

Non-technical summary
Blood flow to muscle increases during exercise in order to deliver more oxygen. When there is less oxygen in the blood, as in systemic hypoxia, blood flow also increases. If exercise occurs during hypoxia, the blood flow response is greater than during normal oxygen conditions, but the mechanisms by which this happens are not clear. We show that two substances that the body produces, nitric oxide and prostaglandins, contribute to this increased blood flow during hypoxic exercise. These results help us better understand how oxygen delivery is regulated and may be especially important for populations which are unable to produce these substances that help increase blood flow.
Abstract
Abstract
Exercise hyperaemia in hypoxia is augmented relative to the same level of exercise in normoxia. At moderate exercise intensities, the mechanism(s) underlying this augmented response are currently unclear. We tested the hypothesis that endothelium-derived nitric oxide (NO) and vasodilating prostaglandins (PGs) contribute to the augmented muscle blood flow during hypoxic exercise relative to normoxia. In 10 young healthy adults, we measured forearm blood flow (FBF; Doppler ultrasound) and calculated the vascular conductance (FVC) responses during 5 min of rhythmic handgrip exercise at 20% maximal voluntary contraction in normoxia (NormEx) and isocapnic hypoxia (HypEx; O2 saturation ∼85%) before and after local intra-brachial combined blockade of NO synthase (NOS; via NG-monomethyl-l-arginine: l-NMMA) and cyclooxygenase (COX; via ketorolac). All trials were performed during local α- and β-adrenoceptor blockade to eliminate sympathoadrenal influences on vascular tone and thus isolate local vasodilatation. Arterial and deep venous blood gases were measured and oxygen consumption (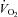 ) was calculated. In control (saline) conditions, FBF after 5 min of exercise in hypoxia was greater than in normoxia (345 ± 21 ml min−1vs. 297 ± 18 ml min−1; P < 0.05). After NO–PG block, the compensatory increase in FBF during hypoxic exercise was blunted ∼50% and thus was reduced compared with control hypoxic exercise (312 ± 19 ml min−1; P < 0.05), but this was not the case in normoxia (289 ± 15 ml min−1; P = 0.33). The lower FBF during hypoxic exercise was associated with a compensatory increase in O2 extraction, and thus
) was calculated. In control (saline) conditions, FBF after 5 min of exercise in hypoxia was greater than in normoxia (345 ± 21 ml min−1vs. 297 ± 18 ml min−1; P < 0.05). After NO–PG block, the compensatory increase in FBF during hypoxic exercise was blunted ∼50% and thus was reduced compared with control hypoxic exercise (312 ± 19 ml min−1; P < 0.05), but this was not the case in normoxia (289 ± 15 ml min−1; P = 0.33). The lower FBF during hypoxic exercise was associated with a compensatory increase in O2 extraction, and thus 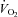 was maintained at normal control levels (P = 0.64–0.99). We conclude that under the experimental conditions employed, NO and PGs have little role in normoxic exercise hyperaemia whereas combined NO–PG inhibition reduces hypoxic exercise hyperaemia and abolishes hypoxic vasodilatation at rest. Additionally,
was maintained at normal control levels (P = 0.64–0.99). We conclude that under the experimental conditions employed, NO and PGs have little role in normoxic exercise hyperaemia whereas combined NO–PG inhibition reduces hypoxic exercise hyperaemia and abolishes hypoxic vasodilatation at rest. Additionally, 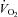 of the tissue was maintained in hypoxic conditions at rest and during exercise, despite attenuated oxygen delivery following NO–PG blockade, due to an increase in O2 extraction at the level of the muscle.
of the tissue was maintained in hypoxic conditions at rest and during exercise, despite attenuated oxygen delivery following NO–PG blockade, due to an increase in O2 extraction at the level of the muscle.
