. 1996). Second, there is an induction of transcription of a large group of genes that are involved in DNA replication and repair (Aboussekhra et al. 1996; Kiser and Weinert 1996; Navas et al. 1996). However, not much is known with regard to the mechanisms of the G1, S, or G2 cell cycle arrests in response to the DNA damage.
Recently it has been shown that yeast cells irradiated by UV or X rays in early G1 can delay the onset of Start in a checkpoint-dependent manner (Siede et al. 1993, 1994). Start, in this context, is operationally defined as the point at which cells have committed to the mitotic cell cycle and are resistant to arrest by α-factor. This commitment process involves accumulation of threshold levels of G1 cyclin proteins, Cln1 and Cln2, which in turn triggers destruction of B-type cyclin inhibitor Sic1 and thereby irreversibly commits cells to S phase and mitosis (for review, see Cross 1995; King et al. 1996; Nasmyth 1996). The rate of Cln1 and Cln2 accumulation is regulated primarily at the transcript level, which can vary depending on growth conditions and affect the length of the G1 phase (Baroni et al. 1994; Tokiwa et al. 1994; Willems et al. 1996).
In this work we show that MMS, a DNA-damaging agent, can induce a Mec1-, Rad53-, and Rad9-dependent delay before Start that is similar to the one observed by Siede and coworkers after UV or X-ray irradiation (Siede et al. 1993, 1994). The MMS-induced delay is characterized by a prolonged inhibition of CLN1 and CLN2 transcription, followed by a gradual reaccumulation of these transcripts and resumption of the cell cycle. The reaccumulation of CLN mRNA is faster in a rad53-11 checkpoint mutant and in a swi6 mutant strain than in the wild type. This is not an indirect consequence of the fact that these mutants progress through G1 faster, because it can also be observed in G1-arrested cells. Overproduction of a truncated form of Swi4, which activates CLN1 and CLN2 transcription but is independent of Swi6 in its activity, can also reduce the delay of S phase upon MMS treatment. This suggests the possibility that Rad53 might act on Swi6 to inhibit CLN transcription in damaged G1 cells. We have found that Swi6 undergoes a Rad53-dependent phosphorylation that is induced in the presence of MMS in vivo. The kinase responsible is likely to be Rad53 or an associated kinase because Swi6 can be phosphorylated by immunoprecipitated Rad53 in vitro in a pattern resembling the damage-induced pattern in vivo. On the basis of these observations, we suggest that reduced transcription of CLN1 and CLN2 owing to an inhibitory phosphorylation of Swi6 contributes to the checkpoint-mediated delay of Start.
Results
MMS damage in early G1 delays the onset of CLN transcription
MMS is a DNA-alkylating agent that methylates bases and can induce a variety of lesions including strand breaks (Dhillon and Hoekstra 1994). We have used this agent to study the G1 checkpoint that delays the onset of S phase in the presence of DNA lesions. Figure 1 shows the behavior of wild-type cells that were arrested in early G1 by α-factor, treated or not treated with 0.1% MMS for 30 min, and then allowed to progress through the cell cycle by removal of MMS and α-factor. Samples were taken to determine budding index and DNA content. As can be seen from Figure 1A, the MMS-treated cells remain unbudded for at least 45 min, whereas the untreated cells bud between 15 and 30 min after release from the α-factor. FACS profiles of these cultures are consistent with the budding indexes and show (Fig. 1B) that the MMS-treated cells maintain a 1N DNA content and do not enter S phase for ∼45 min. In contrast, the untreated cells are already well into S phase 30 min after the release from α-factor. In addition to the slow entry into S phase, a considerable fraction of the MMS-treated cells are still in S phase 105 min after release. This very slow S phase is consistent with previous observations that MMS can induce the S-phase checkpoint (Paulovich and Hartwell 1995).
Figure 1.

Exposure of G1 cells to MMS causes a delay of S phase. Wild-type (BY2235) cells were arrested by α-factor in G1, and one-half of the culture was incubated with 0.1% MMS for 30 min. Then MMS was inactivated as described in Materials and Methods, and both cultures were filtered out of α-factor, resuspended in the fresh media, and allowed to progress into the cell cycle. These cells are 25%–50% viable after such treatment (data not shown). Aliquots of cultures were taken before the release (time point 0) and every 15 min after to determine the percentage of budded cells (A) and DNA content (B). For the latter, samples were fixed, stained with propidium iodide as described in Materials and Methods, and subjected to FACS analysis. (C) A separate culture was treated as described for A and B, and aliquots for RNA isolation were taken before the release from α-factor and every 10–15 min after. These RNAs were analyzed by S1 protection with radiolabeled probes against CLN1 and SIR3 RNAs. The protected fragments corresponding to these RNAs are marked. (D) The S1 protection data presented in C were quantitated using PhosphorImager software. CLN1 mRNA levels were normalized to the internal control mRNA (SIR3) levels and plotted.
UV irradiation in early G1 can transiently arrest cells before Start (Siede et al. 1993, 1994), as judged by the fact that the arrested cells are sensitive to α-factor. Cells arrested in G1 by MMS treatment are also α-factor sensitive (see below). Moreover, this pre-Start arrest is reflected in CLN transcript levels. As seen in Figure 1, C and D, whereas untreated cells rapidly induce a normal burst of CLN1 transcript 10 min after the α-factor release, MMS-treated cells are greatly delayed in their accumulation of CLN1 transcript. Two simple possibilities could account for this. First, MMS treatment could inhibit some early event and prevent progression to the time at which CLN transcription commences. Alternatively, MMS could directly inhibit CLN transcription, and that could be responsible for delaying Start.
CLN1 and CLN2 transcripts are reversibly down-regulated upon MMS treatment in G1
To address in more detail whether CLN1 and CLN2 transcripts are directly affected by MMS addition, wild-type yeast cultures were arrested in G1 with α-factor and released into fresh media containing increasing doses of MMS. These cells were incubated for 20 min and harvested, and then CLN1 and CLN2 transcript levels were measured. We found that the CLN transcripts were specifically down-regulated in response to MMS addition in a dose-dependent manner (data not shown). To see if cell cycle progression is required to see this down-regulation, we used a cdc4 strain, which arrests at the G1/S transition upon incubation at nonpermissive temperature (37°C). This strain was first incubated at 37°C for 3 hr to establish a uniform arrest. Then MMS was added for 30 min to an aliquot of these arrested cells, and samples were harvested for mRNA measurements. CLN1 levels were reduced in the MMS-treated cells by 5- to 10-fold compared with untreated controls (Fig. 2, lanes 1,2). The same result was obtained with cdc28 cells arrested in G1 (data not shown). This indicates that MMS treatment significantly inhibits CLN transcription or decreases mRNA stability. We then followed CLN transcript levels for 2 hr after MMS was removed and found that these transcripts remain low for up to 60 min, after which they start to reaccumulate (Fig. 2B). Therefore, down-regulation of CLN transcription by MMS is prolonged but reversible and can occur independently of cell cycle progression.
Figure 2.
