

Non-technical summary
The kidney plays a critical role in regulating body fluid volume and blood pressure by conserving ions, solutes and water. Knowing the processes that underpin the handling of ions, solutes and water by the kidney is essential to our understanding of fluid and blood pressure regulation. Movement of ions is mediated by specific transport proteins found in the membranes of kidney cells. These proteins are regulated by additional proteins, called accessory proteins. In the current study, we have examined the role of the accessory protein KCNE1 in regulating a channel, KCNQ1, which is important in kidney function. We have observed that in the absence of KCNE1 the kidney has difficulty conserving sodium, chloride and water. However, by using specific inhibitors of these proteins we have also determined that although KCNE1 has a role in kidney function, the mechanism of its action is unlikely to be by regulating the protein KCNQ1.
Abstract
Abstract
KCNE1 is a protein of low molecular mass that is known to regulate the chromanol 293B and clofilium-sensitive K+ channel, KCNQ1, in a number of tissues. Previous work on the kidney of KCNE1 and KCNQ1 knockout mice has revealed that these animals have different renal phenotypes, suggesting that KCNE1 may not regulate KCNQ1 in the renal system. In the current study, in vivo clearance approaches and whole cell voltage-clamp recordings from isolated renal proximal tubules were used to examine the physiological role of KCNE1. Data from wild-type mice were compared to those from KCNE1 knockout mice. In clearance studies the KCNE1 knockout mice had an increased fractional excretion of Na+, Cl−, HCO3− and water. This profile was mimicked in wild-type mice by infusion of chromanol 293B, while chromanol was without effect in KCNE1 knockout animals. Clofilium also increased the fractional excretion of Na+, Cl− and water, but this was observed in both wild-type and knockout mice, suggesting that KCNE1 was regulating a chromanol-sensitive but clofilium-insensitive pathway. In whole cell voltage clamp recordings from proximal tubules, a chromanol-sensitive, K+-selective conductance was identified that was absent in tubules from knockout animals. The properties of this conductance were not consistent with its being mediated by KCNQ1, suggesting that KCNE1 regulates another K+ channel in the renal proximal tubule. Taken together these data suggest that KCNE1 regulates a K+-selective conductance in the renal proximal tubule that plays a relatively minor role in driving the transport of Na+, Cl− and HCO3−.
Introduction
KCNE1 (also known as minK and IsK) is a protein of molecular mass approximately 14.5 kDa that belongs to the KCNE family. These proteins act as β subunits, regulating the pore-forming subunits of ion channels (reviewed by Pongs & Schwarz, 2010). KCNE1 was first identified in rat kidney (Takumi et al. 1988), but most subsequent work has concentrated on the role it plays in the heart. KCNE1 regulates the voltage-gated K+ channel KCNQ1, which is found in both excitable and non-excitable tissues. In the presence of KCNE1, Q1-mediated currents increase in magnitude, and demonstrate strong voltage- and slow time-dependent activation, which typically takes several seconds to reach steady state (Sanguinetti et al. 1996). In the heart the KCNQ1–E1 complex conducts the delayed rectifier current IKS (Barhanin et al. 1996). KCNE-mediated current (IKS) plays a critical role in repolarisation of cardiac myocytes, and mutations in Q1 or E1 cause some forms of long QT syndrome, a disease associated with prolonged cardiac action potentials and an increased risk of sudden death (Seebohm et al. 2008). KCNQ1–E1-mediated forms of long QT syndrome are also associated with deafness, as the channel complex also plays a critical role in the formation of the K+-rich endolymph in the ear (Casimiro et al. 2001). KCNE1 also plays a role in regulatory volume decrease in murine tracheal cells and renal epithelial cells (Lock & Valverde, 2000; Belfodil et al. 2003; Millar et al. 2004), while KCNQ1 is important in alveolar cell repair (Trinh et al. 2007) and the regulation of glucose metabolism (Boini et al. 2009).
Although there are a number of studies suggesting that KCNE1 is important in the regulation of cell volume of renal epithelial cells (Barriere et al. 2003b; Belfodil et al. 2003; Millar et al. 2004), there is still uncertainty about the physiological role of KCNE1 and whether it regulates KCNQ1 in renal epithelial cells. This uncertainty is due to differences in data related to protein expression, and from functional studies in KCNE1 and KCNQ1 knockout mice. For example, some studies suggest that KCNQ1 is expressed strongly in the distal tubule, connecting tubule and cortical and medullary collecting ducts (Demolombe et al. 2001; Zheng et al. 2007), with weaker expression in the late proximal tubule and none in the early proximal tubule (Zheng et al. 2007). In contrast Vallon et al. (2001) reported strong expression in late parts of the proximal tubule. KCNE1, on the other hand, is expressed at higher levels in the cortex, with one study providing evidence for the distal tubule and cortical collecting duct (Demolombe et al. 2001) and another study indicating expression in the apical membrane of proximal tubule cells (Vallon et al. 2001). It is clear, therefore, that although there is some overlap between the localization of KCNQ1 and KCNE1, there are other nephron segments where each protein is expressed independently. Differences in expression are also implicated by differences in the phenotypes of KCNQ1 and KCNE1 knockout mice. A previous study has shown that under control conditions KCNE1 knockout mice display a reduced plasma glucose concentration and increased urinary loss of Na+, Cl− and glucose (Vallon et al. 2001). It should, however, be noted that the plasma glucose levels reported in this study were high, around 120 mm. In contrast, KCNQ1 knockout mice maintained on a normal dietary intake of food and water show normal urinary loss of Na+ and glucose (Vallon et al. 2005). Data from isolated proximal tubule in this later study did indicate that the loss of KCNQ1 appeared to impact on the membrane potential of these cells and thereby reduced the driving force for Na+ and glucose cotransport. However, the lack of effect on glucose handling in the in vivo clearance studies would suggest that its physiological contribution to the regulation of glucose handling is minor.
These previous studies suggest that KCNE1 may not regulate KCNQ1 in the nephron, and they also raise questions as to the importance of KCNE1 under normal physiological conditions. The aim of the current study was twofold, to examine whether KCNQ1 is the channel regulated by KCNE1 under in vivo conditions by examining the effects on electrolyte balance and renal function of KCNQ1 inhibitors in wild-type and KCNE1 knockout mice, and secondly to determine whether KCNE1 plays an important role in renal proximal tubule function by determining the effects of ablation of KCNE1 on conductance properties of isolated proximal tubules.
Methods
Animal model
Adult wild-type (WT) and KCNE1 knockout (KO) mice (129-KCNE1tm1shf/J) (129/SV) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and used to breed mice for use in experimental procedures. Animals were genotyped according to standard Jackson Laboratory procedures (http://jaxmice.jax.org/strain/003009.html). For experimental procedures data from adult KO animals were compared to adult WT littermates. Mice were typically used between 6 and 10 weeks of age, had free access to food and water and were housed under a 12 h light–dark cycle. All experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 and local ethical review.
In vivo studies
Adult mice were weighed and then anaesthetized with a combination of sodium thiopentone (i.p. injection 100 mg kg−1; Thiovet, C-Veterinary Products, Leyland, UK), ketamine (i.p. injection 10 mg kg−1) and xylazine (i.p. injection 1.5 mg kg−1; Sigma-Aldrich, Poole, UK). Animals were placed on a thermostatically controlled heated blanket to maintain body temperature at 37°C (Harvard Apparatus, Edenbridge, UK) and surgically prepared for renal clearance experiments. Polyethylene cannulae (Portex, outer diameter 0.63 mm, bore 0.50 mm) were placed in the left jugular vein for intravenous infusion and the left carotid artery for continuous blood pressure monitoring and blood sampling. The bladder was also cannulated via a suprapubic incision to allow urine collection. A tracheotomy maintained a clear airway, with pure oxygen blown over the neck area.
