

Non-technical summary
Plasma potassium concentration is a major determinant of muscle contractility and nerve conduction. The maintenance of plasma potassium concentration depends on the ability of kidneys to daily secrete in the urine the exact quantity of potassium ingested in the food. We show that in nephrotic syndrome, a common disease featuring abnormal urinary protein excretion and sodium retention, the membrane protein called ROMK channel responsible for kidney potassium secretion is inhibited. Thus, nephrotic rats are unable to excrete a dietary load of potassium and develop hyperkalaemia. Based on these findings, we would recommend not only a low sodium diet but also a controlled potassium diet for patients with nephrotic syndrome.
Abstract
Abstract
Nephrotic syndrome features massive proteinuria and retention of sodium which promotes ascite formation. In the puromycin aminonucleoside-induced rat model of nephrotic syndrome, sodium retention originates from the collecting duct where it generates a driving force for potassium secretion. However, there is no evidence for urinary potassium loss or hypokalaemia in the nephrotic syndrome. We therefore investigated the mechanism preventing urinary potassium loss in the nephrotic rats and, for comparison, in hypovolaemic rats, another model displaying increased sodium reabsorption in collecting ducts. We found that sodium retention is not associated with urinary loss of potassium in either nephrotic or hypovolaemic rats, but that different mechanisms account for potassium conservation in the two models. Collecting ducts from hypovolaemic rats displayed high expression of the potassium-secreting channel ROMK but no driving force for potassium secretion owing to low luminal sodium availability. In contrast, collecting ducts from nephrotic rats displayed a high driving force for potassium secretion but no ROMK. Down-regulation of ROMK in nephrotic rats probably stems from phosphorylation of ERK arising from the presence of proteins in the luminal fluid. In addition, nephrotic rats displayed a blunted capacity to excrete potassium when fed a potassium-rich diet, and developed hyperkalaemia. As nephrotic patients were found to display plasma potassium levels in the normal to high range, we would recommend not only a low sodium diet but also a controlled potassium diet for patients with nephrotic syndrome.
Introduction
Nephrotic syndrome, which is defined by massive proteinuria and hypoalbuminaemia, is always associated with the retention of sodium which promotes the formation of ascites and/or oedema (Doucet et al. 2007). The mechanism of sodium retention has been deciphered using the puromycin aminonucleoside (PAN)-induced rat model of nephrotic syndrome that reproduces the biological and clinical signs of the human disease (Frenk et al. 1955; Pedraza-Chaverri et al. 1990). Sodium retention in PAN nephrotic (PN) rats originates from the aldosterone-sensitive distal nephron (ASDN), and stems from the marked stimulation of the basolateral Na+,K+-ATPase and the apical sodium channel ENaC in principal cells (Ichikawa et al. 1983; Deschenes et al. 2001; Lourdel et al. 2005). Principal cells also secrete K+ and thereby regulate plasma K+ concentration. K+ secretion in principal cells depends on the presence of active potassium channels at the apical membrane, mainly the renal outer medullary K+ channel (ROMK), and on a lumen-negative transepithelial voltage (Vte). The PDte is generated by electrogenic Na+ reabsorption and therefore depends on the presence of ENaC at the apical cell membrane and on the availability of Na+ in the luminal fluid, i.e. on the load of Na+ delivered to the ASDN. PN rats display hyperaldosteronaemia (Pedraza-Chaverri et al. 1990; Deschenes & Doucet, 2000), a high PDte in their cortical collecting duct (CCD) (Deschenes et al. 2001) and normal Na+ delivery to ASDN (Ichikawa et al. 1983). They should therefore increase their secretion of K+ and develop hypokalaemia. However, even though plasma K+ levels in either PN rats or nephrotic patients have not been rigorously documented to our knowledge, our current clinical experience with nephrotic patients suggests that their plasma K+ concentration remains within normal range. Furthermore, we analysed data available from the Robert Debré hospital and found that the potassium concentration in plasma varies within a normal range in nephrotic children, with a tendency to be high rather than low (Fig. 1).
Figure 1. Plasma potassium concentration in nephrotic children.
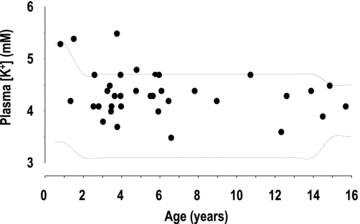
K+ concentration was measured in the plasma of children (age 3 months to 16 years) with idiopathic nephrotic syndrome at the time of their admission to the nephrology department at Robert Debré children's hospital (Paris), before onset of steroid therapy. The dotted lines limit the range of variation of plasma K+ concentration (mean ± 2SD) in age-matched non-nephrotic children admitted for other pathologies in the same department during the same period.
If confirmed, the inhibition of K+ secretion in the ASDN in nephrotic syndrome would suggest that apical K+-secreting channels are down-regulated. Several mechanisms have been reported to inhibit ROMK activity. In the presence of high aldosterone plasma levels, inhibition of ROMK activity may be mediated by with-no-lysine-kinase 4 (WNK4), whose mutations are responsible for pseudohypoaldosteronism type II (PHAII), a Mendelian disease featuring hypertension and hyperkalaemia. WNK4 is a molecular switch that modulates the Na+–K+ exchange ratio in the ASDN (Kahle et al. 2008), in part through differential regulation of ENaC and ROMK. In its conformational state induced by PHAII mutations, but also thought to be induced in states such as hypovolaemia that associate high plasma levels of both aldosterone and angiotensin 2, WNK4 stimulates ENaC and inhibits ROMK. In the case of K+ depletion, a state with low aldosterone plasma levels, ROMK inhibition is mediated by multiple pathways involving WNK1, MAP kinases p38 and ERK, and Src family protein tyrosine kinases (Wang & Giebisch, 2009).
The aim of this study was therefore to confirm that K+ excretion is not increased in PN rats and to elucidate the underlying mechanism. For this purpose: (a) we compared K+ handling by in vitro microperfused cortical collecting ducts (CCD) and in vivo in PN and Na+-depleted (LN) rats, two models displaying high aldosterone and angiotensin 2 levels, but differing by the load of Na+ delivered to the CCD (normal or reduced in PN and LN rats, respectively), and by the presence of proteinuria in PN rats; (b) we analysed the functional expression of ROMK in the CCDs of these rats and the mechanism of its inhibition in PN rats; and (c) we evaluated the ability of PN rats to adapt to an increased dietary input of K+.