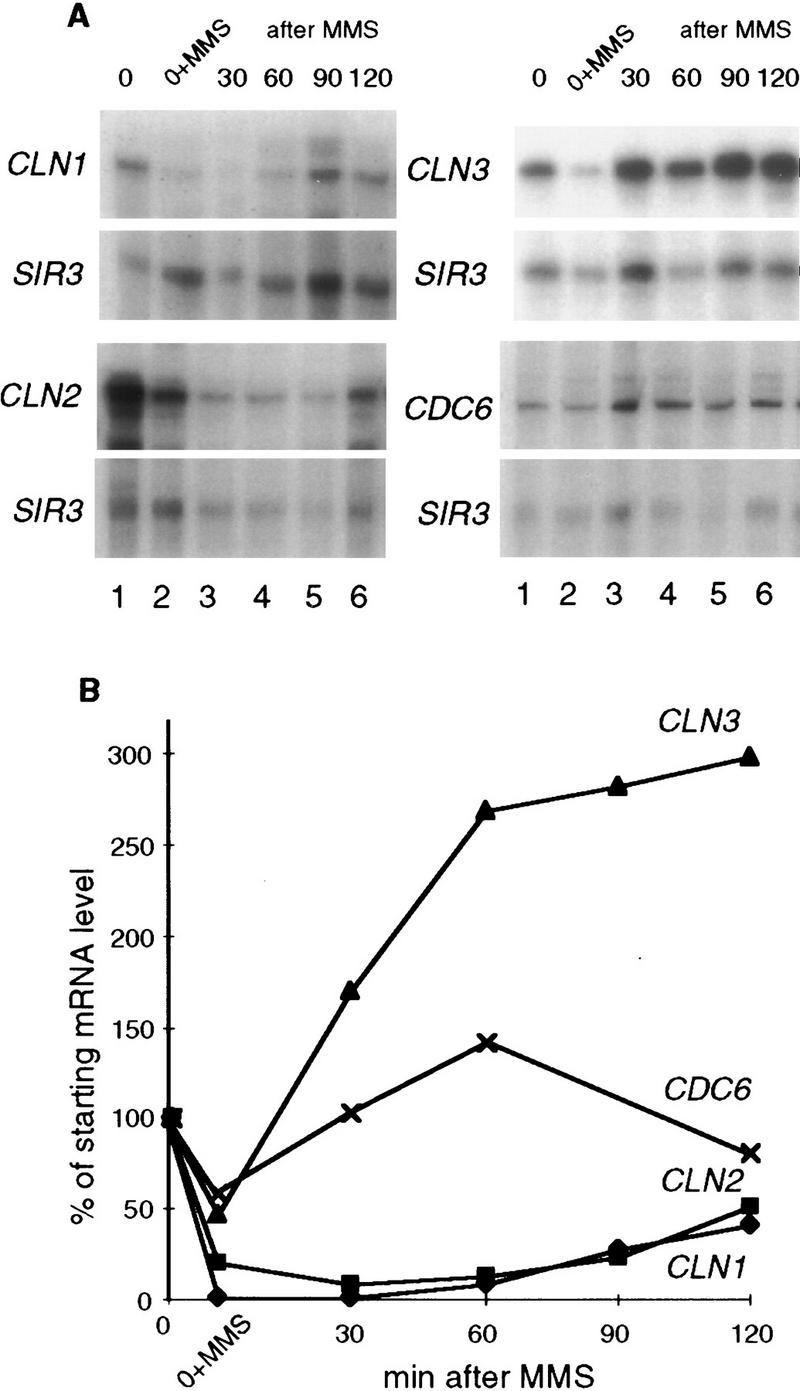
MMS causes reversible down-regulation of CLN1 and CLN2 mRNAs. (A) The cdc4-1 (BY665) strain was arrested as described above and divided in two cultures, and 0.1% MMS was added to one part for 30 min, whereas the other part was incubated without MMS for 30 min and harvested (0). An aliquot was harvested out of the MMS-containing culture (0+MMS), and in the rest of the culture, MMS was inactivated and the culture was filtered into the fresh 37°C media and allowed to recover from MMS at this temperature. Aliquots were taken throughout the time of recovery (30, 60, 90, and 120 min after the release from MMS). CLN1, CLN2, CLN3, CDC6, and SIR3 mRNA levels were measured by S1 protection in these aliquots. (B) mRNA levels shown in A were quantitated, normalized, and plotted. The mRNA levels seen in MMS and after MMS removal are expressed as the percentage of the starting normalized mRNA level seen at 37°C in the untreated culture. Note that the 90-min time point for CDC6 mRNA could not be reliably normalized to its internal control, because the latter was apparently lost from this sample on the gel.
Importantly, other cell cycle-dependent transcripts that peak during G1 phase did not show the same response to MMS addition that was observed for CLN1 and CLN2. For example, the CDC9 transcript, which is known to be DNA damage-inducible (McClanahan and McEntee 1984; Ruby and Szostak 1985), underwent virtually no reduction in MMS and was elevated after MMS removal (data not shown). The CDC6 transcript level was reduced <50% in MMS and was rapidly restored after MMS was removed (Fig. 2). The CLN3 transcript also dropped by 50% in the presence of MMS but was actually induced to a reproducibly higher level shortly after MMS removal. We have noted the presence of STRE elements and three short stretches of homology to the DRE (damage response element) of RNR2 (Elledge and Davis 1989) in the CLN3 promoter, which could be responsible for this damage-induced increase. In addition, the HIS3, MATa1, and SIR3 mRNAs, which were used as internal controls, displayed little or no sensitivity to MMS within the range of MMS concentrations and incubation times applied in our study. Therefore, although high levels of MMS and/or prolonged exposure to it eventually eliminate all transcripts (data not shown), the CLN1 and CLN2 transcripts are consistently more sensitive to it than the other transcripts we have monitored.
Start delay in MMS-treated cells can be reduced by deregulation of CLN transcription
The experiments shown so far suggest that CLN transcript levels may be directly affected by MMS treatment. To see whether this down-regulation of CLN levels is sufficient to explain the observed delay of S phase in damaged cells, we asked whether modest increases in CLN expression could shorten the delay. To do this, we first tried a strain producing elevated levels of the Swi4 transcription factor, which is a primary activator of CLN1 and CLN2 transcription (for review, see Breeden 1996). We found that simply overproducing Swi4 from the GAL promoter does not noticeably reduce the G1 delay after MMS as compared with the wild-type strain (data not shown). However, GAL induction of carboxy-terminally truncated Swi4 (GAL::SWI4-t) can reduce the delay of S phase after MMS treatment. The Swi4-t strain deregulates CLN transcription such that CLN1 message is easily detectable in G1 cells treated with MMS, and then it increases modestly during the recovery period to a peak level that is about twofold higher than that of the wild-type cells (Fig. 3A). Consistent with the deregulated CLN1 levels, Swi4-t cells bud and become α-factor resistant more rapidly than wild-type cells after MMS treatment (Fig. 3B,C). Thus, it appears that increased CLN levels allow a partial bypass of the MMS-induced delay of Start. This is not particularly surprising in view of the observed reduction of the CLN transcripts in response to MMS (Fig. 1), but it argues against an alternative model, which is that the G1 delay mechanism in response to DNA damage operates exclusively via CDK inhibition.
Figure 3.

Overproduction of the carboxy-terminally truncated Swi4, Swi4-t, diminishes the MMS sensitivity of CLN1 mRNA and shortens the delay of budding and of the S-phase entry after MMS treatment. (A) The wild-type W303-1a strains transformed with the vector (pBD860) or with the GAL::SWI4-t plasmid (pBD1168) were arrested by α-factor in rich media with raffinose. Galactose was added to induce Swi4-t overexpression 1 hr before the release, and in 30 min 0.1% MMS was added. After an additional 30 min, cells were released both from MMS and α-factor and allowed to grow in rich media with galactose. Aliquots were collected before the release and in 15-min intervals after it to perform S1 protection measurements of CLN1 and SIR3 RNA levels. These levels were then quantitated, and CLN1 was normalized to SIR3 and plotted. (B) The same two strains were arrested by α-factor as before, and a half of the culture of each strain was treated with 0.1% MMS for 30 min before the release from α-factor arrest. Aliquots were taken to count the percentage of budded cells. Note that the budding results shown in B are the average of two independent experiments. (C) The wild-type W303-1a strain with vector (pBD860) or with the GAL::SWI4-t plasmid (pBD1168) were arrested and treated with MMS as described in B. After the release from α-factor alone or MMS and α-factor, the cultures were allowed to grow in rich media with galactose and aliquots were removed and placed into microtiter plate wells. To these aliquots, α-factor (at 10 μg/ml) was added. α-Factor samples were incubated for 30–45 min, then formaldehyde was added to 4% to fix the cells, and the percentage of budded cells was counted.
Checkpoint mutants have a shorter G1 delay after MMS treatment than wild-type cells
Mutations in MEC1, RAD53, and RAD9 make cells defective in G1 delay upon UV and ionizing radiation damage (Siede et al. 1993, 1994, 1996), and they are sensitive to MMS (Paulovich and Hartwell 1995; Paulovich et al. 1997a). These mutants are also defective in their G1 delay after MMS treatment. Figure 4 shows the kinetics of budding and DNA synthesis in isogenic wild type, rad9Δ, rad53-11, and mec1-1 checkpoint-defective mutants. In the absence of damage, these mutants and the wild-type cells have a similar rate of progression through the G1/S transition (Fig. 4A). By 30 min after release, all three of the untreated strains have budded and entered S phase. However, after treatment with a pulse of MMS damage, (Fig. 4A,B), the wild-type cells delay entry into S phase and are still primarily in G1 as judged by budding (Fig. 4A) and FACS analysis (Fig. 4B) at the 150-min time point, whereas the rad9Δ, rad53-11, and mec1-1 strains go through this transition ∼30 min faster. The checkpoint-defective strains also go through Start earlier than wild type, as judged by α-factor resistance (Fig. 4C). Interestingly, the mec1-1 mutant appears to bud and become α-factor resistant more rapidly than the others. In summary, just as was observed for GAL::SWI4-t, in the absence of checkpoint function, the MMS-induced delay of Start is clearly diminished but not completely eliminated.