Once the venous cannula was implanted, mice received an intravenous infusion of 2.25% bovine serum albumin (BSA) in 0.9% NaCl at 0.6 ml h−1, which was maintained until the end of surgery. After surgery was completed there was an equilibration period of 45 min, followed by urine collection over a 60 min experimental period. In the first 15 min of equilibration mice received 0.6 ml of infusate over the first 15 min and 0.6 ml h−1 maintenance infusion thereafter. To determine the rate of glomerular filtration (GFR), [3H]inulin (GE Healthcare, Little Chalfont, UK) was infused at a 1.5 μCi priming dose (bolus) during the first 15 min of equilibration and then a 4.5 μCi h−1 maintenance dose thereafter. Previous experiments have established that these protocols produce stable plasma [3H]inulin activity, and ion levels (Na+, Cl−, K+ and HCO3−) during the 60 min experimental period (Kibble et al. 2001). For this reason, a terminal plasma sample was used for calculation of renal clearance. After the terminal blood sample had been taken, animals were killed by anaesthetic overdose according to UK legislation. [3H]inulin was assayed by liquid scintillation counting (Packard Tricarb 2100TR), Na+ and K+ concentrations by single channel flame photometry (Sherwood model 410, Scientific Laboratory Supplies, Nottingham, UK), Cl− by electrometric titration (Jenway PCLM3), HCO3− (i.e. total CO2) by carbon dioxide analyser (Corning 965, Corning Medical and Scientific, Medfield, MA, USA), glucose by microfluorometry (Nanoflo microfluorometer, World Precision Instruments, Sarasota, FL, USA) and osmolality by freezing point depression (Roebling osmometer, Camlab, Cambridge, UK).
The KCNQ1–KCNE1 K+ channel complex is inhibited by chromanol 293B (Lerche et al. 2007). Experiments therefore examined whether infusion of chromanol 293B altered renal function in WT and KO mice. If KCNE1 regulates a chromanol-sensitive channel in the kidney, addition of chromanol to WT animals would be expected to mimic data obtained in the KO animals. In contrast chromanol would be expected to have little effect on KCNE1 KO animals. Mice infused intravenously with chromanol 293B (Tocris Bioscience, Bristol, UK; a dimethyl sulfoxide (DMSO) stock added to the NaCl/BSA infusion solution) were given an initial bolus of 8 mg kg−1 h−1, followed by a 4 mg kg−1 h−1 maintenance dose. Previous studies in dogs have used bolus doses of between 1 and 10 mg kg−1 (Bauer et al. 1999; Varro et al. 2000). All control mice received an equivalent amount of DMSO vehicle. As a control, a second K+ channel inhibitor, clofilium, was also used. Clofilium also inhibits KCNQ1 (Yang et al. 1997), although the KCNQ1–KCNE1 complex is less sensitive to this inhibitor. In the current study, if clofilium acts on the KCNQ1–KCNE1 complex then the same effect as chromanol should be observed, i.e. an effect in WT mice but no effect in KCNE1 KO animals. In contrast, if clofilium is inhibiting another channel then the same response should be observed in both WT and KO animals. Mice infused intravenously with clofilium tosylate (ICN Biomedicals Inc., Aurora, OH, USA; a DMSO stock added to the NaCl/BSA infusion solution) were given 76 μg kg min−1 (both bolus and maintenance). This dose was equivalent to 150 nmol kg−1 min−1, within the range utilised in previous in vivo studies (63–200 nmol kg−1 min−1) (Carlsson et al. 1992; Farkas et al. 2008). Again, control animals were infused with an equivalent amount of DMSO.
Given the proximal tubule location for KCNE1, it would be expected that the loss of KCNE1 would impact on proximal tubule function. Therefore the handling of HCO3− was also examined in both WT and KO mice, in the absence and presence of chromanol or clofilium. To assess the overall magnitude of HCO3− reabsorption, animals were treated with acetazolamide (Sigma-Aldrich), a carbonic anhydrase inhibitor (Burg & Green, 1977). Animals were infused with a 20 mg kg−1 h−1 bolus, followed by a 10 mg kg−1 h−1 maintenance dose (Fransen et al. 1993), all in the standard NaCl/BSA infusion solution. Control mice were infused with an equivalent amount of infusion solution.
Tubule isolation
Whole cell clamp studies were performed in isolated proximal tubules in order to identify chromanol-sensitive currents. Tubules were isolated using a previously published modified protocol from Schafer et al. (1997). Adult mice were killed by cervical dislocation according to UK legislation. The kidneys were removed and placed in ice-cold modified Eagle's minimum essential medium (MEM). After peeling off the renal capsule, thin (< 0.5 mm) tangential slices were taken from the cortex. These slices were torn with watchmakers' forceps and placed in a conical flask containing MEM, type II collagenase (0.5 mg ml−1), protease E (0.1 mg ml−1), soyabean trypsin/chymotrypsin inhibitor (50 μg ml−1) and glycine (5 mm). The flask was incubated at 37°C without agitation, and at 10 min intervals the digestion medium was decanted, the incubation medium recharged and digestion continued. Decanted medium was placed on ice and tubules were allowed to settle to the bottom of the tube. The supernatant was gently removed and replaced with ice-cold MEM plus 1% BSA. Tubules were stored on ice until required. Proximal tubules were identified by their diameter (approximately 40 μm), the presence of convolutions and, when observed, an attached glomerulus (Schafer et al. 1997).
Whole cell recordings
Proximal tubules were placed in a Perspex bath on the stage of an inverted microscope (Olympus IX70) and held in place using two holding pipettes (manufactured using a Narishige MF900 microforge). Once tubules were clamped in place, whole-cell recordings were obtained via the basolateral aspect of the cell and currents recorded in a variety of different extracellular solutions (see below) at room temperature. Whole cell recordings were typically obtained at a site distant from the glomerulus; the exact recording location along the proximal tubule could not be determined precisely since full length tubules were not obtained by this preparation method. Solutions were changed using a small volume perfusion system (Automate, Digitimer Ltd, Welwyn Garden City, UK), which typically took around 10–15 s to exchange solution bathing the tubule.
Voltage protocols were driven from a Dell computer equipped with a Digidata interface (Axon Instruments, Union City, CA, USA) using the pCLAMP software, Clampex 8, and sampled at 15 kHz (Axon Instruments). Recordings were made using a List EPC-7 amplifier, with currents saved directly onto the computer hard disk following low-pass filtering at 5 kHz. Two different voltage protocols were used. In the first protocol, cells were held at –80 mV and then stepped to between +140 and –80 mV in –20 mV steps, with each step lasting 5 s. In the second protocol cells were held at –40 mV, and then stepped to between +100 and –80 mV in –20 mV steps, again with each step lasting for 5 s. Mean current at each potential was measured using Clampfit (Axon Instruments) either initially on stepping the potential or within 50 ms of the end of the potential step, as specified in Results. The reversal potential (Vrev) of these currents was determined using polynomial regression. Whole cell slope and chord conductances were calculated by taking the slope of the line over the potential ranges −80 to +140 mV or −80 to +100 mV, except where stated. Cell area was calculated from the capacity transients seen in response to a 20 mV potential step, with membrane capacitance assumed to be 1 μF cm−2. The pipette solution contained (in mm): 145 KCl, 1 MgCl2, 0.5 ethylene glycol tetraacetic acid (EGTA), 10 Hepes (titrated to pH 7.4 with KOH) and 1 Na2ATP. The control bath solution contained (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes (titrated to pH 7.4 with NaOH), 10 mannitol and 100 μm gadolinium chloride (to block non-selective cation channels). To determine relative permeability the Vrev of currents was determined in control bath solution (high NaCl) and high extracellular K+ (NaCl in the control bath was replaced with KCl, giving a final extracellular K+ concentration of 135 mm). Relative permeability was then determined using the Goldman–Hodgkin–Katz equation. To examine the effect of the general K+ channel inhibitor barium (Ba2+), 10 mm mannitol in the NaCl control solution was replaced with 5 mm BaCl2. When the effect of chromanol was examined, 100 μm was added directly to the extracellular solution. A previous study has shown that the KCNQ1–KCNE1 complex is activated by cAMP (Kurokawa et al. 2003). Therefore, tubules were incubated for 30 min in the presence of forskolin (FSK, 10 μm) and 3-isobutyl-1-methylxanthine (IBMX, 100 μm) to increase cAMP levels before whole cell recordings were obtained.
