

Abstract
A role for the membrane/cytoskeleton interface in the development and progression of cancer is established, yet poorly understood. The neurofibromatosis type II (NF2) tumor suppressor gene encodes a member of the ezrin/radixin/moesin (ERM) family of membrane/cytoskeleton linker proteins thought to be important for cell adhesion and motility. We report that in contrast to the narrow spectrum of benign tumors in human NF2 patients, Nf2 heterozygous mice develop a variety of malignant tumors. Using the fact that Nf2 is linked to the p53 tumor suppressor locus in the mouse we have also investigated the effects of genetic linkage of cancer-predisposing mutations on tumorigenesis and examined the genetic pathway to tumor formation involving Nf2 loss. Importantly, we observed a very high rate of metastasis associated with Nf2 deficiency, with or without loss of p53 function, and we provide experimental evidence supporting a role for Nf2 loss in metastatic potential. Together, our results suggest an important role for the NF2 tumor suppressor, and perhaps the ERM family in tumor formation and metastasis.
Keywords: Merlin, NF2, tumor suppressor, cytoskeleton, osteosarcoma, metastasis
Neurofibromatosis type II (NF2) is a dominantly inherited disorder featuring the predisposition to develop multiple benign tumors of the central nervous system. The hallmark feature of NF2 is the development of bilateral schwannomas of the eighth cranial (auditory) nerves; NF2 patients are also predisposed to the development of spinal schwannomas, meningiomas, and ependymomas at rates much higher than that of the normal population (Huson 1994). The NF2 tumor suppressor gene was identified by positional cloning and loss of heterozygosity (LOH) studies and found to encode a member of the band 4.1 family of cytoskeletal-associated proteins thought to be involved in the organization of the actin cytoskeleton (Rouleau et al. 1993; Trofatter et al. 1993). The NF2 gene product shares closest similarity to ezrin, radixin, and moesin (the ERM proteins), which comprise a subset of this family, and thus was given the name merlin (moesin-, ezrin-, and radixin-like protein) (Trofatter et al. 1993). The amino-terminal halves of these proteins share the greatest similarity (the band 4.1 domain), with ∼85% amino acid identity among the ERMs (for review, see Tsukita et al. 1997; Vaheri et al. 1997).
The ERM proteins localize to cortical actin structures, particularly specialized or dynamic regions, such as membrane ruffles, microvilli, or the cleavage furrow and can bind directly to actin through a highly conserved motif at their extreme carboxyl terminus (for review, see Tsukita et al. 1997; Vaheri et al. 1997). The ERM proteins may be rendered inactive by an intramolecular association; certain stimuli such as phosphorylation or phosphatidyl-4,5-bis-phosphate (PIP2) binding may serve to “open up” the protein conformation, allowing homo- or heterodimerization, which has been shown to occur under certain conditions (Gary and Bretscher 1993). All three ERM proteins have been shown to bind to the transmembrane protein CD44, providing a direct link between the actin cytoskeleton and the membrane (Tsukita et al. 1994). Although ERM function has been linked to a number of cellular activities, including the motility, cell/substrate, and cell/cell adhesion of epithelial cells; immortalization of fibroblasts; and the sensitization of target cells to killing by natural killer cells; there remains no consensus concerning the molecular function of these proteins (Helander et al. 1996). Recent evidence suggests that they may participate in the Rho GTPase signaling network that controls such diverse cellular activities as cytoskeletal reorganization, cell motility, cell proliferation, and membrane trafficking (Hirao et al. 1996; Mackay et al. 1997; Takahashi et al. 1997; Matsui et al. 1998).
In contrast, much less is known about merlin, which does not contain the carboxy-terminal actin-binding motif found in the ERM proteins, but does localize to cortical actin structures and is particularly enriched in membrane ruffles (Gonzalez-Agosti et al. 1996; R.J. Shaw, A.I. McClatchey, T. Jacks, in prep.). The mouse and human NF2 proteins are highly related, sharing 98% amino acid identity (Haase et al. 1994; Claudio et al. 1997). Moreover, a Drosophila homolog of the NF2 protein, dmerlin, shares 55% amino acid identity with human merlin and localizes to endocytic vesicles, implying a role for merlin in the formation or trafficking of those structures (McCartney and Fehon 1996). It has been reported that reduction of merlin expression by antisense oligonucleotides reduces the adhesion and increases the proliferation of Schwann-like cells, and that overexpression of merlin leads to growth arrest of fibroblasts (Lutchman and Rouleau 1995; Huynh and Pulst 1996). In addition, we have determined that merlin is a phosphoprotein; serine/threonine phosphorylation of merlin is modulated by a number of stimuli in cell culture, including the availability of growth factors, confluency, and by cell adhesion (Shaw et al. 1998). Despite these observations, the molecular nature of merlin function remains poorly understood. However, given its identity as a tumor suppressor protein, the study of the NF2 gene product represents an avenue into the poorly understood interface between the proliferative state of the cell and the cytoskeleton, which must reorganize during the processes of cell division and differentiation, as well as during the transformation and invasion stages of malignancy.
To develop a system through which to study the function of merlin, we have targeted the disruption of the mouse Nf2 gene and investigated the consequences of merlin loss in mouse tumorigenesis and development. Previously, we have reported a requirement for merlin function at the initiation of gastrulation during embryogenesis (McClatchey et al. 1997). Here, we describe dramatic tumorigenic and metastatic consequences of loss of merlin function in adult animals. These results are surprising given the rather limited association between merlin loss and cancer development in humans and this implies a very important role for this pathway specifically, and the membrane/cytoskeletal interface generally, in cancer development and progression.
Results
Nf2 heterozygous mice are cancer prone
Previously, we have described the generation of a targeted mutation at the mouse Nf2 locus by homologous recombination in ES cells (McClatchey et al. 1997). This mutation, designed to mimic germ-line mutations identified in human NF2 patients (for review, see Gusella et al. 1996), was introduced into D3 129/SvPas ES cells by homologous recombination (Simpson et al. 1997). Neither full-length nor aberrantly sized Nf2 protein was detected by Western blot analysis of cell extracts from Nf2 homozygous mutant ES cells or tumor cells displaying loss of the wild-type Nf2 allele (McClatchey et al. 1997; see below for derivation of tumor cells). Furthermore, a homozygous mutation at the mouse Nf2 locus leads to embryonic failure immediately before gastrulation, indicating that merlin function is critical at a very early stage in development (McClatchey et al. 1997).
One of the original motivations for targeting the mouse Nf2 locus was to attempt to create an animal model for human NF2. We generated 99 Nf2 +/− and 23 wild-type F1 (C57BL/6 × 129/Sv) siblings, and 37 inbred 129/Sv Nf2 +/− animals by breeding chimeras derived from each of three original targeted ES cell clones to wild-type C57BL/6 or 129/Sv animals (identical results were obtained for animals derived from each of the three ES cell clones; see Materials and Methods for a description of the 129Sv substrain used). We also generated 45 Nf2 +/− F2 animals by intercrossing Nf2 +/− F1 mice. Nf2 heterozygous mice were monitored closely for the development of tumors over the course of nearly 3 years and found to be cancer prone. Figure 1A illustrates the decreased survival of Nf2 heterozygous F1 mice compared to their wild-type siblings. Fifty percent of F1 Nf2 heterozygotes died or were sacrificed by the age of 22.4 months (672 days), whereas 50% of their wild-type siblings survived to 27.3 months (818 days). Inbred 129/Sv Nf2 heterozygotes exhibit an additional decrease in survival (median, 20.3 months or 608 days; data not shown). Interestingly, we found a statistically significant decrease in the survival of females compared to males, which is most pronounced on the inbred 129/Sv background (one-tailed t test; t = 3.199; P = 0.0008 for total males vs. females). This may be explained at least in part by tissue-specific biases in tumor development (see below).
Figure 1.