Figure 4.

The delay of budding and S-phase entry is shorter in mec1-1, rad53-11, and rad9Δ strains than in the wild type. (A,B) The isogenic wild type (BY2006), mec1-1 (BY2226), rad53-11 (BY2007), and rad9Δ (BY2227) strains were arrested by α-factor and released into fresh media with or without 0.2% MMS. The “no MMS” cultures were allowed to progress into the cell cycle. In the “+MMS” cultures, after incubation for 30 min, MMS was inactivated and the cultures were filtered, resuspended in another change of media, and allowed to grow. (A) Aliquots were taken from treated and untreated cultures to count the percentage of budded cells. Note that the 30-min time point for MMS-treated cultures corresponds to the moment of release from MMS. In B, the DNA content for the cells shown in A was determined by FACS and an example of DNA content distribution for the 150-min time point is presented. The 150-min time point for which FACS data are shown is marked by a box in A. (C) The same four strains were arrested by α-factor, and 0.2% MMS was added for the last 30 min before the release from the pheromone. After that, MMS was inactivated and the cultures were filtered into the fresh media. Samples were taken in 15-min intervals and transferred to microtiter plates with 10 μg/ml of α-factor, as described in the legend to Fig. 3. Percentage of budded cells was scored after 30 to 45-min incubation in α-factor. Presented are the average values of two independent experiments.
A RAD53 checkpoint mutant treated with MMS recovers CLN1 transcription faster than wild type
In the signal transduction cascade from DNA damage to the cell cycle machinery targets, Rad53 is thought to be the most proximal of all the known checkpoint genes to the targets (Navas et al. 1996; Sanchez et al. 1996; Sun et al. 1996). Because mec1-1, rad9Δ, and rad53-11 mutants exhibit a similarly shortened G1 delay after MMS treatment, we chose to characterize the rad53-11 mutant in more detail. As expected on the basis of Figure 4A, control and rad53-11 cells express comparable levels of CLN1 mRNA after release from α-factor arrest in the absence of DNA damage (Fig. 5A, B). When these cells are treated with MMS, the initial down-regulation of CLN1 mRNA is about the same in the rad53-11 and wild-type cells (Fig. 5C,D; data not shown). However, the recovery of high CLN1 mRNA levels is more rapid in the rad53-11 mutant than in the wild-type cells (Fig. 5C, D).
Figure 5.

The rad53-11 strain has a faster rate of CLN1 transcript recovery after MMS than the wild type. (A) The isogenic wild-type (BY2006) and rad53-11 (BY2007) strains were arrested by α-factor and released as usual. Aliquots were taken in 10-min intervals for 50 min, RNAs were isolated, and the levels of CLN1 and SIR3 were measured by S1 protection. (B) Data obtained in A were quantitated as above. (C) The same two strains were treated with MMS in the same way as described in the legend to Fig. 4A. Aliquots were taken before the release from α-factor (0), with 0.2% MMS (30+MMS) and at intervals after MMS was removed. Levels of CLN1 and SIR3 were measured by S1 protection. (D) The CLN1 and SIR3 levels shown in C were quantitated, and CLN1 was normalized to SIR3 and plotted.
Because we have shown that CLN mRNA down-regulation and recovery are not dependent on cell cycle progression, we were interested to see whether the same is true for the fast recovery of CLN transcripts in the rad53-11 strain. cdc4 rad53-11 double mutants were constructed (see Materials and Methods), and the cdc4 arrests and MMS treatments were repeated as described for Figure 2. In this case, the results of two experiments with two independent cdc4 rad53-11 isolates were quantitated and used to produce the plot shown in Figure 6A. It is clear from this analysis that cdc4 rad53-11 strains arrested in G1 recover their CLN1 message levels faster than the cdc4 RAD strain. During the cdc4 arrest, CLN1 transcript levels are slightly lower in the cdc4 rad53-11 strain as compared with the cdc4 strain (data not shown); so its ability to recover CLN1 mRNA after MMS treatment cannot be a result of the unusually high CLN1 levels.
Figure 6.
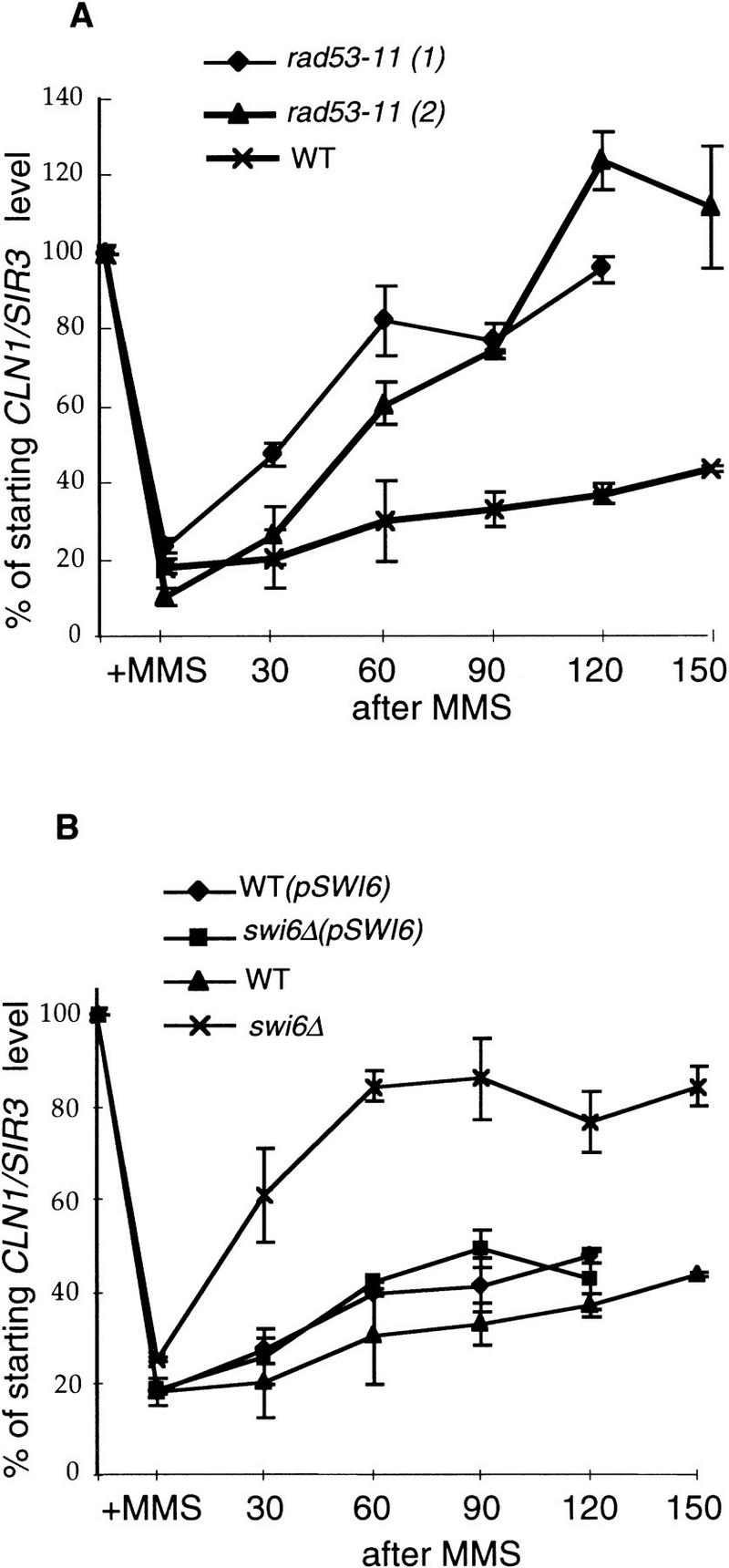
The CLN1 transcription is recovered faster in the cdc4 rad53-11 and cdc4 swi6Δ strains than in the cdc4 strain upon MMS treatment during the arrest at nonpermissive temperature. The cdc4 (BY2240), two isolates of cdc4 rad53-11 (BY2243 and BY2287) and cdc4 swi6::TRP1 (BY2241) strains, BY2240 transformed with pBD1265 (with SWI6 gene), and BY2241 transformed with pBD1265 were treated the same way as described in the legend to Fig. 2. They were first arrested at 37°C for 3 hr, and then an aliquot from both cultures was collected to serve as control (0), and to the rest of the cultures, 0.1% MMS was added for 30 min (+MMS). Then MMS was inactivated and removed by filtration, and the cultures were resuspended in the fresh 37°C media and incubated at this temperature for up to 150 min. Aliquots were taken throughout the time of recovery from MMS. RNAs were isolated, and CLN1 and SIR3 transcript levels were measured. In each case, data from two to three experiments were quantitated and their average plotted. The mRNA levels seen in MMS and after MMS removal are expressed as the percentage of the starting normalized mRNA level seen at 37°C in the untreated culture. wt, cdc4 BY2240, wt(pSWI6), cdc4 BY2240 transformed with pSWI6 pBD1265, rad53-11 (1) and rad53-11 (2), cdc4 rad53-11 BY2287 and BY2243, respectively, swi6Δ, cdc4 swi6::TRP1 BY2241, swi6Δ(pSWI6) cdc4 swi6::TRP1 BY2241 transformed with pSWI6 pBD1265.