Results show that both nephrotic and Na+-depleted rats maintain a normal K+ balance despite high plasma aldosterone levels and vanishingly low Na+ excretion. However, the mechanism of K+ conservation is different in the two models: CCDs from LN rats display high expression of ROMK at the apical membrane but no driving force for K+ secretion owing to low luminal Na+ availability. In contrast, CCDs from nephrotic rats display a high driving force for K+ secretion but no ROMK. Down-regulation of ROMK in nephrotic rats is probably accounted for by phosphorylation of ERK arising from the presence of proteins in the luminal fluid. Accordingly, nephrotic rats display a blunted capacity to excrete K+ and develop hyperkalaemia when fed a high K+ diet.
Methods
Animals
Male Sprague–Dawley rats (Charles Rivers, L'Abresles, France) weighing 150–170 g at the onset of the experimentation were housed and handled according to French legislation and the principles of UK regulations, and under the responsibility of an authorized experimenter (A.D., license no. 75-699 renewal). Unless indicated otherwise, animals were fed a standard laboratory chow (A04, Safe, Augy, France) containing 2.5 g of Na+ and 6.7 g K+ kg−1 with free access to deionised water. For surgery, animals were anaesthetized by intraperitoneal injection of a mix including Domitor (Pfizer, 0.5 μg (g body wt)−1), Climasol (Graeub, 2 μg (g body wt)−1) and Fentanyl Jansen (Janssen Cilag Lab, 5 ng (g body wt)−1). Animals were awakened by a subcutaneous injection of a mix containing Antisedan (Pfizer, 750 ng (g body wt)−1), Sarmasol (Graeub, 200 ng (g body wt)−1) and Narcan (Aguettant, 133 ng (g body wt)−1). Before killing, animals were anaesthetized with pentobarbital (Sanofi, France, 50 mg (kg body wt)−1, i.p.). Nephrotic syndrome was induced by a single intra-jugular injection of aminonucleoside puromycine (PAN) (Sigma–Aldrich, 150 mg (kg body wt)−1). Control rats received a single injection of isotonic NaCl (1 ml (100 g body wt)−1). To induce Na+ depletion, rats received a single dose of furosemide (Roche, 100 mg (kg body wt)−1) orally, and thereafter were fed a Na+-depleted diet (Safe, synthetic diet containing 0.11 g of Na+ and 6.7 g K+ kg−1). K+ loading was induced by feeding the rats the A04 diet supplemented with potassium gluconate (final K+ content, 50 g kg−1). For in vitro studies (microperfusion, immunoblotting, immunohistology), animals were studied 6 days after vehicle or PAN injection (maximum of sodium retention and proteinuria) or after the onset of Na+ depletion or K+ loading.
Metabolic studies
Animals were housed in individual metabolic cages, starting 3 days before the experimentation. Daily food intake was measured and 24 h urine was collected starting 1 day before the onset of the experimental period. In one experimental series, we studied a recovery period following Na+ depletion: after 7 days of Na+ depletion, rats were switched back to the standard diet and studied in metabolic cages for two additional days.
Urine creatinine and protein concentrations were measured in an automatic analyser (Konelab, Thermo, France). Urine sodium and potassium was measured by flame spectrophotometry (Instrumentation Laboratory). Blood samples were collected before killing to determine plasma aldosterone level (RIA Aldosterone, Immunotech, Marseille, France), Na+ and K+ concentrations (flame spectrophotometry), bicarbonate concentration and pH (ABL77, Radiometer). Ascites was measured by moistening and weighing an absorbent paper. Urinary excretion of sodium, potassium and protein were expressed as a function of creatinine excretion.
Microdissection of CCDs
CCDs were dissected either from fresh kidney slices (for microperfusion) or after a treatment with collagenase (for immunoblotting, RT-PCR and immuno-histochemistry). For RT-PCR experiments, microdissection was performed under RNase-free conditions. For collagenase treatment, left kidneys of pentobarbital-anaesthetized rats (see above) were infused via the abdominal aorta with incubation solution (Hank's solution supplemented with 1 mm pyruvate, 0.1% bovine serum albumin (BSA), 0.5 mm MgCl2, 1 mm glutamine and 20 mm Hepes, pH 7.4) containing collagenase (Worthington, 337 UI mg−1, 0.18% wt/vol). Kidneys were cut into small pieces which were incubated for 25 min at 30°C in oxygenated incubation solution containing 0.1% collagenase. CCDs were dissected under stereomicroscopic observation in incubation solution supplemented with antiproteases (Protease inhibitor cocktail tablets, Roche) at 4°C.
In vitro microperfusion
The left kidney was removed rapidly from pentobarbital-anaesthetised rats and coronal slices were prepared and placed in bath solution (see below) containing 6% BSA at room temperature. Single CCDs dissected from corticomedullary rays were transferred to a perfusion chamber mounted on the stage of an inverted microscope, and perfused by a gravity-driven system at a rate of ∼2 nl min−1. The bath flow rate was ∼12 ml min−1, to ensure a rapid renewal of bath solutions, and its temperature was maintained at 37°C. CCDs were perfused under symmetrical conditions, with bath and perfusate containing (in mm): 118 NaCl, 23 NaHCO3, 1.2 MgSO4, 2 K2HPO4, 2 calcium lactate, 1 sodium citrate, 5.5 glucose, 5 alanine and 12 creatinine, pH 7.4 (bath continuously gassed with 95% O2–5% CO2).
The PDte was recorded at the tip of the perfusion pipette and referred to the bath with microelectrodes made of Ag–AgCl half cell connected to salt–agar bridges (0.16 m NaCl, 3% agar) through a 1 m KCl bath. Four 20–30 min collection periods were performed on each tubule. The collected volume was determined under water-saturated mineral oil with calibrated pipettes. Concentrations of Na+, K+ and creatinine were determined by HPLC (Dionex DX500), and ions fluxes (J) were calculated as:
where [X]p and [X]c are the ion concentrations in the perfusate and collection respectively, 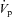 and
and 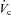 are the perfusion and collection rates respectively, L is the tubule length and t is the collection time. Therefore, positive values indicate net absorption, whereas negative values indicate secretion.
are the perfusion and collection rates respectively, L is the tubule length and t is the collection time. Therefore, positive values indicate net absorption, whereas negative values indicate secretion.
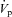 was calculated as:
was calculated as:
where [creat]c and [creat]p are the concentrations of creatinine in the collection and perfusate, respectively. For each tubule, fluxes were calculated as the mean of the four collection periods.