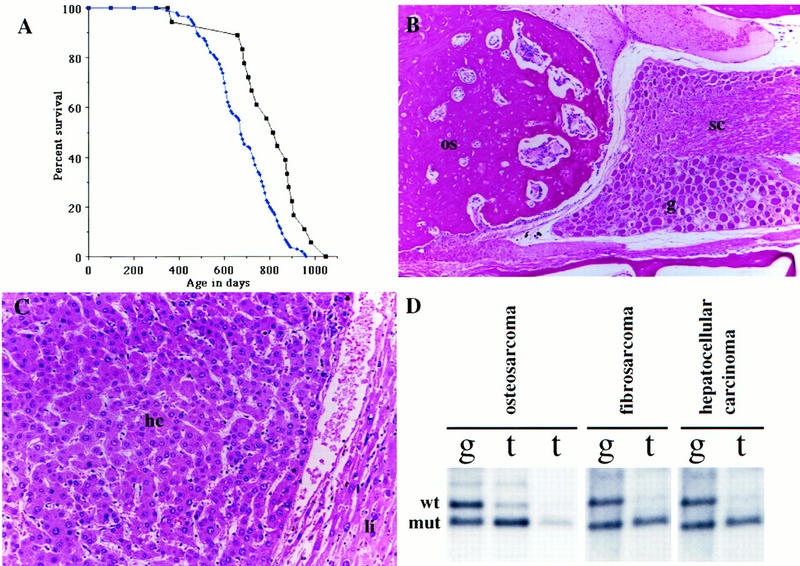
Nf2 heterozygous mice are cancer prone. (A) Survival curve showing the decreased survival of Nf2 heterozygotes on an F1 (C57BL/6 × 129/Sv) background (blue diamonds), compared to that of their wild-type F1 siblings (black squares). (B) Histological section through an osteosarcoma in a Nf2 heterozygote. The tumor (os) is growing out of a vertebral bone and pressing on the spinal cord (sc) and associated spinal ganglion (g). This tumor is very well differentiated, with a high ratio of calcified bone matrix to cell nucleus (100×). (C) Histological section of a hepatocellular carcinoma in an Nf2 heterozygote. Note the trabecular nature of the tumor itself (hc), manifested as nests of cells with spaces in between. Normal liver on the right (li) is being compressed by the tumor (200×). (D) Loss of Nf2 heterozygosity in tumors from Nf2 +/− animals. Southern blotting of paired tail [(g) germ line] and tumor (t) samples. (Top band) The wild-type allele; (bottom band) the mutant allele.
In contrast to human NF2 patients, Nf2 +/− mice developed a variety of malignant tumors later in life (10–30 months). The spectrum of tumors observed in Nf2 +/− F1 (C57BL/6 × 129Sv) mice is depicted in Table 1; 63% developed osteosarcoma, followed by lymphoma (15%; see legend to Table 1), lung adenocarcinoma (10%), hepatocellular carcinoma (9%), and fibrosarcoma (9%; see legend to Table 1). The tumor spectrum in inbred Nf2 +/− 129/Sv animals was very similar to that of F1 animals (not shown). Lymphoma and lung adenocarcinoma were common tumors that arose in wild-type F1 control animals (30% and 44%, respectively). As discussed below, these cancers are likely to be spontaneous background tumors in Nf2 heterozygotes whose occurrence is unrelated to the loss of Nf2 function. The frequency of osteosarcoma was significantly higher in Nf2 +/− F1 and 129/Sv females than in males (F1: 77% vs. 56%; 129/Sv: 84% vs. 67%), whereas fibrosarcomas arose predominantly in males (Table 1). The average age of death of 129/Sv females with osteosarcomas was 18.7 months, compared to 23 months for males, suggesting an earlier onset or more rapid growth of osteosarcomas in females. This difference may reflect known effects of estrogen on bone growth (for review, see Rizzoli and Bonjour 1997). In contrast, a strong bias for hepatocellular carcinoma development in males was observed, consistent with chemically induced hepatocarcinogenesis in mice, which predominantly affects males (Frith and Ward 1979). The reduced frequency of lung adenocarcinoma and lymphoma in Nf2 +/− mice compared to controls probably reflects the development of earlier onset osteosarcomas in these animals.
Table 1.
Frequency of the occurrence of various tumor types in Nf2 +/− mice
| Genetic background/gender
|
Osteosarcoma
|
Lymphomaa
|
Lung adenocarcinoma
|
Hepatocellular carcinoma
|
Fibrosarcomab
|
Hepatocellular adenoma
|
|---|---|---|---|---|---|---|
| Wild-type F1 | ||||||
| male (n = 11) | 0% | 0% | 46% (5) | 0% | 18% (2) | 18% (2) |
| female (n = 12) | 0% | 58% (7) | 42% (5) | 8% (1) | 0% | 8% (1) |
| Total (n = 23) | 0% | 30% (7) | 44% (10) | 4% (1) | 9% (2) | 13% (3) |
| Nf2 +/− f1 | ||||||
| male (n = 68) | 56% (38) | 12% (8) | 13% (9) | 13% (9) | 10% (7) | 4% (3) |
| female (n = 31) | 77% (24) | 23% (7) | 0% | 0% | 7% (2) | 0% |
| Total (n = 99) | 63% (62) | 15% (15) | 9% (9) | 9% (9) | 9% (9) | 3% (3) |
| Nf2 +/− F2 | ||||||
| male (n = 15) | 60% (9) | 0% | 13% (2) | 33% (5) | 13% (2) | 13% (2) |
| female (n = 30) | 47% (14) | 17% (5) | 3% (1) | 3% (1) | 0% | 7% (2) |
| Total (n = 45) | 51% (23) | 11% (5) | 7% (3) | 13% (6) | 4% (2) | 9% (4) |
| Nf2 +/− 129Sv | ||||||
| male (n = 18) | 67% (12) | 0% | 11% (2) | 28% (5) | 6% (1) | 11% (2) |
| female (n = 19) | 84% (16) | 11% (2) | 0% | 0% | 0% | 5% (1) |
| Total (n = 37) | 76% (28) | 5% (2) | 5% (2) | 14% (5) | 3% (1) | 8% (3) |
| Total NF2 +/Ün = 181) | 63% (114) | 12% (22) | 8% (14) | 11% (19) | 7% (12) | 6% (10) |
This category includes lymphoblastic, lymphocytic, and follicular center cell lymphoma.
These tumors exhibit a range of features consistent with both fibrosarcomas and rhabdomyosarcomas, often within the same primary tumor.
To determine whether tumor formation in the Nf2 heterozygous mutant mice was dependent on the somatic mutation of the wild-type Nf2 allele, we performed Southern blotting on a panel of tumor DNAs. Nearly all of the osteosarcomas and fibrosarcomas analyzed displayed loss of the wild-type Nf2 allele (LOH; 11/12 and 11/11, respectively; Fig. 1D), consistent with a role for loss of Nf2 function in the etiology of those tumor types. In addition, hepatocellular carcinomas, which were markedly more frequent in our Nf2 heterozygotes compared to wild-type controls (Table 1), usually displayed LOH at the Nf2 locus (78%; 7/9 analyzed by Southern blotting). Hepatocellular adenoma of the liver was also observed in wild-type control mice (13%; 3/23); however, these tumors apparently did not progress to high-grade malignant hepatocellular carcinoma. The Nf2 heterozygotes also developed hepatocellular adenomas (6%; 10/181), which failed to undergo Nf2 LOH. The reduced frequency of hepatocellular adenoma and increased frequency of hepatocellular carcinoma in Nf2 heterozygotes suggests that Nf2 loss contributes to the progression to a highly malignant lesion. However, it is also possible that Nf2 mutation leads to the development of an inherently more aggressive tumor type de novo. In contrast, lung adenocarcinoma and lymphoma (including lymphoblastic, lymphocytic, and follicle center cell), which were frequent tumors in both wild-type and Nf2 +/− animals, do not display loss of the wild-type Nf2 allele, as would be expected for a background tumor.
Osteosarcomas in Nf2 +/− mice were histologically consistent with osteoblastic or osteogenic sarcomas and arose predominantly within the craniofacial bones and vertebral column, often causing paralysis. These tumors were often highly differentiated, containing large amounts of mineralized bone forming mature trabeculae, and exhibiting fairly low cellularity (Fig. 1B). Fibrosarcomas in Nf2 heterozygotes exhibited a spectrum of features consistent with fibrosarcomas and/or rhabdomyosarcomas, including a range of nuclear morphology (spindly to plump), cytoplasm (limited to extensive eosinophilic), and the presence or absence of strap-like cells. Given that we observed this range of features when comparing individual tumors or regions within the same tumor, we have not attempted to subclassify them and will refer to them collectively as fibrosarcomas. Although the frequency of this tumor type in Nf2 +/− F1 mice does not appear to be markedly increased (9%; Table 1), the frequency of fibrosarcomas seen in our wild-type animals (9%, 2/23; see Table 1) is higher than that seen in other F1 control populations from our laboratory (not shown) and is likely to be an overrepresentation of the true background frequency in F1 animals. The hepatocellular carcinomas that exhibited loss of the wild-type Nf2 allele were of the high grade, trabecular form (Fig. 1C; Frith and Ward 1979).
Importantly, we did not detect any schwannomas, meningiomas, or ependymomas in the Nf2 +/− animals, despite examining a sagittal section of the spinal cord and head of each mouse and serial sections (4 μm) through the entire length of both eighth cranial nerves of eight animals. We also examined the lenses of 7 Nf2 +/− and 10 Nf2 +/−;p53 +/− animals (see below) at high power on a dissecting microscope. Although examples of lens fiber disorganization were seen, we did not observe consistently obvious cataracts analogous to those common in human Nf2 patients (D.C. Beebe, pers. comm.). Thus, these animals do not represent a histopathologically accurate model for human NF2, but they reveal other cell types requiring the growth suppressive properties of merlin, and confirm the function of merlin as a tumor suppressor in the mouse.
