

Abstract
Cyclin-dependent kinase inhibitors (CKIs) play key roles in controlling the eukaryotic cell cycle by coordinating cell proliferation and differentiation. Understanding the roles of CKIs requires knowledge of how they are regulated both through the cell cycle and in response to extracellular signals. Here we show that the yeast CKI, Far1p, is controlled by ubiquitin-dependent proteolysis. Wild-type Far1p was stable only in the G1 phase of the cell cycle. Biochemical and genetic evidence indicate that its degradation required the components of the G1–S ubiquitination system, Cdc34p, Cdc4p, Cdc53p, and Skp1p. We isolated a mutant form of Far1p (Far1p-22) that was able to induce cell cycle arrest in the absence of α-factor. Cells that overexpress Far1-22p arrested in G1 as large unbudded cells with low Cdc28p–Clnp kinase activity. Wild-type Far1p, but not Far1-22p, was readily ubiquitinated in vitro in a CDC34- and CDC4-dependent manner. Far1-22p harbors a single amino acid change, from serine to proline at residue 87, which alters phosphorylation by Cdc28p–Cln2p in vitro. Our results show that Far1p is regulated by ubiquitin-mediated proteolysis and suggest that phosphorylation of Far1p by the Cdc28p–Clnp kinase is part of the recognition signal for ubiquitination.
Keywords: Ubiquitin, CKI, degradation, cell cycle, pheromone response, Far1p
The eukaryotic cell cycle is driven by the sequential activation and inactivation of cyclin-dependent kinases (Cdks) (Nigg 1995). Cdks are composed of a catalytic subunit (Cdc28p in budding yeast) and a cyclin regulatory subunit (Nasmyth 1993). The protein kinase activity of the complex is further regulated by specific phosphorylation of the catalytic subunit and by association with Cdk inhibitors (Morgan 1995). CKIs have been identified in a variety of organisms, playing key roles during development, cell cycle regulation, and as effectors of checkpoint mechanisms (Sherr and Roberts 1995; Harper and Elledge 1996). Some of these inhibitory proteins are also implicated in the development of cancer (Hunter and Pines 1994; Sherr 1996). Two CKIs have been identified in budding yeast Saccharomyces cerevisiae: p40Sic1 regulates entry into S phase by inhibiting the Cdc28p–Clbp kinases (Mendenhall 1993; Schwob et al. 1994) and Far1p is specifically required to arrest the cell cycle in response to pheromones (Peter and Herskowitz 1994a). Mating pheromones activate a mitogen-activated protein (MAP) kinase signal transduction pathway that induces changes in gene transcription, alterations of cellular morphology, and cell cycle arrest in G1 (Sprague and Thorner 1992; Herskowitz 1995; Leberer et al. 1997). Cell cycle arrest is mediated by Far1p, which functions as a CKI to inhibit the Cdc28p–Clnp kinase (Peter and Herskowitz 1994b).
Despite the importance of CKIs, little is known about their regulation (Peter 1997). In the case of Far1p, the mRNA levels fluctuate through the cell cycle, peaking in G1 (McKinney et al. 1993). Far1p levels increase severalfold in response to mating pheromones, and the protein becomes rapidly phosphorylated by the MAP kinase Fus3p (Peter et al. 1993). Although transcriptional induction of FAR1 in response to pheromones is necessary to cause cell cycle arrest, it is not sufficient (Chang and Herskowitz 1992; Peter and Herskowitz 1994a), indicating that post-translational modification is also required to activate Far1p. The CKI p40Sic1 appears to be regulated through the cell cycle by ubiquitin-dependent degradation (Schwob et al. 1994). Likewise, the mammalian inhibitor p27Kip1 has been shown to be ubiquitinated in vitro and in vivo (Pagano et al. 1995).
Ubiquitin-dependent degradation of proteins has emerged as a key mechanism for regulating cell cycle transitions. Destruction is triggered by the covalent attachment of ubiquitin, which targets proteins for degradation by the proteasome (Ciechanover 1994; Hochstrasser 1996). At least two distinct ubiquitination complexes regulate cell cycle-dependent destruction of target proteins. Cdc16p, Cdc23p, and Cdc27p are part of a large multisubunit complex, termed anaphase promoting complex (APC), which is required for the onset of anaphase by degrading mitotic cyclins and as yet unknown proteins (King et al. 1996). Cdc34p, Cdc4p, Cdc53p, and Skp1p are required for cells to enter S phase, and cells lacking the function of any of these proteins arrest with a G1 DNA content and an elongated bud (Nasmyth 1996). CDC34 encodes a ubiquitin-conjugating enzyme (Goebl et al. 1988), whereas little information is available on the molecular function of the other proteins. Cdc4p contains two sequence motifs found in many unrelated proteins: an F-box, which mediates the interaction with Skp1p, and several WD40 repeats that may be important for the interaction with target proteins (Bai et al. 1996). Ubiquitin-dependent proteolysis of p40Sic1 appears to be crucial for cells to enter S phase (Schwob et al. 1994; Deshaies 1997). Although it is not yet understood how p40Sic1 is specifically recognized by the ubiquitination machinery, recent experiments suggest a role for phosphorylation by the Cdc28p–Clnp kinase (Schneider et al. 1996; Tyers 1996).
To study the post-translational regulation of the CKI Far1p, we have isolated a mutant Far1p (Far1-22p), which arrests cells even in the absence of mating pheromones (Peter and Herskowitz 1994a). Whereas wild-type Far1p was only present in the G1-phase, Far1-22p was found in the nucleus throughout the cell cycle. Our results strongly suggest that Far1-22p arrests cells in G1 because it can no longer be ubiquitinated and degraded by the G1–S degradation system. Sequencing of Far1-22p revealed that the stabilizing mutation changed Serine 87 (Ser-87) to a proline residue. Interestingly, Ser-87 was located in a consensus phosphorylation site for Cdc28p kinase and could be specifically phosphorylated by Cdc28p–Cln2p in vitro. Our results show that Far1p is regulated by ubiquitin-dependent proteolysis, and suggest that an important determinant of the recognition signal for ubiquitination is a specific phosphorylation of Far1p by the Cdc28p–Clnp kinase.
Results
Cells producing Far1-22p arrest their cell cycle by inhibiting the Cdc28p–Clnp kinase in the absence of the mating pheromone pathway
Transcription of the CKI Far1p is induced severalfold in response to mating pheromones by a process that requires the transcriptional activator Ste12p (Chang and Herskowitz 1990; Oehlen et al. 1996). Transcriptional induction of Far1p, however, is not sufficient to cause cell cycle arrest: cells that produce wild-type Far1p from the inducible GAL1 promoter grow normally (Fig. 1; Chang and Herskowitz 1992). In an attempt to investigate the mechanism of the post-transcriptional activation of Far1p, we isolated an allele of FAR1 (FAR1-22) that was able to inhibit cell division in the absence of α-factor (Fig. 1A). Cells expressing Far1-22p from the GAL1 promoter were unable to form colonies in the presence of galactose (which activates the GAL1 promoter), but grew normally on medium containing glucose (Fig. 1A,B; data not shown). To determine whether the function of Far1-22p requires basal activity of the mating pathway, growth inhibition was examined in a/α diploid cells that are unable to respond to pheromones (Johnson 1995): Like haploid cells, a/α diploids were inhibited by Far1-22p (Fig. 1A) showing that Far1-22p was able to prevent cell proliferation, not only in the absence of α-factor, but also in cells lacking a functional pheromone response. Consistent with these results, Far1-22p inhibited proliferation of cells deleted for various components of the mating pathway such as FUS3 (Fig. 1B), STE11, or STE7 (data not shown).
Figure 1.

Cells producing Far1-22p arrest their cell cycle in G1 independently of the mating pathway. (A) Haploid a (K699), α (K700), or a/α diploid cells (YMP562) were transformed with a plasmid expressing either wild-type Far1p or Far1-22p from the inducible GAL promoter and grown on medium containing galactose. Note that cells expressing Far1-22p were unable to form colonies. (B) fus3Δ cells (K2297) that carry a plasmid coding for either wild-type Far1p or Far1-22p from the inducible GAL promoter were grown on media containing galactose (top, GAL promoter on) or glucose (bottom; GAL promoter off). (C,D) Cells expressing wild-type Far1p (YMP128, right panels) or Far1-22p (YMP126, left panels) from the inducible GAL promoter were analyzed 6 hr after addition of galactose. (C) (Top) Phase-contrast photographs; (bottom) actin visualized after staining with rhodamine phalloidin. (D) Flow cytometric DNA quantification.
Next, we examined the phenotype of cells arrested by Far1-22p. After synthesis of Far1-22p was induced for 6 hr, ∼90% of the cells accumulated as large unbudded cells, indicating that they arrested in the G1 phase of the cell cycle before bud emergence (Fig. 1C, top left). These cells contained predominantly one nucleus as visualized by 4′, 6-diamidino-2-phenyl-inadole (DAPI) (data not shown) and a 1N DNA content as determined by fluorescence-activated cell sorting (FACS) analysis (Fig. 1D). Staining the cells with rhodamine phalloidin revealed that the actin cytoskeleton was unpolarized, indicative of cells arrested at Start (Fig. 1C, bottom left panel). In contrast, cells producing wild-type Far1p continued to bud normally (Fig. 1C; top right panel) and displayed polarized actin (Fig. 1C, bottom right panel; Lew and Reed 1995). In addition, cells arrested by overexpression of Far1-22p were able to fully induce a FUS1–lacZ reporter construct in response to α-factor (data not shown), suggesting that they were blocked in a mating-competent state (Oehlen and Cross 1994). Taken together, these observations suggest that cells producing Far1-22p arrest at Start in the G1 phase of the cell cycle.