Immunoblotting
Pools of 50–60 CCDs were solubilized at 95°C for 5 min in Laemmli buffer and stored at −20°C until use. Proteins were separated by SDS–PAGE on 10% polyacrylamide gels and electro-transfered to Hybond-P membrane (GE Healthcare). After blocking in TBS–Nonidet P40 buffer (50 mm Tris base, 150 mm NaCl, 0.2% Nonidet P40) containing 5% non-fat dried milk, blots were successively incubated with either an anti-ROMK antibody recognizing all ROMK isoforms (Alomone Labs; dilution 1:500) or an anti-GAPDH antibody (Abcam; dilution 1:1000), an anti-ERK or an anti-phosphoERK antibody (Cell Signalling; dilution 1:1000), and horseradish peroxidase-linked anti-rabbit antibody (Promega France, Charbonnières, France) and revealed by an enhanced chemiluminescence light detecting kit (Amersham, Arlinghton Heights, IL, USA). The membrane was stripped (4 times in 25 mm glycine, pH 2, 0.2% SDS buffer) between uses with the different primary antibodies. Densitometry of the different bands was quantified with imageJ software.
Immunohistochemistry
Microdissected CCDs were transferred to Superfrost Gold+ glass slides, rinsed twice with PBS and fixed for 20 min with paraformaldehyde (4% in PBS). Afterwards, they were incubated 20 min at room temperature in 100 mm glycine in PBS, rinsed three times in PBS, permeabilized for 30 s with 0.1% Triton in PBS, and rinsed with PBS. After blocking in PBS containing 0.5% BSA (except for experiments with anti-albumin antibody) and 5% goat serum for 30 min at room temperature, slides were incubated with primary antibodies: anti-ROMK (Alomone Labs; dilution 1:500, 1 h at room temperature), anti-anion exchanger 1 (AE1) used as a specific marker of CCDs (gift of Dr Eladari, 1:1000, 1 h at room temperature) or FITC conjugated anti-albumin (DakoCytomotion, 1:100, 1 h at room temperature). After rinsing with PBS–Tween 0.05% (once) and PBS (twice), slides were incubated with the secondary antibody (1:500, 1 h at room temperature): TRITC-coupled anti-mouse IgG (for AE1) or FITC-coupled anti-rabbit IgG (for ROMK). After rinsing once with PBS–Tween and twice with PBS, slides were mounted and observed on a confocal microscope (×40, Zeiss observer.Z1, LSM710).
RNA extraction and RT-PCR
RNAs were extracted from pools of 40–60 CCDs using the RNeasy micro kit (Qiagen, Hilden, Germany) and reverse transcribed using a first strand cDNA synthesis kit for RT-PCR (Roche Diagnostics), according to the manufacturers' protocols. Real-time PCR was performed using a cDNA quantity corresponding to 0.1 mm of CCD with LightCycler 480 SYBR Green I Master qPCR kit (Roche Diagnostics) according to the manufacturer's protocol. Specific primers (available upon request) were designed using ProbeDesign (Roche Diagnostics).
mCCD cell culture
Clones of wild type mCCD cells (provided by Dr Rossier) were grown on collagen-coated transwell filter cups in DMEM–F12 (Invitrogen, Cergy Pontoise, France) supplemented with 10 ng ml−1 EGF, 1 nm T3, 50 nm dexamethasone, 5 μg ml−1 apo-transferrin, 0.9 μm insulin, 100 μg ml−1 penicillin, 100 μg ml−1 streptomycin and 5% FCS at 37°C in a 5% CO2–95% O2 mix. Growth medium was changed every 48 h. After 5 days, confluent cells were grown for another 5 days in DMEM–F12 supplemented with 3 nm dexamethasone and thereafter they were starved for 24 h in DMEM–F12. After washing three times with dexamethasone-supplemented DMEM–F12, BSA (1–10 mg ml−1) was added to either apical or basolateral or both sides of filter cups. After 6 h incubation, mCCD cells viability was evaluated by measuring the transepithelial potential.
For immunoblotting, cells were rinsed three times with PBS and were solubilized in lysis buffer containing 150 mm NaCl, 50 mm Tris/HCl (pH 7.5), 1% Triton X-100 and 5 mm EDTA, with antiproteases inhibitor (Protease inhibitor cocktail, Roche). Cell lysates were processed as described above. For immunocytochemistry, cells were washed three times with PBS containing 1 mm MgCl2 and 0.1 mm CaCl2, and incubated for 1 h at 4°C in PBS with 1 mg ml−1 EZ-Link sulfo-N-hydroxysuccinimido-LC-LC-biotin (Pierce). After three washes with PBS, cells were fixed for 20 min with paraformaldehyde (4% in PBS) at room temperature, rinsed three times with PBS and permeabilized with 0.1% Triton X-100 for 3 min. Cells were blocked for 30 min with PBS containing 5% goat serum and thereafter incubated for 1 h with FITC-conjugated anti-albumin antibody (1:100) in PBS containing 5% goat serum. After three washes with PBS, cells were incubated with Cy5-conjugated streptavidin (1:500, Sigma–Aldrich). Filters were excised from the filter cup and mounted with Vectashield mounting medium containing DAPI (Vector Laboratories). Slides were visualized with a confocal microscope (LSM 520, Zeiss).
Statistics
Results are expressed as means ± SEM from several animals. Comparison between groups was performed either by unpaired Student's t test or by variance analysis followed by a PLSD Fisher test, as appropriate.
Results
Handling of K+ in nephrotic and Na+-depleted rats
After 6 days of treatment, PN and LN rats displayed similar plasma concentrations of Na+, K+, Cl−, HCO3− and blood pH as controls (Table 1). Plasma Ca2+ concentration was slightly but significantly lower in PN rats than in the other two groups. Plasma aldosterone was high in PN rats and even higher in LN rats.
Table 1.