Nf2-deficient tumors are highly metastatic
A striking feature of the tumors that developed in the Nf2 +/− mice was their high frequency of metastasis to distant sites such as the lung and liver. This was unexpected given the generally low rate of metastasis associated with endogenously arising tumors in the mouse (Frith et al. 1981). Histologically, we found that nearly all of the osteosarcomas (95% or 61/64 in F1 animals; 90% or 104/115 overall) in Nf2 +/− mice metastasized. Although the primary sites of metastasis were the lung and liver, we also frequently found pockets of tumor cells in the kidney and occasionally in the spleen. Many of the osteosarcomas that metastasized in these mice were relatively small, well-differentiated primary tumors exhibiting a high ratio of extracellular matrix (calcified bone) to cell nuclei, such as the one in Figure 1B. The metastases were also often highly differentiated (Fig. 2, cf. B and C with A), frequently more so than the primary tumor. In addition, 64% (7/11) of the fibrosarcomas that exhibited LOH at the Nf2 locus, and 57% (4/7) of the hepatocellular carcinomas that exhibited LOH at the Nf2 locus, metastasized (neither of the 2/9 hepatocellular carcinomas found to retain the wild-type allele metastasized). For comparison, one study revealed that 13% (4/30) of the osteosarcomas and 0% (0/36) of the fibrosarcomas arising in p53 +/− or wild-type animals on a similar genetic background metastasized (Taverna et al. 1998). Osteosarcomas and fibrosarcomas in wild-type mice have been reported to metastasize with frequencies of 4%–46% and 18%, respectively, depending on the method of induction and the genetic background (Frith et al. 1981; Luz et al. 1991). Notably, two of the wild-type F1 control mice developed fibrosarcomas, neither of which metastasized. Taken together, these observations strongly suggest that Nf2-deficient tumor cells possess a marked propensity to metastasize and raise the possibility that loss of merlin function is somehow increasing metastatic potential in this tumor model (see below).
Figure 2.
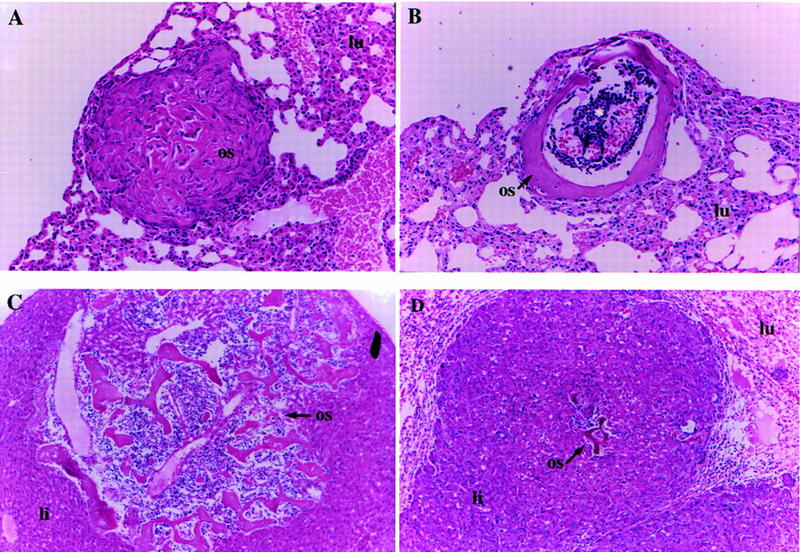
Tumors that arise in Nf2 heterozygous mice exhibit a very high rate of metastasis. (A) A fairly undifferentiated osteosarcoma metastasis (os) in the lung (lu) of a Nf2 +/− mouse. Some mineralization is present, but there is abundant cellularity. This is the typical histological appearance of an osteosarcoma metastasis (200×). (B) Highly differentiated metastasis in a Nf2 +/− mouse. This is an unusual form of metastasis frequently seen in Nf2 heterozygotes. The mineralized bone has formed a collar as in a long bone, and some lymphocytes appear to have homed to the “pseudobone marrow cavity” in the center (asterisk) (100×). (C) A highly differentiated osteosarcoma metastasis (os) in the liver (li) of a Nf2 +/− mouse. Again, there appears to be lymphocyte homing and formation of “bone marrow.” Two large blood vessels are also present within the metastasis and probably provided the route of entry for the tumor cells into the lung (100×). (D) A metastasis from a hepatocellular carcinoma (li) to the lung. This mouse also had an osteosarcoma that metastasized to the liver metastasis (os; center). The normal lung tissue is compressed in the upper corners (lu) (100×).
Cooperativity between an Nf2 mutation and a mutation in the p53 tumor suppressor gene
Given the relatively late onset of tumorigenesis in Nf2 heterozygotes, we investigated the possibility that mutations in other tumor suppressor genes might cooperate with an Nf2 mutation to accelerate or alter the spectrum of tumorigenesis in these mice. Nf2 heterozygous mice were mated to mice carrying a mutation in the p53 tumor suppressor gene (Jacks et al. 1994). p53 heterozygous mice develop a number of sarcomas including osteo-, fibro-, and hemangiosarcomas between the ages of 9 and 24 months (Donehower et al. 1992; Jacks et al. 1994). In contrast to humans, the mouse Nf2 and p53 loci are linked, residing at a significant genetic distance from one another on chromosome 11 (∼40 cM; Dietrich et al. 1996; Fig. 3A). Given that the loss of an entire chromosome is a relatively frequent event during mouse tumorigenesis (Luongo et al. 1994), we investigated the tumorigenic phenotype of mice that carry mutations in the Nf2 and p53 genes on the same chromosome 11 (in cis) and on opposite chromosomes 11 (in trans). This allowed us to address the importance of the configuration of the two mutations, in addition to the overall genetic load.
Figure 3.
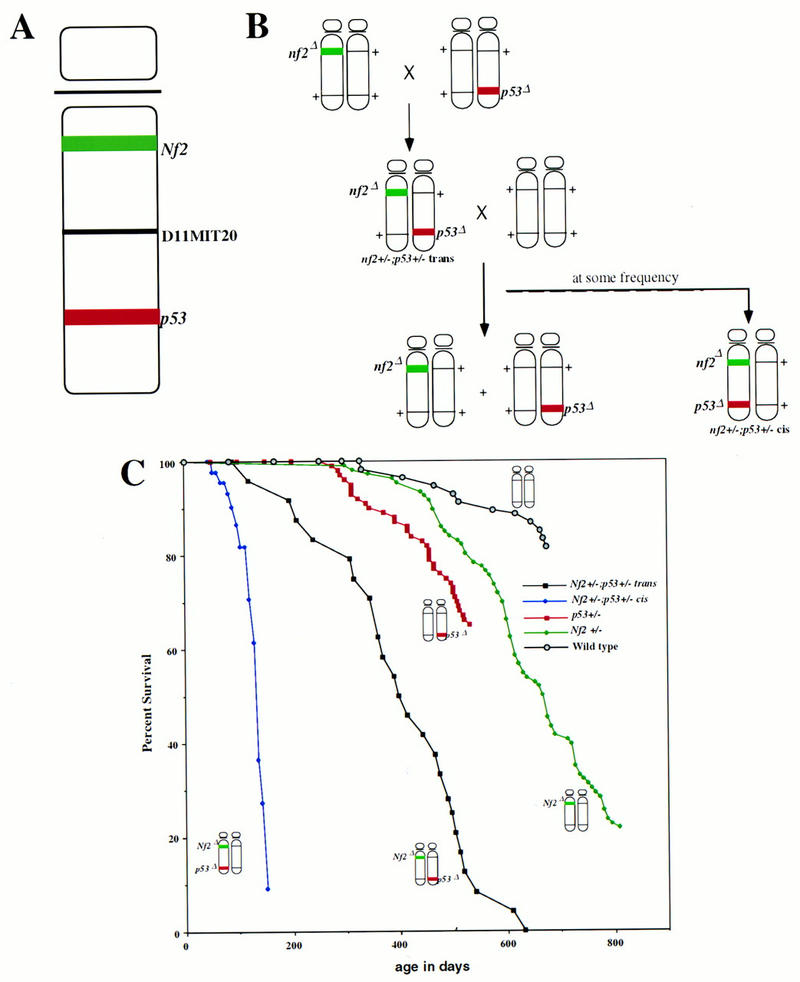
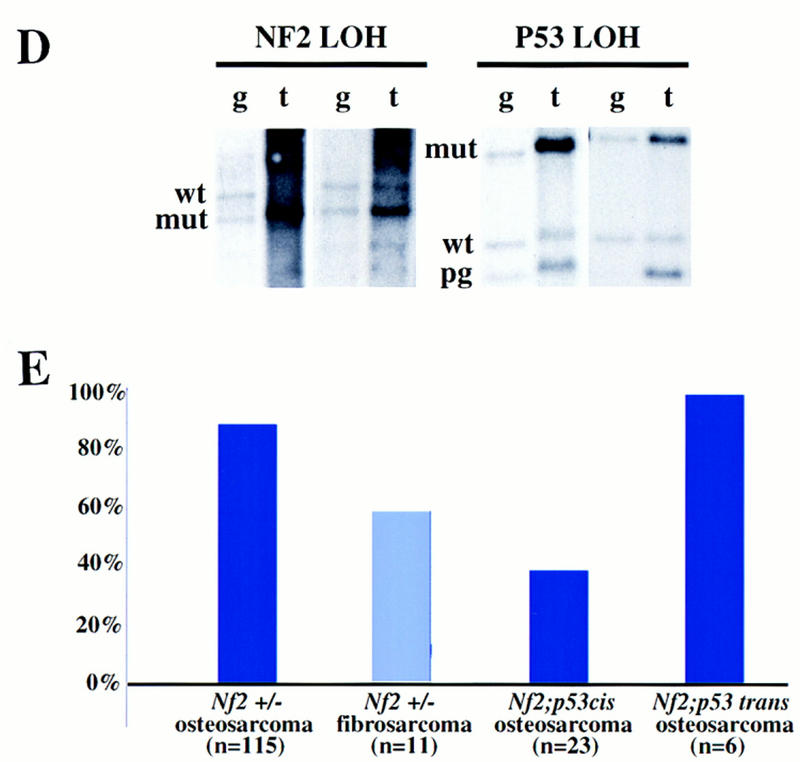
Generation of Nf2 +/−;p53 +/− compound heterozygotes in cis and in trans. (A) Map of mouse chromosome 11, depicting the locations of the Nf2 and p53 loci, as well as the D11MIT20 SSLP marker used for allelotyping. (B) Scheme for the generation of Nf2 +/−;p53 +/− trans and cis mice. (C) Survival of mice carrying both Nf2 and p53 mutations either in cis on the same chromosome 11, or in trans, on opposite chromosomes 11, compared to the survival of either singly heterozygous parental strain. (D) Absence of both the Nf2 and p53 wild-type alleles in tumors from Nf2 +/−;p53 +/− cis mice. The same DNAs were digested with either StuI (for Nf2 LOH) or StuI plus EcoRI (for p53 LOH) restriction enzymes, and probed with a Nf2-specific probe (left) or a p53-specific probe [right; (pg) pseudogene]. Residual wild-type signals present in the tumor samples are likely the result of contaminating normal tissue. The intensities of residual wild-type p53 vs. wild-type Nf2 signals appear unequal here; however, most tumors exhibit equivalent levels of contaminating tissue. (E) Frequency of metastasis associated with Nf2-deficient or Nf2 plus p53-deficient osteosarcoma or fibrosarcoma.