The transition from G1 to S phase requires activation of the G1 cyclin-dependent kinase Cdc28p-Clnp (Nasmyth 1993). Whereas the Cdc28p catalytic subunit is present at constant levels throughout the cell cycle, the levels of the G1 cyclins Cln1p and Cln2p peak as cells pass through Start (Koch and Nasmyth 1994). Cells arrested by overexpression of Far1-22p contained high levels of Cln2p (Fig. 2B), showing that Far1-22p did not prevent synthesis of Cln2p. In contrast, cells arrested by α-factor repress transcription of CLN1 and CLN2 (Wittenberg et al. 1990). Thus, our results indicate that repression of CLN1 and CLN2 transcription is not directly caused by Far1p, but rather by an α-factor-induced event. In response to α-factor, Far1p has been shown previously to bind directly and thereby inhibit the activity of Cdc28p associated with the G1-cyclins Cln1p and Cln2p (Peter and Herskowitz 1994b). As shown in Figure 2A, the histone H1 kinase activity associated with Cln2p was reduced in cells arrested with Far1-22p when compared with cells expressing either wild-type Far1p (data not shown) or an inactive mutant of Far1-22p (Far1-22/Δp). The Cln2p-associated kinase activity was not reduced when synthesis of Far1-22p was not induced (Fig. 2A). Binding of Far1p and Cdc28p requires a segment between amino acids 285 and 350 of Far1p (Peter et al. 1993). Cells producing Far1-22p deleted for this segment (Far1-22/Δp) failed to inhibit cell division (Fig. 2C), suggesting that binding of Far1-22p and Cdc28p kinase was required to arrest the cell cycle. Finally, the effect of Far1-22p could be suppressed by high levels of G1-cyclins: Cells overexpressing a truncated form of Cln3p (Cross 1988) were able to grow in the presence of Far1-22p (Fig. 2D). Taken together, these results suggest that Far1-22p, at least in part, arrests cells at Start by inhibiting the Cdc28p–Clnp kinase in the absence of α-factor.
Figure 2.

Cells producing Far1-22p arrest in G1 by inhibition of the G1 kinase, Cdc28p–Clnp. (A,B) Cln2p tagged at its carboxyl terminus with three copies of the HA epitope (Cln2–HA) was immunoprecipitated from extracts prepared from cells (YMT263) expressing either Far1-22p or for control, an inactive mutant form of Far1p, Far1-22/Δ285–350, from the inducible GAL promoter. Cells were grown in media containing galactose (GAL, GAL promoter on) or glucose (GLU, GAL promoter off). Cln2–HAp-associated kinase activity was measured with histone H1 as a substrate and quantified (A). The kinase activity associated with Cln2–HAp from cells grown in glucose was normalized to 100%. Similar amounts of Cln2–HAp were immunoprecipitated in each assay as shown by immunoblotting (B). (C) Growth inhibition caused by production of Far1-22p was dependent on the ability of Far1p to bind to the Cdc28p–Clnp kinase. The following Far1p proteins were analyzed: wild-type Far1p, Far1-22p, Far1-Δ285–350, which is unable to bind to Cdc28p–Clnp (Peter et al. 1993), and Far1-22/Δ285–350. (D) Growth inhibition caused by expression of Far1-22p was rescued by co-overexpression of a stable G1-cyclin, Cln3p (DAF1). DAF1-1 cells (IH2517), which express a stable form of the G1 cyclin Cln3p or isogenic wild-type cells (IH2518), were transformed with plasmids expressing either wild-type Far1p, Far1-22p, or Far1-22/Δ285–350p from the inducible GAL promoter.
Far1-22p has an increased half-life and is stable throughout the cell cycle
The presence of wild-type Far1p is restricted to the G1 phase of the cell cycle. This pattern is ensured by regulated transcription of FAR1 and by proteolysis (McKinney and Cross 1995). Because we found that steady-state levels of Far1-22p were higher than wild-type Far1p (data not shown), we compared the stability of wild-type Far1p with Far1-22p. Both proteins were expressed from the GAL1 promoter, at which time synthesis was turned off by addition of glucose. We observed rapid degradation of wild-type Far1p with a half-life of ∼30 min (Fig. 3A,B). In contrast, Far1-22p remained stable throughout the time course (half-life ∼120 min), showing that Far1-22p harbors a specific alteration that stabilizes the protein in vivo.
Figure 3.

Far1-22p has an increased half-life and is present throughout the cell cycle. (A) Cells (K699) that carry a plasmid coding for either wild-type Far1–GFPp or Far1-22p–GFP from the inducible GAL promoter were grown in raffinose and expression was induced by addition of galactose for 5 hr. Glucose was then added to shut off the GAL promoter, and samples were taken every 30 min as indicated and immunoblotted for the presence of Far1–GFP fusion protein (lanes 1–8). The specificity of the GFP antibodies was confirmed by including cells expressing an HA-tagged version of Far1p (lane 1). (B) Quantitation of the GAL shutoff experiments. The degradation of wild-type Far1p-GFP and Far1-22-GFP proteins was quantified by PhosphorImager and plotted against the time after repressing the GAL promoter. (C) Far1–22p, but not wild-type Far1p, is present throughout the cell cycle. EY957 cells producing wild-type Far1p–GFP (lanes 9–11) or Far1–22p-GFP (lanes 12–14) from the inducible GAL promoter were grown in medium containing raffinose (GAL promoter off), arrested in S phase with hydroxyurea (HU) or in mitosis with nocodazole (Noc), and then expression was induced by addition of galactose. Control cells were treated identically except that they were not exposed to the drugs (expo). After 5 hr, cells were analyzed for the presence of Far1p–GFP by immunoblotting with antibodies specific for GFP (top panel). Equal loading was confirmed by immunoblotting the samples with antibodies against actin (bottom panel).
Next, we examined whether Far1p was degraded in a cell cycle-dependent manner. To drive FAR1 transcription through the cell cycle, we expressed Far1p from the inducible GAL1 promoter. Cells were arrested in S phase with hydroxyurea (HU) or in mitosis with the microtubule depolymerizing drug nocodazole (Noc). Synthesis of either wild-type Far1p or Far1-22p was then induced by addition of galactose and analyzed by immunoblotting. Little wild-type Far1p could be detected in cells arrested in S phase or in mitosis (Fig. 3C). The remaining low levels of Far1p were most likely the result of incomplete cell cycle arrest (90% arrest in both cases). In contrast, Far1-22p was present at high levels in both S and M phases of the cell cycle (Fig. 3C). The finding that Far1-22p was present in a cell cycle-independent manner also showed that FAR1 RNA was present throughout the cell cycle when driven from the GAL1 promoter. Thus, we conclude that wild-type Far1p, but not Far1-22p, was rapidly degraded at cell cycle stages other than G1. The degradation system for Far1p appears, therefore, to be active once cells pass through Start and remains active throughout the cell cycle until the cells exit from mitosis.
We have also studied the presence of Far1p during the cell cycle by use of fusions of either wild-type Far1p or Far1-22p to the green fluorescent protein (GFP). The Far1p–GFP fusion protein was functional and able to restore both cell cycle arrest and mating competence to cells lacking Far1p (data not shown). As shown in Figure 4, wild-type Far1–GFP protein was readily visualized in the nucleus of unbudded G1 cells, but very little Far1p was detected in cells that had passed Start (Fig. 4, top row). These results confirmed that wild-type Far1p was degraded after Start even when continuously expressed from the GAL1 promoter. Prolonged induction of Far1-22p–GFP resulted in accumulation of the cells at Start. Far1-22p–GFP was visualized in the nucleus, showing that the stabilizing mutation that affects Far1-22p did not interfere with the subcellular localization of the protein. Interestingly, however, Far1-22p–GFP was present in the nucleus of cells at all stages of the cell cycle (Fig. 4, bottom row), confirming that Far1-22p was not degraded after cells passed Start.
Figure 4.

Wild-type Far1p, but not Far1–22p, is expressed in a cell cycle-dependent manner. Wild-type Far1p or Far1-22p was fused at its carboxyl terminus to GFP and produced in yeast (EY957) from the inducible GAL promoter. Note that wild-type Far1p–GFP (top row) was found in the nucleus of unbudded cells (G1), but staining disappeared in small budded cells (G1/S), large budded cells (G2/M), and cells in mitosis (M). In contrast, Far1-22p–GFP (bottom row) could be detected in the nucleus of cells at all stages of the cell cycle.
Degradation of Far1p is required to efficiently re-enter the cell cycle after α-factor arrest
To address the physiological significance of Far1p degradation, we asked whether degradation of Far1p was required for cells to re-enter the cell cycle after arrest by α-factor. Cells deleted for FAR1, but producing Far1-22p from its own promoter, were able to grow normally because α-factor is required for full transcriptional induction of FAR1 (Chang and Herskowitz 1990; Oehlen et al. 1996). These cells were hypersensitive to α-factor, forming a halo that was larger than that of cells expressing wild-type Far1p (Fig. 5A, top panels), showing that Far1-22p was functional. Interestingly, halos formed by cells expressing Far1-22p failed to fill in (Fig. 5A, bottom panel), indicating that these cells were unable to efficiently resume cell division. To further examine recovery from α-factor arrest, far1Δ cells producing either wild-type Far1p or Far1-22p were monitored microscopically as they synchronously re-entered the cell cycle. As shown in Figure 5 (B, open circles), cells producing wild-type Far1p started to bud after an initial lag phase, indicating that they resumed cell division and entered S phase. In contrast, cells producing nondegradable Far1-22p failed to re-enter the cell cycle and remained unbudded (Fig. 5B, filled squares). Determination of Far1p levels by immunoblotting revealed that wild-type Far1p rapidly disappeared at the time of bud emergence (Fig. 5B), whereas Far1-22p remained constant and accumulated in slower migrating forms (Fig. 5C). This shift in mobility of Far1p was a consequence of phosphorylation, because incubation with alkaline phosphatase (CIP) eliminated the slower migrating forms (data not shown). Taken together, these results thus confirmed that Far1p was degraded when cells pass through Start and further indicated that degradation of Far1p is required for cells to efficiently re-enter the cell cycle after α-factor arrest.