Chemicals and solutions
The osmolality of all solutions was measured (Roebling osmometer) and the final osmolality was adjusted to 300 ± 1 mosmol kg−1 by the addition of water or mannitol as required. Chemicals were obtained from Sigma-Aldrich except where stated and were of analytical grade.
Statistics
Results are presented as means ± 1 SEM, with the number of experiments in parentheses (n). Significance was tested using ANOVA or Student's t test as appropriate, and significance assumed at the 5% level.
Results
In vivo studies
In vivo studies were utilised to examine whether KCNQ1 is the channel regulated by KCNE1 under in vivo conditions by examining the effects on electrolyte balance and renal function of KCNQ1 inhibitors chromanol 293B and clofilium in wild-type and KCNE1 knockout mice. Chromanol 293B inhibits the KCNQ1–KCNE1 K+ channel complex (Lerche et al. 2007). Clofilium also inhibits KCNQ1 (Yang et al. 1997), although the KCNQ1–KCNE1 complex is less sensitive to this inhibitor. It also inhibits HERG (Perry et al. 2004) and the two-pore domain K+ channel TASK-2 (Niemeyer et al. 2001), which is known to be involved in volume regulation of mouse proximal tubules (Barriere et al. 2003a).
Effect of chromanol and clofilium
The majority of haemodynamic parameters and plasma concentrations recorded in WT and KO animals in the absence or presence of chromanol or clofilium were not significantly different between each treatment group (online Supplemental Material, Tables S1 and S2). There were significant differences in renal parameters between WT and KO mice. KO mice demonstrated an increased fractional excretion (FE) of Na+ (P < 0.01), Cl− (P < 0.04) and water (P < 0.006) (Fig. 1A, B and E) and urine flow rate (P < 0.01) (Fig. 1F). The renal handling of K+ (P = 0.3) and glucose (P = 0.19) was not different in KO animals (Fig. 1C and D). These differences were mimicked by exposing WT mice to chromanol. Like KO animals, chromanol-treated WT mice also had increased urine flow rate (P < 0.001) and FE of Na+ (P < 0.001), Cl− (P < 0.002) and water (P < 0.002) (Fig. 1A, B, E and F). However, the addition of chromanol to KO animals was without effect on these parameters (P > 0.5) (Fig. 1A, B, E and F). In contrast, infusion of another K+ channel inhibitor, clofilium, produced a different response. In both WT and KO animals clofilium was associated with an increase in the FE of Na+ (P < 0.01), Cl− (P < 0.03) and H2O (P < 0.04) (Fig. 1A, B and E). Unlike chromanol, the pattern of response to clofilium was the same for both WT and KO animals.
Figure 1. In vivo renal parameters in WT and KO mice infused with DMSO vehicle, chromanol or clofilium.
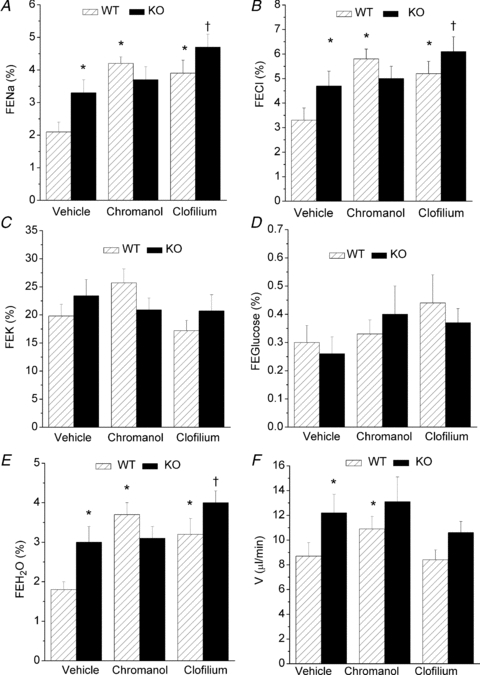
WT mice, hatched bars; KO mice, filled bars. Chromanol, 4 mg kg−1 h−1 maintenance dose; clofilium, 76 μg kg−1 min−1 maintenance dose. FE, fractional excretion (Na+, Cl−, K+, glucose, water); V, urine flow rate. *Significant difference from WT vehicle; †significant difference from KO vehicle. n = 8 for WT vehicle, n = 9 for KO vehicle, n = 6 for WT chromanol, n = 9 for KO chromanol, n = 10 for WT clofilium, n = 9 for KO clofilium.
Renal glucose responses
A previous study on KCNE1 KO mice provided evidence for an increase in the FE of glucose (Vallon et al. 2001). However, in the current study no effect was observed. The FEglucose was 0.30 ± 0.06% (n = 8) and 0.26 ± 0.06% (n = 9) in WT and KO animals, respectively (P = 0.19) (Fig. 1D). To determine whether an effect of glucose handling in KO animals could be induced, both WT and KO animals were infused with a 5% glucose solution. Data from these experiments are shown in Table 1. In both WT and KO animals, plasma glucose was increased compared to control infusion, 8.4 ± 2.0 mm (n = 8) and 10.8 ± 1.5 mm (n = 9) for control WT and control KO animals (supplemental Table S1), respectively, compared to the glucose infusion data shown in Table 1. There was also an expected increase in FEglucose (compare Fig. 1D to Table 1). However, there was no difference in plasma glucose (P = 0.53), glucose clearance (P = 0.86), urinary glucose excretion (P = 0.74) or FE of glucose (P = 0.87) between WT and KO animals.
Table 1.
Effect of glucose infusion
| Treatment | n | GFR (μl min−1) | Pglucose (mm) | Cglucose (μl min−1) | UVglucose (μmol min−1) | FEglucose (%) |
|---|---|---|---|---|---|---|
| WTglucose | 8 | 372 ± 26 | 34.6 ± 2.80 | 49 ± 22 | 1.90 ± 0.88 | 12.3 ± 5.2 |
| KOglucose | 8 | 406 ± 31 | 32.4 ± 2.5 | 45 ± 14 | 1.56 ± 0.51 | 10.6 ± 3.3 |
n, number of animals; Pglucose, plasma glucose; Cglucose, glucose clearance; UVglucose, urinary glucose loss; FEglucose, fractional excretion of glucose. WTglucose and KOglucose indicate glucose treated animals.
Renal HCO3− responses
In contrast to the lack of effect on glucose handling, there were small but significant effects on HCO3− handling (Fig. 2). As expected, the carbonic anhydrase inhibitor acetazolamide (ACTZ) increased the renal excretion of HCO3− (P < 0.001), and this was associated with a fall in plasma HCO3− levels (P < 0.001) (Fig. 2A). There was no effect of absence of KCNE1 on plasma HCO3− (P > 0.4), although both clofilium (P < 0.001) and chromanol (P < 0.03) did produce small but significant reductions in plasma HCO3− in both WT and KO mice. In terms of HCO3− clearance and urinary HCO3− loss, only ACTZ had an effect (P < 0.0001) (Fig. 2B and C). However, the fractional excretion of HCO3− was increased in KO mice versus WT (P < 0.02), and chromanol in WT mimicked the increase (P < 0.01) observed in KO mice (Fig. 2D). The chromanol induced loss of HCO3− in WT mice was absent in KO mice (P > 0.6) (Fig. 2D). Clofilium was without effect on the fractional excretion of HCO3− in both WT and KO mice (P > 0.24) (Fig. 2D). It should be noted that although the fractional excretion of HCO3− in KO animals was double that of WT, the increase was small in comparison to the effect of ACTZ.