Blood parameters in control, nephrotic and sodium-depleted rats
| Control | PN | LN | |
|---|---|---|---|
| Na+ (mm) | 139.5 ± 0.9 (4) | 142.3 ± 0.8 (8) | 138.6 ± 0.7 (7) |
| K+ (mm) | 4.23 ± 0.14 (6) | 4.53 ± 0.16 (9) | 3.76 ± 0.20 (7) |
| Cl− (mm) | 109.5 ± 1.4 (6) | 118.3 ± 4.8 (9) | 110.1 ± 0.7 (7) |
| Ca2+ (mm) | 1.35 ± 0.03 (6) | 1.24 ± 0.03 (9)* | 1.26 ± 0.04 (7) |
| HCO3− (mm) | 21.9 ± 0.3 (6) | 21.8 ± 0.6 (9) | 20.1 ± 0.7 (7) |
| pH | 7.35 ± 0.06 (6) | 7.40 ± 0.02 (9) | 7.35 ± 0.01 (7) |
| Aldosterone (pm) | 349 ± 86 (5) | 7229 ± 891 (7)** | 22319 ± 2357 (6)** |
Parameters were determined in control rats, nephrotic rats at day 6 after PAN injection (PN), and sodium-depleted rats at day 6 after treatment onset (LN). Values are means ± SEM; the number of animals is shown in parentheses.
P < 0.025
P < 0.001 as compared to controls.
In control animals, urinary excretion of Na+ and K+ remained constant throughout the experimental period (Fig. 2A), indicating that animals were fully adapted to the metabolic cages. As previously described (Deschenes & Doucet, 2000), urinary excretion of Na+ increased at day 1 following PAN administration and thereafter decreased by ∼50% at days 2–4 and down to ∼5% of its control value at days 5–6. Proteinuria appeared at day 4. Urinary excretion of K+ decreased by ∼25% as early as day 1 following PAN administration and remained at that level throughout the experiment (Fig. 2B). In LN rats, urinary excretion of Na+ increased at day 1, as a consequence of furosemide administration, and thereafter decreased to vanishingly low levels (∼1%). Urinary excretion of K+ peaked at day 1, and thereafter returned to its basal level (Fig. 2C). Thus, neither PN nor LN rats increased their urinary excretion of K+.
Figure 2. Renal excretion of sodium, potassium and protein.
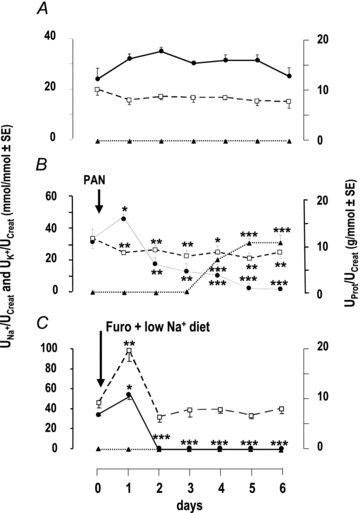
Time course of urinary excretion of sodium (filled circles, continuous line), potassium (open squares, dashed lines) and protein (filled triangles, dotted lines) in control (A), nephrotic (B) and sodium-depleted rats (C). Nephrotic syndrome was induced by a single injection of PAN, and sodium depletion was induced by a single injection of furosemide and feeding a sodium-depleted diet at times indicated by arrows. Results for day 0 correspond to the 24 h urine samples collected the day before the injection of PAN or the onset of sodium depletion. Sodium, potassium and protein excretion are expressed as a function of creatinine excretion. Data are means ± SEM from 6 animals in each group. *P < 0.05, **P < 0.005 and ***P < 0.001 as compared to day 0.
As previously reported (Tomita et al. 1985), in vitro microperfused CCDs from control rats displayed no significant transport of Na+ and K+ (JNa and JK, respectively) and their PDte was not different from zero. In contrast, CCDs from PN rats displayed a lumen negative PDte and reabsorbed Na+, but they did not secrete K+. CCDs from LN rats displayed similar JNa values as PN rats but secreted K+. As a consequence their PDte was lower than that of PN rats (P < 0.01) because it was partially shunted by K+ secretion (Fig. 3A–C). Actually, secretion of K+ was linearly correlated with PDte in LN but not in PN rats (Fig. 3D).
Figure 3. Ion transport in cortical collecting ducts.
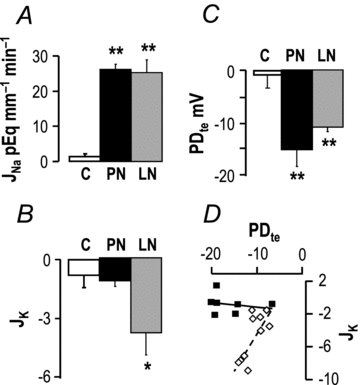
The fluxes of Na+ (A, JNa) and K+ (B, JK) and the transepithelial voltage (C, PDte) were measured in in vitro microperfused CCDs from control (C), nephrotic (PN) and sodium-depleted rats (LN). Positive and negative fluxes indicate reabsorption and secretion, respectively. JNa and JK are in pEq mm−1 min−1, and PDte is in mV. Values are means from 6 control, 7 nephrotic and 10 sodium-depleted CCDs, and data are means ± SEM. *P < 0.005 and **P < 0.001 as compared to CCDs from normal rats. D, relationship between JK and PDte in CCDs from nephrotic (filled symbols) and sodium-depleted rats (open symbols).
The absence of K+ secretion by CCDs from PN rats in vitro is consistent with the in vivo observation that K+ excretion did not increase despite marked Na+ retention. In contrast, some mechanism present in vivo but not in vitro must curtail the ability of CCDs from LN rats to secrete K+ in order to account for the absence of kaliuresis. We hypothesized that the luminal Na+ availability, which was high in vitro but supposedly low in vivo, might be the limiting factor. To test this hypothesis, we measured the urinary excretion of K+ upon reversion from the Na+-depleted diet to the normal diet, a manoeuver expected to increase distal Na+ delivery. As anticipated, K+ excretion increased immediately after the diet reversion whereas normal Na+ excretion resumed only 1 day later (Fig. 4).
Figure 4. Excretion of sodium and potassium during reversal of sodium depletion.
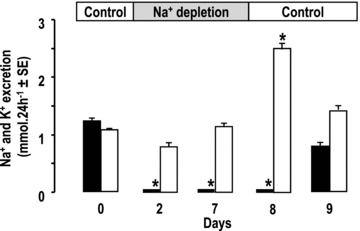
Excretion of sodium (filled columns) and potassium (open columns) in rats successively submitted to control feeding, sodium depletion and back to control feeding, as indicated in the bar on top. Results for day 0 correspond to the 24 h urine samples collected the day before the onset of sodium depletion. Values are means ± SEM from 5 animals. *P < 0.005 as compared to day 0.