Figure 5.



Degradation of Far1p is required for cells to recover efficiently from α-factor induced cell cycle arrest. (A) cell cycle arrest was assayed by halo formation of far1Δ cells transformed with single-copy plasmids carrying wild-type FAR1, FAR1-22, FAR1-60F3 (Peter et al. 1993), or no insert. (Top two rows) bar1Δ cells (K2180), (bottom row) BAR1+ cells (YMP1054). Filter disks contain 0.1 μg (top two rows) and 10 μg of α-factor (bottom row). Note that cells producing Far1-22p are hypersensitive to pheromone and that halos fill in inefficiently. (B,C) Cells producing Far1-22p are unable to re-enter the cell cycle. Cells deleted for FAR1 (K2180) producing wild-type Far1p or Far1-22p from the GAL promoter were grown in raffinose medium to an OD600 of 0.4, at which time cells were arrested by the simultaneous addition of α-factor and galactose. After 3 hr, cells were washed and reinoculated in fresh medium, without α-factor, but containing galactose. Every 30 min, samples were harvested. Re-entry into the cell cycle was monitored microscopically by analyzing the percentage of unbudded cells (B) Levels of wild-type Far1p (bottom panel) and Far1-22p (top panel) were determined after immunoblotting extracts with Far1p antibodies (C, lanes 1–7). Note that in contrast to cells producing wild-type Far1p, Far1-22p is accumulating in G1 and cells are unable to form a bud.
Far1p is stabilized in cells harboring temperature-sensitive alleles in components of the G1–S ubiquitination system
Next, we examined whether Far1p degradation requires the G1–S degradation system. Because overexpression of Far1-22p, which cannot be degraded, causes cell cycle arrest, we reasoned that overexpression of wild-type Far1p in a cell that exhibited a reduced ability to degrade Far1p might similarly cause cell cycle arrest in G1. Consistent with this hypothesis, we found that overexpression of Far1p in a cdc34 mutant caused inviability at the semipermissive temperature (Fig. 6A). This effect was specific as wild-type cells or cells harboring a temperature-sensitive allele of CDC16, which is required to degrade mitotic cyclins (Irniger et al. 1995), grew normally when overexpressing Far1p. Examination of cdc34 cells overexpressing Far1p revealed that they arrested as large, unbudded cells (∼90% after 6 hr of induction of Far1p by addition of galactose). In contrast, cdc34 mutant cells harboring control constructs accumulated prior to DNA replication with large elongated buds (Fig. 6B, top row). Staining of the actin cytoskeleton with rhodamine phal
Figure 6.

Overexpression of wild-type Far1p is lethal in cdc34 cells at the semipermissive temperature because of stabilization of the Far1 protein. (A) Temperature-sensitive cdc16 (K4102) and cdc34 (YMT670) mutants, as well as isogenic wild-type cells (K699), were transformed with a plasmid carrying wild-type FAR1 under the control of the inducible GAL promoter or an empty vector (vec) for control. Transformants were plated on media containing galactose and incubated at 30°C. Wild-type and cdc16 mutant strains tolerate overexpression of Far1p, whereas cdc34 mutants do not. All strains grew normally when plated on medium containing glucose (data not shown). (B) cdc34 cells (YMT670) carrying a plasmid expressing Far1p from the inducible GAL promoter (pFar1; left panels) or control plasmid with no insert (vec; right panels) were grown in raffinose at 30°C, at which time expression of Far1p was induced by addition of galactose. After 5 hr, cells were fixed and analyzed by phase contrast microscopy (Phase, top panels) or after actin staining with rhodamine phalloidin (Actin, bottom panels). cdc34 cells producing Far1p arrest with a morphology indistinguishable from that of wild-type cells producing Far1-22p (Fig. 1C). (C) The half-life of wild-type Far1p was determined in cdc34 far1Δ (top panel; YMP1056) and far1Δ cells (bottom panel; YMP1054), which express Far1p from the GAL promoter. After repression of the GAL promoter at the indicated times (minutes), aliquots were removed and examined by immunoblotting as described. The specificity of the Far1p antibodies was confirmed by analysis of cells carrying an empty vector (vec; lane 1). The position of Far1p is marked by an arrowhead; the asterisks mark an unspecific protein recognized by the Far1p antiserum.
loidin showed that cdc34 cells producing Far1p were unpolarized, indicating that they were arrested at Start (Fig. 6B, bottom row). Thus, the morphology of these cells appeared indistinguishable from wild-type cells arrested by stabilized Far1-22p (Fig. 1C), suggesting that Far1p is degraded by a Cdc34p-dependent mechanism.
In addition to Cdc34p, the CDC53, CDC4, and SKP1 gene products have also been implicated in the ubiquitin-dependent degradation of target proteins (Nasmyth 1996; Deshaies 1997). Overexpression of Far1p was lethal in any of these backgrounds at the semi-permissive temperature, suggesting that these components were required for degradation of Far1p (Fig. 7A; data not shown). Specific temperature-sensitive alleles of SKP1 that arrest cells either before DNA replication in G1 or before mitosis in G2 (Bai et al. 1996; Connelly and Hieter 1996) have been identified. Interestingly, overexpression of Far1p was able to arrest cells harboring the G1-specific allele of SKP1 but had no effect on cells harboring the G2-specific allele of SKP1 (Fig. 7A). Taken together, these results indicate that Far1p is degraded by the G1–S ubiquitination system involving the ubiquitin-conjugating enzyme Cdc34p and the proteins Cdc53p, Cdc4p, and Skp1p.
Figure 7.

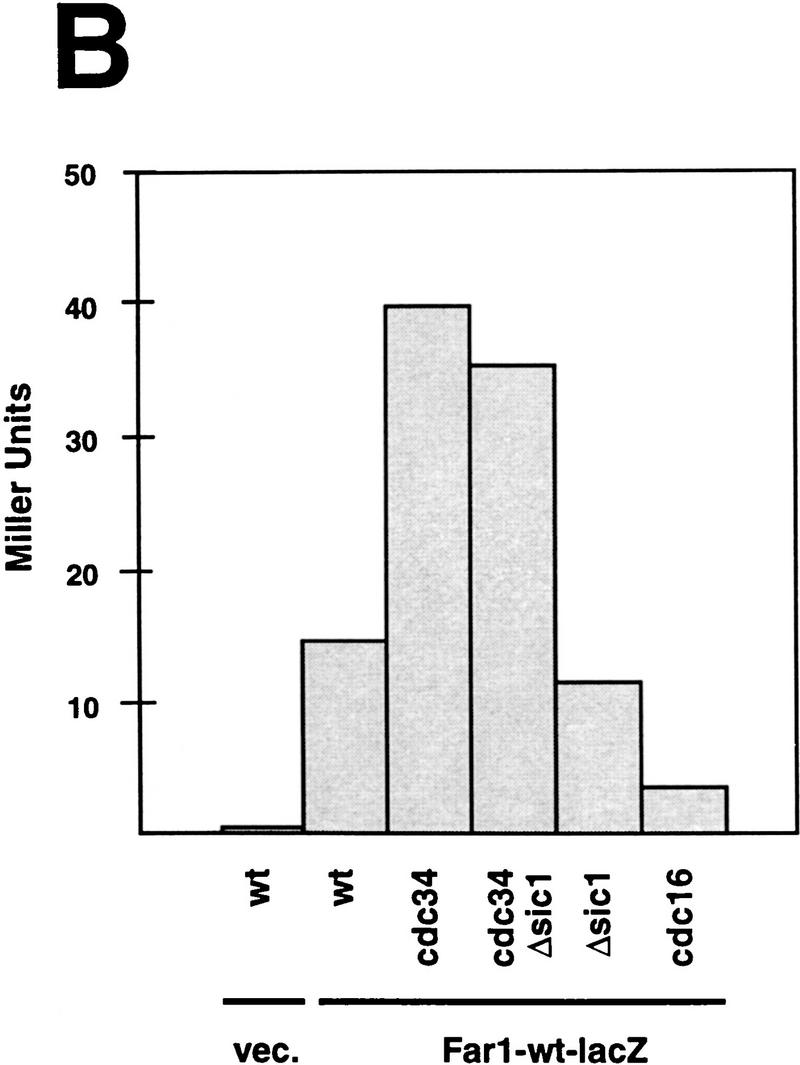
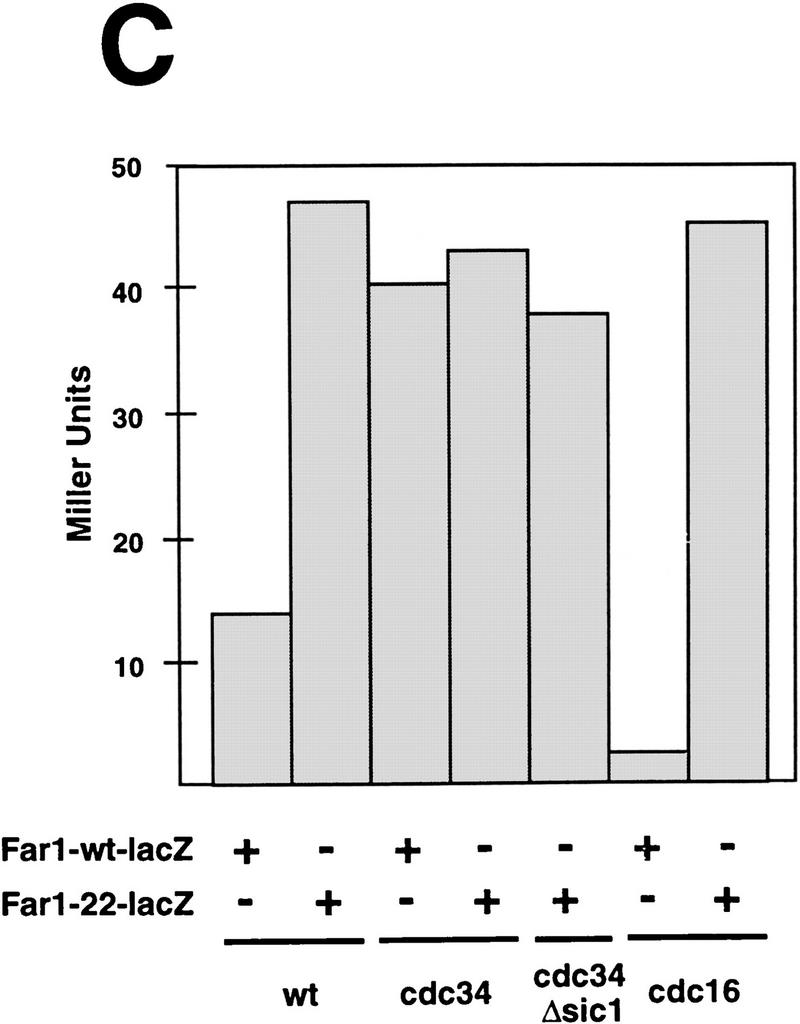
Overexpression of Far1p is lethal in skp1 mutants in an allele-specific fashion and Far1p accumulates in cdc34 mutants independent of the cell cycle stage. (A) The effect of overexpression of Far1p was tested in specific temperature-sensitive alleles of SKP1 that either cause arrest in G1 (skp1-11, Y552) or in G2 (skp1-12, Y554) as described in Fig. 6. Only cells harboring the G1-specific allele of SKP1 were arrested by overexpression of wild-type Far1p. (B) Far1p accumulates in cdc34 (YMT670) and cdc34 Δsic1 cells (ES464). The indicated strains were arrested at 37°C and the accumulation of a Far1–LacZ fusion protein was quantified and plotted as Miller Units deduced from three independent experiments. Note that Far1–LacZp accumulates in cdc34 mutants independently of the cell cycle arrest point; no accumulation was observed in cdc16 cells or cells deleted for SIC1. (C) Far1-22–LacZp accumulates in wild-type (K699) and cdc16 (K4102) mutant cells but is produced at levels similar to wild-type Far1–LacZp in cdc34 (YMT670) and cdc34 Δsic1 (ES464) cells.