Swi6 mutants treated with MMS also recover CLN1 transcription faster than wild type
GAL::SWI4-t is capable of reducing the MMS-induced delay of Start, but GAL-induced wild-type Swi4 is not. Swi4-t is more stable than wild-type Swi4, but both proteins are expressed at such high levels under these conditions that this difference is unlikely to be the reason for the selective deregulation of CLN transcripts by Swi4-t. The other important difference between Swi4 and Swi4-t is that Swi4-t has lost the carboxy-terminal domain necessary for association with Swi6 (Sidorova and Breeden 1993). Thus, we wondered whether Swi6 could be the target of negative regulation by the Rad53 pathway under conditions of DNA damage. swi6 mutants grow slowly and do not recover synchronously from α-factor arrest; so to avoid these complications, we generated cdc4 swi6Δ and cdc4 SWI6 strains. These cells were arrested at the G1/S transition, and CLN1 transcript was monitored after cells were treated with a pulse of MMS. As can be seen in Figure 6B, the absence of Swi6 does not eliminate the drop of CLN1 transcript levels upon MMS treatment, but, like rad53-11, it allows a faster recovery of CLN1 transcription after MMS (Fig. 6A). Thus, the maintenance of low CLN1 mRNA levels in response to DNA damage is Rad53 dependent and Swi6 dependent. The requirement for Swi6 to inhibit CLN expression is surprising because it was first identified as an activator of transcription. However, this result could be explained if Swi6 is modified in response to DNA damage to a form that causes the Swi4/Swi6 complex to repress rather than activate transcription.
Swi6 undergoes MMS-inducible Rad53-dependent phosphorylation in vivo
Rad53 is a dual-specificity protein kinase whose consensus phosphorylation site is not known (Zheng et al. 1993). Upon treatment with DNA-damaging agents such as hydroxyurea and MMS, Rad53 undergoes phosphorylation, which alters its mobility in SDS-PAGE (Sanchez et al. 1996; Sun et al. 1996). This phosphorylation is performed by an upstream kinase, possibly Mec1, rather than by Rad53 itself (Sanchez et al. 1996; Sun et al. 1996). In vitro Rad53 can phosphorylate itself (Zheng et al. 1993; Fay et al. 1997) and histone H1 (Sun et al. 1996), and Rad53 kinase activity is increased upon treatment with a checkpoint-activating agent, hydroxyurea (Sun et al. 1996). One potential target of Rad53 in living cells is the Dun1 kinase, which is phosphorylated in a Rad53-dependent, MMS-inducible manner (Allen et al. 1994). To see whether MMS also affects the Swi6 phosphorylation pattern, we monitored Swi6 phosphorylation by peptide mapping using the same MMS treatment conditions we used to follow CLN transcription. Wild-type cells were arrested in G1 and released into radioactive orthophosphate-containing media, and MMS was added to one-half of the culture for 40 min. Extracts were made, Swi6 was immunoprecipitated from these cells, and its phosphorylation state was determined by peptide mapping. Figure 7A shows that upon MMS treatment Swi6 gains phosphates on at least two new peptides (designated a and b). This phosphorylation can be acquired as early as 20 min after introduction of MMS, and it persists for at least 90 min after MMS is eliminated (data not shown). Thus, these phosphorylations are temporally correlated with the interval during which CLN mRNA levels remain low.
Figure 7.
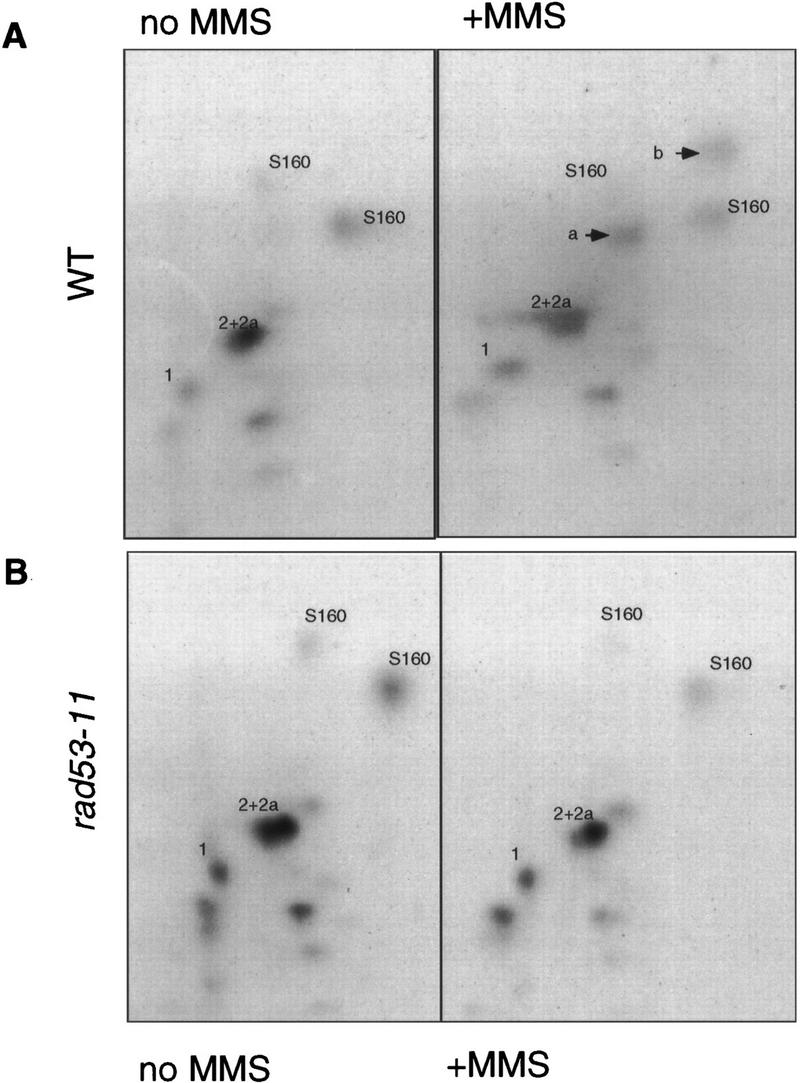
The Swi6 protein undergoes MMS-inducible RAD53-dependent phosphorylation. (A) The wild-type (BY2006) strain was arrested in G1 by α-factor and released into radiolabeled orthophosphate-containing media. MMS (0.1%) was added to one-half of this culture for 40 min, after which time both cultures were harvested. Swi6 was immunoprecipitated out of extracts made from harvested cells, digested with trypsin, and resolved on TLC plates as described in Materials and Methods. MMS-inducible peptides a and b are marked by arrows. (B) The rad53-11 (BY2007) strain was arrested by α-factor and released as in A, 0.1% MMS was added to one-half for 40 min, and then both halves were harvested and treated as above. S160 stands for serine-160-containing peptides (Sidorova et al. 1995) that are marked here for the reference. Other major constitutive phosphopeptides of Swi6 are numbered.
We then asked if this phosphorylation of Swi6 occurs in a checkpoint-defective RAD53 mutant, rad53-11, which is incapable of delaying G1 or of prolonging the inhibition of CLN1 transcription upon MMS treatment. The rad53-11 strain was synchronized and labeled as above, and the Swi6 phosphorylation pattern was analyzed (Fig. 7B). In contrast to the isogenic wild-type cells, there was no MMS-inducible phosphorylation of Swi6 isolated from the MMS-treated rad53-11 strain. Therefore, this phosphorylation of Swi6 requires the checkpoint-proficient Rad53 kinase.