We intercrossed Nf2 and p53 singly heterozygous mice, generating Nf2;p53 double heterozygotes carrying the mutations in trans (Fig. 3B). These trans mice were then mated with wild-type mice. Compound heterozygous progeny from this cross reflect the occurrence of meiotic recombination between the two loci, placing the mutations on the same chromosome 11 (in cis). We generated Nf2 +/−;p53 +/− cis mice with a frequency of 18% from this cross, reflecting the genetic distance between the two loci (37.6% of the mice from this cross were compound heterozygous or wild type, reflecting the total number of recombination events and agreeing roughly with the predicted genetic distance of ∼40 cM between Nf2 and p53 in the mouse; Dietrich et al. 1996). Fifty-three Nf2 +/−;p53 +/− cis and 31 Nf2 +/−;p53 +/− trans mice representing both C57BL/6:129/Sv F1 and inbred 129/Sv genetic backgrounds were then aged and monitored for signs of disease. No obvious differences between the two genetic backgrounds were observed.
Mice carrying mutations at the Nf2 and p53 tumor suppressor loci in cis rapidly developed multiple tumors and died by 5 months of age, exhibiting dramatically reduced survival compared to that of Nf2 or p53 singly heterozygous mutant animals or to that of mice carrying Nf2 and p53 mutations in trans (Fig. 3C). Using histological examination, we found that nearly all of these mice developed osteosarcomas (77%) and/or fibrosarcomas (32%). Both tumor types reproducibly displayed loss of both the Nf2 and p53 wild-type alleles (6/6 tested by Southern blot analysis; Fig. 3D,E). We also examined the status of a polymorphic marker between the Nf2 and p53 loci (D11MIT20; Fig. 3A); 2/2 of the germ-line/tumor DNA pairs that were informative for the D11MIT20 polymorphism showed loss of the C57BL/6 allele and retention of the 129/Sv-derived allele in the tumor. Taken together, these results suggest that loss of the entire wild-type chromosome 11 occurs frequently during the etiology of these tumors. Interestingly, although mice that developed fibrosarcomas usually developed one to two individual tumors, we often identified as many as 10 osteosarcoma lesions in each Nf2 +/−;p53 +/− cis mouse, reflecting either metastatic spread of an individual primary tumor through the bone or multiple independent tumors. Most of these osteosarcomas arose within the spinal column or the craniofacial bones, as in Nf2 heterozygotes; however, the predominant site of osteosarcoma formation in Nf2 +/−;p53 +/− cis mice was within the thin bones lining the nasal passages, a site rarely affected in Nf2 heterozygous mice. These tumors reached only a very small size before they blocked the airways completely. The rate of metastasis associated with these tumors was also high (∼40% overall), although not as high as that observed in Nf2 heterozygotes alone, probably because of the large number of small individual tumors per mouse and their frequent localization to the nasal passages, leading to rapid mortality.
Unexpectedly, mice carrying Nf2 and p53 mutations in trans also showed a significant decrease in survival (Fig. 3C). Given the genotype of tumors arising in Nf2 +/−;p53 +/− cis mice, we had expected these mice to exhibit the same tumor spectra and survival rate as that of Nf2 or p53 heterozygous mice alone, and to detect loss of either the wild-type Nf2 or p53 allele but not both in their tumors. Instead, we found that Nf2 +/−;p53 +/− trans mice predominantly developed osteosarcomas (74%) or fibrosarcomas (8%) between the ages of 4 and 21 months (mean, 13.3 months; Fig. 3C). Importantly, when we investigated the status of the Nf2 and p53 loci in tumors in these mice, we found that most of them had lost both the Nf2 and p53 wild-type alleles (6/9 analyzed by Southern blotting). A number of mechanisms could have led to this outcome: (1) somatic recombination, placing the mutations in cis, followed by loss of the other chromosome 11; (2) small deletions involving the Nf2 and p53 loci separately; or (3) gene conversion, resulting from recombination that occurs distal to p53 or proximal to Nf2. In each scenario, two events are required to inactivate both tumor suppressor genes, in contrast to the single event required in Nf2 +/−;p53 +/− cis mice, or three events required in Nf2 or p53 singly heterozygous animals. Two Nf2 +/−;p53 +/− trans mice developed tumors exhibiting loss of the wild-type p53 allele and loss of the mutant Nf2 allele, again implying loss of an entire chromosome and suggesting normal expression of merlin in these tumors. Importantly, these mice developed a fibrosarcoma and an osteosarcoma, respectively, neither of which metastasized, whereas 100% (6/6) of the Nf2 +/−;p53 +/− trans tumors exhibiting loss of both wild-type alleles did metastasize.
By crossing Nf2 +/−;p53 +/− cis mice to p53 −/− mice, which are prone to developing thymic lymphoma, we also generated 14 Nf2 +/−;p53 −/− mice. We found that the survival of these mice was very similar to that of Nf2 +/−;p53 +/− cis animals. Nf2 +/−;p53 −/− mice developed predominantly osteosarcomas, which exhibited Nf2 LOH, and thymic lymphomas, which did not (data not shown).