Consistent with the proposed role of Cdc34p in the degradation of Far1p, the half-life of wild-type Far1p was dramatically increased in cdc34 and cdc53 cells at the nonpermissive temperature (Fig. 6C; data not shown). To determine whether the cell cycle stage, rather than lack of Cdc34p function, accounted for the accumulation of Far1p, we analyzed Far1p levels in cdc34 cells that were simultaneously deleted for SIC1. These cells are able to replicate normally but arrest subsequently in G2, presumably because they fail to degrade an as yet unknown target (Schwob et al. 1994). We expressed Far1p fused at its carboxyl terminus to LacZ from the inducible GAL promoter, which facilitated quantification of Far1p levels by a color assay (Barral et al. 1995). The Far1–LacZ fusion protein, like wild-type Far1p, was degraded in a cell cycle-dependent manner (Fig. 7B,C; B. Catarin and M. Peter, unpubl.). As expected, Far1p acccumulated in cdc34 cells, and was present at levels approximately three- to fourfold higher than in isogenic wild-type cells or cells lacking SIC1 (Fig. 7B; McKinney et al. 1993). Importantly, Far1p levels remained high in cdc34 sic1Δ cells, whereas low levels of Far1p were detected in cdc16 cells, which arrest in mitosis. In contrast, the Far1-22–LacZ fusion protein was present at higher levels than wild-type Far1–LacZ protein in both wild-type and cdc16 cells (Fig. 7C) whereas the levels of Far1–LacZp and Far1-22–LacZp were comparable when produced in cdc34 or cdc34 sic1Δ cells (Fig. 7C). Taken together, these results show that lack of Cdc34p itself, rather than arrest at a particular stage of the cell cycle, was responsible for the accumulation of Far1p.
In vitro ubiquitination of Far1p depends upon Cdc34p, Cdc4p, and cyclins
To determine whether Far1p is ubiquitinated by the G1–S ubiquitination system, we in vitro-translated Far1p in rabbit reticulocyte lysates (Fig. 8A) or wheat germ extracts (Fig. 8B) in the presence of [35S]methionine and added DEAE-fractionated yeast extracts (Verma et al. 1997a). These extracts are devoid of the G1 cyclins Cln1p, Cln2p, and Cln3p and are depleted of Cdc34p (Fig. 8A) or lack functional Cdc4p (Fig. 8B). Addition of Far1p, purified GST–Cln2p, ubiquitin (Ub), and Cdc34p resulted in accumulation of slower migrating forms of Far1p (Fig. 8A, lane 2). Methylated ubiquitin (meUb), which blocks formation of multiubiquitin chains by preventing Ub–Ub ligation (Hershko and Heller 1985), prevented accumulation of these high molecular weight forms of Far1p (Fig. 8, lane 3), showing that they were ubiquitinated. Ubiquitination in vitro was dependent on Cdc34p (Fig. 8A, lane 4) and Cdc4p (Fig. 8B). DEAE-fractionated extracts prepared from cdc4-ts mutants failed to ubiquitinate wild-type Far1p (Fig. 8B, lane 1). Addition of purified Cdc34p (Fig. 8, lane 1) or baculovirus-infected insect cell lysate containing Cdc4p (Fig. 8A, lane 2) alone did not restore activity of the extract. However, addition of Cdc34p and Cdc4p together restored Far1p ubiquitination (Fig. 8A, lane 3). As a control, insect lysate containing Cdc28p and Cdc34p did not lead to ubiquitination of Far1p (Fig. 8A, lane 4). Ubiquitination of Far1p was also dependent on the addition of the G1 cyclin Cln2p (Fig. 8A, lane 5), indicating that Far1p may need to be phosphorylated by Cdc28p–Cln2p to be recognized by the Cdc34p-ubiquitination system. Alternatively, Cdc28p–Cln2p may be necessary to activate a component of the ubiquitination machinery. Importantly, no ubiquitination could be detected on Far1-22p (Fig. 8A, lane 7), suggesting that the increased half-life of Far1-22p in vivo results from its failure to be ubiquitinated. These results show that wild-type Far1p, but not Far1-22p, is ubiquitinated in a Cdc34p- and Cdc4p-dependent manner in vitro and further suggest that ubiquitination of Far1p may require a phosphorylation event governed by the Cdc28p–Cln2p kinase.
Figure 8.

Wild-type Far1p but not Far1-22p is ubiquitinated in vitro. Wild-type Far1p (A, lanes 1–5; B, lanes 1–4) or Far1-22p (A, lanes 6–10) synthesized in vitro in the presence of [35S]methionine was incubated in fractionated extracts prepared from cells lacking CLN1,2, and 3 (A) or cells deleted for CLN1,2, and 3, which are also temperature-sensitive for cdc4 (B). These extracts are devoid of Cdc34p and contain low levels of ubiquitin (Verma et al. 1997a). (A) Wild-type Far1p was ubiquitinated if the extract was supplemented with the addition of Cdc34p, GST–Cln2p, and ubiquitin (lane 2). Reduced accumulation of high molecular weight ubiquitin conjugates was observed if methyl-ubiquitin was added to block chain extension (lane 3). No ubiquitination was observed with Far1-22p (lanes 6–10). (B) Ubiquitination of wild-type Far1p was dependent on Cdc4p (lanes 1 and 4). Ubiquitination in cdc4-extracts was restored only by the simultaneous addition of purified Cdc34p and baculo-infected insect lysate containing Cdc4p (lane 3). The addition of insect lysate containing Cdc28p serves as a specificity control (lane 4).
Wild-type Far1p, but not Far1p-22p, is phosphorylated on Ser-87 by Cdc28p–Cln2p
We have shown previously that Far1p is phosphorylated in vivo in a Cdc28p-dependent manner (Peter et al. 1993; Tyers and Futcher 1993). To determine whether Far1p was directly phosphorylated by Cdc28p–Cln2p kinase in vitro, Far1p fused to glutathione S-transferase (GST) was expressed in Escherichia coli and phosphorylated with Cdc28p–Cln2p immunoprecipitated from yeast (Fig. 9A, lane 3). As shown in Figure 9A, full-length Far1p was readily phosphorylated, whereas no phosphorylation could be detected on GST alone (Fig. 9, lane 2) or in the absence of substrate (Fig. 9, lane 1). A fragment encompassing amino acids 50–340 of wild-type Far1p (Far1p50-340) fused to GST was phosphorylated (Fig. 9A, lane 4), whereas little phosphate was incorporated into the same segment of Far1-22p (Fig. 9A, lane 5), showing that Far1-22p could not be fully phosphorylated by Cdc28p–Cln2p kinase. Subcloning and sequencing of the FAR1-22 allele revealed that Ser-87 was mutated to a proline residue (Fig. 9B). In the wild-type Far1p, Ser-87 is followed by a proline residue and, therefore, conforms to the minimal consensus sequence for both CDKs and MAP kinases (Nigg 1995). Therefore, we tested whether Ser-87 was indeed a phosphorylation site for Cdc28p–Cln2p in vitro. Phosphoamino acid analysis confirmed that in wild-type Far1p50-340 phosphate was predominantly incorporated on a serine residue (Fig. 9C, top panel). In contrast, when Ser-87 was mutated to a threonine residue (S87T), phosphorylation occured predominantly on threonine (Fig. 9C, bottom panel), showing that Ser-87 was the major phosphorylation site for Cdc28p–Cln2p in vitro. Full-length Far1-22p still migrated slower on SDS gels, however, when compared with unphosphorylated Far1p, indicating that Ser-87 was not the only phosphorylation site on Far1p in vivo. Nevertheless, mutation of Ser-87 to a nonphosphorylatable residue resulted in a nonubiquitinated and stable Far1 protein in vivo. Taken together, we propose that phosphorylation of Ser-87 and possibly other residues by Cdc28p–Clnp kinase serves as a recognition signal for ubiquitination and subsequent degradation of Far1p (Fig. 10).