Rad53 or an associated kinase phosphorylates Swi6
To begin to address whether this phosphorylation of Swi6 is performed directly by Rad53, we sought to reconstitute the MMS-inducible, Rad53-dependent kinase reaction in vitro. First, Rad53 was immunoprecipitated out of untreated and MMS-treated wild-type (WT) or rad53-11 cells, and radioactive ATP was added to monitor phosphorylation. As seen in Figure 8A, the wild-type Rad53 immunoprecipitate incorporates phosphate into a single polypeptide of ∼90 kD that has been shown previously to be Rad53 itself (Zheng et al. 1993; Sun et al. 1996; Fay et al. 1997). This basal level of Rad53 kinase activity is relatively low but is markedly stimulated by MMS. In contrast, we observe a very low kinase activity in immunoprecipitates of the checkpoint-defective Rad53 isolated from the isogenic rad53-11 strain. Moreover, this low kinase activity cannot be stimulated by MMS (Fig. 8A, lane 4), despite the fact that Rad53-11 undergoes the mobility shift that is correlated with the Mec1-dependent phosphorylation and activation (data not shown). This could indicate that the rad53-11 defect is in its catalytic function, though more complicated scenarios could be proposed. It is of interest because RAD53 is an essential gene (Zheng et al. 1993), and rad53-11 is a viable, checkpoint-deficient allele (Weinert et al. 1994) that has not been characterized in vitro previously. Here, it serves as the ideal negative control, allowing us to conclude that the kinase activity we observe in this assay is Rad53 dependent.
Figure 8.

The MMS-inducible, Rad53-dependent phosphorylation of Swi6 can be reconstituted in vitro. (A) Rad53 was immunoprecipitated out of lysates of the wild-type BY2006 and rad53-11 BY2007 cells that were treated (M) or not treated (−) with 0.1% MMS for 40 min. Radiolabeled ATP was added to immunoprecipitates to allow phosphorylation, and reactions were stopped with SDS loading buffer, boiled, and resolved on SDS-PAGE. The arrows mark the position of Rad53, both the steady-state form (Rad53) and the damage-modified form (Rad53-P), and positions of protein molecular weight markers are shown at left. (B) Rad53 was immunoprecipitated as in A, and recombinant Swi6 was added along with radiolabeled ATP. Reactions were stopped by addition of AB buffer (see Materials and Methods), and Swi6 was released by incubation at 4°C for 30 min and immunoprecipitated with Swi6 antibodies. These immunoprecipitates were resolved on SDS-PAGE. Swi6 position is marked by an arrow. (C) Wild-type W303 1A strain transformed with pGAL::RAD53 pBD2146 was grown either in raffinose (RAF) or galactose (GAL) to induce overexpression of RAD53 (see Materials and Methods). Protein extracts were prepared out of these cells, Rad53 was immunoprecipitated, and kinase assays with recombinant Swi6 (Swi6) or mock preparation (mock) (see Materials and Methods) were performed as described above. Products of kinase reactions were directly loaded onto SDS PAGE. Swi6 and Rad53 positions are marked by arrows.
We then performed a Rad53 kinase reaction with exogenously added recombinant Swi6 (Sidorova and Breeden 1993). Because Swi6 and the phosphorylated form of Rad53 migrate very closely in SDS-PAGE, Swi6 was immunoprecipitated from each reaction before electrophoresis (see Materials and Methods for details). As seen with Rad53, we observed highly efficient phosphorylation of Swi6 that was MMS induced and Rad53 dependent (Fig. 8B). The checkpoint-defective Rad53-11, as well as wild-type Rad53 from untreated cells, incorporated about an order of magnitude less phosphate into Swi6 than the wild-type kinase precipitated from MMS-treated cells. Two possibilities could account for this effect. The kinase activity of the Rad53 from undamaged cells could be too low to incorporate substantial radioactivity into Swi6. Alternatively, MMS could change the specificity of Rad53. We believe that the former possibility is more likely the case, as Rad53 isolated from undamaged cells that overexpresses Rad53 by ∼10-fold (Zheng et al. 1993; data not shown) is capable of incorporating substantial amounts of phosphate into Swi6 (Fig. 8C). Thus, the ability to phosphorylate Swi6 is not restricted to the MMS-activated form of Rad53.
To see whether the pattern of Rad53-dependent phosphorylation of Swi6 was similar in vivo and in vitro, we used the higher resolution method of peptide mapping. Figure 9, A and B, shows peptide maps of in vivo labeled Swi6, and in this experiment three Rad53-dependent, MMS-inducible phosphopeptides can be readily detected (marked a, b, c). Figure 9C shows the peptide map of Swi6 phosphorylated in vitro by Rad53 that has been immunoprecipitated from the cells overproducing Rad53 from a GAL promoter. Among the products of this reaction are three peptides whose positions of migration are indistinguishable from those of the inducible, Rad53-dependent peptides a, b, and c seen in vivo (Fig. 9B). Figure 9D confirms that these prominent peptides are derived from Swi6, as they are not detectable among the products of the kinase reaction between Rad53 and a mock substrate, which has been prepared from bacteria harboring vector instead of the SWI6-expressing plasmid. In summary, Swi6 undergoes MMS-induced and Rad53-dependent phosphorylations in vivo, and these phosphorylations appear to be reproducible in vitro with immunoprecipitated Rad53.
Figure 9.

Rad53-dependent phosphopeptides of Swi6 generated in vivo and in vitro migrate to similar positions on phosphopeptide maps. (A,B) Wild-type (BY2286) strain was synchronized in G1 by α-factor and released into radiolabeled orthophosphate-containing media, and 0.1% MMS was added to one-half of this culture for 40 min, after which time both halves were harvested. Phosphopeptide maps of Swi6 isolated out of these two cultures were generated as described for Fig. 7 (see Materials and Methods for details). MMS-inducible peptides are marked by arrows. Three of such peptides could be detected (a, b, c). Serine 160-containing peptides (S160) are marked for the reference. (C,D) Kinase assays were performed as in Fig. 8C with Rad53 isolated out of the wild-type strain overexpressing RAD53 from the GAL promoter (BY2292) with Swi6 (C) or mock preparation (D) as a substrate. Radioactively labeled products were resolved on SDS-PAGE as in Fig. 8C. Swi6 and a corresponding region out of the SDS-PAGE lane where mock preparation was run were excised, and proteins were eluted and subjected to phosphopeptide analysis as in Figs. 7 and 9A,B. Rad53-dependent in vitro peptides that migrate similar to the MMS-inducible Rad53-dependent peptides generated in vivo are marked by arrows. (x) Map origins.
Discussion
MMS-induced G1 delay
Previous studies have established that a yeast cell that receives treatment with UV or ionizing irradiation in G1 can undergo a transient and checkpoint-dependent arrest in G1 (Siede et al. 1993, 1994). In this paper we show that a short pulse of MMS, administered either during an α-factor-induced G1 arrest or immediately after release from the pheromone treatment, can produce a similar effect. With MMS doses of 0.1%–0.2%, these cells remain in G1 and do not enter S phase for up to 90 min after withdrawal of the genotoxic agent. The MMS-damaged G1 cells show a prolonged sensitivity to α-factor that indicates that they pause at or before Start. As with the UV studies, this G1 delay is also dependent on the RAD53, MEC1, and RAD9 genes (Siede et al. 1993, 1994, 1996; Weinert et al. 1994). However, in our studies, we have found that these checkpoint mutants only shorten but do not eliminate the delay of Start, suggesting that there may be other contributing factors. In addition, we observe that this G1 delay is correlated with a delay in the accumulation of CLN1 and CLN2 transcripts.
Recently, Paulovich and Hartwell (1995) showed that continual treatment of G1 cells with low levels of MMS (0.03%) induces a profound slowing of S phase, which is dependent on RAD53 and MEC1 and to a lesser degree, on RAD9, RAD17, and RAD24 (Paulovich et al. 1997a). We have reproduced this finding and observed a dosage dependence to the cell’s response. For example, administering 0.05% MMS to cells that were released from α-factor-induced G1 arrest causes a <50% decrease in CLN mRNA levels (data not shown) and no significant G1 delay because these cells will traverse to S phase almost as fast as untreated controls (J. Sidorova, unpubl.). MMS (0.1%) is sufficient to induce a G1 delay if cells are treated during the α-factor arrest (Figs. 1 and 3), but 0.2% MMS is required if it is administered immediately after release (Fig. 4; J. Sidorova, unpubl.). These observations may be explained by the fact that cells continue to grow during an α-factor arrest and they attain the critical size required to go through to S phase very rapidly upon release. In this context, a low dose of MMS may not cause enough damage to trigger a response before cells exit G1. This is consistent with the possibility that there is a threshold of damage needed to trigger a slowdown in G1 or that the delay can only be triggered at an early step in G1 progression. Alternatively, MMS-induced lesions may be generated too slowly to occur in this time frame or they may be less readily recognized in G1 than in S and G2 cells (Paulovich et al. 1997b).