Figure 2. In vivo HCO3− handling data in WT and KO mice infused with DMSO vehicle, chromanol or clofilium.
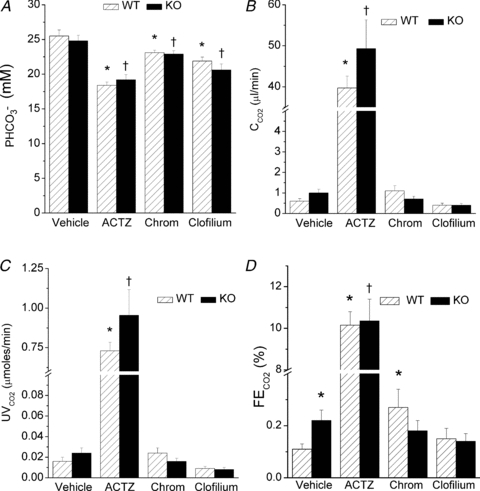
WT mice, hatched bars; KO mice, filled bars. Chromanol, 4 mg kg−1 h−1 maintenance dose; clofilium, 76 μg kg−1 min−1 maintenance dose. 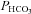 , plasma HCO3− concentration;
, plasma HCO3− concentration; 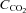 , HCO3− clearance;
, HCO3− clearance; 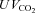 , urinary excretion HCO3−;
, urinary excretion HCO3−; 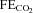 , fractional excretion of HCO3−. n = 14 for WT vehicle, n = 6 for WT ACTZ, n = 16 for KO vehicle, n = 6 for KO ACTZ, n = 6 for WT chromanol, n = 9 for KO chromanol, n = 10 for WT clofilium, n = 9 for KO clofilium. *Significant difference from WT vehicle; †significant difference from KO vehicle.
, fractional excretion of HCO3−. n = 14 for WT vehicle, n = 6 for WT ACTZ, n = 16 for KO vehicle, n = 6 for KO ACTZ, n = 6 for WT chromanol, n = 9 for KO chromanol, n = 10 for WT clofilium, n = 9 for KO clofilium. *Significant difference from WT vehicle; †significant difference from KO vehicle.
These in vivo clearance studies demonstrated that infusion of chromanol 293 to WT mice was able to mimic the effects observed in KCNE1 KO mice. In contrast chromanol was without effect in the KCNE1 KO mice, suggesting that KCNE1 regulates a chromanol-sensitive pathway in renal tubules.
Whole cell recordings
Whole cell recordings were used to determine whether KCNE1 plays an important role in renal proximal tubule function by determining the effects of ablation of KCNE1 on conductance properties of cells in whole isolated renal proximal tubules.
Total current properties
The capacitance of the cells in WT tubules was 11.7 ± 0.74 pF (n = 41). This was not significantly different to the cells in KO tubules (P = 0.1) (13.5 ± 0.84 pF, n = 30). Cells were initially clamped at –80 mV, and then stepped to between +140 and –80 mV in –20 mV steps. Cells were clamped at each test potential for 5 s and mean currents recorded initially on stepping the potential and again at the end of the voltage step. In both WT and KO tubules, total currents were voltage independent, but demonstrated a small amount of time-dependent inactivation. Figure 3A shows typical current recordings from cells in a WT and KO tubule, while Fig. 3B shows the mean steady state currents recorded from WT and KO tubules. In both WT (P = 0.026) and KO (P = 0.006) tubules the whole cell conductance (determined between −80 and +140 mV) decreased from the start of the potential step compared to the end of the potential step (Fig. 3C). This appeared to be a consequence of a small fall in outward currents (Fig. 3A). The Vrev values of the currents were not significantly different between WT and KO tubules, −32.2 ± 3.62 mV (n = 15) versus−36.5 ± 4.16 mV (n = 11), respectively (P = 0.42). There was also no significant difference between the magnitude of the cellular conductances (P = 0.38) (Fig. 3C).
Figure 3. Total steady state whole cell currents in WT and KO proximal tubule cells.
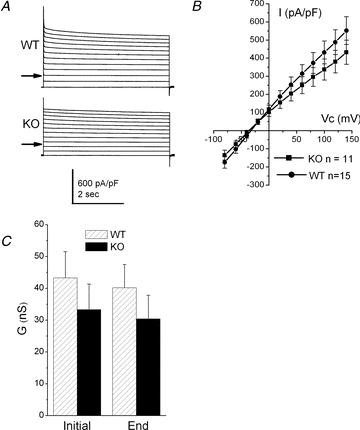
A, typical traces obtained from WT (upper) and KO (lower) recordings. Clamp potential was stepped from −80 mV to between +140 and −80 mV. The arrows indicate the zero current level. B, mean I–V curve of total current in WT (▪, n = 15) and KO recordings (•, n = 11). C, mean conductance in WT and KO recordings initially on stepping the potential and at the end of the potential step.
To examine the relative permeability of the total current, the NaCl bath solution (130 mm NaCl and 5 mm KCl) was replaced by one containing KCl (135 mm KCl). Cells were held at −40 mV and then clamped to between +100 and −80 mV, in −20 mV steps. In WT cells, the mean Vrev of total currents in NaCl was −34.5 ± 5.07 mV (n = 9; Fig. 4A). Replacement of Na+ by K+ shifted the Vrev by 31.7 ± 5.18 mV (P < 0.001) to −2.80 ± 0.99 mV, corresponding to a Na+: K+ permeability ratio of 0.30 ± 0.07. In KO cells the mean Vrev of total currents in NaCl was −39.0 ± 6.48 mV (n = 7; Fig. 4B). Replacement of Na+ by K+ shifted the Vrev by 35.8 ± 6.65 mV (P < 0.002) to −3.20 ± 2.32 mV, corresponding to a Na+:K+ permeability ratio of 0.26 ± 0.08. There was no significant difference in either the shift in Vrev (P = 0.61) or the permeability ratio (P = 0.54) between WT and KO cells.
Figure 4. Relative permeability of total currents.
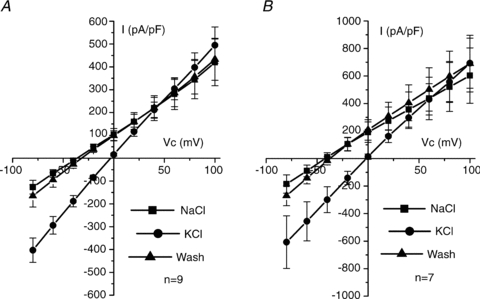
A, mean I–V curve of total current in WT recordings (n = 9) exposed to bath NaCl (▪), bath KCl (•) and then NaCl wash (▴). B, mean I–V curve of total current in KO recordings (n = 7) exposed to bath NaCl (▪), bath KCl (•) and then NaCl wash (▴). Mean currents were recorded at the end of the potential step.
Effect of barium
With NaCl in the bath, cells were initially clamped at −80 mV, and then stepped to between +100 and −80 mV in −20 mV steps for 5 s. Currents were recorded under control conditions and then with 5 mm Ba2+ in the bath. Addition of Ba2+ inhibited whole cell currents in both WT and KO (Fig. 5A). In WT cells whole cell conductance (determined between +100 and −80 mV) was 40.0 ± 5.3 nS and 30.7 ± 4.36 nS, in the absence and presence of Ba2+, respectively, a decrease of 23% (P < 0.001) (n = 13; Fig. 5B). Ba2+ shifted the Vrev of the total current from −30.1 ± 2.50 mV to −17.2 ± 3.08 mV (P < 0.001), consistent with inhibition of a K+ conductance. The Vrev of the Ba2+-sensitive conductance was −65.1 ± 5.17 mV (Fig. 5D), which corresponded to a Na+:K+ permeability ratio of 0.07 ± 0.01. The conductance of KO cells (determined between +100 and −80 mV) was 40.5 ± 7.87 nS and 28.5 ± 6.45 nS, in the absence and presence of Ba2+, respectively, a decrease of 30% (P < 0.01) (n = 6; Fig. 5C). Ba2+ shifted the Vrev of the total current from −38.7 ± 5.13 mV to −23.5 ± 3.75 mV (P < 0.001), consistent with inhibition of a K+ conductance. The Vrev of the Ba2+-sensitive conductance was −69.5 ± 8.61 mV (Fig. 5D), which corresponded to a Na+:K+ permeability ratio of 0.06 ± 0.04. There was no difference between the magnitude of the Ba2+-sensitive conductance in WT and KO cells (9.34 ± 1.57 nS versus 11.5 ± 3.12 nS, respectively, P = 0.52), the Vrev of the Ba2+-sensitive conductance (P = 0.67), nor the Na+:K+ permeability (P = 0.83) (Fig. 5D).
