

Abstract
Summary: Here, we present ContEst, a tool for estimating the level of cross-individual contamination in next-generation sequencing data. We demonstrate the accuracy of ContEst across a range of contamination levels, sources and read depths using sequencing data mixed in silico at known concentrations. We applied our tool to published cancer sequencing datasets and report their estimated contamination levels.
Availability and Implementation: ContEst is a GATK module, and distributed under a BSD style license at http://www.broadinstitute.org/cancer/cga/contest
Contact: kcibul@broadinstitute.org; gadgetz@broadinstitute.org
Supplementary information: Supplementary data is available at Bioinformatics online.
1 INTRODUCTION
Next-generation sequencing methods are generating vast amounts of short sequence reads for the purpose of studying DNA sequence variations and identifying those that affect human disease. Many novel methods allow for the interrogation of the structure of the genome with unprecedented sensitivity due to the digital nature of the data (Trapnell and Salzberg, 2009). Rare events present in only a fraction of the sequenced material, as is the case in somatic mutation discovery in cancer genome studies (Berger et al., 2011; Chapman et al., 2011), can be accurately detected by sequencing to greater read depth. Moreover, genome partitioning techniques (Gnirke et al., 2009) allow for even greater sensitivity at a lower cost by targeting only regions of interest.
However, these methods can be heavily compromised by contamination. Three major classes of DNA contamination exist: cross-individual, within-individual and cross-species. Cross-individual is the most critical to control, as even small levels of contamination can cause many false positives, particularly in contrastive tumor versus normal cancer studies (Fig. 1A). Within-individual contamination, such as normal DNA contamination of tumor DNA in cancer studies, typically leads to decreased sensitivity. Cross-species contamination is easily detected by aligning to unique regions of potentially contaminating species. In order to address the most critical need, we developed ContEst within the GATK (McKenna et al., 2010) to accurately estimate the cross-individual contamination level in next-generation sequencing data.
Fig. 1.

(A) False positive somatic mutations detected per megabase on in silico contaminated data; most cancers have ~1 true event per megabase (B) accuracy with single contaminating sample (C) accuracy with multiple contaminating samples (D) accuracy with respect to read depth; shaded areas indicate 95% confidence interval (E) contamination estimates of TCGA Ovarian dataset.
2 METHODS
Given genotype information about the sequenced sample from a genotyping array in VCF format (http://www.1000genomes.org), general population frequency information (provided with ContEst) and the sequencing data in BAM format (Li et al., 2009), we use a Bayesian approach to calculate the posterior probability of the contamination level and determine the maximum a posteriori probability (MAP) estimate of the contamination level.
The method first identifies the homozygous single nucleotide polymorphism (SNP) sites based on the array data, S≡{si}, i=1,…, N, and the alleles at these sites, A≡{Ai}. For each site, si, we denote the probability in the contaminating population to observe Ai at that site by fi, and therefore the probability to see the other allele is 1 − fi. In addition, we denote by bij and eij the called base of the j−th read that covers si and its quality (represented by its probability of being incorrect), respectively. The number of reads that cover si, i.e. the depth at that site, is denoted by di. For a contamination fraction c, we can now calculate the posterior probability using the Bayes rule:
Using a uniform prior on c, i.e.P(c)=1, and assuming that the reads (and noise) are independent and equivalent for all three types of substitutions and discarding sites suspected to be genotyping array data errors (Supplementary Material), we obtain:
where
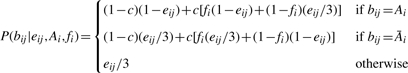 |
The qualities of bases are typically represented using a Phred-like Q-scores, i.e. e=10−q/10. Finally, we evaluate the above equation for c∈[0, 1] and normalize to 1 in order to get the posterior probability. The MAP estimate of c is the mode of this distribution, and a confidence interval can be calculated using the minimal interval containing 95% of the posterior probability. Note that reads that do not support a known allele at S contribute a factor that is independent of c, hence we can ignore them in the calculation.
For tumor samples, we recommend using the genotypes of the patient-matched normal when available instead of the tumor, since homozygous SNPs in regions of loss of heterozygosity in the tumor will interpret contamination with normal cells from the same patient as foreign DNA since they have different genotypes.
3 RESULTS
Using next-generation sequencing data from the TCGA Ovarian publication (TCGA Research Network, 2011), we identified 12 exome-capture BAMs with low contamination, having very few reads that do not match the homozygous calls from their genotyping arrays (Supplementary Table S1). Next, we created in silico datasets by mixing a primary sample with one or more contaminants at specific contamination levels (Supplementary Material). Reassuringly, the estimate of the contamination level of the primary sample alone was 0.08%. ContEst was able to accurately predict the level of contamination across a wide range of conditions including more than a single contaminating sample. (Fig. 1B and C)
In order to assess the accuracy as a function of sequencing depth, we downsampled the depth of the sequencing data (Fig. 1D), and demonstrated that ContEst produces accurate estimates even with average coverage <5×.
Applying the method to data obtained from the TCGA Ovarian publication (Supplementary Table S2) indicates that low levels of physical contamination are common (Fig. 1E). Independent validation of all somatic events likely ensured that this contamination did not cause false positives in the publication. However, given a distribution of contamination as seen in TCGA (Fig. 1E), and an estimated error rate at non-dbSNP sites from contamination as shown in Figure 1A, a typical cancer project might expect >10% of the samples to have >1.5% contamination, causing ~0.2 errors/Mb per sample, which is a significant fraction of the typical somatic mutation rate of 1/Mb per sample.
In addition, ContEst has proven to be essential in lab quality control to identify and monitor sources of contamination, which has helped decrease contamination at the Broad Institute.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge our colleagues from the Broad Sequencing Platform, Genetics Analysis Platform and The Cancer Genome Atlas Project who supported the development of ContEst, as well as Rameen Beroukhim for valuable discussions.
Funding: National Human Genome Research Institute (grant number U24 CA126546).
Conflict of Interest: none declared.
REFERENCES
- Berger M.F., et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman M.A., et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Salzberg S.L. How to map billions of short reads onto genomes. Nat. Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.