Introduction
It is well established that during exercise under hypoxic conditions, muscle blood flow is augmented relative to the same level of exercise in normoxia in order to maintain oxygen delivery to the active tissue (Hartley et al. 1973; Rowell et al. 1986; Wilkins et al. 2008). This augmentation of blood flow occurs in spite of an increase in sympathetic activity and α-adrenoceptor-mediated vasoconstriction (Hanada et al. 2003; Wilkins et al. 2008). During exercise, similar to at rest, systemic hypoxia does not alter the responsiveness of directly stimulated α-adrenoceptors as compared to normoxia, suggesting that the augmented hyperaemic response during exercise is not due to a blunting of the vasoconstrictor signal (Dinenno et al. 2003; Wilkins et al. 2006). Recent experimental evidence suggests at moderate-intensity forearm exercise, local blockade of β-adrenoceptors does not impact the augmentation of muscle blood flow during hypoxia whereas at lower intensities muscle blood flow is reduced by ∼50% (Wilkins et al. 2008). Collectively, these data imply a role for other local vasodilator signals to the enhanced hypoxic exercise blood flow response at greater exercise intensities.
A variety of substances either in circulation, produced by the blood vessels or produced by the muscle tissue have been proposed to be involved in the local regulation of skeletal muscle vascular tone during systemic hypoxia with or without superimposed exercise (Marshall, 1999). In regards to systemic hypoxia in resting muscle, we recently employed regional (forearm) sympathoadrenal blockade to determine the independent and interactive roles of nitric oxide (NO) and prostaglandins (PGs) to hypoxic vasodilatation in resting humans to isolate local vasodilator mechanisms without concomitant α- and β-adrenoceptor influences on vascular tone. Our findings indicated that neither NO nor PGs were obligatory for hypoxic vasodilatation; however, combined inhibition of these putative endothelium-derived substances abolished the response (Markwald et al. 2011). These data are the first to demonstrate an interaction between NO and PGs in the regulation of vascular tone under hypoxic conditions in humans.
In regards to exercise, the local signals involved in the augmented blood flow responses to contracting muscle during hypoxia have recently received much attention. Studies by Casey and colleagues (2009, 2010) indicate that adenosine is not involved but that acute inhibition of NOS significantly reduces the compensatory vasodilatation observed during hypoxic exercise (Casey et al. 2010). These studies were performed under normal conditions in which sympathetic vasoconstriction was intact, and given that sympathetic restraint of muscle blood flow during hypoxic exercise is greater than that during normoxia (Wilkins et al. 2008), these observations reflect an interaction between NO-mediated vasodilatation and sympathetic α-adrenoceptor vasoconstriction. Although this is one way to study hypoxic exercise hyperaemia, our laboratory has been interested in isolating the local vasodilators during hypoxic conditions (Markwald et al. 2011), and as such, we have performed studies utilizing pharmacological inhibition of both α- and β-adrenoceptors to eliminate sympathoadrenal influences on vascular tone.
Given our recent findings under these experimental conditions that combined inhibition of NO and vasodilating PGs abolished hypoxic vasodilatation in resting muscle, whereas single inhibition of NO or PGs alone did not impact the response (Markwald et al. 2011), we now question whether these two endothelium-derived substances play a significant role in augmenting the hyperaemic response during hypoxic exercise. In normoxia, there is evidence to suggest that (1) NO or PGs are not obligatory for exercise hyperaemia (Boushel et al. 2002; Mortensen et al. 2007; Schrage et al. 2007, 2010), (2) NO independently contributes ∼20% to exercise hyperaemia whereas the independent PG contribution is modest and transient (Schrage et al. 2004), and (3) NO and PGs act synergistically in the regulation of muscle blood flow during exercise (Schrage et al. 2004; Mortensen et al. 2007, 2009b). Further, Mortensen and colleagues (2007, 2009b) demonstrated a significant reduction in muscle blood flow with combined NO and PG inhibition during knee extensor exercise that was associated with a lower oxygen consumption of the exercising muscle. With respect to oxygen consumption, in our recent study on the roles of NO and PGs in mediating hypoxic vasodilatation in resting muscle (Markwald et al. 2011), venous blood samples were not taken and thus it is unknown whether tissue oxygen consumption was reduced during combined NO–PG blockade. Additionally, if combined NO–PG blockade reduces exercise hyperaemia in hypoxia, it is currently unknown whether this would impair oxygen consumption of the active muscle.
With this information as background, the primary purpose of the present investigation was to test the hypothesis that NO and PGs contribute to the augmented muscle blood flow observed during hypoxic exercise as compared to normoxic exercise at the same moderate workload. Further, we hypothesized that even with potential reductions in muscle blood flow and thus oxygen delivery, oxygen extraction would increase in a compensatory manner to maintain oxygen consumption of both the resting and active skeletal muscle tissue.