Expression of ROMK in nephrotic and Na+-depleted rats
We evaluated whether differences in the K+ secreting ability of CCDs from PN and LN rats were accounted for by differential expression of ROMK, the main K+ secreting channel under normal conditions (Wang & Giebisch, 2009). CCDs from normal rats expressed ROMK1, ROMK2 and ROMK6 (which encodes the same protein as ROMK2) mRNAs, the latter two being present at ∼3-fold lower levels than ROMK1 mRNA. These three transcripts were less abundant in CCDs from PN and LN rats than in controls, except for ROMK2, the level of which was not changed in LN rats (Fig. 5A). As compared to control rats, the amount of ROMK protein (including ROMK1 and ROMK2) in isolated CCDs was reduced by ∼40% in PN rats whereas it was higher (∼150%) in LN rats, although this increase did not reach statistical significance (Fig. 5B). Immunohistochemistry on isolated CCDs confirmed the changes in ROMK expression in CCD of PN and LN rats, respectively (Fig. 5C), and showed that ROMK staining was mostly diffuse within the cytoplasm in control animals and mainly at the cell border in LN rats.
Figure 5. Expression of ROMK.
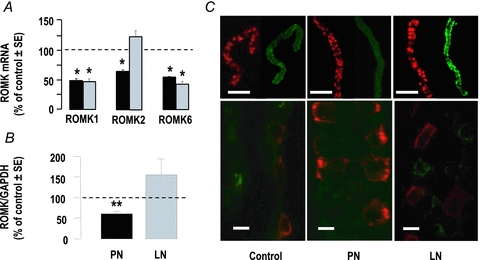
A, quantitative RT-PCR analysis of ROMK1, ROMK2 and ROMK6 mRNA expression in CCDs from nephrotic (black columns) and sodium-depleted rats (grey columns). Results were calculated as a function of tubular length and were expressed as per cent of mean controls. Values are means ± SEM from 11 animals; *P < 0.005 as compared to controls. B, immunoblot analysis of ROMK in CCDs from control, nephrotic (PN) and sodium-depleted (LN) rats. Results were calculated as ratios of ROMK over GAPDH and were expressed as per cent of mean controls in each experiment. Values are means ± SEM from 7 animals; **P < 0.001 as compared to controls. C, immuno-labelling of ROMK (green) and anion exchanger AE1 utilized as a specific marker of collecting ducts (red) in CCDs isolated from control, PN and LN rats. Scale bars: 100 μm and 10 μm for top and bottom images, respectively.
Proteinuria shown by NP but not LN rats may account for the differential regulation of ROMK. Actually, albumin has been reported to activate ERK in renal tubular cells (Reich et al. 2005; Pearson et al. 2008) and phosphorylation of ERK participates in the down-regulation of ROMK during K+ depletion (Wang & Giebisch, 2009). Therefore, we investigated whether albumin also stimulates ERK in collecting duct cells and whether ERK is activated in CCDs from PN rats.
Addition of albumin (1–10 g l−1) to the apical side of mCCD cells markedly increased the phosphorylation of ERK within 6 h whereas addition to the basolateral side had no effect (Fig. 6A and B). Dynasore, a membrane-permeable inhibitor of dynamin (Macia et al. 2006), blocked albumin endocytosis and prevented activation of ERK by albumin (Fig. 6C and D). Immunohistochemistry revealed the presence of intracellular albumin in CCDs from PN rats but not in control or LN rats (Fig. 7A). Accordingly, ERK was activated in the CCDs of PN but not LN rats (Fig. 7B).
Figure 6. Effect of albumin on ERK activation in mCCD cells.
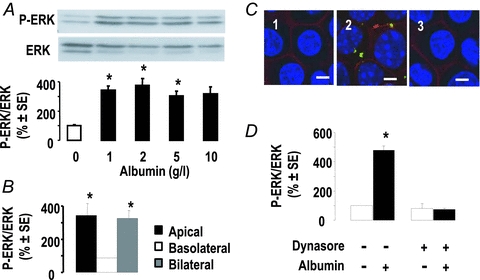
A, representative immunoblot and mean densitometric analysis of ERK activation in mCCD cells 6 h after apical addition of various concentrations of albumin. Results were calculated as ratios of phospho-ERK over total ERK and were expressed as per cent of mean controls in each experiment. Values are means ± SEM from 4 experiments. *P < 0.05 as compared to controls. B, effect of albumin (1 g l−1, 6 h) added on either side of mCCD cells on ERK phosphorylation. Data, calculated as above, are means ± SEM from 2 experiments. *P < 0.05 as compared to controls. C, effect of dynasore on albumin endocytosis. mCCD cells were incubated in the absence (1) or presence of apical albumin (1 g l−1, 6 h) after treatment without (2) or with (3) dynasore (80 μm, 1 h). Cells were immuno-labelled for biotin (red) to show the membranes and albumin (green); nuclei were stained with DAPI. D, effect of dynasore on albumin-induced activation of ERK. After treatment as above, ERK and phospho-ERK were quantified by immunoblot. Data are means ± SEM from 6 experiments. *P < 0.05 as compared to controls. Scale bars, 10 μm.
Figure 7. Albumin endocytosis and ERK phosphorylation in CCDs.
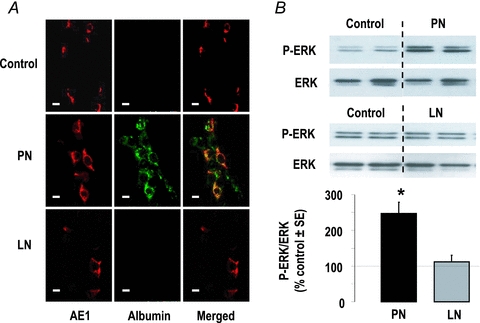
A, immuno-labelling of CCDs from control, nephrotic (PN) and sodium-depleted rats (LN) with anti-anion exchanger AE1 (red) and anti-albumin (green) antibodies. B, phosphorylation of ERK in control, PN and LN rats. Top image shows representative blots and bottom graph shows densitometric analysis. Results were calculated as ratios of phospho-ERK over total ERK and were expressed as per cent of mean controls in each experiment. Values are means ± SEM from 4–6 experiments. *P < 0.01 as compared to controls.
