

Abstract
IFN-γ–producing Th1 and IL-17–producing Th17 cells are the key participants in various autoimmune diseases, including multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis (EAE). Although both of these T cell subsets are known to be regulated by specific transcription factors and cytokines, the role of microRNAs that control these two inflammatory T cell subsets and whether targeting microRNAs can have therapeutic effects are not known. In this study, we show that microRNA-155 (Mir-155) expression is elevated in CD4+ T cells during EAE, and Mir-155−/− mice had a delayed course and reduced severity of disease and less inflammation in the CNS. The attenuation of EAE in Mir-155−/− mice was associated with a decrease in Th1 and Th17 responses in the CNS and peripheral lymphoid organs. The T cell-intrinsic function of Mir-155−/− was demonstrated by the resistance of Mir-155−/− CD4+ T cell-repleted Rag-1−/− mice to EAE. Finally, we found that anti–Mir-155 treatment reduced clinical severity of EAE when given before and after the appearance of clinical symptoms. These findings demonstrate that Mir-155 confers susceptibility to EAE by affecting inflammatory T cell responses and identify Mir-155 as a new target for therapeutic intervention in multiple sclerosis.
CD4+ T cell-mediated autoimmunity has been accepted as one of the most important aspects of multiple sclerosis (MS) pathogenesis. IFN-γ–producing Th1 cells have been considered the type of effector T cells that mediate the pathogenesis of MS and its animal model, experimental autoimmune encephalomyelitis (EAE) (1–3). However, recent studies have indicated that IL-17–producing Th17 cells are involved and are as critical as Th1 cells in this pathogenesis (4). The development of autoreactive T cells is largely determined by their cytokine milieu. Cytokines involved in the Th1 and Th17 axes of inflammation are detected in the CNS of mice with EAE and active lesions of MS patients (2–6). Moreover, adoptive transfer of both Th1 and Th17 cells can initiate the autoimmune cascade in this disease (7, 8). Distinct signaling pathways govern the differentiation of Th1 versus Th17 cells. IL-12 and IFN-γ signals are important for Th1 cell differentiation. The Th1 cytokine IFN-γ signals through STAT-1, which in turn activates the T-box transcription factor T-bet, which is the key inducer of IFN-γ and Th1 cell differentiation (9). Several cytokines, such as TGF-β, IL-6, IL-1β, and IL-21, have been shown to regulate and induce the differentiation of naive T cells toward the Th17 phenotype (10). The differentiation of Th17 cells requires the expression of transcription factor retinoid orphan nuclear receptor (RORγt, an orphan nuclear hormone receptor). Although the cytokines described above positively regulate Th17 differentiation, other cytokines in the immune system have been shown to negatively regulate differentiation of Th17 cells. The cytokines IL-4, IFN-γ, IL-2, and IL-27 have been shown to inhibit Th17 cell differentiation (11). Although differentiation and function of Th1 and Th17 cells are regulated through specific set of cytokines and transcription factors, the role of microRNAs (miRNA) that target these two pathogenic T cell subsets during autoimmune inflammation is not known.
miRNA are small endogenous noncoding RNAs that post-transcriptionally repress the expression of genes. Dysregulation of miRNA expression and function is associated with a variety of human diseases, including cancer and many inflammatory diseases (12). The enzyme responsible for regulatory RNA biogenesis, Dicer, is required for lymphocyte function, which suggests regulatory role for miRNAs in the immune system. miRNAs can affect developmental outcomes in thymic T cell precursors, influence T regulatory (Treg) cell development, and affect the production of Abs to thymic-dependent Ags (12–15). However, the role of miRNAs in the regulation and control of autoimmune disease is unclear. In this study, we demonstrate that microRNA-155 (Mir-155) influences both Th1 and Th17 effector subsets and contributes to autoimmune pathology.
Materials and Methods
Mice
C57BL/6 wild-type (WT), miRNA-155−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Animals were maintained in a specific pathogen-free condition in the animal facility of Harvard Institutes of Medicine. All mice were 6–8 wk old at the beginning of experiments. All experiments were in accordance with guidelines from the committee on animals at Harvard Medical School.
Induction and evaluation of EAE
Mice were injected s.c. in both flanks with 100 μg myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) dissolved in PBS emulsified in an equal volume CFA-CFA (Difco) supplemented with 5 mg/ml Mycobacterium tuberculosis H37Ra and injected twice i.p. with 200 ng pertussis toxin (List Biological Laboratories) administered on the day of immunization and 48 h later. Clinical assessment of EAE was performed daily after disease induction, according to the following criteria: 0, no disease; 1, tail paralysis; 2, hindlimb weakness or partial paralysis; 3, complete hindlimb paralysis; 4, forelimb and hindlimb paralysis; 5, moribund state. Mean clinical scores on separate days were calculated by adding scores of individual mice and dividing total number of mice in each group, including mice that did not develop signs of EAE. For histopathological studies, spinal cords were dissected from female mice (n = 5/group), fixed in 10% formalin in PBS, and embedded in a single paraffin block. The 6- to 10-μm–thick sections were stained with H&E and luxol fast blue, and stained sections were evaluated for immune cell infiltration and demyelination.
Generation and isolation of dendritic cells
Dendritic cells (DCs) were derived from bone marrow progenitor cells using a previously described method (16). In brief, the femoral and tibial cells were harvested in DC culture medium (RPMI 1640 medium, 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 ng/ml GM-CSF, and 10 ng/ml IL-4) and seeded in 24-well plates at a density of 1 × 106 cells/ml/well. Culture medium was replaced with fresh medium every 3 d. At day 6, dislodged cells were used as bone marrow-derived DCs. Splenic DCs were isolated using CD11c beads (Miltenyi Biotec).