Methods
Subjects
With Institutional Review Board approval and after written informed consent, a total of 10 young healthy adults (8 men, 2 women; age, 21 ± 1 years; weight, 72.5 ± 2.3 kg; height, 178 ± 2 cm; body mass index, 22.7 ± 0.6 kg m−2; means ± SEM) participated in the present study. All subjects were non-smokers, non-obese, normotensive (resting blood pressure <140/90) and not taking any medications. Studies were performed in the Human Cardiovascular Physiology Laboratory at Colorado State University (altitude: ∼1500 m) after a 4 h fast and 24 h abstention from caffeine and exercise, with subjects in the supine position. Female subjects were studied during the early follicular phase of their menstrual cycle to minimize any potential cardiovascular effects of sex-specific hormones. All studies were performed according to the Declaration of Helsinki.
Arterial and venous catheterization
A 20-gauge, 7.6 cm catheter was placed in the brachial artery of the non-dominant arm under aseptic conditions after local anaesthesia (2% lidocaine) for local administration of study drugs and blood sampling. The catheter was connected to a 3-port connector as well as a pressure transducer for mean arterial pressure (MAP) measurement and continuously flushed at 3 ml h−1 with heparinized saline. The two side ports were used for drug infusions (Kirby et al. 2009; Crecelius et al. 2010; Markwald et al. 2011). In addition, an 18-gauge, 5.1 cm catheter was inserted in retrograde fashion into an antecubital vein of the experimental arm for deep venous blood samples (Dinenno et al. 2002). Heparinized saline was continuously infused through this catheter at a rate of approximately 3 ml min−1 for the duration of the study to keep it patent.
Forearm blood flow and vascular conductance
A 12 MHz linear-array ultrasound probe (Vivid 7, General Electric, Milwaukee, WI, USA) was used to determine brachial artery mean blood velocity (MBV) and brachial artery diameter. The probe was securely fixed to the skin over the brachial artery proximal to the catheter insertion site as previously described (Crecelius et al. 2010). For blood velocity measurements, the probe insonation angle was maintained at <60 deg and the frequency used was 5 MHz. The Doppler shift frequency spectrum was analysed via a Multigon 500V TCD (Multigon Industries, Mt Vernon, NY, USA) spectral analyser from which mean velocity was determined as a weighted mean of the spectrum of Doppler shift frequencies. Brachial artery diameter measurements were made in duplex mode at end-diastole and between contractions (in triplicate) during steady-state conditions. Forearm blood flow (FBF) was calculated as:
where the FBF is in ml min−1, the MBV is in cm s−1, the brachial diameter is in centrimetres, and 60 was used to convert from ml s−1 to ml min−1. Forearm vascular conductance (FVC) was calculated as (FBF/MAP) × 100, and expressed as ml min−1 (100 mm Hg)−1. All studies were performed in a cool temperature-controlled environment with a fan directed toward the forearm to minimize the contribution of skin blood flow to forearm haemodynamics.
Blood gas analysis
Brachial artery and deep venous blood samples were immediately analysed with a clinical blood gas analyser (Siemens Rapid Point 400 Series Automatic Blood Gas System, Los Angeles, CA, USA) for partial pressures of oxygen and carbon dioxide (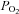 and
and 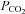 ), haemoglobin concentration ([Hb]), oxygen content (
), haemoglobin concentration ([Hb]), oxygen content (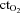 ), pH and oxygen saturation (
), pH and oxygen saturation (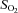 ).
).
Rhythmic handgrip exercise
Maximal voluntary contraction (MVC; mean 44.1 ± 2.8 kg, range 28.3–57.3 kg) was determined for the experimental arm as the average of three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL, USA) that were within 3% of each other. Forearm exercise during the trials was performed with weight corresponding to 20% MVC (mean 8.6 ± 0.6 kg, range 5.6–11.5 kg) attached to a pulley system and lifted 4–5 cm over the pulley at a duty cycle of 1 s contraction–2 s relaxation (20 contractions per minute) using both visual and auditory feedback to ensure the correct timing as described previously (Kirby et al. 2008; Crecelius et al. 2010). We chose this workload based on recent data that suggests a greater potential role for local vasodilators in muscle blood flow regulation during hypoxia at this relative intensity (Wilkins et al. 2008). We also aimed to minimize sympathetically mediated increases in heart rate and mean arterial blood pressure due to exercise (Victor & Seals, 1989).