Figure 9.

Far1p is phosphorylated by Cdc28p–Cln2p kinase on Ser-87 in vitro. (A) Full-length wild-type Far1p was produced in E. coli, purified as a GST fusion protein and phosphorylated in vitro with Cdc28p–Cln2p kinase immunoprecipitated from yeast extracts (lane 3). As a control, the kinase assays were performed with GST alone (lane 2) or without substrate (lane 1). Phosphorylated proteins were analyzed by autoradiography (top left panel) and immunoblotting with specific antibodies against Far1p (bottom left panel). The arrowhead marks the position of GST-Far1 protein. A fragment containing amino acids 50–340 of either wild-type Far1p (lane 4) or Far1-22p (lane 5) was expressed as a GST fusion protein in E. coli and phosphorylated in vitro by immunoprecipitated Cln2–HAp-associated kinase. Kinase reactions were also carried out with histone H1 (lane 7) or in the absence of substrate (lane 6). The specificity of the immunoprecipitated Cln2p-associated kinase was confirmed by including an untagged Cln2p (lane 8). The addition of equal amounts of GST–Far1p in the kinase reactions was verified by immunoblotting with antibodies against Far1p (bottom right panel). The arrowhead points to the position of the GST-Far150-340 protein; the bracket marks the position of phosphorylated histone H1. (B) Sequencing of the FAR1-22 allele uncovered a single mutation that changes Ser-87 to proline. Note that Ser-87 is followed by a proline residue and is, therefore, a potential CDK phosphorylation site (Nigg 1995). (C) Ser-87 is phosphorylated by Cdc28p–Cln2p kinase in vitro. A fragment containing amino acids 50–340 of either wild-type Far1p (top panel) or Far1p-S87T, in which Ser-87 was replaced by a threonine residue (bottom panel), was phosphorylated in vitro by immunoprecipitated Cln2–HAp-associated kinase and phosphoamino acid analysis was performed. (S) The position of phosphoserine; (T) The position of phosphothreonine.
Figure 10.

A model for the degradation of Far1p. Far1p binds to the Cdc28p–Clnp kinase, which in turn phosphorylates Far1p on Ser-87. Phosphorylated Far1p is recognized by the ubiquitination machinery composed of Cdc34p, Cdc53p, Cdc4p, and Skp1p, which ubiquitinates Far1p. Ubiquitinated Far1p is then degraded by the proteasome.
Discussion
Far1p is degraded in a cell cycle-dependent manner
Far1p accumulates during the G1 phase of the cell cycle. This expression pattern is achieved in part by transcriptional regulation of FAR1, which is restricted to G1 and late M phase of the cell cycle (McKinney et al. 1993). We have found that even when Far1p is expressed from constitutive promoters, however, it is still detectable only in G1 cells, because Far1p is rapidly degraded at stages other than G1. The observation that a nondegradable mutant form of Far1p is present throughout the cell cycle excludes the possibility that translation of FAR1 RNA is restricted to G1, as is the case for the mammalian CKI p27 (Hengst and Reed 1996). We show that Far1p is degraded at the G1 to S transition and remains unstable until cells exit from mitosis. Cells overexpressing a form of Far1p that fails to be degraded accumulate predominantly at Start and not in S phase or mitosis, which is consistent with previous in vitro assays indicating that Far1p inhibits Cdc28p complexed with G1 cyclins but is a poor inhibitor of S phase or M phase Cdks (Peter and Herskowitz 1994b).
Far1p is a yeast CKI that is required to arrest the cell cycle in the presence of mating pheromones but is not necessary for normal cell cycle progression (Chang and Herskowitz 1990). The degradation of Far1p is physiologically relevant during yeast mating. Cells arrested with α-factor that are unable to find a mating partner adapt to the presence of mating pheromones and resume cell division (Sprague and Thorner 1992). Re-entry into the cell cycle requires the degradation of Far1p: Cells that produce nondegradable Far1p were unable to properly activate the Cdc28p–Clnp kinase after release from α-factor and, as a consequence, failed to recover efficiently.
Far1p degradation by the G1–S ubiquitination system requires phosphorylation of Ser-87
Two ubiquitin-dependent degradation systems trigger key transitions during the eukaryotic cell cycle. The onset of S phase is initiated by the proteolysis of p40Sic1, which involves the G1–S ubiquitination system, whereas the onset of anaphase requires the anaphase-promoting complex (APC), which degrades inhibitors of sister chromatid separation (Nasmyth 1996). Substrates for the APC typically contain a short sequence termed the destruction box, which is both necessary and sufficient for degradation (King et al. 1996). Substrates for the G1–S ubiquitination system do not contain such a destruction box, and it is currently unknown how these proteins are recognized. The amino-terminal 50 amino acid residues of Far1p are required for its degradation in vivo (McKinney and Cross 1995). It is unlikely, however, that these 50 amino acid residues comprise a destruction signal per se, but they contain a functional nuclear localization signal and a truncated Far1 protein lacking these 50 amino acids found predominantly in the cytoplasm (M. Peter, unpubl.). Because Cdc34p is localized in the nucleus (Goebl et al., 1994), cytoplasmic localization may result in stabilization of Far1p. In contrast, failure to degrade Far1-22p is not caused by mislocalization, because Far1-22p still localized to the nucleus. Recent evidence suggests that phosphorylation might provide a signal for ubiquitination and degradation by the G1–S destruction system (Deshaies 1997). Mutations that inactivate phosphorylation sites of the G1 cyclin Cln2p in yeast or mammalian cyclin E stabilize these proteins in vivo (Clurmann et al. 1996; Lanker et al. 1996; Won and Reed 1996). Furthermore, phosphorylated, but not unphosphorylated, Cln2p specifically interacts with Cdc53p (Willems et al. 1996). Several lines of evidence indicate that degradation of Far1p also requires phosphorylation that may be achieved by the Cdc28p–Clnp kinase. First, degradation of Far1p occurs when cells progress through Start, concomitant with the activation of the Cdc28p–Clnp kinase. Second, Far1p is phosphorylated in vitro and in vivo by Cdc28p–Clnp kinase (Peter et al. 1993; Tyers and Futcher 1993). Third, Far1p is stable in cells harboring a temperature-sensitive mutation of CDC28 (Oehlen and Cross 1994; M. Peter and S. Henchoz, unpubl.). Fourth, we have shown that ubiquitination of Far1p in vitro requires active Cdc28p–Clnp kinase. Finally, mutation of a site phosphorylated by Cdc28p–Cln2p kinase stabilizes Far1p in vivo and prevents its ubiquitination. Taken together, we propose the following model for the degradation of Far1p (Fig. 10). Far1p binds to the Cdc28p–Clnp kinase, which in turn phosphorylates Far1p on Ser-87 and possibly other residues. Phosphorylated Far1p is then recognized by the G1–S ubiquitination system composed of Cdc34p, Skp1p, Cdc53p, and Cdc4p, which ubiquitinates Far1p. Ubiquitinated Far1p is subsequently degraded by the 26S proteasome. It remains to be shown whether Far1p is ubiquitinated in vivo. Despite the use of a mutant ubiquitin (UbK48R, G76A), which cannot be cleaved by ubiquitin-deconjugating enzymes (Hodgins et al. 1992; Willems et al. 1996), we were unable, so far, to convincingly show covalently attached ubiquitin on Far1p in vivo. We cannot, therefore, rigorously exclude the possibility that degradation of Far1p in vivo does not require ubiquitination of Far1p itself but might involve a ubiquitinated protein that targets Far1p to the proteosome.
A similar pathway has been proposed for controlling degradation of the CKI p40Sic1 (Deshaies 1997). Analogous to Far1p, an essential requirement for ubiquitination and degradation of p40Sic1 appears to be phosphorylation by the Cdc28p–Clnp kinase, thereby ensuring that S phase cannot be initiated before the G1-specific kinase Cdc28p–Cln1p (or Cln2p) has been activated (Tyers 1996; Schneider et al. 1996; Verma et al. 1997b). Whereas available results strongly support a function of the Cdc28p–Clnp kinase in the recognition of the substrate, they do not exclude the possibility that Cdc28p–Clnp kinase might also directly regulate the activity of the G1–S ubiquitination system. At least two lines of evidence, however, argue against the latter interpretation. First, it is unlikely that Cdc28–Cln2p is required to activate a component in the extract other than the substrate because p40Sic1 is ubiquitinated in vitro in the absence of Cln2p provided that p40Sic1 is phosphorylated by Cdc28p–Cln2p prior to addition to the extract (R. Verma and R.J. Deshares, unpubl.). Second, mutations in Far1p that prevent phosphorylation by Cdc28p–Cln2p kinase also disrupt ubiquitination, suggesting that phosphorylation of the substrate is essential for ubiquitination.