Adaptation to K+ loading
Metabolic studies (Fig. 8A–C and Table 2) showed that normal rats fed a K+-enriched diet initially reduced their food intake by 75% and increased their urinary excretion of K+ so as to maintain their K+ balance. Thereafter, their food intake progressively increased back to 50% of control and their K+ excretion increased proportionally. Consequently, their plasma K+ level remained normal after 1 week on the high K+ diet (Table 2). PN rats reduced their food intake more drastically (by 90–95%) and lastingly, increased their urinary excretion of K+ less and developed hyperkalaemia. As a consequence of their dietary restriction, nephrotic rats were Na+ deprived and developed fewer ascites than PN rats fed the standard diet (1.9 ± 0.2 and 9.4 ± 0.8 ml; mean ± SEM, P < 0.001, in K+-loaded and control PN rats, respectively).
Figure 8. Handling of potassium in potassium-loaded rats.
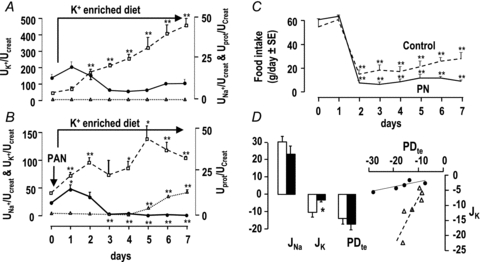
A and B, time course of urinary excretion (U) of sodium (filled circles, continuous line), potassium (open squares, dashed lines) and protein (filled triangles, dotted lines) in control (A) and nephrotic rats (B) fed a potassium-enriched diet. Nephrotic syndrome was induced by a single injection of PAN at the beginning of day 1, and potassium loading was started on the same day by feeding a K+-enriched diet. Sodium, potassium and protein excretion is expressed as a function of creatinine excretion. Data are means ± SEM from 6 animals in each group. C, daily food intake in control (dashed line) and nephrotic rats (continuous line) fed a potassium-enriched diet. *P < 0.05 and **P < 0.005 as compared to day 0. D, fluxes of Na+ (JNa) and K+ (JK), transepithelial voltage (PDte), and relationship between JK and PDte in microperfused CCDs from control (open columns and symbols) and nephrotic rats (filled columns and symbols) fed a potassium-enriched diet. JNa and JK are in pEq mm−1 min−1, and PDte is in mV. Values are means ± SEM from 6 control and 5 nephrotic rats. *P < 0.05 as compared to controls.
Table 2.
Blood parameters in control and nephrotic potassium-loaded rats
| HK-Control | HK-PN | |
|---|---|---|
| Na+ (mm) | 140.0 ± 0.5 (5) | 135.3 ± 0.9 (4)* |
| K+ (mm) | 4.30 ± 0.09 (5) | 6.90 ± 0.65 (4)* |
| Cl− (mm) | 100.8 ± 1.1 (5) | 99.0 ± 0.4 (4) |
| Ca2+ (mm) | 1.19 ± 0.01 (5) | 1.00 ± 0.02 (4)** |
| HCO3− (mm) | 28.7 ± 0.7 (5) | 33.2 ± 2.5 (4) |
| pH | 7.32 ± 0.1 (5) | 7.40 ± 0.20 (4) |
| Aldosterone (pm) | 10565 ± 1818 (4) | 16693 ± 3330 (4) |
Parameters were determined in control and nephrotic rats (PN) 6 days after the onset of K+ loading (HK). Values are means ± SEM; the number of animals is shown in parentheses.
P < 0.005
P < 0.001 as compared to HK-controls.
CCDs from normal and PN rats fed a high K+ diet for 1 week reabsorbed Na+ and displayed a high PDte. CCDs from both groups of rats secreted K+, but at a much lower rate in PN than in control rats. Secretion of K+ was correlated with PDte in both groups of rats, but the regression line was steeper in control than in PN rats (Fig. 8D and Table 3), indicating that the latter displayed a blunted capacity to secrete K+.
Table 3.
Summary of in vitro microperfusion data
| JNa (pEq mm−1 min−1) | JK (pEq mm−1 min−1) | PDte (mV) | JK/PDte | |
|---|---|---|---|---|
| Control | −0.5 ± 1.6 | −0.5 ± 0.4 | 3.5 ± 1.7 | NS |
| PN | 27.6 ± 1.0 | −0.9 ± 0.4 | −16.2 ± 1.8 | NS |
| LN | 28.6 ± 4.0 | −4.8 ± 0.9 | −10.7 ± 0.8 | 0.89 |
| HK-Control | 30.1 ± 3.5 | −10.3 ± 2.7 | −13.9 ± 2.6 | 1.61 |
| HK-PN | 23.1 ± 4.8 | −3.2 ± 0.7 | −17.0 ± 3.5 | 0.17 |
Discussion
Extracellular fluid K+ concentration is a critical determinant of resting membrane potential, and hence of the activity of excitable cells. Extracellular K+ homeostasis is tightly dependent on the ability of the kidneys to daily excrete a load of K+ matching its ingestion. Early studies have shown the tubular origin of K+ excreted in the urine (Morel & Guinnebault, 1956), and later studies have shown that K+ secretion takes place along what is now known as the ASDN (Hierholzer, 1961; Malnic et al. 1964), mainly in the connecting tubule and the collecting duct. Through stimulation of ENaC and Na+,K+-ATPase activities, aldosterone increases K+ secretion unless an additional mechanism inhibits ROMK activity. As ROMK is constitutively in an open state (Ho et al. 1993), regulation of its activity relies on that of its density at the apical membrane, and hence on the regulation of its synthesis and trafficking between the cell membrane and intracellular pools. Transcriptional regulation of ROMK expression by aldosterone is controversial (Beesley et al. 1998; Wald et al. 1998; Fodstad et al. 2009), and in recent years, the consensus has arisen that most regulation of ROMK activity takes place at the level of membrane trafficking, in particular through endocytosis (Wang & Giebisch, 2009).
During hypovolaemia, stimulation of the renin–angiotensin–aldosterone system increases ENaC activity (Palmer et al. 1994) but this is not associated with increased K+ secretion, allowing for the restoration of extracellular fluid volume without alterations in plasma K+ levels. Inhibition of ROMK during hypovolaemia is thought to result from alterations of WNKs activity which stimulates ROMK endocytosis (Kahle et al. 2003; Lazrak et al. 2006; Wade et al. 2006). However, present results suggest that the maintenance of normal K+ excretion during hypovolaemia is not due to a decrease in the membrane expression of ROMK, since we observed instead an increase in its density (Fig. 5) and a high K+-secreting capacity in CCDs from Na+-depleted rats (Fig. 3 and Table 3). The association between decreased ROMK mRNA levels and increased protein abundance (Fig. 5) suggests that endocytosis and degradation of ROMK are decreased, which may be accounted for by phosphorylation of the channel by the aldosterone-induced kinase Sgk1 (Yoo et al. 2003). Alternately, our data suggest that the functional inhibition of ROMK observed in vivo (Fig. 2) stems from the absence of driving force, namely from the absence of depolarisation of the apical membrane (Gray et al. 2005), brought about by the low luminal concentration of Na+ probably prevailing in the collecting duct of Na+-depleted rats. Supporting this hypothesis, we observed that re-feeding Na+-depleted rats a Na+-containing diet increased rapidly their K+ excretion, before increasing their Na+ excretion (Fig. 4).