Analysis of Mir-155 expression
For analysis of Mir-155 expression, real-time RT-PCR analyses were carried out using TaqMan miRNA assays (Applied Biosystems) and relative expression was calculated using the CT method, as described, and normalized to uniformly expressed snoRNA135 (Applied Biosystems). Mir-155 values were expressed relative to the expression of snoRNA135. The levels of Mir-155 in CNS-infiltrating DCs during EAE are normalized to DCs isolated from spleen of naive mice.
RNA isolation, cDNA synthesis, and real-time PCR
Total RNA was isolated from cell pellets using RNA easy micro kit (Qiagen). RNA was stored at −80°C. First-strand cDNA synthesis was performed for each RNA sample from 0.5–1 μg total RNA using TaqMan reverse-transcription reagents. cDNA was amplified using sequence-specific primers (probes used were identified by Applied Biosystems assay identification number: IFN-γ, Mm01168134_m1; T-bet, Mm00450960_m1; IL-17, Mm99-999062_m1; RORγt, Mm01261019-m1; IL-17F, Mm00521423_m1; Foxp3, Mm00475151_m1; IL-4, Mm00445259_m1; IL-23R, Mm00519942_m1; IL-23, Mm01160011_m1; IL-12, Mm99999066_m1; IL-6, Mm99999064_m1; IL-1β, Mm01336189_m1; TNF-α, Mm00443258_m1 [Applied Biosystems]) and real-time PCR mix (Applied Biosystems) on ABI7500 cycler. GAPDH gene was used as an endogenous control to normalize for differences in the amount of total RNA in each sample. All values were expressed relative to the expression of GAPDH.
Anti–mir-155 treatment
An antisense oligonucleotide modified by locked nucleotide acid (LNA; Exiqon) was synthesized to inhibit Mir-155. For in vivo anti–mir-155 treatment, 30 μl lipofectamine 2000 (Invitrogen) was mixed with anti–mir-155 (20 μg/mouse) dissolved in 170 μl PBS, and the liposome complexes were administered i.v. to MOG-immunized mice on day 5, 7, 9, and 11 postimmunization. For EAE reversal, Mir-155 inhibitor was administered when a clinical score of ≥1.5 was observed.
Proliferative responses of T cell and cytokine analysis
Spleens or draining lymph nodes (LN; inguinal regions) were harvested and pooled from EAE mice, and single-cell suspensions were prepared. Cells were cultured at 0.5 million/well in 96-well U-bottom plates with a range of concentrations of MOG35–55 peptide in RPMI 1640 medium supplemented with 10% FCS. Plates were pulsed after 60 h of culture with [3H] thymidine at 1 μCi/well for the final 18 h, harvested, and assayed for proliferation. Mean incorporation of thymidine in DNA was measured in triplicate wells and is indicated as cpm. For ELISA, supernatants were harvested at 72 h of culture. The concentrations of indicated cytokines were measured by quantitative capture ELISA, according to the guidelines of the manufacturers (BD Biosciences).
Cell culture
Total CD4+ T cells were cultured with plate-coated anti-CD3 and anti-CD28 mAb (2 μg/ml) for 72 h. For in vitro Th17 cell differentiation, naive CD4+ CD62LhighCD44low T cells from WT and Mir-155−/− mice were sorted by flow cytometry and activated with plate-bound anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) in the presence of TGF-β (3 ng/ml), IL-6 (20 ng/ml), with or without IL-23 (10 ng/ml), IL-1β (10 ng/ml), and TNF-α (10 ng/ml). For Th1 polarization, naive CD4+CD62LhighCD44low T cells were activated with IL-12 (20 ng/ml) in the presence of anti–IL-4 (20 μg/ml). Supernatants from cultures were harvested 4 d after initiation of cultures. IFN-γ and IL-17 in the supernatants were assayed by ELISA kits (BD Biosciences).
CD4+ T cell transfer
CD4+ T cells were prepared from the spleens of WT and Mir-155−/− mice using the CD4+ T cell isolation kit (Miltenyi Biotec) (purity was >95%). CD4+ T cells (2 × 107 per mouse) were injected i.p. into Rag1−/− mice. Five d later, the recipient mice were subjected to EAE induction.
Preparation and evaluation of CNS cells
Animals were perfused with cold PBS. Brains and spinal cords were dissected and incubated in 2.5 mg/ml collagenase D for 30 min at 37°C. Single-cell suspensions were prepared by passing through 70-μm strainer. Cells were washed in RPMI 1640 medium, and mononuclear cells were isolated using a discontinuous Percoll gradient (Pharmacia, Piscataway, NJ). Cells were washed twice, and CD11c+ cells were isolated from this suspension by magnetic separation using microbeads (Miltenyi Biotec).
Statistical analysis
Statistical analysis was performed using the unpaired t test. A p value <0.05 was considered significant. Data are presented as mean SEM. For EAE, groups were compared using linear regression analysis.
Results
Mir-155−/− mice develop attenuated EAE
To study the role of Mir-155 in EAE development, we first sought to analyze the expression of Mir-155 in mice during the course of EAE. We found that Mir-155 expression was increased in spleen, LN, and CNS during EAE (Supplemental Fig. 1). Because CD4+ T cells are the key mediators of pathogenesis in EAE, we investigated whether CD4+ T cells express Mir-155 during EAE. Mir-155 expression was analyzed in CD4+ T cells (spleen, LN, and CNS) from C57BL/6 mice immunized for EAE with MOG35–55 peptide. We found that Mir-155 expression was markedly increased in CD4+ T cells isolated in spleen, LN, and CNS of EAE mice compared with naive mice (Fig. 1A). The potential role of Mir-155 in EAE was next tested using Mir-155–deficient mice (Mir-155−/− mice). Compared with WT mice, Mir-155−/− mice showed delayed onset of neurologic impairment and markedly less severe disease (Fig. 1B). The attenuation of paralytic symptoms in the Mir-155−/− mice was associated with decreased inflammation and demyelination ((Fig. 1C, 1D). Although we found that EAE was associated with demyelination and infiltration of CD4+ T cells and CD11c+ cells from peripheral lymphoid organs to the CNS in both Mir-155−/− and WT mice, the number of CD4+ T cells and CD11c+ cells was markedly lower in the CNS of Mir-155−/− mice at the peak of disease (Fig. 1E). Thus, Mir-155 significantly influenced the course of progressive EAE induced by MOG 35-55.