The activity of the Cdc28p–Cln1(2)p kinase is restricted to an interval between Start and the initiation of S phase, and thus correlates with the abrupt disappearance of Far1p and p40Sic1 (Nasmyth 1993). The degradation machinery for Far1p and p40Sic1, however, remains fully active throughout the G2 and M phases of the cell cycle, as indicated by the G1-specific presence of Far1p expressed from a constitutive promoter (Figs. 3C and 4). How is degradation maintained during phases at which Cdc28p–Cln1(2)p kinase activity is low? The stability of Far1-22p throughout the cell cycle indicates that degradation of Far1p in G2 is still dependent on phosphorylation of Ser-87. We propose that in G2, a kinase other then Cdc28p–Cln1(2)p—perhaps Cdc28p complexed with Cln3p or Clbp cyclins—can phosphorylate Far1p on this site.
Far1p and Cdc28p–Clnp kinase antagonize each other
Far1p was identified because it is able to inhibit the activity of the Cdc28p–Clnp kinase (Chang and Herskowitz 1990; Peter and Herskowitz 1994a). Our results now show that Cdc28p–Cln2p, in turn, inhibits Far1p by triggering its degradation, thus establishing a negative feedback loop between Far1p and the Cdc28p–Clnp kinase (Cross 1995; Peter 1997). Such a negative feedback loop might serve to amplify small differences between the activities of Far1p and Cdc28p–Clnp kinase. It appears, therefore, that the balance between the activity of the G1 kinase Cdc28p–Clnp and its inhibitor Far1p determines whether a cell divides or arrests. The activity of the G1 kinase Cdc28p–Clnp sets a threshold that inhibitors such as Far1p must overcome to arrest cells at Start. In the absence of pheromones, Far1p is turned over rapidly by ubiquitin-dependent proteolysis, thereby preventing accumulation of Far1p to levels sufficient to inhibit Cdc28p–Clnp and allowing cell cycle progression. The balance of Far1p relative to Cdc28p–Cln1(2)p can be tipped by production of a stable Far1p or by activation of the pheromone response pathway. The pheromone response pathway decreases the levels of the G1-cyclins Cln1p and Cln2p (Wittenberg et al. 1990) and increases the levels of Far1p (Chang and Herskowitz 1990; Oehlen et al. 1996). Overexpression of the G1-cyclin Cln2p, either by strong promoters or by stabilization of the protein, increases the threshold level set by the G1 kinase, causing cells to arrest poorly in response to pheromones (Lanker et al. 1996). Cell cycle arrest can be fully restored by simultaneous overexpression of Far1p (S. Henchoz and M. Peter, unpubl.).
Far1p levels are increased in response to mating pheromones by transcriptional activation of the FAR1 gene (Chang and Herskowitz 1990) and possibly by preventing Far1p degradation. Preliminary results indicate that the half-life of Far1p is increased after treating cells with α-factor by a mechanism that is dependent on the MAP kinase Fus3p (S. Henchoz and M. Peter, unpubl.). It is interesting to note that other MAP kinase signal transduction pathways might similarly regulate ubiquitin-dependent proteolysis. For example, activation of a MAP kinase-signaling cascade in Xenopus eggs prevents degradation of B-type cyclins, which leads to cell cycle arrest in metaphase of meiosis II (Minshull et al. 1994). Likewise, the spindle checkpoint activates a MAP kinase cascade resulting in cell cycle arrest by preventing the scheduled degradation of mitotic cyclins and possibly other destruction box substrates (Hardwick et al. 1996). Finally, the proto-oncogene c-Jun is protected from ubiquitin-dependent degradation after phosphorylation by MAP kinases (Musti et al. 1997). It appears, therefore, that several signaling pathways might control the stability of downstream regulators by preventing their ubiquitin-dependent degradation.
Ubiquitin-dependent degradation: a general mechanism to regulate CKIs?
CKIs from other species might similarly be regulated by ubiquitin-dependent proteolysis (Peter 1997). The half-life of p27Kip1 increases upon contact inhibition of cells and decreases when HeLa cells are arrested in S phase (Hengst and Reed 1996). Consistent with these results, S phase extracts prepared from Swiss 3T3 cells degrade p27Kip1 in vitro whereas mid-G1 extracts do not (Brandeis and Hunt 1996). Furthermore, p27Kip1 protein levels decrease when cells enter the cell cycle from quiescent state or in response to rapamycin (Nourse et al. 1994). Extracts prepared from proliferating cells ubiquitinate p27Kip1 protein at higher rates than extracts prepared from quiescent cells (Pagano et al. 1995). Recent studies indicate that the signal for ubiquitination and subsequent degradation of p27Kip1 might also be phosphorylation. Mutations that affect putative sites of phosphorylation by associated Cdk2-cyclin E kinase stabilize p27Kip1 in vivo (Sheaff et al. 1997; Vlach et al. 1997). Finally, increased proteosome-mediated degradation of p27Kip1 has been observed in certain tumors and correlates with poor prognosis (Loda et al. 1997), suggesting that the ubiquitin-dependent degradation pathway may be involved in tumorogenesis. Ubiquitin-dependent degradation triggered by phosphorylation may represent a conserved mechanism to regulate the activity of CKIs through the cell cycle and in response to extracellular signals.
Materials and methods
Yeast strains
Yeast strains are described in Table 1. Standard yeast growth conditions and genetic manipulations were used as described (Rose and Fink 1990). Yeast transformations were performed by the lithium acetate procedure (Ito et al. 1983).
Table 1.
Yeast strains
| Strain
|
Genotype
|
Source
|
|---|---|---|
| IH1792 | MATa cry1 lys1 | collection |
| IH1793 | MATα lys1 | collection |
| IH2518 | MATa ura3 leu2 trp1 his4 cyh2 bar1 | F. Cross (Rockefeller University, New York, NY) |
| IH2517 | MATa ura3 leu2 trp1 his4 cyh2 bar1 daf1::8xDAF1-1–URA3 | F. Cross |
| K699 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3, GAL+, psi+, ssd1-d2 | K. Nasmyth (IMP, Vienna, Austria) |
| K700 | MATα ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 | K. Nasmyth |
| YMP562 | MATa/α ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 | this study |
| YMP1055 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 FUS1–lacZ::LEU2 | this study |
| YMP126 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 trp1::GAL–FAR1-22–TRP1 bar1-1 | this study |
| YMP127 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 trp1::GAL–FAR1-22/Δ–TRP1 bar1-1 | this study |
| YMP128 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 trp1::GAL–FAR1–TRP1 bar1-1 | this study |
| K4102 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cdc16 | K. Nasmyth |
| YMT670 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cdc34-2 | M. Tyers (Samuel Lunenfeld Research Institute, Toronto Ontario, Canada) |
| ES464 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cdc34-2 sic1::HIS3 | C. Mann (Centre d’Etudes de Saclay, Gif-sur-Yvette CEDEX, France) |
| YMT668 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cdc4-1 | M. Tyers |
| YMT740 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cdc53-1 | M. Tyers |
| YMT263 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 cln2::CLN2–HA–LEU2 | M. Tyers |
| Y80 | MATa can1-100 ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | S. Elledge (Baylor College of Medicine, Houston, TX) |
| Y552 | MATa can1-100 ade2-1 his3-11,15 leu2,3,112 trp1-1 ura3-1 skp1-11 | S. Elledge |
| Y554 | MATa can1-100 ade2-1 his3-11,15 leu2,3,112 trp1-1 ura3-1 skp1-12 | S. Elledge |
| EY957 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 bar1-1 | E. Elion (Harvard University, Boston, MA) |
| K2149 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 bar1::HISG | G. Ammerer (University of Vienna, Austria) |
| K2180 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 bar1::HISG far1::HISG | G. Ammerer |
| K2297 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ dds1-d2 bar1::HISG fus3::LEU2 | G. Ammerer |
| YMP1054 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 far1::LEU2 | this study |
| YMP1056 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 far1::LEU2 cdc34-2 | this study |
| YMP1057 | MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+ psi+ ssd1-d2 far1::LEU2 cdc53-1 | this study |
DNA manipulations
Standard procedures were used for recombinant DNA manipulations (Ausubel et al. 1991; Sambrook et al. 1989). PCR was performed with Vent polymerase (New England Biolabs). PCR products were purified with the Wizard PCR purification kit according to the instructions of the manufacturer (Promega). Site directed mutagenesis was carried out by use of M13 mutagenesis as recommended by the manufacturer (Amersham). DNA sequencing was performed on both strands with the Sequenase kit (United States Biochemical Corp.).
Isolation of FAR1-22 (pTP63)
An amino-terminal fragment of FAR1 was mutagenized by PCR (Leung et al. 1989) by use of the primers oTP52 (5′-GCGCTCGAGATGAAGACACCAACAAGAGTTTCG-3′) and oTP55 (5′-CCGCACGGTACCCATAACATGTTCACCACC-3′) and plasmid pTP62 (pGAL–FAR1) as a template. The PCR product was digested with XhoI and KpnI, ligated into the vector pTP62, which had been digested with XhoI and KpnI, and transformed into DH5α. Transformants were pooled, and the isolated plasmid DNA transformed into the yeast strain K699. Individual yeast transformants were patched on SD–URA plates and subsequently replica-plated onto GAL–URA medium. Plasmids from transformants unable to grow on GAL–URA were isolated and the FAR1 coding sequence subcloned into Bluescript KS+ vector for sequencing. The XhoI–SphI fragment of FAR1-22 was ligated into the XhoI–SphI sites of pTP62 to yield plasmid pTP63.