Figure 5. Effect of bath 5 mm Ba2+ on WT and KO recordings.
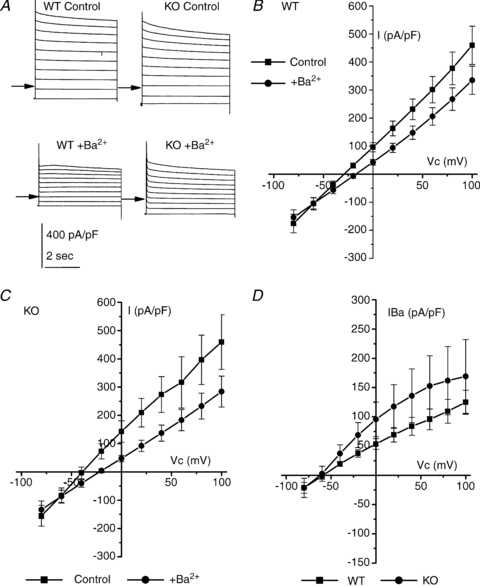
A, typical traces obtained from WT (left) and KO (right) recordings under the control condition (upper) and in the presence of Ba2+ (lower). Clamp potential was stepped from −80 mV to between +100 and −80 mV. B, mean I–V curve in WT recordings (n = 13) under the control condition (▪) and in the presence of Ba2+ (•). C, mean I–V curve in KO recordings (n = 6) under the control condition (▪) and in the presence of Ba2+ (•). D, mean Ba2+-sensitive currents taken from data in B and C in WT (▪) and KO (•) recordings. Mean currents were recorded at the end of the potential step.
Effect of chromanol
Given that the in vivo data showed that there was an effect of chromanol 293B that was lost in KO animals, the effect of chromanol 293B on cellular currents was examined. Cells were clamped initially at −80 mV, and then stepped to between +100 and −80 mV in −20 mV steps for 5 s. Currents were recorded under the control condition (high NaCl) and then with 100 μm chromanol 293B added to the bath (Fig. 6A). In WT recordings chromanol had a small but significant effect on current magnitude (Fig. 6B). Whole cell conductance (determined between +100 and −80 mV) was 42.1 ± 7.50 nS and 37.8 ± 6.48 nS (n = 13), in the absence and presence of chromanol, respectively, a mean fall of 10% (P = 0.04). This fall in conductance was associated with a significant positive shift in Vrev from −37.3 ± 2.27 mV to −33.1 ± 2.44 mV (P < 0.001). The chromanol-sensitive current had a Vrev of −73.9 ± 4.29 mV, corresponding to a Na+:K+ permeability ratio of 0.03 ± 0.01 (Fig. 6D). In KO recordings chromanol was without effect (Fig. 6C). Whole cell conductance was 38.0 ± 6.97 nS and 35.9 ± 6.28 nS in the absence and presence of chromanol, respectively (n = 13) (P = 0.1). There was no effect of chromanol on the Vrev of whole cell currents (–29.2 ± 3.49 mV versus−28.7 ± 3.44 mV, P = 0.55). Figure 6D shows the chromanol-sensitive current in WT recordings in contrast to the lack of current observed in KO recordings.
Figure 6. Effect of 100 μm chromanol on WT and KO recordings.
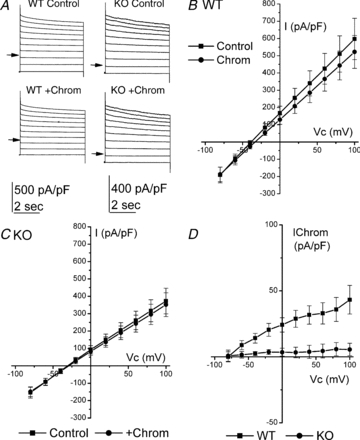
A, typical traces obtained from WT (left) and KO (right) recordings under the control condition (upper) and in the presence of chromanol (lower). Clamp potential was stepped from −80 mV to between +100 and −80 mV. B, mean I–V curve in WT recordings (n = 13) under the control condition (▪) and in the presence of chromanol (•). C, mean I–V curve in KO recordings (n = 13) under the control condition (▪) and in the presence of chromanol (•). D, mean chromanol-sensitive currents taken from data in B and C in WT (▪) and KO (•) recordings. Mean currents were recorded at the end of the potential step.
In WT cells the relative permeability of the chromanol-sensitive conductance was determined with 130 mm NaCl plus 5 mm KCl in the bath. After wash-out of chromanol, bath NaCl was replaced with KCl, giving a 135 mm KCl and 0 mm NaCl bath solution. The chromanol-sensitive conductance with this high K+ solution was then determined (Fig. 7A). Cells were held at −40 mV and then clamped to between +100 and −80 mV, in −20 mV steps. In NaCl, the Vrev of the chromanol-sensitive conductance was −74.1 ± 3.01 mV (n = 7: Fig. 7B). Replacing bath Na+ with K+ produced a shift in Vrev of 69.3 ± 3.40 mV, to −4.76 ± 1.66 mV (Fig. 7B) (P < 0.001). This corresponded to a Na+:K+ permeability ratio of 0.03 ± 0.01. In essentially symmetrical K+ solutions (135 mm outside and 145 mm inside) the chromanol-sensitive conductance was non-rectifying, exhibiting ohmic currents over the potential range −60 to +80 mV. Gout (measured between +80 and +20 mV) was 5.93 ± 1.87 nS, while Gin (measured between −20 and −60 mV) was 6.57 ± 2.32 nS. There was no significant difference between these (P = 0.41).
Figure 7. Relative permeability of 100 μm chromanol-sensitive conductances and effect of cAMP activation.
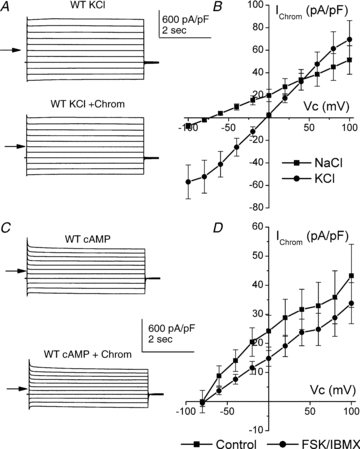
A, typical traces from a WT recording in the presence of bath KCl (upper) and KCl plus chromanol (lower). B, mean I–V curve of chromanol-sensitive currents in paired cells in the presence of Na+ bath solution (▪) or K+ bath solution (•) (n = 7). C, typical traces from a WT tubule previously stimulated with FSK (10 μm) and IBMX (100 μm). In the upper trace the bath contained high Na+, while in the lower trace it contained high Na+ plus chromanol. D, mean I–V curve of chromanol-sensitive currents in cells under the control condition (▪) (n = 13) or after cells had been incubated with FSK (10 μm) and IBMX (100 μm) for 30 min (•) (n = 15). Under both conditions the bath contained high Na+. Mean currents were recorded at the end of the potential step.