FIGURE 1.
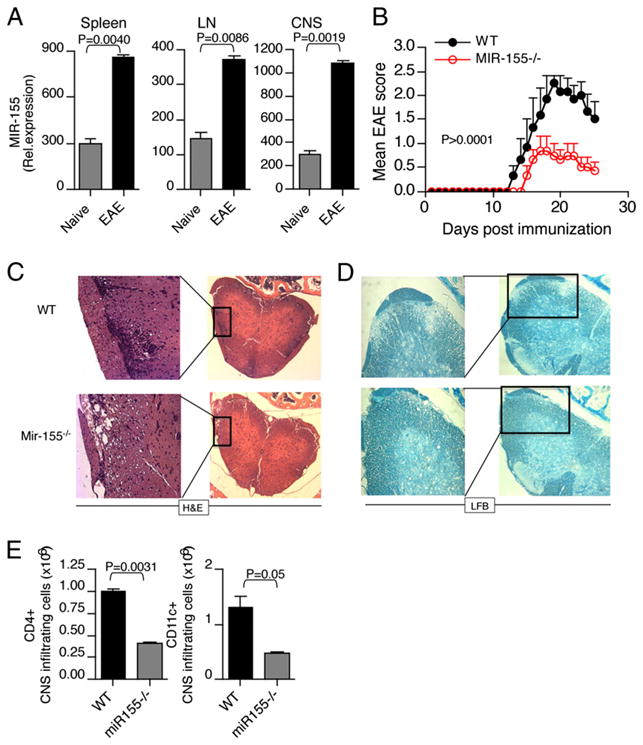
Mir-155−/− mice develop attenuated EAE. Elevated Mir-155 expression in CD4+ T cells during EAE. A, Mir-155 mRNA was determined by quantitative real-time PCR analysis in CD4+ T cells from spleen, LN, and CNS from naive and EAE mice. B, Mir-155−/− mice develop attenuated EAE. Clinical scores of WT mice (WT; n = 8) and Mir-155−/− mice (Mir-155−/−; n = 8) at various times after immunization with MOG(33–55) in CFA. Results are representative of three independent experiments. C and D, Histopathological analysis of spinal cord sections from representative of WT and Mir-155−/− mice (n = 3/group) at 20 d after immunization. Spinal cord sections were stained with H&E and luxol fast blue to access inflammation and demyelination. Original magnification ×20 (C) and ×40 (D). E, Number of CNS-infiltrating CD4+ T cells and CD11c+ cells. Mononuclear cells were isolated from spinal cords of WT and Mir-155−/− mice (n = 3/group) at 20 d after immunization, and then they were pooled and counted. Cells were stained with Abs specific for cell surface markers, as indicated, and the percentage of each subset is shown.
Mir-155 favors T cell-mediated autoimmunity
The candidate contributors to EAE pathology are IFN-γ–secreting Th1 and IL-17–secreting Th17 CD4+ T cells. To explore the impact of Mir-155 on the development of pathogenic Th cell subsets, we isolated spleens from WT and Mir-155−/− mice at 10 d after immunization and examined the recall proliferation and cytokine production in response to MOG p35-55 stimulation. We found that splenocytes from WT mice proliferated more robustly and produced higher levels of IFN-γ and IL-17 than splenocytes from Mir-155−/− mice (Fig. 2A–C). Consistent with the clinical scores, ex vivo CD4+ T cells isolated from spleen and LN of WT mice expressed higher levels of IFN-γ and IL-17 in comparison with CD4+ T cells from Mir-155−/− mice (Fig. 2D, 2E). We then tested ex vivo CD4+ T cells isolated from LN and splenocytes from WT and Mir-155−/− mice for the presence of IL-17 and IFN-γ by intracellular staining. Consistent with the RNA data, Mir-155−/− mice had reduced numbers of Th17 cells in both their LN and spleens compared with WT mice (Fig. 2D, 2E). In addition, we found moderately reduced amounts of Th1 cells in the spleens, but not LN of Mir-155−/− mice (Fig. 2D, 2E). Because Th1 and Th17 cells can initiate and perpetuate the CNS inflammatory response in EAE, we next examined the cytokine expression by CNS-infiltrating CD4+ T cells during the course of the disease. We found an increased expression of IFN-γ and IL-17 in CD4+ T cells isolated from CNS of WT mice compared with Mir-155−/− mice (Fig. 2F). Both Th1 and Th17 cells have been shown to develop under strict influence by Th2 and Treg cells, respectively (10). Mir-155 has been shown to regulate both Th2 and Foxp3 expressing Treg cells. Reduced numbers of Treg cells and enhanced Th2 differentiation were observed from Mir-155−/− mice (13–15). However, Mir-155−/− Treg cells are equally suppressive as WT Treg cells on a per cell basis (15). Consistent with previous studies, we also observed reduced Foxp3 expression in CD4+ T cells isolated from Mir-155−/− mice with EAE in comparison with WT controls (Supplemental Fig. 2A). However, the Th2 cytokine IL-4 expression was not altered between WT and Mir-155−/− mice with EAE (Supplemental Fig. 2B). Thus, the observed difference in CNS inflammatory response was not a reflection of skewing toward a Treg phenotype, but rather due to an increased expression of IFN-γ and IL-17 in CNS-infiltrating CD4+ T cells of WT mice.