Experimental investigation of metastasis
To address the role of Nf2 mutation in metastasis more directly, we examined the capacity of tumor cell lines derived from Nf2-deficient tumors to metastasize with injection into the tail vein of a syngeneic recipient. We used osteosarcoma or fibrosarcoma cell lines derived from p53-deficient tumors as controls for the Nf2-deficient tumor cell lines. High levels of merlin expression were detected in these cell lines by Western blot analysis (data not shown). We were unable to reexpress merlin stably in Nf2-deficient tumor cells despite several attempts, suggesting that high levels of Nf2 expression lead to either growth arrest or cell death. Successful reintroduction of the expression of other tumor suppressor genes has met with the same difficulty, probably reflecting the inherent growth suppressive function of their products. We injected 1–2 million tumor cells of either genotype into the tail vein of syngeneic C57BL/6 × 129Sv F1 recipient animals and monitored the mice for up to 3 weeks postinjection (in three cases nude mice were used with similar results; Table 2). Although the Nf2- and p53-deficient tumor cells were derived from primary tumors of comparable anatomical location and histological appearance (Fig. 4), their ability to colonize the lungs of recipient animals in experimental metastasis assays were quite different (Table 2). All of the mice injected with three different Nf2-deficient fibrosarcoma cell lines became moribund and were sacrificed by or before 3.5 weeks, whereas animals injected with nearly all (3/4) of the p53-deficient fibrosarcoma cell lines survived and appeared to be healthy 3.5 weeks postinjection. Upon dissection, we found that all three Nf2-deficient fibrosarcoma cell lines reproducibly formed hundreds of metastases covering the lungs of syngeneic recipients. Histologically, these metastases had invaded the lung tissue from all vascular regions (Fig. 4A,B). Importantly, we chose to use two fibrosarcoma cell lines for which metastasis from the primary tumor was not detected (NfFB1 and NfFB3; Table 2). In contrast, 2/4 of the p53-deficient fibrosarcoma cell lines did not metastasize at all. One cell line produced some metastases that appeared histologically as round nests of cells that had not appreciably invaded the surrounding tissue (not shown). Only 1/4 of the p53-deficient fibrosarcoma cell lines tested metastasized to the same extent as the Nf2-deficient cells. In fact, these cells were derived from the most pleiomorphic and vascularized tumor used in this study (not shown).
Table 2.
Experimental investigation of the metastatic potential of Nf2-deficient tumor cells by tail vein injection
| Tumor cell linea
|
Tumor type
|
Recipient
|
No. of cells injected
|
Length of experimentb
|
No. of lung metastases
|
|---|---|---|---|---|---|
| NfFB1 | fibrosarcoma | F1 | 1 × 106 | 21 | >200c |
| nude | 2 × 106 | 24 | >200c | ||
| NfFB2 | fibrosarcoma | F1 | 2 × 106 | 12 | >200c |
| F1 | 2 × 106 | 13 | >200c | ||
| NfFB3 | fibrosarcoma | F1 | 2 × 106 | 17 | >200c |
| p53FB1 | fibrosarcoma | F1 | 1 × 106 | 21 | 0 |
| F1 | 2 × 106 | 21 | 0 | ||
| p53FB2 | fibrosarcoma | F1 | 2 × 106 | 21 | 0 |
| F1 | 2 × 106 | 21 | 0 | ||
| p53FB3 | fibrosarcoma | F1 | 1 × 106 | 21 | ∼10 |
| nude | 2 × 106 | 25 | ∼10 | ||
| F1 | 1 × 106 | 21 | >100d | ||
| p53FB4 | fibrosarcoma | F1 | 1 × 106 | 16 | >200c |
| F1 | 2 × 106 | 17 | >200c | ||
| NfOS1 | osteosarcoma | F1 | 1 × 106 | 21 | ∼20 |
| F1 | 1 × 106 | 31 | ∼50 | ||
| nude | 2 × 106 | 24 | >200c | ||
| NfOS2 | osteosarcoma | F1 | 2 × 106 | 26 | ∼5 |
| F1 | 1.4 × 106 | 21 | ∼5 | ||
| p53OS1 | osteosarcoma | F1 | 2 × 106 | 21 | 0 |
| F1 | 2 × 106 | 21 | 0 | ||
| F1 | 0.5 × 106 | 21 | 0 | ||
| F1 | 2 × 106 | 21 | 1 | ||
| p53OS2 | osteosarcoma | F1 | 1 × 106 | 21 | 0 |
| F1 | 1 × 106 | 31 | 0 |
Nf and p53 designations indicate Nf2 and p53 deficiency, respectively.
In days.
The animal was moribund and was sacrificed before the endpoint of the experiment, apparently because of a large metastatic load.
Shortly after injection of the tumor cells, we discovered that this mouse was pregnant; pups were delivered 2 weeks postinjection. Pregnancy may have had a positive effect on the growth of tumor cells in this mouse.
Figure 4.
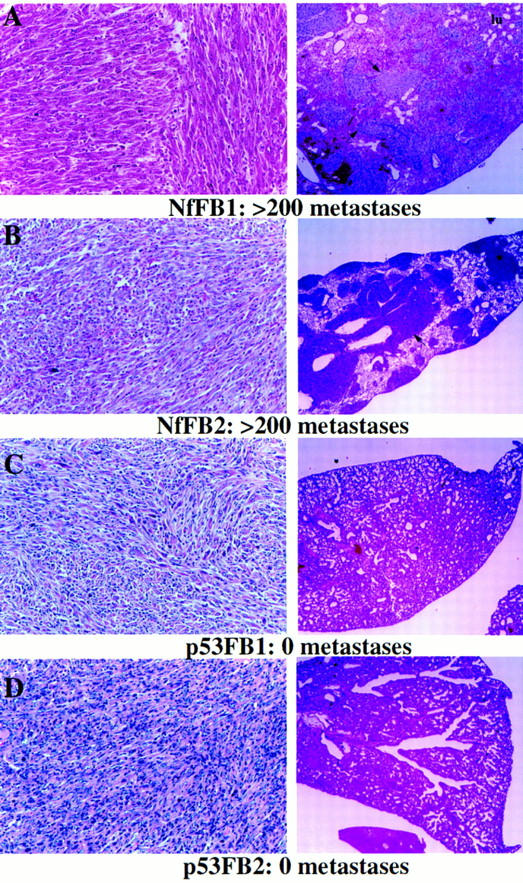
Histological appearance of the primary tumors from which Nf2- or p53-deficient fibrosarcoma cell lines were derived (left) and histological sections of the lungs of recipient animals into which each of these cell lines was injected (right). The total number of metastases produced by each cell line is indicated below. The lungs of animals injected with Nf2-deficient tumor cells (A,B) are covered with metastases (arrows indicate selected metastases), whereas none are detected in the lungs of animals injected with p53-deficient tumor cells (C,D). Note that the lung in A has hemorrhaged; blood fills much of the alveolar spaces (200×).
None of the mice injected with osteosarcoma cells, either Nf2 deficient or control, were moribund at 3 weeks. However, upon dissection, we found that both of the Nf2-deficient osteosarcoma cell lines did metastasize, whereas the two p53-deficient cell lines that we tested did not (Table 2). (We observed one tiny pocket of abnormal cells in the lung of one animal; however, it was not possible to tell whether they were tumor cells because there were so few). The metastases derived from Nf2-deficient osteosarcoma cells were often highly differentiated, similar to the endogenously arising metastases in Nf2 heterozygous mutant animals (not shown). Together, we found that 100% of the Nf2-deficient tumor cell lines tested were capable of surviving in the bloodstream and colonizing a secondary site, whereas most Nf2-expressing tumor cell lines were not. This behavior is not simply attributable to differences in the growth properties of these cells, as their growth rates in vitro did not differ dramatically. In addition, two fibrosarcoma lines, Nf2FB1 and p53FB3, formed tumors at equivalent and rapid rates when injected subcutaneously into nude mice; the tumors did not metastasize in these animals, apparently because of the very rapid increase in tumor volume. These results provide direct support for a role for the loss of merlin function in metastatic potential.
Discussion
In contrast to the limited spectrum of benign tumors associated with NF2 in humans, mice that are the genetic analogs of human NF2 patients develop a variety of malignant tumor types and do not model human NF2. Human NF2 patients do not develop osteosarcomas, fibrosarcomas, or hepatocellular carcinomas (Huson 1994). Whether mutations at the NF2 locus are involved in the development of these tumor types or the metastatic transition of any tumor type in humans is currently unknown. Both the spectrum and the aggressive nature of the tumors associated with Nf2 loss in the mouse is surprising, as is the high rate of metastasis observed in association with Nf2-deficient tumors. Although the reason for the strikingly different phenotype in the mouse is not clear, information from several other systems has provided some insight. For example, considerable evidence suggests that members of the retinoblastoma (Rb) family of proteins can compensate for the loss of one another in certain contexts in vivo (Lee et al. 1996; Hurford et al. 1997). It is possible that the closely related ERM proteins can compensate similarly for merlin loss in mouse Schwann cells or the human osteoblast lineage. Indeed, we have determined that there is considerable overlap in the mRNA expression patterns of the ERMs and Nf2 in the developing mouse embryo (McClatchey et al. 1997; unpubl.). Moreover, the partial sequences of two Nf2-related transcripts from mouse have been reported (Takeshima et al. 1994). Alternatively, there may be marked species-specific differences in the rate of loss of the wild-type allele in different cell types (i.e., Schwann cells vs. osteoblasts). Mounting evidence suggests that this may account at least partially for phenotypic differences between mice and humans carrying mutations in the NF1 gene (S. Shih and T. Jacks, unpubl.). Finally, it is conceivable that synteny differences between the mouse and human genomes may contribute to phenotypic differences, espcially considering the apparently frequent loss of an entire chromosome during mouse tumorigenesis. For example, a growth factor, growth factor receptor, or compensating family member might be linked to Nf2 in one species and not in another; therefore, its expression during tumorigenesis might be affected differentially.