A previous study has shown that the KCNQ1–KCNE1 complex is activated by cAMP (Kurokawa et al. 2003). Therefore, if the chromanol-sensitive conductance observed in WT renal proximal tubule cells is mediated by this complex we would predict that activation of cAMP should increase current magnitude. However, incubating tubules with FSK/IBMX (10 μm/100 μm), which increases cAMP levels, was without effect on the chromanol-sensitive conductance (Fig. 7C and D). The chromanol-sensitive conductance in WT non-FSK/IBMX-treated tubules was 2.33 ± 0.54 nS (n = 13), while in WT FSK/IBMX-treated tubules the chromanol-sensitive conductance was 2.14 ± 0.48 nS (n = 15) (P = 0.79). The Vrev values of these were also not significantly different (−73.9 ± 4.29 mV versus−77.8 ± 4.36 mV, under control and FSK/IBMX conditions, respectively, P = 0.51).
These data provide evidence for the presence of a chromanol-sensitive, KCNE1-regulated K+ conductance in mouse renal proximal tubules that showed no voltage dependence and was not activated by cAMP stimulation.
Discussion
Previous work by Vallon et al. (2001, 2005) has suggested that KCNE1 and KCNQ1 play a role in renal proximal tubule function. In their studies KCNE1 KO mice demonstrated increased urinary loss of glucose, Na+, Cl− and H2O. In addition, it has been suggested that KCNE1 couples with KCNQ1 in the kidney to contribute to renal K+ handling. However, in KCNQ1 KO mice under control conditions there was no renal phenotype, although changes in proximal tubule function were observed on glucose loading. The differences in these two studies suggest that KCNE1 and KCNQ1 have different roles in the nephron, and challenge the idea that these two proteins form a complex that plays an important role in renal K+ handling. In the current study data from KCNE1 knockout mice suggest that it has a relatively minor role in maintaining normal renal function. In addition, the pharmacological, clearance and whole cell conductance data indicate that the channel regulated by KCNE1 in the proximal tubule is unlikely to be KCNQ1.
Where is KCNE1 functionally expressed?
Although the previous functional work has suggested a role for KCNE1 in the proximal tubule (Vallon et al. 2001), data from localization studies are conflicting, with evidence for KCNE1 expression in the distal tubule and cortical collecting duct (Demolombe et al. 2001) and another study indicating a proximal tubule apical membrane location (Vallon et al. 2001). In addition, there are no reports of direct electrophysiological measurement of KCNE1 function in the renal proximal tubule in native tissue. The current study provides the first electrophysiological data that support a role for KCNE1 in regulating K+ channels in renal proximal tubule. In WT tubules, whole cell voltage clamp approaches clearly showed a small but significant effect of the K+ channel inhibitor chromanol 293B. This inhibition was absent in KCNE1 KO mice, indicating that the loss of KCNE1 was impacting on a chromanol-sensitive channel. The conductance sensitive to KCNE1-knockout and chromanol (IE1) was K+ selective, as shown by the negative Vrev under physiological conditions, and the large positive shift in Vrev in response to an increase in extracellular K+ (Na+:K+ permeability ratio of 0.03). IE1 was relatively small, comprising only 10% of the total current observed. Given the high concentration of chromanol used in the present study, this small degree of inhibition could reflect either (1) a small contribution to the total current or (2) a possible issue with the access of the chromanol to the channel. Inhibition by chromanol took around 30 s to reach steady-state (data not shown), suggesting that drug access was not an issue. This compares to around 18 s for Ba2+ inhibition to reach steady state (data not shown). Additionally, it is clear that the total cellular current is not purely due to K+-selective channels. For example, the Vrev of the control current was on the order of −30 to −40 mV, well away from the Nernst potential for K+. Consistent with this, a typical maximal inhibitory concentration of the generic K+ channel inhibitor Ba2+ only blocked control currents by around 30%. This suggests that additional channel types may be present, for example Cl− channels (Rubera et al. 1998). Therefore it is likely that IE1 only plays a small contribution to the cellular K+ currents.
Where is IE1 located? When using single cells the whole cell clamp technique typically records the total current passing across all of the cell membrane, and therefore specific membrane location in epithelial cells cannot be determined. Using tubules introduces another consideration: the issue of the tight junctions and whether the apical and basolateral membranes are electrically isolated. Finally, when clusters of cells are used then measurements could be taken from more than one cell. The capacitance measurements support the idea that currents are recorded from both the apical and basolateral membranes and probably from single cells. Mean capacitance measurements were on the order of 10–13 pF. In previous studies on single mouse proximal tubule cells, capacitance was around 10 pF (Kibble et al. 2001; Balloch et al. 2003). Therefore the capacitance of the cells utilised in the current study are consistent with measuring currents from both the apical and basolateral membranes. This means that IE1 could be on either the basolateral or the apical membrane. In previous single channel work on mouse proximal tubules we have observed a Ba2+-sensitive, inwardly rectifying K+ channel on the basolateral membrane that was not regulated by KCNE1 (Millar et al. 2001). Unfortunately, due to the nature of the apical membrane of proximal tubule cells, it has not been possible to consistently achieve a gigaohm-seal via this membrane to ascertain whether IE1 is found apically. Irrespective of in which membrane the K+ channels are located, however, it is clear from their high K+ permeability that they would be expected to contribute to the membrane potential and therefore the driving force for Na+ reabsorption by the proximal tubule.
What is the physiological role of KCNE1 under control conditions?
As shown by Vallon et al. (2001), loss of KCNE1 was associated with increased excretion of Na+ and Cl−, and fluid excretion rose as a consequence of the increased urinary osmotic load. This is consistent with a role for KCNE1 in renal tubules contributing to the driving force for the uptake of Na+ and Cl−. Together with the voltage clamp data described earlier this suggests that KCNE1 plays a role in renal proximal tubule cell function. Of interest, neither glucose handling nor plasma glucose concentration were affected in our present study, which is in direct contrast to the previous study (Vallon et al. 2001), where increased urinary loss of glucose was observed. The reason for this difference is not clear, but may reflect the high plasma glucose concentrations reported in the Vallon study, on the order of 120 mm. In the current study plasma glucose levels were closer to physiological, around 10 mm. However, even when plasma glucose levels were increased to around 35 mm, both WT and KO animals increased their FE by comparable amounts, suggesting that glucose handling was similar in the two genotypes. Therefore, under control and moderate glucose loading conditions, KCNE1 appears to play a minor role in maintaining glucose uptake. Given the fact that most glucose absorption occurs in the first third of the proximal tubule (Frohnert et al. 1970) it would suggest that, functionally, KCNE1 is playing a role in the more distal two-thirds of the proximal tubule.
What about the handling of other ions?
Around 80% of the filtered HCO3− is reabsorbed in the proximal tubule, with a further 10% in the thick ascending limb and 10% in the distal nephron. Within the proximal tubule the reabsorption of HCO3− is driven by the turnover of the Na+–H+ exchanger. In turn the exchanger is driven by the electrochemical driving force for Na+, and therefore by the proximal tubule apical membrane potential. KCNE1 KO proximal tubules have been shown to have a depolarised membrane potential (Vallon et al. 2001). Within the proximal tubule HCO3− reabsorption is high initially, and decreases along the length of the proximal tubule (Liu & Cogan, 1984). Critically, the rate of bicarbonate transport is still relatively high at points along the proximal tubule when glucose transport is minimal (reviewed by Maddox & Gennari, 1987). Therefore, if KCNE1 and its associated K+ channel were playing an important role in the last two-thirds of the proximal tubule it would be expected that there would be a small to moderate increase in the excretion of HCO3− in KCNE1 KO mice. The data presented show that there was an increased loss of HCO3− in the KO animals, with the 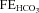 doubled. In contrast, ACTZ caused a much larger increase in HCO3− loss, with the
doubled. In contrast, ACTZ caused a much larger increase in HCO3− loss, with the 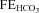 increased 100-fold in WT animals and 50-fold in KO animals. This greater loss is consistent with an effect of ACTZ additionally inhibiting HCO3− handling down the length of the nephron. The loss of HCO3− due to the loss of KCNE1 was relatively small in comparison to the effect of ACTZ, again consistent with a more distal proximal tubule function for KCNE1. Therefore, it is clear that the loss of KCNE1 has an impact on HCO3− handling. This suggests that KCNE1 plays a minor, but significant, role in driving the reabsorption of HCO3−.
increased 100-fold in WT animals and 50-fold in KO animals. This greater loss is consistent with an effect of ACTZ additionally inhibiting HCO3− handling down the length of the nephron. The loss of HCO3− due to the loss of KCNE1 was relatively small in comparison to the effect of ACTZ, again consistent with a more distal proximal tubule function for KCNE1. Therefore, it is clear that the loss of KCNE1 has an impact on HCO3− handling. This suggests that KCNE1 plays a minor, but significant, role in driving the reabsorption of HCO3−.