FIGURE 2.
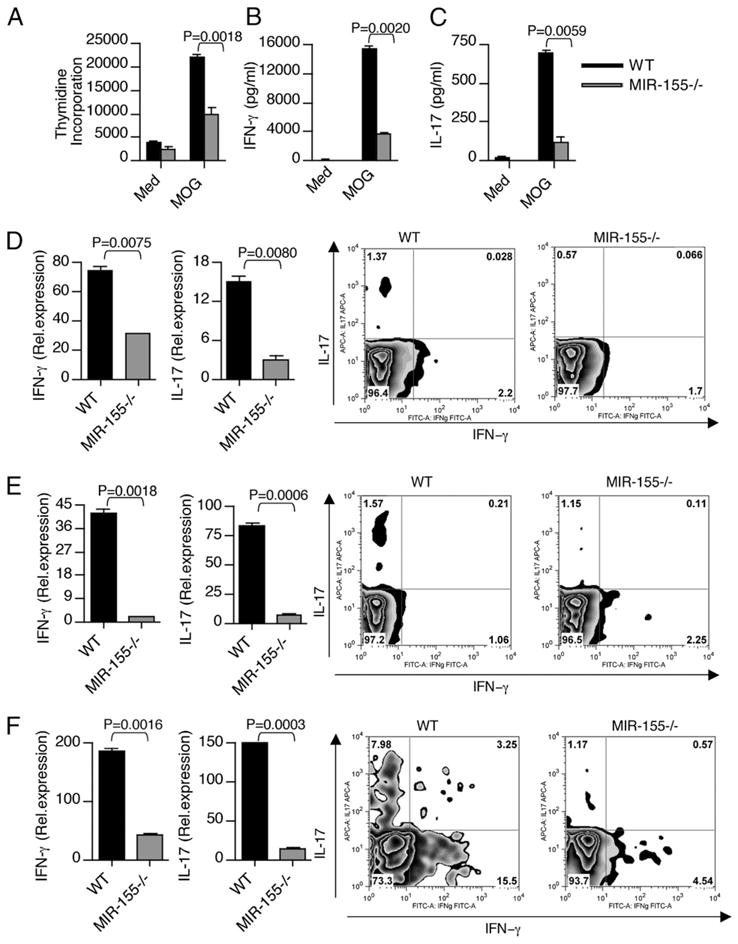
Mir-155−/− mice exhibit lower Th1 and Th17 cytokine production during EAE. Splenocytes from WT and Mir-155−/− mice were harvested 10 d after immunization and were stimulated ex vivo with MOG peptide 35–55. A, In the last 16 h, cells were pulsed with thymidine and assayed for proliferation (cpm). Error bars represent SEM between triplicates. Supernatants from parallel cultures were harvested 72 h after initiation of cultures and assayed by ELISA for IFN-γ (B) and IL-17 (C). Real-time RT-PCR analysis and intracellular staining of IFN-γ and IL-17 in (D) spleen-, (E) LN-, and (F) CNS-derived CD4+ T cells isolated from WT and Mir-155−/− mice with EAE. Data in A–F are representative of three experiments.
Mir-155 regulates CD4+ T cell effector cytokine production
To determine whether the enhanced T cell autoimmunity in WT mice was a result of intrinsic differences in CD4+ T cells, we compared the cytokine profiles of WT and Mir-155−/− total CD4+ T cells in response to ex vivo stimulation with anti-CD3 and anti-CD28. We found higher production of IFN-γ (Fig. 3A, 3C) and IL-17 (Fig. 3B) by WT CD4+ T cells than Mir-155−/− CD4+ T cells. These results indicate that the enhanced cytokine production of IFN-γ and IL-17 that we observed in WT mice was in part a result of intrinsic differences in CD4+ T cells. To further determine the role of Mir-155 on Th1 and Th17 differentiation, WT and Mir-155−/− naive CD4+ T cells were differentiated into Th1 and Th17 cells. When activated under Th1-polarizing condition, Mir-155−/− naive CD4+ T cells produced equal amounts of IFN-γ and expressed normal levels of T-bet comparable to CD4+ T cells from WT mice (Fig. 3D). We then tested whether there is a defect in IFN-γ production in the memory CD4+ T cell compartment that accounted for the reduced IFN-γ levels observed in total CD4+ T cells from Mir-155−/− mice. For this we sorted memory T cells (CD4+CD44highCD62Llow) from WT and Mir-155−/− mice and stimulated with anti-CD3 and CD28 Abs. We did not find significant differences in IFN-γ production in memory T cells between WT and Mir-155−/− mice (Fig. 3E). However, we did find reduced number of memory CD4+ T cells in the spleens of age-matched Mir-155−/− mice compared with WT mice (Fig. 3F). Thus, the increased IFN-γ levels that we observed in total CD4+ T cells from WT mice are due to more memory CD4+ T cells. When naive CD4+ T cells are activated by the combination of IL-6 and TGF-β (Th17-polarizing conditions), IL-17 production in Mir-155−/− T cells was significantly reduced compared with WT-T cells, even when adding IL-23, IL-1β, and TNF-α (Fig. 3G, 3H). In addition, WT-T cells expressed higher levels of Th17-associated molecules such as IL-17F, IL-22, and IL-23R than that of Mir-155−/−-derived Th17 cells (Supplemental Fig. 3A). However, expression of RORγt and IL-21, two other Th17-associated molecules, was not altered between these two groups (Supplemental Fig. 3B), suggesting that Th17 cells are transcriptionally programmed in Mir-155−/− animals even though they do not produce similar levels of effector cytokines. To demonstrate the CD4+ T cell-intrinsic function of Mir-155 in vivo, we transferred WT or Mir-155−/− CD4+ T cells into Rag-1−/− mice and subsequently immunized them with MOG peptide. We found that the T cells both from WT and Mir-155−/− mice are engrafted equally into Rag-1−/− mice and are recovered at normal rates (Fig. 4A). The mice repleted with WT CD4+ T cells developed severe EAE, whereas the mice repleted with Mir-155−/− CD4+ T cells only showed minimal symptoms (Fig. 4B). Consistent with the clinical scores, significant difference in proliferation was observed between WT and Mir-155−/− CD4+ T cells (Fig. 4C). Splenocytes from the Rag1−/− mice reconstituted with WT CD4+ T cells exhibited higher IFN-γ and IL-17 production as compared with those reconstituted with Mir-155−/− CD4+ T cells (Fig. 4D). Reduced IL-17 and IFN-γ mRNA expression in the CNS of mice with Mir-155−/− T cells supports the regulatory role of Mir-155 on inflammatory T cell development (Fig. 4E). Moreover, Rag1−/− mice reconstituted with Mir-155−/− CD4+ T cells had reduced numbers of IL-17 single-positive and IL-17+ IFN-γ+ double-positive cells in their brains compared with WT mice (Fig. 4F). Collectively, these results indicate that Mir-155 deficiency in the CD4+ T cell compartment leads to defective inflammatory T cell development and CNS inflammation during EAE.