Another striking feature of the checkpoint-dependent response to MMS damage in G1 is that G1 cells delay only briefly and then progress into a slow S phase. This slow S is most likely owing to incomplete repair of the damage because it is also checkpoint dependent (Fig. 4; Paulovich and Hartwell 1995). Thus, it appears that the G1 delay is quite transient and not tightly coupled to the presence of damage. This is in contrast to the G2 arrest, which can last for many hours after a single double-strand break introduced by HO endonuclease (Sandell and Zakian 1993). Although the extent of the delay may certainly be affected by the type of DNA damage, one can also hypothesize that a tighter G2 arrest has evolved to protect the cell from an irreversible loss of genetic information that would occur during mitosis of unrepaired chromosomes. As such, the G2 checkpoint arrest may be fundamentally different from the transient delay that occurs in G1 cells, despite the fact that the same checkpoint genes are used to detect the damage.
MMS causes the loss of CLN1 and CLN2 mRNAs
In principle, pausing or slowing down during a given phase of the cycle could be achieved by reducing the amount of the cyclin/Cdk kinase that is active during that phase and/or by preventing the switch from one Cdk form to the other, for example, from Cln/Cdk to Clb/Cdk in the case of the G1/S transition. These effects could be achieved by direct inhibition of kinase activity, depression of cyclin or CDK protein levels, or inhibition of their transcription. In the case of DNA damage-induced delay in G1, we have found that CLN1 and CLN2 message levels are down-regulated by MMS in a dosage-dependent manner and are kept low for some time after the damage. Subsequently, the levels of these messages are restored slowly. The loss of CLN transcripts that occurs after brief exposure to 0.1% MMS cannot be attributed to a global interference with the cell’s transcriptional or post-transcriptional activity. Although this nonspecific interference certainly takes place after prolonged incubation with MMS (J. Sidorova, unpubl.), the CLN1 and CLN2 messages are more sensitive to the short exposures and low dose of MMS used in this study than are several other messages. Moreover, CLN1 sensitivity to MMS can be reduced by overexpression of a truncated form of its transcriptional activator, Swi4-t.
The MMS-induced loss of CLN1 and CLN2 messages is reversible, and the rate of recovery of these messages is dependent on Rad53 function. Rad53 slows the recovery of CLN transcript levels both in cycling cells and in G1-arrested cells. Because this effect is observed in arrested cells, it cannot be an artifact of the differences in the rate of G1 progression. Loss of Rad53 function speeds the recovery of CLN transcript levels and reduces the delay of Start. Deregulated expression of CLN1 and CLN2 can also reduce the G1 delay in response to DNA damage. Thus, the simplest interpretation of these findings is that Rad53-dependent inhibition of CLN transcription contributes to the delay of Start in MMS-treated cells. Rad53 is known to play a role in transcriptional activation of genes required for DNA repair (Aboussekhra et al. 1996; Kiser and Weinert 1996; Navas et al. 1996), and our data indicate that this kinase may have an additional role in repressing transcription of genes that promote progression through the cell cycle.
The role of Swi4 and Swi6 in MMS-induced down-regulation of CLNs
The Swi4/Swi6 complex induces G1/S-specific transcription of CLN1 and CLN2. Swi4 is the DNA-binding subunit of the complex, and in the absence of Swi4, CLN transcription is greatly reduced (Nasmyth and Dirick 1991; Ogas et al. 1991; Cross et al. 1994; Stuart and Wittenberg 1994; Partridge et al. 1997). Lack of Swi6 leads to a constitutive intermediate level of CLN transcription (Dirick et al. 1992; Lowndes et al. 1992). These data have led to the view that Swi4 is the primary activator of CLN transcription and Swi6 plays a regulatory role, both enhancing and repressing CLN transcription depending on the phase of the cell cycle. It is therefore possible that Swi6 may also regulate the activity of the Swi4/Swi6 complex in response to DNA damage. Swi6 could be modified by a DNA damage-dependent mechanism and shift from an activating to a repressing component of the Swi4/Swi6 complex. This would repress CLN transcription and delay the G1/S transition. In agreement with this idea, the absence of Swi6, but not Swi4, causes cells to lose viability rapidly in the presence of MMS (Johnston and Johnson 1995). In addition, we find that the lack of Swi6 can actually increase the rate of CLN1 transcript recovery after MMS, similar to the effect observed in cells with defective Rad53 (Fig. 6). Finally, we have observed that Swi6 undergoes an MMS-dependent change in phosphorylation in wild-type but not rad53-11 cells. In vivo, we observe two to three new phosphorylations on Swi6, which are induced by MMS treatment, and these phosphorylations are present throughout the period when CLN transcription is low. In vitro Rad53, obtained from MMS-treated wild-type yeast cells or from cells overexpressing this kinase from the GAL promoter, can phosphorylate exogenous Swi6, and three phosphopeptides, which appear to comigrate with the MMS-inducible in vivo phosphopeptides, are observed. The rad53-11 strain is unable to promote MMS-inducible phosphorylation of Swi6 in vivo or in vitro. This mutant kinase has a dramatically reduced activity as judged by its decreased ability to phosphorylate itself, and it does not support phosphorylation of the exogenous Swi6. Taken together, all these observations strongly indicate that Swi6 is either a direct substrate of a damage-activated Rad53 in living cells or it is phosphorylated by a Rad53-activated kinase that is associated with Rad53.
These results give rise to the following model for the checkpoint induced by DNA damage (Fig. 10). When G1 cells are subjected to DNA damage by MMS, there is an immediate drop in CLN1 and CLN2 messages that is Rad53-independent. This drop could be mediated by specific promoter elements or by alterations in mRNA stability, and this would be expected to rapidly reduce the rate of G1 progression. This delay is extended by a Rad53- and Swi6-dependent inhibition of further CLN transcription. Temporally correlated with this inhibition, there is a Rad53-dependent change in the phosphorylation state of Swi6, which we speculate may inhibit Swi6 function and provide the mechanism for repressing CLN transcription.
Figure 10.

A model for the MMS-induced changes in transcription. As observed previously, MMS-induced DNA damage activates transcription of many genes involved in DNA repair and replication. In addition, it inhibits transcription of the G1/S-specific cyclins CLN1 and CLN2. MMS causes a rapid and prolonged repression of CLN1 and CLN2 transcription, owing to at least two pathways of regulation. The immediate drop in CLN1 and CLN2 transcription is Rad53 and Swi6 independent. Subsequent maintenance of this low level requires Rad53 and may involve Rad53-mediated inactivation of the Swi4/Swi6 complex by phosphorylation of Swi6. In this way, Rad53 plays a central role in both inducing repair of DNA damage and in delaying entry into S phase.
The inhibition of CLN expression that we observe in response to MMS may not be universal for every kind of damage but is likely to be one of the mechanisms that delay Start in response to DNA damage. Moreover, the recent finding that ectopic overproduction of CLN1 can increase genomic instability in wild-type cells and leads to even more instability and cell death in checkpoint-deficient mec1-1 cells (Vallen and Cross 1995) suggests that the tight control over CLN levels may be crucial not only for cells with damaged DNA but also for undamaged cells. At the same time it is clear that the loss of CLN transcripts is certainly not the only mechanism involved in delaying Start, because ectopic expression of CLNs does not eliminate the delay completely. It is ultimately possible that there are several levels of regulation within G1, both cyclin-mediated and independent, that help to protect cells from DNA damage.
Materials and methods
Strains and plasmids
The yeast strains used in this study are listed in Table 1. The plasmid pBD1168 is a YCp50 vector with GAL::SWI4-t and was described previously (Sidorova and Breeden 1993). The pBD860 is YCp50 with no insert. The plasmid pBD1265 has been described previously (Sidorova and Breeden 1993) and is a 2μ vector with the SWI6 gene. The plasmid pBD2146 is a kind gift of David Stern and has been described by Zheng et al. (1993) as pNB187–SPK1.
Table 1.