Nephrotic rats also display high plasma aldosterone levels but, unlike Na+-depleted rats, their CCDs were not able to secrete K+ despite high transepithelial voltage and Na+ reabsorption rate (Fig. 3 and Table 3), indicating overall inhibition of ROMK. This inhibition cannot be solely accounted for by decreased synthesis of ROMK but should also involve channel endocytosis, since mRNA and protein abundance were reduced only by ∼40% (Fig. 5). As activation of ERK participates in endocytosis-mediated down-regulation of ROMK in response to K+ depletion (Babilonia et al. 2006) or prostaglandin E2 (Jin et al. 2007), it is likely that it is responsible for the inhibition of remnant ROMKs during nephrosis. During K+ depletion, activation of ERK results from superoxide anion-induced activation of MEK. In turn, phosphorylated ERK induces ROMK endocytosis through the expression of tyrosine kinase activity of the Src family (Babilonia et al. 2006). In the context of the nephrotic syndrome, our findings suggest that activation of ERK might be triggered by endocytosis of proteins abnormally present in the tubular fluid during nephrotic syndrome (Figs 6 and 7). Interestingly, it has been reported that albumin-induced capacitation of spermatozoa is mediated through the production of reactive oxygen species, the phosphorylation of ERK and, in turn, the activation of tyrosine kinases (O'Flaherty et al. 2006). Thus, endocytosis of ROMK during K+ depletion and nephrotic syndrome is probably mediated by the same signalling cascade. Our data also show that, as previously demonstrated for sodium retention (Lourdel et al. 2005; de Seigneux et al. 2006), regulation of K+ transport in nephrotic rats is independent of aldosterone.
Increasing dietary K+ intake stimulates K+ secretion along the distal nephron via aldosterone-dependent and -independent mechanisms. Through induction of ENaC and Na+,K+-ATPase, aldosterone increases the electrochemical gradient favourable to K+ exit across the apical membrane. Aldosterone-independent mechanisms include inhibition of the K+-reabsorbing H+,K+-ATPase, as well as activation of ROMK and of large-conductance Ca2+-activated K+ channels (BK) (Wang & Giebisch, 2009). In other words, aldosterone-dependent and -independent adaptations modulate the driving force and the apical membrane K+ conductance, respectively. Considering that aldosterone induces the same effects on ENaC and Na+,K+-ATPase in Na+-depleted and in K+-loaded rats, as supported by the fact that their CCDs displayed similar rates of Na+ reabsorption (Fig. 8 and Table 3), the difference in the K+ secretion rate between these two groups (Fig. 8 and Table 3) might be accounted for by aldosterone-independent mechanisms. Data show that the regression line between K+ secretion and the transepithelial voltage, an index of the driving force for K+ secretion, was two times steeper in CCDs from K+-loaded than Na+-depleted rats (Table 3), indicating that aldosterone-dependent and -independent mechanisms contribute equally to increasing K+ secretion in K+-loaded rats. CCDs from nephrotic rats fed a K+-enriched diet secreted K+ but the regression line between the rate of K+ secretion and the voltage was quite flat (Table 3), suggesting that both aldosterone-dependent and -independent mechanisms of K+ adaptation were blunted. Interestingly, it has been shown that inhibition of ERK stimulates BK activity in CCD (Li et al. 2006). Thus, albumin-induced activation of ERK might be responsible for inhibition of both ROMK and BK in nephrotic rat CCDs.
The retrospective analysis of plasma K+ levels in nephrotic children admitted to the pediatric nephrology department of the Robert Debré hospital confirmed the current clinical observation that nephrotic syndrome does not alter K+ balance in humans (Fig. 1) or in rats. If the mechanisms responsible for resistance to the kaliuretic effect of aldosterone are similar in human and PAN nephrotic rats, our study suggests that nephrotic patients might be at risk of developing hyperkalaemia under a K+-rich diet. Therefore, we would recommend not only a low-sodium diet for patient with nephrotic syndrome, as is usually done, but also a controlled potassium diet, even in patients with a conserved glomerular filtration rate.
Acknowledgments
This work was supported in part by grants from the Agence nationale de la recherche (ANR-06-PHYSIO-035-01), the Fondation Leducq (Transatlantic Network on Hypertension) and the Fondation pour la Recherche Médicale (to M.F.).
Glossary
Abbreviations
- AE1
anion exchanger 1
- ASDN
aldosterone-sensitive distal nephron
- CCD
cortical collecting duct
- ENaC
epithelial sodium channel
- LN
sodium depleted
- PAN
puromycin aminonucleoside
- PHAII
pseudohypoaldosteronism type II
- PN
PAN nephrotic
- WNK4
with-no-lysine-kinase 4
Author contributions
All experiments were performed at the Centre de Recherche des Cordeliers. Authors contributed to the work as follows: Conception and design of the experiments: M.F., G.D. and A.D. Collection, analysis and interpretation of data: M.F., G.B., L.M. and L.C. Drafting the article: A.D. All authors approved the final version of the manuscript.