FIGURE 3.
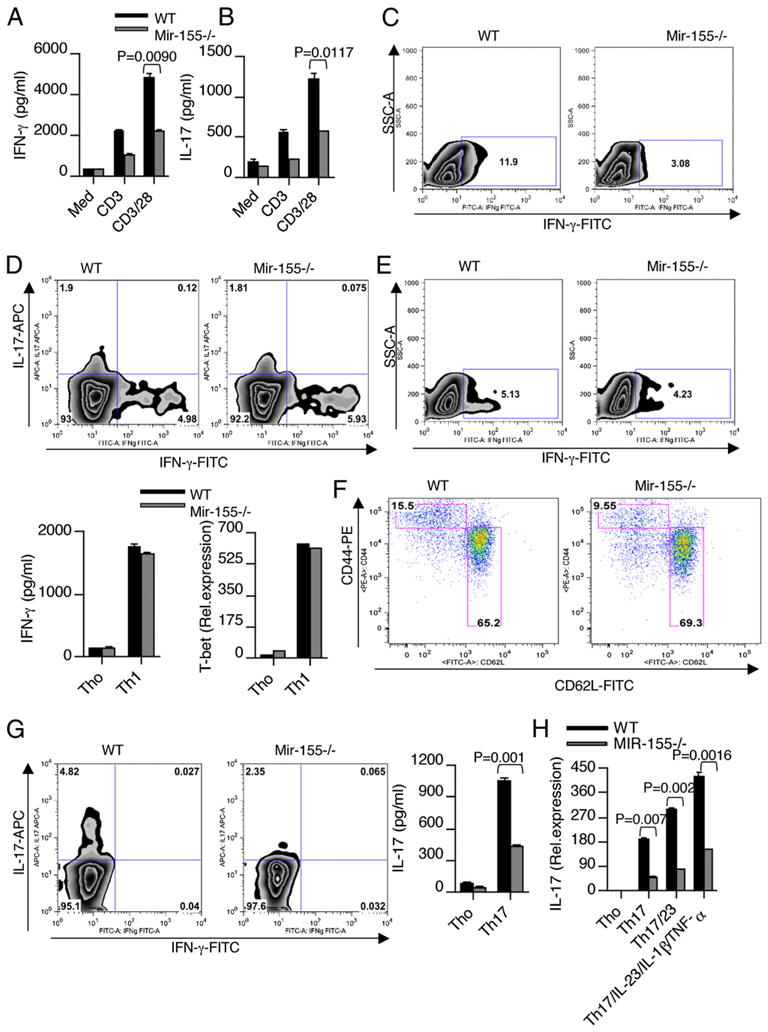
Mir-155 regulates CD4+ T cell effector cytokine production. Total CD4+ T cells were isolated from the spleens of WT and Mir-155−/− mice and stimulated with anti-CD3 and anti-CD28. Supernatants from cultures were harvested 72 h after initiation of cultures and analyzed for cytokines (A, B) IFN-γ and IL-17. Intracellular staining was used to determine the number of IFN-γ–producing cells (C). Data are representative of three independent experiments. D, Naive CD4+ T cells (CD4+CD62LhighCD44low) from spleens were purified by flow cytometry and stimulated with plate-bound Ab against CD3 (2 μg/ml) and CD28 (2 μg/ml) under Th1-skewing condition. Intracellular staining was used to determine the number of Th1 cells. Supernatants from cultures were harvested 4 d after initiation of cultures and analyzed for cytokines IFN-γ by ELISA. Similarly, stimulated cells were harvested 48 h poststimulation for mRNA and analyzed for T-bet by quantitative real-time PCR. Data are representative of five independent experiments. E, Memory CD4+ T cells (CD4+CD62LlowCD44high) from spleens were purified by flow cytometry and stimulated with plate-bound Ab against CD3 (2 μg/ml) and CD28 (2 μg/ml). Intracellular staining was used to determine the number of IFN-γ–producing cells. Data are representative of three independent experiments. F, Flow cytometry analysis of CD44high memory CD4+ T cells in the spleens of WT and Mir-155−/− mice. Naive CD4+ T cells (CD4+CD62LhighCD44low) from spleens were purified by flow cytometry and stimulated with plate-bound Ab against CD3 (2 μg/ml) and CD28 (2 μg/ml) under Th17-skewing condition. G, Intracellular staining was used to determine the number of Th17 cells. H, IL-17 mRNA was determined by quantitative real-time PCR analysis. Data are representative of three to five independent experiments.
FIGURE 4.