We identified three tumor types that arose frequently in Nf2 heterozygous mice and exhibited loss of the wild type Nf2 allele—osteosarcomas, fibrosarcomas, and hepatocellular carcinomas. We also identified other tumor types that arose at a low frequency in Nf2 heterozygotes and did not appear in our wild-type controls. For example, several chondrosarcomas and uterine sarcomas developed in Nf2 +/− mice and in some cases loss of the wild-type Nf2 allele was detected. In addition, several Nf2 +/− animals developed bile duct carcinomas, which were never seen in wild-type animals; however, we did not obtain enough tissue from these lesions for analysis of the status of the Nf2 allele. Together, these observations suggest that Nf2 loss can contribute to the formation of a broad spectrum of tumor types in the mouse.
Although osteosarcomas are rare in wild-type mice, they are commonly induced by radiation, viruses, or viral oncoproteins (for review, see Michiels and Merregaert 1993). Thus, polyomavirus (Py), the Py early region, simian virus SV40, the SV40 large T-antigen, the Finkel–Biskis–Jinkins (FBJ) and Finkel–Biskis–Reilly (FBR) retroviruses, the FBJ-encoded oncoprotein v-fos or its cellular homolog c-fos can all induce the formation of osteosarcomas in mice upon infection or when expressed transgenically (Py and SV40 can also induce the formation of fibrosarcomas). When reported, only a small percentage of the osteosarcomas in these mice were found to metastasize (0%–33%; Finkel et al. 1966; Ruther et al. 1989; Knowles et al. 1990; Wilkie et al. 1994). Finally, as discussed earlier, p53 heterozygous mutant mice develop osteosarcomas and fibrosarcomas that metastasize at low frequency (Taverna et al. 1998; A.I. McClatchey and T. Jacks, unpubl.). Hepatocellular carcinoma is also rare in wild-type mice (Bronson 1990); mouse models of hepatocellular carcinoma have been generated historically by administration of chemical carcinogens (i.e., ethylnitrosourea, benzo(a)pyrene, etc.; Vesselinovitch et al. 1978).
We observed marked cooperativity between a Nf2 mutation and a heterozygous mutation at the p53 locus, which resides on the same mouse chromosome, in the development of a subset of the tumor types that either of the singly heterozygous mutant strains develop. By manipulating the configuration of the two mutations with respect to the two chromosomes 11, we were able to investigate the effects of mutational linkage upon tumorigenesis. The result is illustrated most dramatically by the phenotype of Nf2 +/−;p53 +/− mice that carry the mutations in cis on the same chromosome 11. These mice survive only to the age of 5 months, developing multiple tumors that show loss of both the Nf2 and p53 wild-type alleles, and LOH at a locus between the two. Furthermore, we noted a surprisingly reduced survival of Nf2 +/−;p53 +/− trans mice compared to Nf2 or p53 singly heterozygous mice and observed the loss of both Nf2 and p53 wild-type alleles in those tumors. The frequent loss of an entire chromosome during mouse tumorigenesis has been described previously (Luongo et al. 1994). Although our results appear to provide dramatic support for this, we cannot rule out the possibility that large interstitial deletions occur, leaving a small fragment of the distal end of chromosome 11 fused to the telocentric centromere behind. These results also illustrate the strong selection for loss of both Nf2 and p53 function in the development of osteosarcomas and fibrosarcomas. Whether this reflects a functional relationship between the two proteins or the fact that loss of Nf2 function in the osteoblast lineage normally leads to p53-dependent apoptosis, is unclear.
Our results suggest a role for Nf2 mutation in metastatic potential in the mouse. We have observed a greatly elevated frequency of metastasis associated with tumors that have lost Nf2 function, with or without concomitant loss of p53 function. In fact, the anatomical locations and histological profiles of many of these tumors are identical to those that arise in p53 singly heterozygous mice and metastasize at a much lower frequency. Although the observed cooperation between Nf2 and p53 loss in osteosarcoma and fibrosarcoma formation supports a common cellular origin for these tumors, it is possible that the particular cell type affected by Nf2 mutation is fundamentally different and may be particularly metastatically competent. Alternatively, it is possible that the increased rate of metastasis in Nf2 +/− mice reflects their advanced age. In this light it is important to note that we saw no bias in the frequency of metastasis occurring in younger (10–19 months) versus older (20+ months) Nf2 +/− animals (data not shown). Moreover, tumors occurring in Nf2 +/−;p53 +/− trans mice that lose heterozygosity at both the Nf2 and p53 loci also exhibited a high rate of metastasis, yet their average age of death is younger than that of p53 singly heterozygous animals whose tumors metastasized infrequently (Fig. 3C). The lower frequency of metastasis identified in young (3- to 5-month-old) Nf2 +/−;p53 +/− cis animals most likely reflects the rapid lethality associated with a large number of primary tumors in each animal and their frequent localization to the nasal passages. In an effort to begin to address the role of merlin loss in metastatic potential directly, we characterized the properties of Nf2- and p53-deficient osteosarcoma and fibrosarcoma cells in an experimental metastasis assay. We found Nf2-deficient tumor cells to be much more proficient in colonizing the lungs of syngeneic recipent animals than their p53-deficient counterparts, strongly supporting a role for merlin loss in promoting metastatic potential.
The mechanism by which loss of merlin function contributes to tumor formation could in fact be related to a role for its loss in promoting metastasis. The physical location of merlin at the membrane/cytoskeletal interface suggests that merlin is somehow involved in the integration of extracellular signals with those involved in reorganizing the cytoskeleton and controlling cell cycle entry. Upon loss of adhesion, fibroblasts undergo reversible growth arrest, whereas epithelial or endothelial cells undergo apoptosis (or anoikis; for review, see Frisch and Ruoslahti 1997). This regulatory system probably underlies the phenomenon of anchorage dependence for normal cell growth and its failure leads to the anchorage-independent growth of tumor cells. We have determined that merlin protein levels are up-regulated upon certain growth arrest stimuli including loss of adhesion, confluence, or serum deprivation, suggesting that merlin may normally participate in the receipt of or response to such growth arrest cues (Shaw et al. 1998). Perhaps in the absence of merlin function, such cues are misinterpreted or not received, resulting in inappropriate cell-cycle entry and continued proliferation. Successful metastasis further requires migration of tumor cells through blood vessel walls (intravasation), anchorage-independent survival in the circulation, exit from the blood vessels (extravasation), and survival and invasion of a secondary tissue where the extracellular microenvironment will be foreign (i.e., lung or liver). Nf2-deficient tumor cells may fail to properly receive growth arrest signals in suspension as they travel through the bloodstream, or misinterpret growth factor signals in the lung or liver microenvironment as survival signals. The unusually differentiated nature of many of the osteosarcoma metastases in Nf2 +/− mice suggests that Nf2-deficient tumor cells may actually interpret signals in the lung or liver as differentiation signals. Alternatively, this may reflect the survival of Nf2-deficient tumor cells in a foreign microenvironment prior to sustaining the full repertoire of genetic mutations that normally accompanies full transformation. It has been suggested that the rate-limiting step in a successful metastatic event is survival and invasion of a distant site; tumor cells that are able to survive in the bloodstream are often unable to colonize a secondary tissue (for review, see Chambers et al. 1995). We characterized a number of low passage Nf2-deficient tumor cell lines in an experimental metastasis assay and found that 100% of them are capable of colonizing the lungs of recipient animals. Moreover, most Nf2-expressing tumor cell lines derived from histologically matched p53-deficient tumors were unable to colonize the lungs of recipient animals. The inability to stably reintroduce Nf2 expression into the tumor cells and the early lethality of Nf2 homozygous mutant embryos makes it difficult to investigate directly merlin’s role in these processes. However, the continued characterization of the behavior of these cells and ultimately of primary Nf2-deficient cells both in vitro and in vivo will be invaluable in addressing these issues.