Strains used in this study
| Strain
|
Genotype
|
Source
|
|---|---|---|
| W303-1a | MATa ade2 his3 leu2-3,112 trp1-1 ura3 | Sidorova and Breeden (1993) |
| BY600 | MATa swi6::TRP1 ade2 ho::lacZ ura3 his3 leu2-3,112 trp1-1 can1-100 met2 | Sidorova and Breeden (1993) |
| BY665 | MATα cdc4-1 ade1 ade2 leu2 lys2 ura1 | L. Hartwell (FHCRC, Seattle, WA) |
| BY1365 | MATa cdc28-13 ade2 ade3 leu2 trp1-1 ura3 | J. Roberts (FHCRC, Seattle, WA) |
| BY1699 | MATα cdc28-13 ade2 ura3 trp1 leu2 ho::lacZ | BY1365 × BY600 |
| BY1956 | MATa swi6::TRP1 swi6-38::LEU2 ade2 ho::lacZ ura3 his3 leu2-3,112 trp1-1 can1-100 met2 | Breeden collection |
| BY2006 | MATa ura3 leu2 trp1 his3 | Paulovich and Hartwell (1995) |
| BY2007 | MATa ura3 leu2 trp1 his3 rad53-11::URA3 | Paulovich and Hartwell (1995) |
| BY2181 | MATα cdc4-1 leu2 rad53-11::URA3 | BY665 × BY2007 |
| BY2226 | MATa ura3 leu2 trp1 his3 mec1-1::HIS3 | Paulovich and Hartwell (1995) |
| BY2227 | MATa ura3 leu2 trp1 his3 rad9Δ::LEU2 | Paulovich et al. (1997a) |
| BY2235 | MATa ura3 leu2-3,112 trp1 his3 | Breeden collection |
| BY2240 | MATα cdc4-1 leu2 | BY1956 × BY2181 |
| BY2241 | MATα cdc4-1 leu2 trp1 swi6::TRP1 | BY1956 × BY2181 |
| BY2243 | MATα cdc4-1 leu2 rad53-11::URA3 | BY1956 × BY2181 |
| BY2286 | MATa ura3 leu2-3,112 trp1 his3 | Breeden collection |
| BY2287 | MATα cdc4-1 leu2 trp1 rad53-11::URA3 | BY1956 × BY2181 |
| BY2292 | MATa leu2-3,112 ura3-52 (pNB187-SPK1) | Zheng et al. (1993) |
Growth conditions
All rich (YEP) and minimal (YC) media and growth conditions were as described before (Breeden and Mikesell 1991). Cultures used for labeling with 32PO4 were allowed to double at least once in low phosphate YEP media with 2% glucose (Rubin 1975) before the experiment. For synchrony experiments, cultures were grown to an OD660 of 0.2 and arrested by addition of α-factor or by shifting to high temperature. Arrest with α-factor was performed with 5 mg/liter of this pheromone in low phosphate or YEP media with appropriate carbon source typically for 90–120 min. Cells were released from the arrest by filtration. cdc strains were arrested at 37°C for 3 hr. MMS was added from a 100% or a 10% solution directly to the cultures and the cultures were shaken vigorously to ensure resuspension of MMS and incubated for times indicated in the figure legends (typically 30 min). As a rule, immediately before washing away by filtration, MMS was inactivated by the addition of an equal volume of freshly made 10% sodium thiosulfate solution to the culture. Thiosulfate addition does not affect the delay of S phase or CLN levels (J. Sidorova, unpubl.). In experiments involving induction of the GAL promoter, cultures were grown in selective media with 2% raffinose for 12–20 hr, switched to the rich media with raffinose, and synchronized by α-factor. Galactose was added 30–60 min before the release from α-factor. GAL induction of RAD53 expression was performed in selective media for 3–4 hr.
RNA isolation and S1 protection
These procedures were performed as described previously (Breeden and Mikesell 1991), except for the annealing of radiolabeled probes to RNAs, which was done in formamide buffer (80% formamide, 40 mm PIPES at pH 6.4, 400 mm NaCl, 1 mm EDTA at pH 8.0) at 30°C. The probes used for detection of CLN1 and CLN2, CLN3, CDC6, MATa1, HIS3, and SIR3 transcripts are described (Breeden and Mikesell 1991; Foster et al. 1993; McInerny et al. 1997). mRNA levels were measured with a PhosphorImager 400A (Molecular Dynamics, Sunnyvale, CA), and the signals were quantitated using ImageQuant software.
FACS analysis
For FACS analysis of DNA content, cells were fixed in 70% ethanol for at least 1 hr, washed twice in 50 mm Tris-HCl (pH 7.8), resuspended in the same buffer, and digested with RNase (20 μg/ml) for 4 hr at 37°C. Cells were pelleted and resuspended in 125 mm Tris-HCl (pH 7.8), 105 mm NaCl, 39 mm MgCl2, and 20 μg/ml of propidium iodide. Cells were analyzed on Becton Dickinson FACScan, and the obtained information was presented using CellQuest software.
Protein analysis
The 32PO4 labeling conditions, extract preparation, Swi6 immunoprecipitation, and phosphopeptide mapping procedures were performed exactly as described (Sidorova et al. 1995). Immunoprecipitation of Rad53 for kinase assays was performed as described (Zheng et al. 1993; Sun et al. 1996) with previously characterized polyclonal antibodies, which were kindly provided by David Stern and Steve Elledge (Zheng et al. 1993; Allen et al. 1994), with the following modifications: The extract preparation and immunoprecipitation buffer [20 mm Tris-HCl (pH 8.0), 10 mm MgCl2, 1 mm EDTA, 5% glycerol, 0.3 m (NH4)2SO4, 1 mm DTT, 1 mm PMSF, 1 mm benzamidine, 1 μg/ml of leupeptin, 1 μg/ml of pepstatin A, 1 μg/ml of sodium orthovanadate] was supplemented with 0.1% NP-40. Immunoprecipitates were washed three times with the original washing buffer (PBS, 1% Triton X-100, 10% glycerol, 100 μm sodium orthovanadate), three times with washing buffer with 0.8 m NaCl, and once with the same buffer with 1.6 m NaCl. Rad53 kinase reaction was performed as described (Zheng et al. 1993; Sun et al. 1996). Recombinant Swi6 (100–200 ng) (Sidorova and Breeden 1993) was used per reaction. In some cases Swi6 was substituted with the mock preparation, which is a protein extract fraction obtained using Swi6 purification protocol from Escherichia coli harboring an empty vector instead of the Swi6-expressing plasmid. Products of the kinase reaction were either loaded directly onto SDS-PAGE or Swi6 was reprecipitated out of reaction mixture. In the latter case, 24 volumes of AB buffer (20 mm Tris-HCl at pH 7.5, 50 mm NaCl, 0.5% NP-40, 0.5% DOC, 0.3% SDS) were added to the reactions and Swi6 was eluted for 30 min at 4°C. This step was necessitated by the fact that after a kinase reaction, Swi6 is found predominantly associated with beads. After elution was performed as described, Rad53 remained associated with agarose beads, whereas Swi6 was released (data not shown). These eluates were then separated from the protein–A agarose beads (GIBCO BRL) and incubated with anti-Swi6 polyclonal antibodies (Sidorova and Breeden 1993) and a fresh portion of protein A–agarose beads (Sidorova and Breeden 1993). Precipitates were washed with RIPA and high salt buffers (10 mm Tris-HCl at pH 7.5, 2 m NaCl, 1% NP-40, 0.5% DOC) and resolved on SDS-PAGE.
Acknowledgments
We gratefully acknowledge members of the laboratory for support and helpful discussions. Special thanks are also due to David Stern for a gift of the GAL::SPK1 strain and for the Rad53 antibodies, to Steve Elledge for Rad53 antibodies, and to Lee Hartwell and members of his laboratory for providing the strains and for helpful critiques. This research was funded by a grant (GM41073) from the National Institutes of Health to L.L.B.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lbreeden@fhcrc.org; FAX (206) 667-6526.