T cell-intrinsic effect of Mir-155 in EAE. Rag-1−/− mice (n = 5/group) were injected with either WT or Mir-155−/− CD4+ T cells. Seven d after transfer, mice were harvested, and engraftment of CD3+CD4+ T cells was assayed by flow cytometry with splenocytes and LN (A). Rag-1−/− mice (n = 5/group) were injected with either WT or Mir-155−/− CD4+ T cells. Seven d after transfer, mice were immunized with MOG peptide emulsified in CFA and were monitored for EAE (B). Results are representative of two independent experiments. C, Splenocytes from these mice were activated in vitro with MOG p35–55 (20 μg/ml). Proliferation was measured by [3H]thymidine incorporation assay. Error bars represent SEM between triplicates. D, Cytokines were measured in culture supernatants using ELISA. Data are representative of two experiments. E and F, Real-time RT-PCR and flow cytometry analysis of IFN-γ and IL-17 in CNS-derived CD4+ T cells isolated from Rag-1−/− mice reconstituted with T cells from WT and Mir-155−/− mice.
Mir-155 positively regulates Th1- and Th17-polarizing cytokine expression in DCs
Polarization of T cells to Th1, Th2, or Th17 phenotypes is a critical feature of cell-mediated immunity, and is influenced by production of cytokines by DC. For example, DC-secreted IL-12 has been shown to favor Th1 development, whereas DC-secreted IL-10 or IL-27 has been shown to induce IL-10–producing Tr1 cells (17, 18). Likewise, DC-secreted IL-1β and IL-6 appear to synergize with IL-23 in the induction of IL-17 by CD4+ T cells (19). To further understand the nature of defective Th1 and Th17 immune responses in vivo, we explored the possibility of an intrinsic requirement for Mir-155 in DC function. Therefore, we tested whether Mir-155 modulates the expression of Th1- and Th17-polarizing cytokine by DCs. For this, we stimulated bone marrow-derived DCs from WT and Mir-155−/− mice with the TLR ligand LPS and analyzed cytokine expression. In agreement with previous study in macrophages (20), LPS stimulation induced Mir-155 expression in DCs (Fig. 5A). Although LPS stimulation induced the expression of Th1-polarizing cytokine IL-12 and Th17-polarizing cytokines IL-1β, IL-6, IL-23, and TNF-α both from WT and Mir-155−/− DCs, significant differences were observed between DCs from Mir-155−/− mice compared with WT mice. In Mir-155−/− DCs, decreased levels of both Th1- and Th17-polarizing cytokines were observed in response to LPS stimulation (Fig. 5B, 5C), although we did not observe a difference in Th2-polarizing cytokine. Furthermore, Mir-155 deficiency did not alter DC development in vitro, as we do not see any difference in DC numbers from WT and Mir-155−/− mice-derived BM-DC culture (Supplemental Fig. 3C). Similarly, reduced expression of both Th1- and Th17-polarizing cytokines was observed from macrophages in response to LPS stimulation (Supplemental Fig. 3D, 3E). Indeed, in a DC–T cell coculture system, Mir-155 deficiency in DCs resulted in significantly less IFN-γ and IL-17 production from T cells (Fig. 5D). In EAE, the disease is induced peripherally by injection of self-Ag in association with a strong adjuvant containing killed whole mycobacteria. Thus, TLRs may play an important role by inducing expression of Mir-155 and inflammatory cytokines from DCs, which may promote Th1 and Th17 development and contribute to the pathogenesis of autoimmune disease. To investigate whether Mir-155 expression is induced in DCs after EAE induction, we measured Mir-155 levels in DCs isolated from mice with EAE and compared with DCs isolated from naive mice. We found that during EAE, Mir-155 expression is elevated in DCs both in the periphery and in the CNS (Fig. 5E). To test whether Mir-155 modulates Th1- and Th17-polarizing cytokine expression by DCs in vivo, we induced EAE in WT and Mir-155−/− mice. We found that both Th1- and Th17-polarizing cytokine expression was markedly higher in WT-DCs than that of Mir-155−/− DCs at the peak of disease in spleen (Fig. 5F) and LN (Fig. 5G). These results suggest that Mir-155 in DCs plays a significant role in expression of cytokines required for Th1 and Th17 cell responses.
FIGURE 5.

Mir-155 positively regulates Th1-and Th17-polarizing cytokine expression in DCs. Bone marrow-derived DCs isolated from WT mice (n = 5/group) were stimulated with LPS. A, Mir-155 mRNA was determined by quantitative real-time PCR analysis in DCs in response to LPS. B and C, Bone marrow-derived DCs were isolated from WT and Mir-155−/− mice and were cultured with 100 ng/ml LPS. Cell-free supernatants were measured for Th1- and Th17-polarizing cytokine by ELISA. D, CD4+ T cells isolated from WT mice were cocultured with CD11c+ DCs derived from WT and Mir-155−/− mice. Supernatants from cultures were harvested 72 h after initiation of cultures and assayed by ELISA for IFN-γ and IL-17. E, Mir-155 mRNA was determined by quantitative real-time PCR analysis in DCs isolated from LN and CNS of mice with and without EAE (n = 4/group). Data are representative of two independent experiments. Real-time PCR measurement of Th1-and Th17-polarizing cytokines in DCs isolated from spleen (F) and LN (G) of WT and Mir-155−/− mice with EAE. Data are representative of three experiments.