In summary, these data are consistent with the idea that KCNE1 regulates a K+ channel in the nephron at the level of the proximal tubule. This KCNE1–K+ channel complex (IE1) appears to play a minor role in driving ion transport in the proximal segment, as loss of KCNE1 only had a small effect on Na+, Cl−, water and HCO3− handling. The differential effect on reabsorption of HCO3− and glucose suggests that IE1 is functionally important in the latter two-thirds of the proximal tubule. It is likely that the KCNE1–K+ channel complex contributes to the resting membrane potential in these cells and therefore influences ion transport. When KCNE1 is absent, the driving force for ion uptake is reduced, leading to the renal phenotype observed.
Is KCNE1 regulating KCNQ1?
KCNQ1 is inhibited by chromanol 293B (Lerche et al. 2007) and clofilium (Yang et al. 1997). Therefore sensitivity to chromanol would be consistent with a role for KCNQ1. Both electrophysiological and clearance data suggest that KCNE1 is regulating a chromanol-sensitive K+ channel. In the voltage clamp studies, a chromanol-sensitive K+ current was observed that was absent in the KCNE1 KO animals. In addition, in WT animals infusion of chromanol was able to mimic the phenotype of the KCNE1 KO mice, i.e. increased loss of Na+, Cl−, HCO3− and water. The chromanol effect was absent in the KCNE1 KO animals, consistent with absence of KCNE1 causing loss of K+ channel function. Given that this loss of function was also observed directly in the proximal tubule voltage clamp studies, this suggests that the main site of function for KCNE1 may be the proximal tubule, although a role in other segments cannot be excluded. Therefore these data strongly support a proximal tubule role for IE1. However, the electrophysiological evidence would argue against a role for KCNQ1. IE1 in WT cells demonstrated several properties that suggest it is not attributable to KCNQ1. In the first instance the current–voltage profile of IE1 was not consistent with KCNQ1, with IE1 showing an ohmic profile, while the KCNQ1–KCNE1 complex typically shows outward rectification (Yang et al. 1997). In addition, the Q1–E1 complex is not open at negative potentials, while IE1 was active. IE1 also did not demonstrate the slow time-dependent activation typical of the Q1–E1 complex (at +100 mV 73.2 ± 9.68 pA pF−1versus 47.7 ± 9.49 pA pF−1 (n = 12), on initially stepping the potential and at the end of the potential step, respectively). Moreover, increasing cAMP levels, which is known to activate the Q1–E1 complex (Kurokawa et al. 2003; Nicolas et al. 2008), was without effect on IE1.
The renal effects of clofilium, which inhibits KCNQ1 (Yang et al. 1997), were more complex. At the level of the tubule, clofilium had similar effects on the renal clearance profile to chromanol. However, with clofilium infusion there was an increase in Na+, Cl− and water excretion, but there was no impact on HCO3− excretion and, moreover, these responses to clofilium were intact in KCNE1 KO mice. Therefore, although clofilium also has the ability to disrupt renal function, presumably through inhibition of other K+ channels, these channels are distinct from those mediating IE1. Clofilium also had an impact at the level of the glomerulus, reducing GFR in both wild-type and knockout mice, suggesting that clofilium-sensitive channels play a role in renal haemodynamics. Taken together, the chromanol and clofilium data suggest that in the renal proximal tubule the K+ channels regulated by KCNE1 are not KCNQ1.
In summary, The present study supports the idea that KCNE1 plays a role in renal proximal tubule function, contributing to the driving force for Na+ and Cl− transport, and provide the first electrophysiological evidence for the presence of a KCNE1-regulated, chromanol-sensitive K+ channel (IE1) in these cells. The lack of effect of KCNE1 knockout on glucose handling and the magnitude of the impact on HCO3− handling suggest that it is likely to be functionally important in the distal parts of the proximal tubule. Consistent with previous in vivo studies, IE1 is unlikely to be attributable to KCNQ1, as they share few properties. Overall these data suggest that KCNE1 and IE1 play a relatively minor role in proximal tubule reabsorption.
Acknowledgments
This work was supported by the Wellcome Trust.
Glossary
Abbreviations
- ACTZ
acetazolamide
- C
clearance
- FE
fractional excretion
- FSK
forskolin
- GFR
glomerular filtration rate
- HCT
haematocrit
- IBMX
3-isobutyl-1-methylxanthine
- IE1
conductance sensitive to KCNE1 knockout and chromanol
- KO
knockout
- MAP
mean arterial pressure
- P
plasma concentration
- UV
urinary excretion
- Vrev
reversal potential
Author contributions
Conception and design of the experiments: all authors; collection, analysis and interpretation of the data: I.D.M., A.M.N., H.C.T.; interpretation of the data only: L.R., J.D.K. and S.J.W.; drafting the article or revising it critically for important intellectual content: all authors. All authors authorised the final version of the manuscript. All experiments were carried out at the University of Sheffield.
Authors' present addresses
J. D. Kibble: College of Medicine, University of Central Florida, Health Sciences Campus at Lake Nona, 6850 Lake Nona Blvd 409J, Orlando, FL 32827, USA.
I. D. Millar: Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, UK.
S. J. White: Department of Physiology, Ross University School of Medicine, PO Box 266, Portsmouth Campus, Picard, Commonwealth of Dominica, PC 000152.