Construction of integrating plasmids for Far1p and Far1-22p
For integration of wild-type FAR1 and FAR1-22 expressed from the GAL promoter, the entire FAR1 coding sequence with the GAL1 promoter was isolated from pTP62 and pTP63, respectively, after digestion with PvuII. The fragments were then ligated to PvuII-cut pRS304 (Sikorski and Hieter 1989) to yield plasmid pTP69 (wild-type FAR1) or pTP70 (FAR1-22). The plasmids were digested with Bsu36I to target integration to the TRP1 locus.
Construction of epitope-tagged versions of Far1p
An EcoRI site was introduced by PCR at the stop codon of FAR1 by use of pTP62 as a template and primers oTP322 (5′-TGTTTGAATTTGTCGCATGCTGATGGG-3′) and oTP323 (5′-CGAAGAATTCGAGGTTGGGAACTTCCAGGGTCTGATGGATCCTTTGCTG-3′). The PCR fragment digested with EcoRI and SphI was isolated. A fragment of FAR1, which includes the GAL1 promoter was obtained after digesting pTP62 or pTP63 with NotI and SphI. The coding sequence of GFP harboring the S65T mutation (Heim et al. 1995) was isolated as an EcoRI–BamHI fragment. The three fragments were then ligated into the NotI and BamHI sites of pRS316 (Sikorski and Hieter 1989) to yield pTP68 (GAL–FAR1–GFP) and pTP91 (GAL–FAR1-22–GFP). pTP68 complemented the mating and cell cycle arrest defects of far1Δ cells (K2180).
The FAR1–lacZ fusions expressed from the GAL10 promoter were created after ligation of the XhoI–SphI fragment of pTP62 or pTP63 into the Fusionator plasmid (pSJ101, Barral et al. 1995), which was digested with XhoI and SphI to yield pBC1 and pBC2, respectively. pBC1 complemented the cell cycle arrest defect of far1Δ cells (K2180).
Antibodies and Western blots
Cell extracts were prepared as described previously (Peter et al. 1993). Proteins were separated by SDS-PAGE and electroblotted to nitrocellulose (Schleicher & Schuell) by use of the Minigel system (Bio-Rad Labs, Hercules, CA). Blots were probed with monoclonal antibodies against GFP or with affinity-purified antibodies against Far1p (Peter et al. 1993) and developed by use of epichemiluminescence (Amersham Corp., Arlington Heights, IL).
Kinase assays and expression of GST-Far1p
GST-Far1 fusion proteins to be used as substrates in the kinase reactions were expressed in E. coli and purified as described (Peter and Herskowitz 1994a). Histone H1 was purchased from Boehringer Mannheim (No 1004 875). Cdc28p–Cln2p was immunoprecipitated from extracts as described. For in vitro phosphorylation assays, Cdc28p–Cln2p kinase was isolated from cells overexpressing Cln2–HAp or untagged Cln2p as a control from the constitutive ADH1 promoter (Peter and Herskowitz 1994a). Kinase assays were performed as described previously (Peter and Herskowitz 1994a).
Phosphoamino acid analysis was performed according to Peter et al. (1990). Briefly, phosphorylated proteins were eluted from SDS gels, hydrolyzed at 110°C for 1 hr in 6 m HCl, and the resulting mixture was separated in two dimensions on TLC plates.
Determination of half-life and quantification
Cells harboring plasmids encoding FAR1-GFP (pTP68) or FAR1-22-GFP (pTP91) were grown at 30°C to early log phase in raffinose-URA medium (2% raffinose), at which time expression was induced for 4 hr by addition of galactose (2% final concentration). A first aliquot was removed before expression was turned off by addition of glucose to a final concentration of 2%. Aliquots were collected every 30 min, and Far1p levels analyzed by immunoblotting. Western blots were quantified with the Image QuantTM software (Molecular Dynamics, Inc.). The half-life of Far1p in cdc34 Δfar1 (YMP1056), cdc53 Δfar1 (YMP1057) or Δfar1 cells (YMP1054) carrying a plasmid encoding FAR1 (pTP62) from the GAL promoter was determined as follows: cells were grown in raffinose–URA medium (2% raffinose) at 25°C, at which time the expression of Far1p was induced by the addition of galactose (2% final). After 30 min, the culture was shifted to 37°C for 2 hr, at which time the GAL promoter was repressed by the addition of glucose (2% final). Aliquots were collected and analyzed as described above.
Determination of Far1–LacZ levels
Transformants containing either pBC1 (GAL–FAR1–lacZ) or pBC2 (GAL–FAR1-22–LacZ), were grown at 25°C in raffinose–LEU medium to early log phase at which time the temperature was shifted to 37°C to induce cell cycle arrest of the cdc mutants. After 90 min, the GAL promoter was induced by addition of galactose to 2% final concentration. Cells were harvested 2 hr later and β-galactosidase levels were determined as described (Stern et al. 1984), except that because of the different arrest morphology, Miller units were normalized to cell number rather then OD600. Miller units were determined from three independent experiments.
Microscropy and flow cytometry
Cells were grown as indicated, sonicated, fixed with formaldehyde to a final concentration of 3.7%, and viewed by differential interference contrast microscopy. Yeast actin was visualized with rhodamin phalloidin (Molecular Probes Inc., Eugene, OR) as described previously (Peter et al. 1996).
Cells harboring plasmids encoding Far1p–GFP (pTP68) or Far1-22p–GFP (pTP91) expressed from the GAL promoter were grown in raffinose–URA medium (2% raffinose) at 30°C. Expression was induced for 4 hr by addition of galactose to 2% final concentration. GFP fluorescence was visualized with an argon laser at 488 nm with a Zeiss Axiovert 100 microscope (Zeiss laser Scanning Microscope 410) with a 63× Plan-Apochromat objective (1.4 oil). Standardized conditions for the pinhole size, gain, and offset (brightness and contrast) were used for image capture. Each image was the average of eight scans. Image capture and background subtraction were performed to allow direct comparisons.
Flow cytometry was carried out as described in Epstein and Cross (1992). Briefly, cells were fixed in 70% ethanol, washed and digested with RNase A for 5 hr. The DNA was stained then with propidium iodine and analyzed on a FACScan (Becton-Dickinson).
Ubiquitination assays
A cell free in vitro ubiquitination assay system in budding yeast has been described previously (Verma et al. 1997a,c). To summarize, both wild-type and mutant FAR1 (Fig. 8A) transcription templates were generated from pTP62 and pTP63, respectively, by PCR by use of a 5′ oligonucleotide containing a T7 RNA polymerase promoter (Verma et al. 1997a). The 5′ oligonucleotide is (CCCGAATTCTTAATACGACTCACTATAGGATCCTACTTTAACGTCAAGGAG) and the 3′ oligonucleotide is (GCGGGATCCCTAGAGGTTGGGAACTTCCAG). Alternatively, a linearized pET24b plasmid carrying T7-directed wild-type FAR1 with a carboxy-terminal HIS tag (provided by F. Cross) was used (Fig. 8B). The templates were then transcribed and translated in vitro according to the manufacturers instructions (Promega) to generate [35S]methionine-labeled Far1p substrates. Both wild-type (Fig. 8A) and cdc4 (Fig. 8B) crude extracts were made from G1 cyclin-depleted cells and then fractionated on DEAE Sepharose column. The 0.25 m NaCl eluate, which lacked the endogenous Cdc34p, was used in these assays. Cdc34p was purified from E. coli as described (Banerjee et al. 1993). Cdc4p and Cdc28p were expressed in baculovirus-infected insect cells (Verma et al. 1997c). The radiolabeled substrate was incubated in 10 μl of reaction mixture (Verma et al. 1997a) containing yeast extract (100 μg), reaction buffer containing protease inhibitors, ATP mix, ubiquitin (10 μg), and Cdc34p (100 ng). For reactions with cdc4 extracts, 0.5 μl of insect lysate (∼5 mg/ml) containing Cdc4p or Cdc28p was used. Reactions were incubated at 24°C for 60 min, terminated by the addition of SDS-PAGE sample buffer, boiled, and evaluated by SDS-PAGE and autoradiography.
Pheromone response and mating assays
Halo and mating assays were carried out as described previously (Valtz and Peter 1997). To measure induction of the FUS1–lacZ construct, cells (YMP1055) producing either wild-type Far1p or Far1-22p from the inducible GAL promoter for 6 hr were exposed to α-factor (1 μg/ml) for 1 hr. β-galactosidase levels were quantified as described (Stern et al. 1984). For the α-factor release experiments, strain K2180 producing either wild-type Far1p or Far1-22p from the GAL promoter was grown in raffinose-URA medium (2% raffinose) at 30°C, at which time expression of Far1p and cell cycle arrest was induced by addition of galactose (2% final concentration) and α-factor (1 μg/ml). After 3 hr, cells were washed twice with prewarmed GAL–URA medium (2% galactose) and inoculated in GAL–URA medium at 30°C (time 0). Samples were removed every 30 min and protein extracts prepared as described. An aliquot of cells was fixed, sonicated and the percentage of unbudded cells was determined microscopically as described (Valtz and Peter 1997).