Anti–Mir-155 treatment resulted in inhibition of EAE and reduction of CNS inflammation
We then investigated whether systemic administration of anti–Mir-155 in vivo affects the course of EAE. Therapeutic efficacy of LNA-modified oligonucleotide (LNA–anti-miR) technology has recently been reported (21, 22). Administration of LNA-modified anti–Mir-155 during the preclinical stage of EAE (beginning on day 5 after immunization) significantly ameliorated clinical disease (Fig. 6A). In addition, we found that peripheral administration of anti–Mir-155 during EAE affected both CD4+ T cells and APCs, including DCs and macrophages (Supplemental Fig. 4). To investigate whether anti–Mir-155 treatment affected Ag-specific recall responses, we isolated spleen and LN of MOG-immunized mice treated with anti–Mir-155 or scrambled control and stimulated them in vitro with MOG peptide. We found that splenocytes from anti–Mir-155–treated mice had a diminished MOG-specific proliferative response, reduced IFN-γ and IL-17 secretion (Fig. 6B, 6C). In addition, consistent with our data and with the dramatic inhibitory effect on the clinical EAE symptoms by anti–Mir-155, we found that anti–Mir-155 treatment prevented upregulation of IL-17 and IFN-γ transcripts in CD4+ T cells isolated from the CNS of the anti–Mir-155–treated mice (Fig. 6D). Moreover, DCs isolated from LN of anti–Mir-155–treated mice had lower levels of Th1-polarizing cytokine IL-12 and Th17-polarizing IL-1β, IL-6, IL-23, and TNF-α (Fig. 6E). Thus, anti–Mir-155 suppresses EAE in association with a decrease in IL-17 and IFN-γ. In the clinical setting of MS, therapeutic intervention is often started after the onset of the symptoms. Therefore, it is important to investigate whether a treatment regimen, which is effective in EAE prevention, can also reverse established disease. Thus, we tested the efficacy of Mir-155 inhibitor on EAE after the onset of clinical symptoms. We found that treatment with Mir-155 inhibitor after the onset of EAE (score of ≥1.5) enhances clinical recovery from EAE (Fig. 6F).
FIGURE 6.

Anti–Mir-155 treatment reduces the clinical severity of EAE. A, Clinical scores of EAE in WT mice (n = 5/group) treated with anti–Mir-155 or scrambled control on days 5, 7, 9, and 11 postimmunization. Results are representative of two independent experiments. B, Splenocytes obtained from anti–Mir-155 and control mice were restimulated with MOG35–55 (20 μg/ml) for 72 h. Proliferation was measured by [3H]thymidine incorporation assay. Error bars represent SEM between triplicates. C, Cell-free supernatants were assayed for IL-17 and IFN-γ by ELISA. D, Real-time RT-PCR analysis of IFN-γ and IL-17 in CNS-derived CD4+ T cells isolated from mice treated with or without anti–Mir-155. E, Real-time PCR measurement of Th1- and Th17-polarizing cytokines in DCs isolated from LN of control and anti–Mir-155–treated mice (n = 5/group). Data are representative of two independent experiments. F, Mice treated with Mir-155 inhibitor after onset of EAE (score of ≥1.5) resulted in a rapid clinical recovery from EAE.
Discussion
IFN-γ and IL-17 are produced by Th1 and Th17 cells, respectively. They are key proinflammatory mediators of cellular immunity that underlie crucial pathological events during development of EAE. Although genetic studies showed that neither IFN-γ nor IL-17 is essential for the initiation of the disease, multiple studies are in agreement that high levels of IFN-γ and/or IL-17 correlate with severity of the disease. In MS patients, elevated levels of IL-17 and IFN-γ correlated with exacerbation of the disease. The Th1 lineage and chemokine receptor-mediated events can initiate EAE by facilitating entry of Th17 cells into the CNS (23, 24). Furthermore, the ratio of Th1 cells to Th17 cells can affect the CNS lesions and disease severity (8). Our study provides evidence that Mir-155 is an important positive regulator of CNS autoimmune inflammation. We found that mice deficient in Mir-155 exhibit a delayed course and reduced severity of disease and less inflammation in the CNS. The attenuation of EAE in Mir-155−/− mice was associated with a decrease in Th1 and Th17 responses in the CNS and peripheral lymphoid organs. The increased IL-17 and IFN-γ expression in WT mice suggested that Mir-155 favored the emergence of Th17 and Th1 cells, which would sustain inflammation and exacerbate clinical signs of EAE. Thus, the reduction of IFN-γ and IL-17 is most likely a major underlying mechanism for the amelioration of EAE in Mir-155−/− mice. Finally, we found that in vivo silencing of Mir-155 using LNA-modified anti–Mir-155 reduced clinical severity of EAE associated with a decrease in Th1 and Th17 cells. In addition, it was reported by O’Connell et al. (25) that Mir-155−/− mice develop reduced footpad inflammation during delayed-type hypersensitivity. Moreover, a recent study by Blüml et al. (26) suggest that Mir-155−/− mice are resistant to collagen-induced arthritis; this was associated with decreased Th17 cell numbers. Thus, our results together with others suggest that Mir-155 regulates both Th1 and Th17 responses that can markedly affect T cell-mediated autoimmunity.
What is inducing Mir-155?
Mir-155 locus is located within a region known as B cell integration cluster, which was originally thought to be a proto-oncogene associated with lymphoma. Mir-155 was first implicated in the oncogenesis of hematopoietic malignancies based on the finding that B cell integration cluster/Mir-155 expression is upregulated in lymphomas of B cell origin and chronic lymphocytic leukemia (27, 28). Consistent with this observation, transgenic expression of Mir-155 in B cells causes acute lymphoblastic leukemia (29). Although the hazards of unbalanced Mir-155 expression are clearly demonstrated by the diversion of lymphoid and myeloid cells to an oncogenic fate, recent findings suggest that Mir-155 was induced upon activation of myeloid and lymphoid cell types in the mouse. Various TLR ligands have been shown to induce Mir-155 expression in monocyte and macrophage cell lines (20). This induction was dependent on the signaling pathways initiated by TLR activation, implicating Mir-155 as a downstream player in innate immune function. In addition, Mir-155 is induced in macrophages and DCs after exposure to a variety of inflammatory cytokines such as IFN-β, IFN-γ, and TNF-α (20). In addition to innate immune cells, T lymphocytes also display similar induction of Mir-155 in response to activating stimuli (30).