The strong similarity between merlin and the ERM proteins suggests that these proteins function analogously. However, in contrast to the growth and motility-suppressing function of merlin revealed by the tumorigenic and invasive consequences of its loss, several lines of evidence suggest that ERM proteins promote cell proliferation and motility. First, ERM function has been linked recently to the signaling network of the Rho family of small GTPases that promote cytoskeletal reorganization, cell growth, and cell motility (for review, see Van Aelst and D’Souza-Schorey 1997). Tiam-1, a positive regulator of the Rho family member Rac was identified originally as an invasion-promoting protein implicated in metastasis (Habets et al. 1994). Rho promotes the interaction between the ERM proteins and CD44, which has itself been shown to play an important role in invasion and metastasis (for review, see Kincade et al. 1997). The ERM proteins can also bind to and apparently inactivate RhoGDI, a negative regulator of Rho GTPases that also inhibits the motility of fibroblasts (Takahashi et al. 1997). Moreover, it has been demonstrated recently that activation of the Rho pathway leads to phosphorylation of ERM proteins in vivo, perhaps through the Rho effector Rho kinase, which can phosphorylate the carboxyl terminus of radixin in vitro (Matsui et al. 1998). Second, ezrin can be phosphorylated directly by HGF/scatter factor; ezrin is apparently both necessary and sufficient for HGF/scatter factor-induced epithelial cell migration (Crepaldi et al. 1997). Finally, fos, which induces osteosarcomas in mice, also induces the expression, phosphorylation, and relocalization of ezrin (Lamb et al. 1997b). fos-transformed fibroblasts exhibit increased invasiveness in vitro, contingent on fos-induced expression and relocalization of the ERM membrane partner CD44 (Lamb et al. 1997a). Up-regulation of ezrin has also been correlated with the increased proliferation and immortalization of fibroblasts in vitro (Kaul et al. 199). Together, these studies indicate a positive role for the ERM proteins, especially ezrin, in cell growth and motility. Therefore, an intriguing possibility is that merlin may function to regulate or antagonize the function of the other ERM proteins; removal of merlin could constitutively activate these pathways. Merlin is the most distantly related ERM family member and does not contain the carboxy-terminal actin-binding domain present in the ERM proteins. In addition, it is interesting to note that recent evidence suggests that the overexpression of merlin alters the subcellular localization of ezrin in some circumstances (Sainio et al. 1997).
In summary, these studies reveal profound consequences for loss of merlin function in mouse tumorigenesis and suggest a much broader role for merlin and perhaps the other ERM proteins in the development and progression of cancer. We have generated a manipulatable system and a set of tools that can be used to further investigate merlin function and the role of the cytoskeleton more generally in tumorigenesis and metastasis. Importantly, this system can also be used to study the metastatic process and potentially to identify other genes whose function may be perturbed during metastasis.
Materials and methods
Generation and genotyping of Nf2 heterozygous mice
The generation of Nf2 +/− ES cell clones derived from D3 ES cells of the 129Sv/Pas substrain has been described (McClatchey et al. 1997). Three different 129/SvPas Nf2 +/− ES cell clones were used to generate 21 chimeric animals by injection into wild-type C57BL/6 blastocyst stage embryos. Chimeric animals were bred to either wild-type C57BL/6 or 129Sv/Jae animals to produce 99 Nf2 +/− F1, 23 wild-type F1, and 37 129Sv Nf2 +/− animals. The 129Sv/Pas and 129Sv/Jae substrains are nearly identical (Simpson et al. 1997); lineages resulting from the breeding of chimeras and 129Sv/Jae mice are hereafter referred to as 129Sv. In addition, 45 F2 animals were generated by intercrossing of F1 heterozygotes. Tail DNA was isolated and genotyped using a cocktail of primers: a (5′-GGGGCTTCGGGAAACCTGG-3′), b (5′-GTCTGGGAAGTCTGTGGAGG-3′), and c (5′-CTATCAGGACATAGCGTTGG-3′) (McClatchey et al. 1997). Primer pair a-b amplifies a 306-bp product from the wild-type allele, whereas primer pair a-c amplifies a 575-bp product from the mutant allele.
Generation and genotyping of Nf2 and p53 mutant mice
Nf2 and p53 heterozygous mice were intercrossed, producing Nf2 +/−;p53 +/− trans mice, which were then mated to wild-type animals to produce Nf2 +/−;p53 +/− cis animals. Detection of the p53 mutant allele was performed by PCR analysis as described (Jacks et al. 1994).
Analysis of Nf2 and p53 loss of heterozygosity
Nf2 and p53 LOH analysis was evaluated by Southern blotting. Tumor DNA was isolated and extracted once with phenol/chloroform (1:1) and once with chloroform/isoamyl alcohol (24:1) and precipitated with ethanol. Nf2 LOH analysis was performed by Southern blotting of StuI-digested DNA and hybridization to a 233-bp genomic Nf2 probe (McClatchey et al. 1997). Similarly, p53 LOH analysis was performed by Southern blotting of StuI–EcoRI-digested DNA and hybridization to a probe corresponding to exons 7–10 of the p53 cDNA (Jacks et al. 1994).
For allelotyping, simple sequence length polymorphism (SSLP) marker D11MIT20 (Research Genetics) was chosen because of its location approximately midway between the Nf2 and p53 loci, and because it is informative with respect to the C57BL/6 and 129/Sv strains (Y. Chen and T. Jacks, unpubl.). Primers a and b detect a 116-bp band specific to C57BL/6 DNA and a ∼150-bp band specific to 129/Sv DNA by PCR.
Necropsy and histology
Animals were sacrificed upon decline in the health of the animal (i.e., weight loss, paralysis, ruffling of fur, or inactivity) or obvious tumor burden. A full autopsy was performed and tissues were fixed in either Bouin’s fixative (bone) or 10% neutral-buffered formalin (non-bone), dehydrated, and paraffin imbedded. Sections (4 μm) were generated and stained with hematoxylin and eosin. For detection of metastases, a single section through the spleen, each kidney, or each lobe of the lung was examined. The liver was cut into several pieces to fit easily in standard tissue-processing cassettes; therefore, approximately two to three sections through each lobe was examined.
Derivation of tumor cell lines
A small piece of tumor was rinsed in PBS, minced in trypsin/EDTA for 15–30 min, followed by further dissociation and plating in DMEM plus 20% fetal bovine serum (Life Technologies). Tumor cells were passaged twice and frozen at 2 × 106 cells/vial; in general, one vial was thawed and passaged 0–1 time before tail vein injection (see below).
Tail vein injections/metastasis assays
P3-P6 tumor cells (1 × 106 to 2 × 106) were resuspended in 200 μl of PBS and injected into the tail vein of syngeneic F1 C57BL/6:129Sv animals using a 27-gauge needle. The animals were monitored and sacrificed when moribund or upon the sacrifice of matched animals injected simultaneously with p53-deficient tumor cells (2–4 weeks; see Table 2).
Acknowledgments
We thank Jeff Settleman for helpful suggestions and critical reading of the manuscript. T.J. is an Associate Investigator of the Howard Hughes Medical Institute. A.I.M. was supported by a Young Investigator Award from the National Neurofibromatosis Foundation and is a recipient of a Burroughs Wellcome Career Award in the Biomedical Sciences. This work was supported in part by a grant from the Department of the Army.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mcclatch@helix.mgh.harvard.edu; FAX (617) 726-7808.