References
- Aboussekhra A, Vialard JE, Morrison DE, Angeles de la Torre-Ruiz M, Cernakova L, Fabre F, Lowndes NF. A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. EMBO J. 1996;15:3912–3922. [PMC free article] [PubMed] [Google Scholar]
- Al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes & Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- Baroni MD, Monti P, Alberghina L. Repression of growth-regulated G1 cyclin expression by cyclic AMP in budding yeast. Nature. 1994;371:339–342. doi: 10.1038/371339a0. [DOI] [PubMed] [Google Scholar]
- Breeden L. Start-specific transcription in yeast. In: Farnham PJ, editor. Current topics in microbiology and immunology. Berlin, Germany: Springer-Verlag; 1996. pp. 95–127. [DOI] [PubMed] [Google Scholar]
- Breeden L, Mikesell G. Cell cycle-specific expression of the SWI4 transcription factor is required for the cell cycle regulation of HO transcription. Genes & Dev. 1991;5:1183–1190. doi: 10.1101/gad.5.7.1183. [DOI] [PubMed] [Google Scholar]
- Cross FR. Starting the cell cycle: What’s the point? Curr Opin Cell Biol. 1995;7:790–797. doi: 10.1016/0955-0674(95)80062-x. [DOI] [PubMed] [Google Scholar]
- Cross FR, Hoek M, McKinney JD, Tinkelenberg AH. Role of Swi4 in cell cycle regulation of CLN2 expression. Mol Cell Biol. 1994;14:4779–4787. doi: 10.1128/mcb.14.7.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon N, Hoekstra MF. Characterization of two protein kinases from Schizosaccharomyces pombe involved in regulation of DNA repair. EMBO J. 1994;13:2777–2788. doi: 10.1002/j.1460-2075.1994.tb06571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirick L, Moll T, Auer H, Nasmyth K. A central role for SWI6 in modulating cell cycle Start-specific transcription in yeast. Nature. 1992;357:508–513. doi: 10.1038/357508a0. [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. Identification of the DNA Damage-Responsive Element of RNR2 and evidence that four distinct cellular factors bind it. Mol Cell Biol. 1989;9:5373–5386. doi: 10.1128/mcb.9.12.5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay DS, Sun Z, Stern DF. Mutations in SPK1/RAD53 that specifically abolish checkpoint but not growth-related functions. Curr Genet. 1997;31:97–105. doi: 10.1007/s002940050181. [DOI] [PubMed] [Google Scholar]
- Foster R, Mikesell GE, Breeden L. Multiple Swi6-dependent cis-acting elements control SWI4 transcription through the cell cycle. Mol Cell Biol. 1993;13:3792–3801. doi: 10.1128/mcb.13.6.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari KL, Santerre A, Sekelsky JJ, McKim KS, Boyd JB, Hawley RS. The mei-41 gene of D. melanogaster is a structural and functional homolog of the human Ataxia Telangiectasia gene. Cell. 1995;82:815–821. doi: 10.1016/0092-8674(95)90478-6. [DOI] [PubMed] [Google Scholar]
- Hunter T. When is a lipid kinase not a lipid kinase? When it is a protein kinase. Cell. 1995;83:1–4. doi: 10.1016/0092-8674(95)90225-2. [DOI] [PubMed] [Google Scholar]
- Johnston LH, Johnson AL. The DNA repair genes RAD54 and UNG1 are cell cycle regulated in budding yeast but MCB promoter elements have no essential role in the DNA damage response. Nucleic Acids Res. 1995;23:2147–2152. doi: 10.1093/nar/23.12.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters J-M, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Kiser GL, Weinert TA. Distinct roles of yeast MEC and RAD checkpoint genes in transcriptional induction after DNA damage and implications for induction. Mol Biol Cell. 1996;7:703–718. doi: 10.1091/mbc.7.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes NF, Johnson AL, Breeden L, Johnston LH. SWI6 protein is required for transcription of the periodically expressed DNA synthesis genes in budding yeast. Nature. 1992;357:505–508. doi: 10.1038/357505a0. [DOI] [PubMed] [Google Scholar]
- Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: Implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- McClanahan T, McEntee K. Specific transcripts are elevated in Saccharomyces cerevisiae in response to DNA damage. Mol Cell Biol. 1984;4:2356–2363. doi: 10.1128/mcb.4.11.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerny CJ, Partridge JF, Mikesell GE, Creemer DP, Breeden LL. A novel Mcm1-dependent promoter element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes & Dev. 1997;11:1277–1288. doi: 10.1101/gad.11.10.1277. [DOI] [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell. 1995;82:831–840. doi: 10.1016/0092-8674(95)90480-8. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Dirick L. The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell. 1991;66:995–1013. doi: 10.1016/0092-8674(91)90444-4. [DOI] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase ε form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes & Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Ogas J, Andrews BJ, Herskowitz I. Transcriptional activation of CLN1, CLN2, and a putative new G1 cyclin (HCS26) by SWI4, a positive regulator of G1-specific transcription. Cell. 1991;66:1015–1026. doi: 10.1016/0092-8674(91)90445-5. [DOI] [PubMed] [Google Scholar]
- Partridge JF, Mikesell GE, Breeden LL. Cell cycle-dependent transcription of CLN1 involves Swi4 binding to MCB-like elements. J Biol Chem. 1997;272:9071–9077. doi: 10.1074/jbc.272.14.9071. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH. A checkpoint regulates the rate of progression through S Phase in S. cerevisiae in response to DNA damage. Cell. 1995;82:841–847. doi: 10.1016/0092-8674(95)90481-6. [DOI] [PubMed] [Google Scholar]
- Paulovich AG, Margulies RU, Garvik BM, Hartwell LH. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics. 1997a;145:45–62. doi: 10.1093/genetics/145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Toczyski CP, Hartwell LH. When checkpoints fail. Cell. 1997b;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- Pecker I, Avraham KB, Gilbert DJ, Savitsky K, Rotman G, Hamic R, Fukso T, Schrock E, Hirotsune S, Tagle D, Collins FS, Wynshaw-Boris A, Ried T, Copeland NG, Jenkins NA, Shiloh Y, Ziv Y. Identification and chromosomal localization of ATM, the murine homolog of the ataxia-telangiectasia gene. Genomics. 1996;35:39–45. doi: 10.1006/geno.1996.0320. [DOI] [PubMed] [Google Scholar]
- Rubin GM. Preparation of RNA and ribosomes from yeast. Methods Cell Biol. 1975;12:45–64. doi: 10.1016/s0091-679x(08)60951-6. [DOI] [PubMed] [Google Scholar]
- Ruby SW, Szostak JW. Specific Saccharomyces cerevisiae genes are expressed in response to DNA-damaging agents. Mol Cell Biol. 1985;5:75–84. doi: 10.1128/mcb.5.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA. Loss of a yeast telomere: Arrest, recovery and chromosome loss. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1700–1701. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Sidorova J, Breeden L. Analysis of the SWI4/SWI6 protein complex, which directs G1/S-specific transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:1069–1077. doi: 10.1128/mcb.13.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova J, Mikesell G, Breeden L. Cell cycle regulated phosphorylation of Swi6 controls its nuclear localization. Mol Biol Cell. 1995;6:1641–1658. doi: 10.1091/mbc.6.12.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siede W, Friedberg AS, Friedberg EC. RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci. 1993;90:7985–7989. doi: 10.1073/pnas.90.17.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siede W, Friedberg AS, Dianova I, Friedberg EC. Characterization of G1 checkpoint control in the yeast Saccharomyces cerevisiae following exposure to DNA-damaging agents. Genetics. 1994;138:271–281. doi: 10.1093/genetics/138.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siede W, Allen JB, Elledge SJ, Friedberg EC. The Saccharomyces cerevisiae MEC1 gene, which encodes a homolog of the human ATM gene product, is required for G1 arrest following radiation treatment. J Bacteriol. 1996;178:5841–5843. doi: 10.1128/jb.178.19.5841-5843.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart D, Wittenberg C. Cell cycle-dependent transcription of CLN2 is conferred by multiple distinct cis-acting regulatory elements. Mol Cell Biol. 1994;14:4788–4801. doi: 10.1128/mcb.14.7.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes & Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- Tokiwa G, Tyers M, Volpe T, Futcher B. Inhibition of G1 cyclin activity by the Ras/cAMP pathway in yeast. Nature. 1994;371:342–345. doi: 10.1038/371342a0. [DOI] [PubMed] [Google Scholar]
- Vallen EA, Cross FR. Mutations in RAD27 define a potential link between G1 cyclins and DNA replication. Mol Cell Biol. 1995;15:4291–4302. doi: 10.1128/mcb.15.8.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T. and L. Hartwell. 1989. Control of G2 by the RAD9 gene of Saccharomyces cerevisiae. J. Cell Sci. (Suppl.) 12: 145–148. [DOI] [PubMed]
- Weinert T, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes & Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Zheng P, Fay DS, Burton J, Xiao H, Pinkham JL, Stern DF. SPK1 is an essential S-phase specific gene of Saccharomyces cerevisiae that encodes a nuclear serine/threonine/tyrosine kinase. Mol Cell Biol. 1993;13:5829–5842. doi: 10.1128/mcb.13.9.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]