What are the relevant Mir-155 targets?
Many direct targets of Mir-155 in DCs and T cells have been identified. Mir-155 has been shown to limit Th2 cell development through repression of c-Maf (14). It was established that Mir-155 is required for the differentiation and proliferation, but not the immunosuppressive action of CD4+ Treg cells (15). Further studies indicate that Mir-155 downregulates the protein suppressor of cytokine signaling 1 (SOCS1), which promotes competitive fitness and increased proliferation through the IL-2 and STAT5 signaling pathways (13). The essential role of Mir-155 elucidated by targeting SOCS1 expression is not only confined to Treg cells, but also macrophages and DCs. Recently, it has been shown that IL-12 production by DCs is regulated by Mir-155–mediated targeting of SOCS1 (31). Consistent with previous studies, we found activated DCs from Mir-155−/− mice secreted less IL-12 than those from WT mice. Overexpression of Mir-155 in myeloid cells leads to myeloproliferative disorder through suppression of SHIP1 expression (29). Both SHIP1 and SOCS1 have been shown to negatively regulate cytokine expression in DCs (32, 33). It is well known that the functional phenotype of CD4+ T lymphocytes is dictated by the cytokine secretion by DCs. In addition to the Th1-polarizing cytokine IL-12, we found reduced expression of Th17-polarizing cytokines in DCs from Mir-155−/− mice with EAE. Altered expression of Th1- and Th17-polarizing cytokine expression in DCs that influence the differentiation of Th1 and Th17 responses could be an additional mechanism by which Mir-15−/− mice are resistant to EAE. Mir-155 has been shown to target DC-specific intracellular adhesion molecule-3 grabbing nonintegrin and reduces its pathogen-binding ability on maturation after directly targeting the transcription factor PU.1 (34). In addition, Mir-155 has been shown to promote Ig class switching in B cells via targeted repression of activation-induced cytidine deaminase and the transcription factor PU.1 (35).
Increased expression of Mir-155 has been observed in brain lesions from MS patients and in synovial samples from patients with rheumatoid arthritis (36, 37). Given the ameliorating effect of anti–Mir-155 in EAE and the expression profile of Mir-155 in MS patients, silencing Mir-155 may be an effective therapeutic approach in the treatment of MS.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants NS038037, AI043458, and NS23132 and the Nancy Davis Foundation. G.M. is supported by a postdoctoral fellowship from National Multiple Sclerosis Society, New York. N.J. is supported by the Swiss Multiple Sclerosis Society.
Abbreviations used in this article
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- LN
lymph node
- LNA
locked nucleotide acid
- Mir-155
microRNA-155
- miRNA
microRNA
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- ROR
retinoid orphan nuclear receptor
- SOCS1
suppressor of cytokine signaling 1
- Treg
T regulatory
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Diveu C, McGeachy MJ, Cua DJ. Cytokines that regulate autoimmunity. Curr Opin Immunol. 2008;20:663–668. doi: 10.1016/j.coi.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Windhagen A, Newcombe J, Dangond F, Strand C, Woodroofe MN, Cuzner ML, Hafler DA. Expression of costimulatory molecules B7-1 (CD80), B7-2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995;182:1985–1996. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 4.Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, Duquette P, Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 5.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12-and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 12.O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 13.Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 15.Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 16.Murugaiyan G, Agrawal R, Mishra GC, Mitra D, Saha B. Functional dichotomy in CD40 reciprocally regulates effector T cell functions. J Immunol. 2006;177:6642–6649. doi: 10.4049/jimmunol.177.10.6642. [DOI] [PubMed] [Google Scholar]
- 17.Martin E, O’Sullivan B, Low P, Thomas R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity. 2003;18:155–167. doi: 10.1016/s1074-7613(02)00503-4. [DOI] [PubMed] [Google Scholar]
- 18.Murugaiyan G, Mittal A, Weiner HL. Identification of an IL-27/osteopontin axis in dendritic cells and its modulation by IFN-gamma limits IL-17-mediated autoimmune inflammation. Proc Natl Acad Sci USA. 2010;107:11495–11500. doi: 10.1073/pnas.1002099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 20.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 22.Worm J, Stenvang J, Petri A, Frederiksen KS, Obad S, Elmén J, Hedtjärn M, Straarup EM, Hansen JB, Kauppinen S. Silencing of microRNA-155 in mice during acute inflammatory response leads to de-repression of c/ebp Beta and down-regulation of G-CSF. Nucleic Acids Res. 2009;37:5784–5792. doi: 10.1093/nar/gkp577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor RA, Prendergast CT, Sabatos CA, Lau CW, Leech MD, Wraith DC, Anderton SM. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blüml S, Bonelli M, Niederreiter B, Puchner A, Mayr G, Hayer S, Koenders MI, van den Berg WB, Smolen J, Redlich K. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum. 2011;63:1281–1288. doi: 10.1002/art.30281. [DOI] [PubMed] [Google Scholar]
- 27.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–249. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 29.O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, Kroeger P, McWeeny K, Halbert DN, Mollison KW, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 31.Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, Blazar BR, Zeng Y, Zhou X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood. 2011;117:4293–4303. doi: 10.1182/blood-2010-12-322503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 33.An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, Qi R, Cao X. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood. 2005;105:4685–4692. doi: 10.1182/blood-2005-01-0191. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) J Biol Chem. 2009;284:16334–16342. doi: 10.1074/jbc.M109.011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, Di Virgilio M, Reina San-Martin B, Heidkamp G, Schwickert TA, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28:630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, Lassmann H, Wekerle H, Hohlfeld R, Meinl E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132:3342–3352. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 37.Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, Detmar M, Gay S, Kyburz D. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–1009. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.