Supplementary material
Table S1
Table S2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Balloch KJ, Hartley JA, Millar ID, Kibble JD, Robson L. A hypertonicity-activated nonselective conductance in single proximal tubule cells isolated from mouse kidney. J Membr Biol. 2003;192:191–201. doi: 10.1007/s00232-002-1075-8. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barriere H, Belfodil R, Rubera I, Tauc M, Lesage F, Poujeol C, Guy N, Barhanin J, Poujeol P. Role of TASK2 potassium channels regarding volume regulation in primary cultures of mouse proximal tubules. J Gen Physiol. 2003a;122:177–190. doi: 10.1085/jgp.200308820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere H, Rubera I, Belfodil R, Tauc M, Tonnerieux N, Poujeol C, Barhanin J, Poujeol P. Swelling-activated chloride and potassium conductance in primary cultures of mouse proximal tubules. Implication of KCNE1 protein. J Membr Biol. 2003b;193:153–170. doi: 10.1007/s00232-003-2014-z. [DOI] [PubMed] [Google Scholar]
- Bauer A, Becker R, Freigang KD, Senges JC, Voss F, Hansen A, Muller M, Lang HJ, Gerlach U, Busch A, Kraft P, Kubler W, Schols W. Rate- and site-dependent effects of propafenone, dofetilide, and the new IKs-blocking agent chromanol 293b on individual muscle layers of the intact canine heart. Circulation. 1999;100:2184–2190. doi: 10.1161/01.cir.100.21.2184. [DOI] [PubMed] [Google Scholar]
- Belfodil R, Barriere H, Rubera I, Tauc M, Poujeol C, Bidet M, Poujeol P. CFTR-dependent and -independent swelling-activated K+ currents in primary cultures of mouse nephron. Am J Physiol Renal Physiol. 2003;284:F812–F828. doi: 10.1152/ajprenal.00238.2002. [DOI] [PubMed] [Google Scholar]
- Boini KM, Graf D, Hennige AM, Koka S, Kempe DS, Wang K, Ackermann TF, Foller M, Vallon V, Pfeifer K, Schleicher E, Ullrich S, Haring H-U, Haussinger D, Lang F. Enhanced insulin sensitivity of gene-targeted mice lacking functional KCNQ1. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1695–R1701. doi: 10.1152/ajpregu.90839.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg M, Green N. Bicarbonate transport by isolated perfused rabbit proximal convoluted tubules. Am J Physiol Renal Physiol. 1977;233:F307–F314. doi: 10.1152/ajprenal.1977.233.4.F307. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Abrahamsson C, Drews L, Duker G. Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation. 1992;85:1491–1500. doi: 10.1161/01.cir.85.4.1491. [DOI] [PubMed] [Google Scholar]
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange Nielsen Syndrome. Proc Natl Acad Sci U S A. 2001;98:2526–2531. doi: 10.1073/pnas.041398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demolombe S, Franco D, de Boer P, Kuperschmidt S, Roden D, Pereon Y, Jarry A, Moorman AFM, Escande D. Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am J Physiol Cell Physiol. 2001;280:C359–C372. doi: 10.1152/ajpcell.2001.280.2.C359. [DOI] [PubMed] [Google Scholar]
- Farkas A, Dempster J, Coker SJ. Importance of vagally mediated bradycardia for the induction of torsade de pointes in an in vivo model. Br J Pharmacol. 2008;154:958–970. doi: 10.1038/bjp.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen R, Boer WH, Boer P, Dorhout Mees EJ, Koomans HA. Effects of furosemide or acetazolamide infusion on renal handling of lithium: a micropuncture study in rats. Am J Physiol Regul Integr Comp Physiol. 1993;264:R129–R134. doi: 10.1152/ajpregu.1993.264.1.R129. [DOI] [PubMed] [Google Scholar]
- Frohnert PP, Hohmann B, Zwiebel R, Baumann K. Free flow micropuncture studies of glucose transport in the rat nephron. Pflugers Arch. 1970;315:66–85. doi: 10.1007/BF00587238. [DOI] [PubMed] [Google Scholar]
- Kibble JD, Balloch KJD, Neal AM, Hill C, White S, Robson L, Green R, Taylor CJ. Renal proximal tubule function is preserved in Cftrtm2cam delta-F508 cystic fibrosis mice. J Physiol. 2001;532:449–457. doi: 10.1111/j.1469-7793.2001.0449f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa J, Chen L, Kass RS. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc Natl Acad Sci U S A. 2003;100:2122–2127. doi: 10.1073/pnas.0434935100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Bruhova I, Lerche H, Steinmeyer K, Wei AD, Strutz-Seebohm N, Lang F, Busch AE, Zhorov BS, Seebohm G. Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol Pharmacol. 2007;71:1503–1511. doi: 10.1124/mol.106.031682. [DOI] [PubMed] [Google Scholar]
- Liu FY, Cogan MG. Axial heterogeneity in the rat proximal convoluted tubule. I. Bicarbonate, chloride, and water transport. Am J Physiol Renal Physiol. 1984;247:F816–F821. doi: 10.1152/ajprenal.1984.247.5.F816. [DOI] [PubMed] [Google Scholar]
- Lock H, Valverde MA. Contribution of the IsK (MinK) potassium channel subunit to regulatory volume decrease in murine tracheal epithelial cells. J Biol Chem. 2000;275:34849–34852. doi: 10.1074/jbc.C000633200. [DOI] [PubMed] [Google Scholar]
- Maddox DA, Gennari FJ. The early proximal tubule: a high-capacity delivery-responsive reabsorptive site. Am J Physiol Renal Physiol. 1987;252:F573–F584. doi: 10.1152/ajprenal.1987.252.4.F573. [DOI] [PubMed] [Google Scholar]
- Millar I, White S, Kibble J, Robson L. IsK does not regulate a basolateral channel in mouse renal proximal convoluted tubules. J Physiol. 2001;535.P:S002. [Google Scholar]
- Millar ID, Hartley JA, Haigh C, Grace AA, White SJ, Kibble JD, Robson L. Volume regulation is defective in renal proximal tubule cells isolated from KCNE1 knockout mice. Exp Physiol. 2004;89:173–180. doi: 10.1113/expphysiol.2003.026674. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Park K-H, El Harchi A, Camonis J, Kass RS, Escande D, Merot J, Loussouarn G, Le Bouffant F, Baro I. IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc Res. 2008;79:427–435. doi: 10.1093/cvr/cvn085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Barros LF, Sepulveda FV. Modulation of the two-pore domain acid-sensitive K+ channel TASK-2 (KCNK5) by changes in cell volume. J Biol Chem. 2001;276:43166–43174. doi: 10.1074/jbc.M107192200. [DOI] [PubMed] [Google Scholar]
- Perry M, de Groot MJ, Helliwell R, Leishman D, Tristani-Firouzi M, Sanguinetti MC, Mitcheson J. Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol. 2004;66:240–249. doi: 10.1124/mol.104.000117. [DOI] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- Rubera I, Tauc M, Bidet M, Poujeol C, Cuiller Ba, Watrin A, Touret N, Poujeol P. Chloride currents in primary cultures of rabbit proximal and distal convoluted tubules. Am J Physiol Renal Physiol. 1998;275:F651–F663. doi: 10.1152/ajprenal.1998.275.5.F651. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schafer JA, Watkins ML, Li L, Herter P, Haxelmans S, Schlatter E. A simplified method for isolation of large numbers of defined nephron segments. Am J Physiol Renal Physiol. 1997;273:F650–F657. doi: 10.1152/ajprenal.1997.273.4.F650. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Strutz-Seebohm N, Ureche ON, Henrion U, Baltaev R, Mack AF, Korniychuk G, Steinke K, Tapken D, Pfeufer A, Kaab S, Bucci C, Attali B, Merot J, Tavare JM, Hoppe UC, Sanguinetti MC, Lang F. Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ Res. 2008;103:1451–1457. doi: 10.1161/CIRCRESAHA.108.177360. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ohkubo H, Nakanishi S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science. 1988;242:1042–1045. doi: 10.1126/science.3194754. [DOI] [PubMed] [Google Scholar]
- Trinh NTN, Prive A, Kheir L, Bourret J-C, Hijazi T, Amraei MG, Noel J, Brochiero E. Involvement of KATP and KvLQT1 K+ channels in EGF-stimulated alveolar epithelial cell repair processes. Am J Physiol Lung Cell Mol Physiol. 2007;293:L870–L882. doi: 10.1152/ajplung.00362.2006. [DOI] [PubMed] [Google Scholar]
- Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J, Volkl H, Warth R. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol. 2001;12:2003–2011. doi: 10.1681/ASN.V12102003. [DOI] [PubMed] [Google Scholar]
- Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci U S A. 2005;102:17864–17869. doi: 10.1073/pnas.0505860102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varro A, Balati Bt, Lost N, Takacs J, Virag L, Lathrop DA, Csaba L, Talosi L, Papp JG. The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol. 2000;523:67–81. doi: 10.1111/j.1469-7793.2000.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W-P, Levesque PC, Little WA, Conder ML, Shalaby FY, Blanar MA. KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc Natl Acad Sci U S A. 1997;94:4017–4021. doi: 10.1073/pnas.94.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Verlander JW, Lynch IJ, Cash M, Shao J, Stow LR, Cain BD, Weiner ID, Wall SM, Wingo CS. Cellular distribution of the potassium channel KCNQ1 in normal mouse kidney. Am J Physiol Renal Physiol. 2007;292:F456–F466. doi: 10.1152/ajprenal.00087.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.