References
- Babilonia E, Li D, Wang Z, Sun P, Lin DH, Jin Y, Wang WH. Mitogen-activated protein kinases inhibit the ROMK (Kir 1.1)-like small conductance K channels in the cortical collecting duct. J Am Soc Nephrol. 2006;17:2687–2696. doi: 10.1681/ASN.2006050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley AH, Hornby D, White SJ. Regulation of distal nephron K+ channels (ROMK) mRNA expression by aldosterone in rat kidney. J Physiol. 1998;509:629–634. doi: 10.1111/j.1469-7793.1998.629bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Seigneux S, Kim SW, Hemmingsen SC, Frokiaer J, Nielsen S. Increased expression but not targeting of ENaC in adrenalectomized rats with PAN-induced nephrotic syndrome. Am J Physiol Renal Physiol. 2006;291:F208–F217. doi: 10.1152/ajprenal.00399.2005. [DOI] [PubMed] [Google Scholar]
- Deschenes G, Doucet A. Collecting duct (Na+/K+)-ATPase activity is correlated with urinary sodium excretion in rat nephrotic syndromes. J Am Soc Nephrol. 2000;11:604–615. doi: 10.1681/ASN.V114604. [DOI] [PubMed] [Google Scholar]
- Deschenes G, Wittner M, Stefano A, Jounier S, Doucet A. Collecting duct is a site of sodium retention in PAN nephrosis: a rationale for amiloride therapy. J Am Soc Nephrol. 2001;12:598–601. doi: 10.1681/ASN.V123598. [DOI] [PubMed] [Google Scholar]
- Doucet A, Favre G, Deschenes G. Molecular mechanism of edema formation in nephrotic syndrome: therapeutic implications. Pediatr Nephrol. 2007;22:1983–1990. doi: 10.1007/s00467-007-0521-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodstad H, Gonzalez-Rodriguez E, Bron S, Gaeggeler H, Guisan B, Rossier BC, Horisberger JD. Effects of mineralocorticoid and K+ concentration on K+ secretion and ROMK channel expression in a mouse cortical collecting duct cell line. Am J Physiol Renal Physiol. 2009;296:F966–F975. doi: 10.1152/ajprenal.90475.2008. [DOI] [PubMed] [Google Scholar]
- Frenk S, Antonowicz I, Craig JM, Metcoff J. Experimental nephrotic syndrome induced in rats by aminonucleoside; renal lesions and body electrolyte composition. Proc Soc Exp Biol Med. 1955;89:424–427. doi: 10.3181/00379727-89-21833. [DOI] [PubMed] [Google Scholar]
- Gray DA, Frindt G, Palmer LG. Quantification of K+ secretion through apical low-conductance K channels in the CCD. Am J Physiol Renal Physiol. 2005;289:F117–F126. doi: 10.1152/ajprenal.00471.2004. [DOI] [PubMed] [Google Scholar]
- Hierholzer K. Secretion of potassium and acidification in collecting ducts of mammalian kidney. Am J Physiol. 1961;201:318–324. doi: 10.1152/ajplegacy.1961.201.2.318. [DOI] [PubMed] [Google Scholar]
- Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- Ichikawa I, Rennke HG, Hoyer JR, Badr KF, Schor N, Troy JL, Lechene CP, Brenner BM. Role for intrarenal mechanisms in the impaired salt excretion of experimental nephrotic syndrome. J Clin Invest. 1983;71:91–103. doi: 10.1172/JCI110756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Wang Z, Zhang Y, Yang B, Wang WH. PGE2 inhibits apical K channels in the CCD through activation of the MAPK pathway. Am J Physiol Renal Physiol. 2007;293:F1299–F1307. doi: 10.1152/ajprenal.00293.2007. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol. 2008;70:329–355. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Wilson FH, Leng Q, Lalioti MD, O'Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci U S A. 2006;103:1615–1620. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Wang Z, Sun P, Jin Y, Lin DH, Hebert SC, Giebisch G, Wang WH. Inhibition of MAPK stimulates the Ca2+-dependent big-conductance K channels in cortical collecting duct. Proc Natl Acad Sci U S A. 2006;103:19569–19574. doi: 10.1073/pnas.0609555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourdel S, Loffing J, Favre G, Paulais M, Nissant A, Fakitsas P, Creminon C, Feraille E, Verrey F, Teulon J, Doucet A, Deschenes G. Hyperaldosteronemia and activation of the epithelial sodium channel are not required for sodium retention in puromycin-induced nephrosis. J Am Soc Nephrol. 2005;16:3642–3650. doi: 10.1681/ASN.2005040363. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Malnic G, Klose RM, Giebisch G. Micropuncture study of renal potassium excretion in the rat. Am J Physiol. 1964;206:674–686. doi: 10.1152/ajplegacy.1964.206.4.674. [DOI] [PubMed] [Google Scholar]
- Morel F, Guinnebault M. [Tubular origin of potassium excreted by the kidney; experimental study with radiopotassium K42 in rabbits] (in French) Helv Physiol Pharmacol Acta. 1956;14:255–263. [PubMed] [Google Scholar]
- O'Flaherty C, de Lamirande E, Gagnon C. Reactive oxygen species modulate independent protein phosphorylation pathways during human sperm capacitation. Free Radic Biol Med. 2006;40:1045–1055. doi: 10.1016/j.freeradbiomed.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Palmer LG, Antonian L, Frindt G. Regulation of apical K and Na channels and Na/K pumps in rat cortical collecting tubule by dietary K. J Gen Physiol. 1994;104:693–710. doi: 10.1085/jgp.104.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson AL, Colville-Nash P, Kwan JT, Dockrell ME. Albumin induces interleukin-6 release from primary human proximal tubule epithelial cells. J Nephrol. 2008;21:887–893. [PubMed] [Google Scholar]
- Pedraza-Chaverri J, Cruz C, Ibarra-Rubio ME, Chavez MT, Calleja C, Tapia E, del Carmen Uribe M, Romero L, Pena JC. Pathophysiology of experimental nephrotic syndrome induced by puromycin aminonucleoside in rats. I. The role of proteinuria, hypoproteinemia, and renin-angiotensin-aldosterone system on sodium retention. Rev Invest Clin. 1990;42:29–38. [PubMed] [Google Scholar]
- Reich H, Tritchler D, Herzenberg AM, Kassiri Z, Zhou X, Gao W, Scholey JW. Albumin activates ERK via EGF receptor in human renal epithelial cells. J Am Soc Nephrol. 2005;16:1266–1278. doi: 10.1681/ASN.2004030222. [DOI] [PubMed] [Google Scholar]
- Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest. 1985;76:132–136. doi: 10.1172/JCI111935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA. WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci U S A. 2006;103:8558–8563. doi: 10.1073/pnas.0603109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald H, Garty H, Palmer LG, Popovtzer MM. Differential regulation of ROMK expression in kidney cortex and medulla by aldosterone and potassium. Am J Physiol Renal Physiol. 1998;275:F239–F245. doi: 10.1152/ajprenal.1998.275.2.F239. [DOI] [PubMed] [Google Scholar]
- Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch. 2009;458:157–168. doi: 10.1007/s00424-008-0593-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA. Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J Biol Chem. 2003;278:23066–23075. doi: 10.1074/jbc.M212301200. [DOI] [PubMed] [Google Scholar]