Acknowledgments
We thank members of the Peter and Herskowitz laboratories and B. Amati for discussions, M. Tyers, S. Elledge, G. Ammerer, E. Elion, D. Finley, J. Li, K. Nasmyth, F. Cross, C. Mann, S. O’Rourke, M. Jaquenoud, and I. Davis for kind gifts of plasmids, strains, and antibodies. Thanks to R. Feldmann for providing baculovirus-expressed Cdc4p and G. Reynards for providing baculovirus-expressed Cdc28p. Tierry Laroche is acknowledged for help with the confocal microscope. We thank B. Amati, V. Simanis, and J. Philips for critical reading of the manuscript. This work was supported by a grant from the Swiss Cancer league, the Swiss National Science Foundation, and the Roche Foundation to M.P., and a research grant from the U.S. National Science Foundation to I.H., and a grant from the National Institutes of Health to R.J.D. (GM 52466-01). R.J.D. is a Scholar of the Lucille P. Markey Charitable Trust and Searle/Chicago Community Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mathias.peter@isrec.unih.ch; FAX (41) 21 652 6933.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: Greene Publishing Associates and Wiley-Interscience; 1991. [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Banergee A, Gregori L, Xu Y, Chau V. The bacterially expressed yeast Cdc34 gene product can undergo autoubiquitination to form a multiubiquitin chain-linked protein. J Biol Chem. 1993;268:5668–5675. [PubMed] [Google Scholar]
- Barral Y, Jentsch S, Mann C. G1 cyclin turnover and nutrient uptake are controlled by a common pathway in yeast. Genes & Dev. 1995;9:399–409. doi: 10.1101/gad.9.4.399. [DOI] [PubMed] [Google Scholar]
- Brandeis M, Hunt T. The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J. 1996;15:5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. Cell. 1990;63:999–1011. doi: 10.1016/0092-8674(90)90503-7. [DOI] [PubMed] [Google Scholar]
- ————— Phosphorylation of Far1 in response to alpha-factor: A possible requirement for cell cycle arrest. Mol Biol Cell. 1992;3:445–450. doi: 10.1091/mbc.3.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Clurmann BE, Sheaff RJ, Thess K, Groudine M, Roberts JM. Turnover of cyclinE by the ubiquitin-proteosome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes & Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Connelly C, Hieter P. Budding yeast SKP1 encodes an evolutionary conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR. DAF1, a mutant gene affecting size control, pheromone arrest, and cell cycle kinetics of Saccharomyces cerevisiae. Mol Cell Biol. 1988;8:4675–4684. doi: 10.1128/mcb.8.11.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Starting the cell cycle: What’s the point? Curr Opin Cell Biol. 1995;7:790–797. doi: 10.1016/0955-0674(95)80062-x. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ. Phosphorylation and proteolysis: Partners in the regulation of cell division in budding yeast. Curr Opin Genet Dev. 1997;7:7–16. doi: 10.1016/s0959-437x(97)80103-7. [DOI] [PubMed] [Google Scholar]
- Epstein CB, Cross FR. CLB5, a novel B-cyclin from budding yeast with a role in S-phase. Genes & Dev. 1992;6:1695–1703. doi: 10.1101/gad.6.9.1695. [DOI] [PubMed] [Google Scholar]
- Goebl MG, Yochem J, Jentsch S, McGrath JP, Varshavsky A, Byers B. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science. 1988;241:1331–1335. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- Goebl MG, Goetsch L, Byers B. The Ubc3 (Cdc34) ubiquitin-conjugating enzyme is ubiquitinated and phosphorylated in vivo. Mol Cell Biol. 1994;14:3022–3029. doi: 10.1128/mcb.14.5.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6:56–64. doi: 10.1016/s0959-437x(96)90011-8. [DOI] [PubMed] [Google Scholar]
- Hengst L, Reed SI. Translational control of p27(Kip1) accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluoresence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hershko A, Heller H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem Biophys Res Comm. 1985;128:1079–1086. doi: 10.1016/0006-291x(85)91050-2. [DOI] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Hodgins RW, Ellison KS, Ellison MJ. Expression of a ubiquitin derivative that conjugates to protein irreversibly produces phenotypes consistent with a ubiquitin deficiency. J Biol Chem. 1992;268:8807–8812. [PubMed] [Google Scholar]
- Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–278. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD. Molecular mechanisms of cell-type determination in budding yeast. Curr Opin Genet Dev. 1995;5:552–558. doi: 10.1016/0959-437x(95)80022-0. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Koch C, Nasmyth K. Cell cycle regulated transcription in yeast. Curr Opin Cell Biol. 1994;6:451–459. doi: 10.1016/0955-0674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G(1) cyclin Cln2 induced by Cdk-dependent phosphorylation. Science. 1996;271:1597–1601. doi: 10.1126/science.271.5255.1597. [DOI] [PubMed] [Google Scholar]
- Leberer E, Thomas DY, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Cell cycle control of morphogenesis in budding yeast. Curr Opin Genet Dev. 1995;5:17–23. doi: 10.1016/s0959-437x(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique. 1989;1:11–15. [Google Scholar]
- Loda M, Cukor B, Tam SW, Lavin P, Fiortentino M, Draetta GF, Jessup JM, Pagano M. Increased proteosome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nature Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Cross FR. FAR1 and the G1 phase specificity of cell cycle arrest by mating factor in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2509–2516. doi: 10.1128/mcb.15.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney JD, Chang F, Heintz N, Cross FR. Negative regulation of FAR1 at the Start of the yeast cell cycle. Genes & Dev. 1993;7:833–843. doi: 10.1101/gad.7.5.833. [DOI] [PubMed] [Google Scholar]
- Mendenhall MD. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science. 1993;259:216–219. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- ————— At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Cyclin-dependent protein kinases: Key regulators of the eukaryotic cell cycle. Bioessays. 1995;17:471–480. doi: 10.1002/bies.950170603. [DOI] [PubMed] [Google Scholar]
- Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH, Massague J, Crabtree GR, Roberts JM. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- Oehlen LJ, Cross FR. G1 cyclins CLN1 and CLN2 repress the mating factor response pathway at Start in the yeast cell cycle. Genes & Dev. 1994;8:1058–1070. doi: 10.1101/gad.8.9.1058. [DOI] [PubMed] [Google Scholar]
- Oehlen LJ, McKinney JD, Cross FR. Ste12 and Mcm1 regulate cell cycle-dependent transcription of FAR1. Mol Cell Biol. 1996;16:2830–2837. doi: 10.1128/mcb.16.6.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Peter M. The regulation of cyclin-dependent kinase inhibitors (CKIs) Prog Cell Cycle Res. 1997;3:99–108. doi: 10.1007/978-1-4615-5371-7_8. [DOI] [PubMed] [Google Scholar]
- Peter M, Herskowitz I. Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science. 1994a;265:1228–1231. doi: 10.1126/science.8066461. [DOI] [PubMed] [Google Scholar]
- ————— Joining the complex: Cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell. 1994b;79:181–184. doi: 10.1016/0092-8674(94)90186-4. [DOI] [PubMed] [Google Scholar]
- Peter M, Nakagawa J, Dorée M, Labbé JC, Nigg EA. In vitro disassembly of the nuclear lamina and M-phase specific phosphorylation of lamins by cdc2 kinase. Cell. 1990;61:591–602. doi: 10.1016/0092-8674(90)90471-p. [DOI] [PubMed] [Google Scholar]
- Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. FAR1 links the signal transduction pathway to the cell cycle machinery in yeast. Cell. 1993;73:747–760. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- Peter M, Neiman AM, Park H-O, van Lohuizen M, Herskowitz I. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO J. 1996;15:7046–7059. [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Fink GR. Methods in yeast genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. pp. 119–187. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schneider BL, Yang QH, Futcher AB. Linkage of replication to START by the Cdk inhibitor Sic1. Science. 1996;272:560–562. doi: 10.1126/science.272.5261.560. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. CyclinE-CDK2 is a regulator of p27Kip1. Genes & Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague GF, Thorner JW. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In: Jones EW, Pringle JR, Broach JR, editors. The molecular and cellular biology of the yeast Saccharomyces cerevisiae. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 657–744. [Google Scholar]
- Stern M, Jensen R, Herskowitz I. Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol. 1984;178:853–868. doi: 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- Tyers M. The cyclin-dependent kinase inhibitor p40SIC1 imposes the requirement for Cln G1 cyclin function at Start. Proc Natl Acad Sci. 1996;93:7772–7776. doi: 10.1073/pnas.93.15.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M, Futcher B. Far1 and Fus3 link the mating pheromone signal transduction pathway to three G1-phase Cdc28 kinase complexes. Mol Cell Biol. 1993;13:5659–5669. doi: 10.1128/mcb.13.9.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz N, Peter M. Functional analysis of FAR1 in yeast. Methods Enzymol. 1997;283:350–365. doi: 10.1016/s0076-6879(97)83029-7. [DOI] [PubMed] [Google Scholar]
- Verma R, Chi Y, Deshaies RJ. Cell free ubiquitination of cell cycle regulators in budding yeast extracts. Methods Enzymol. 1997a;283:366–376. doi: 10.1016/s0076-6879(97)83030-3. [DOI] [PubMed] [Google Scholar]
- Verma, R., R. Annan, M. Huddleston, S. Carr, G. Reynard, and R.J. Deshaies. 1997b. Phosphorylation of Sic1p by G1 cyclin/Cdk is required for its degradation and entry into S phase. Science (in press). [DOI] [PubMed]
- Verma R, Feldman RM, Deshaies RJ. SIC1 is ubiquinated in vitro by a pathway that requires CDC4, CDC34, and cyclin/CDK activities. Mol Biol Cell. 1997c;8:1427–1437. doi: 10.1091/mbc.8.8.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J. 1997;16:5334–5344. doi: 10.1093/emboj/16.17.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems AR, Lanker S, Patton EE, Craig KL, Nason TF, Mathias N, Kobayashi R, Wittenberg C, Tyers M. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell. 1996;86:453–463. doi: 10.1016/s0092-8674(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Wittenberg C, Sugimoto K, Reed SI. G1-specific cyclins of S. cerevisiae: Cell cycle periodicity, regulation by mating pheromone, and association with the p34CDC28 protein kinase. Cell. 1990;62:225–237. doi: 10.1016/0092-8674(90)90361-h. [DOI] [PubMed] [Google Scholar]
- Won K-A, Reed SI. Activation of cyclinE/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]