References
- Bronson RT. Rate of occurrence of lesions in 20 inbred and hybrid genotypes of rats and mice sacrificed at 6 month intervals during the first years of life. In: Harrison DE, editor. Genetic effects on aging II. Caldwell, NJ: The Telford Press, Inc.; 1990. pp. 279–357. [Google Scholar]
- Chambers AF, MacDonald IC, Schmidt EE, Koop S, Morris VL, Khokha R, Groom AC. Steps in tumor metastasis: new concepts from intravital videomicroscopy. Cancer Metastasis Rev. 1995;14:279–301. doi: 10.1007/BF00690599. [DOI] [PubMed] [Google Scholar]
- Claudio JO, Veneziale RW, Menko AS, Rouleau GA. Expression of schwannomin in lens and Schwann cells. Neuroreport. 1997;8:2025–2030. doi: 10.1097/00001756-199705260-00044. [DOI] [PubMed] [Google Scholar]
- Crepaldi T, Gautreau A, Comoglio PM, Louvard D, Arpin M. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J Cell Biol. 1997;138:423–434. doi: 10.1083/jcb.138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WF, Miller J, Steen R, Merchant MA, Damron-Boles D, Husain Z, Dredge R, Daly MJ, Ingalls KA, O’Connor TJ, Evans CA, DeAngelis MM, Levinson DM, Kruglyak L, Goodman N, Copeland NG, Jenkins NA, Hawkins TL, Stein L, Page DC, Lander ES. A comprehensive genetic map of the mouse genome (see comments) [published erratum appears in Nature (1996) 381: 172]. Nature. 1996;380:149–152. doi: 10.1038/380149a0. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Finkel MP, Biskis BO, Jinkins PB. Virus induction of osteosarcomas in mice. Science. 1966;151:698–701. doi: 10.1126/science.151.3711.698. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Frith CH, Ward JM. A morphologic classification of proliferative and neoplastic hepatic lesions in mice. J Environ Pathol Toxicol. 1979;3:329–351. [PubMed] [Google Scholar]
- Frith CH, Littlefield NA, Umholtz R. Incidence of pulmonary metastases for various neoplasms in BALB/cStCrlfC3H/Nctr female fed N-2-fluorenylacetamide. J Natl Cancer Inst. 1981;66:703–712. [PubMed] [Google Scholar]
- Gary R, Bretscher A. Heterotypic and homotypic associations between ezrin and moesin, two putative membrane-cytoskeletal linking proteins. Proc Natl Acad Sci. 1993;90:10846–10850. doi: 10.1073/pnas.90.22.10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Agosti C, Xu L, Pinney D, Beauchamp R, Hobbs W, Gusella JF, Ramesh V. The merlin tumor suppressor localizes preferentially in membrane ruffles. Oncogene. 1996;13:1239–1247. [PubMed] [Google Scholar]
- Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: Loss of merlin’s protective spell. Curr Opin Genet Dev. 1996;6:87–92. doi: 10.1016/s0959-437x(96)90016-7. [DOI] [PubMed] [Google Scholar]
- Haase VH, Trofatter JA, MacCollin M, Tarttelin E, Gusella JF, Ramesh V. The murine NF2 homolog ue encodes a highly conserved merlin protein with alternative forms. Hum Mol Genet. 1994;3:407–411. doi: 10.1093/hmg/3.3.407. [DOI] [PubMed] [Google Scholar]
- Habets GG, Scholtes EH, Zuydgeest D, van der Kammen RA, Stam JC, Berns A, Collard JG. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–549. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- Helander TS, Carpen O, Turunen O, Kovanen PE, Vaheri A, Timonen T. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 1996;382:265–268. doi: 10.1038/382265a0. [DOI] [PubMed] [Google Scholar]
- Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Tsukita S, Tsukita S. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: Possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford RK, Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes & Dev. 1997;11:1447–1463. doi: 10.1101/gad.11.11.1447. [DOI] [PubMed] [Google Scholar]
- Huson SM. Neurofibromatosis 2: Clinical features, genetic counseling and management issues. In: Huson SM, Hughes RAC, editors. The neurofibromatoses: A practical and clinical overview. London, UK: Chapman and Hall Medical Press; 1994. pp. 211–233. [Google Scholar]
- Huynh DP, Pulst SM. Neurofibromatosis 2 antisense oligodeoxynucleotides induce reversible inhibition of schwannomin synthesis and cell adhesion in STS26T and T98G cells. Oncogene. 1996;13:73–84. [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kaul SC, Mitsui Y, Komatsu Y, Reddel RR, Wadhwa R. A highly expressed 81 kDa protein in immortalized mouse fibroblasts: its proliferative function and identity with ezrin. Oncogene. 1996;13:1231–1237. [PubMed] [Google Scholar]
- Kincade PW, Zheng Z, Katoh S, Hanson L. The importance of cellular environment to function of the CD44 matrix receptor. Curr Opin Cell Biol. 1997;9:635–642. doi: 10.1016/s0955-0674(97)80116-0. [DOI] [PubMed] [Google Scholar]
- Knowles BB, McCarrick J, Fox N, Solter D, Damjanov I. Osteosarcomas in transgenic mice expressing an alpha-amylase-SV40 T- antigen hybrid gene. Am J Pathol. 1990;137:259–262. [PMC free article] [PubMed] [Google Scholar]
- Lamb RF, Hennigan RF, Turnbull K, Katsanakis KD, MacKenzie ED, Birnie GD, Ozanne BW. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol Cell Biol. 1997a;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RF, Ozanne BW, Roy C, McGarry L, Stipp C, Mangeat P, Jay DG. Essential functions of ezrin in maintenance of cell shape and lamellipodial extension in normal and transformed fibroblasts. Curr Biol. 1997b;7:682–688. doi: 10.1016/s0960-9822(06)00295-8. [DOI] [PubMed] [Google Scholar]
- Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: Functional overlap between p107 and Rb. Genes & Dev. 1996;10:1621–1632. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- Lutchman M, Rouleau GA. The neurofibromatosis type 2 gene product, schwannomin, suppresses growth of NIH 3T3 cells. Cancer Res. 1995;55:2270–2274. [PubMed] [Google Scholar]
- Luz A, Gossner W, Murray AB. Osteosarcoma, spontaneous and radiation-induced, mouse. In: Jones TC, Mohr U, Hunt RD, editors. Cardiovascular and musculoskeletal systems. Berlin, Germany: Springer-Verlag; 1991. pp. 202–213. [Google Scholar]
- Mackay DJ, Esch F, Furthmayr H, Hall A. Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: an essential role for ezrin/radixin/moesin proteins. J Cell Biol. 1997;138:927–938. doi: 10.1083/jcb.138.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney BM, Fehon RG. Distinct cellular and subcellular patterns of expression imply distinct functions for the Drosophila homologues of moesin and the neurofibromatosis 2 tumor suppressor, merlin. J Cell Biol. 1996;133:843–852. doi: 10.1083/jcb.133.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Saotome I, Ramesh V, Gusella JF, Jacks T. The Nf2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes & Dev. 1997;11:1253–1265. doi: 10.1101/gad.11.10.1253. [DOI] [PubMed] [Google Scholar]
- Michiels L, Merregaert J. Retroviruses and oncogenes associated with osteosarcomas. Cancer Treat Res. 1993;62:7–18. doi: 10.1007/978-1-4615-3518-8_2. [DOI] [PubMed] [Google Scholar]
- Rizzoli, R. and J.P. Bonjour. 1997. Hormones and bones. Lancet (Suppl. 1) 349: sI20–23. [DOI] [PubMed]
- Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, Pulst SM, Lenoir G, Bijlsma E, Fashold R, Dumanski J, de Jong P, Parry D, Eldridge R, Aurias A, Delatttre O, Thomas G. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Ruther U, Komitowski D, Schubert FR, Wagner EF. c-fos expression induces bone tumors in transgenic mice. Oncogene. 1989;4:861–865. [PubMed] [Google Scholar]
- Sainio M, Zhao F, Heiska L, Turunen O, Bakker M, Zwarthoff E, Lutchman M, Rouleau GA, Jaaskelainen J, Vaheri A, Carpen O. Neurofibromatosis 2 tumor suppressor protein colocalizes with ezrin and CD44 and associates with actin-containing cytoskeleton. J Cell Sci. 1997;110:2249–2260. doi: 10.1242/jcs.110.18.2249. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J., A.I. McClatchey, and T. Jacks. 1998. Regulation of the neurofibromatosis type 2 tumor suppressor protein, marlin, by adhesion and growth arrest stimuli. J. Biol. Chem. (in press). [DOI] [PubMed]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nature Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J Biol Chem. 1997;272:23371–23375. doi: 10.1074/jbc.272.37.23371. [DOI] [PubMed] [Google Scholar]
- Takeshima H, Izawa I, Lee PS, Safdar N, Levin VA, Saya H. Detection of cellular proteins that interact with the NF2 tumor suppressor gene product. Oncogene. 1994;9:2135–2144. [PubMed] [Google Scholar]
- Taverna D, Ullman-Cullerè M, Rayburn H, Bronson RT, Hynes RO. A test for the role of alpha 5 integrin/fibronectin interactions in tumorigenesis. Cancer Res. 1998;58:848–853. [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, Haase VH, Ambrose CM, Munroe D, Bove C, Haines JL, Martuza RL, MacDonald ME, Seizinger BR, Short MP, Buckler AJ, Gusella JF. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor [published erratum appears in Cell (1993) 75: 826]. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Oishi K, Sato N, Sagara J, Kawai A, Tsukita S. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401. doi: 10.1083/jcb.126.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Yonemura S, Tsukita S. ERM proteins: Head-to-tail regulation of actin-plasma membrane interaction. Trends Biochem Sci. 1997;22:53–58. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- Vaheri A, Carpen O, Heiska L, Helander TS, Jaaskelainen J, Majander-Nordenswan P, Sainio M, Timonen T, Turunen O. The ezrin protein family: membrane-cytoskeleton interactions and disease associations. Curr Opin Cell Biol. 1997;9:659–666. doi: 10.1016/s0955-0674(97)80119-6. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes & Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Vesselinovitch SD, Mihailovich N, Rao KV. Morphology and metastatic nature of induced hepatic nodular lesions in C57BL × C3H F1 mice. Cancer Res. 1978;38:2003–2010. [PubMed] [Google Scholar]
- Wilkie TM, Schmidt RA, Baetscher M, Messing A. Smooth muscle and bone neoplasms in transgenic mice expressing SV40 T antigen. Oncogene. 1994;9:2889–2895. [PubMed] [Google Scholar]