Systemic isocapnic hypoxia
To isolate the effects of systemic isocapnic hypoxia, we used a self-regulating partial rebreathe system developed by Banzett et al. (2000) and recently described by our laboratory (Markwald et al. 2011). This system allows for constant alveolar fresh air ventilation independent of changes in breathing frequency or tidal volume (Banzett et al. 2000; Dinenno et al. 2003; Wilkins et al. 2008). Using this system we were able to clamp end-tidal CO2 levels despite the hypoxia-induced increases in ventilation. The level of oxygen was manipulated by mixing nitrogen with medical air via an anaesthesia gas blender. For the hypoxic trials, inspired oxygen was titrated to achieve arterial oxygen saturations of ∼85% as assessed via pulse oximetry. For normoxic trials, subjects were placed on the rebreathe system but inspired ambient air. Subjects breathed through a scuba mouthpiece with a nose-clip to prevent nasal breathing. An anaesthesia monitor (Cardiocap/5, Datex-Ohmeda Louisville, CO, USA) was used to determine heart rate (HR; 3-lead ECG) and expired CO2 sampled at the mouthpiece. Ventilation was measured via a turbine pneumotachograph (model 17125 UVM, Vacu-Med, Ventura, CA, USA).
Pharmacological infusions
Regional sympathoadrenal blockade
Phentolamine mesylate (Bedford Laboratories, Bedford, OH, USA), a non-selective α-adrenoceptor antagonist, and propranolol hydrochloride (Baxter, Deerfield, IL, USA), a non-selective β-adrenoceptor antagonist, were administered via brachial artery catheter to eliminate the sympathoadrenal influences on vascular tone as recently described by our laboratory (Markwald et al. 2011). A loading dose totalling 1000 μg (200 μg min−1 for 5 min) of each drug was infused prior to all experimental trials and a maintenance dose (50 μg min−1) was infused throughout the entire study to ensure continuous blockade. The dose of phentolamine used was twice as great as those previously documented to effectively block α-adrenoceptors (Eklund & Kaijser, 1976; Dietz et al. 1997; Halliwill et al. 1997), and we recently showed that this maintains effective α-blockade for several hours (Markwald et al. 2011). The dose of propranolol used has been shown to inhibit forearm vasodilatation in response to isoproterenol (a non-selective β-adrenoceptor agonist) (Johnsson, 1967) as well as reduce vasodilatation in the resting forearm during contralateral isometric handgrip exercise (Eklund & Kaijser, 1976).
Regional NOS inhibition
NG-monomethyl-l-arginine (l-NMMA; Clinalfa/Bachem, Weil am Rhein, Germany), a non-selective NOS inhibitor, was administered intra-arterially to inhibit the production of NO. A loading dose totalling 25 mg (5 mg min−1 for 5 min) and a maintenance dose (1.0 mg min−1) was infused for the duration of the study to ensure continuous blockade. This dose of l-NMMA has been previously shown to significantly reduce basal tone and also the vasodilatory effects of acetylcholine (Dietz et al. 1994; Eisenach et al. 2002), consistent with effective NOS inhibition (Vallance et al. 1989).
Regional COX inhibition
Ketorolac (trade name Toradol, Hospira, Lake Forest, IL, USA), a non-selective COX inhibitor, was administered intra-arterially to inhibit the production of PGs (Markwald et al. 2011). A loading dose totalling 6 mg (600 μg min−1 for 10 min) and a maintenance dose (120 μg min−1) was infused for the duration of the study to ensure continuous blockade. This dose of ketorolac is twice that which was previously demonstrated to transiently (but consistently) reduce forearm blood flow during exercise (Schrage et al. 2004), as well as that which reduced circulating PGF1α (a stable breakdown product of PGs) at rest and during handgrip exercise (Dinenno & Joyner, 2004).
Experimental protocol
A timeline for the overall study and each trial is depicted in Fig. 1. After catheter placement and experimental set-up, resting haemodynamics were determined. Loading doses of phentolamine and propranolol to inhibit sympathoadrenal stimulation of α- and β-adrenoceptors, respectively, were then administered and all experimental trials followed. Maintenance doses of phentolamine and propranolol were infused throughout the remainder of the experiment. Normoxic trials consisted of a 3 min baseline and a 5 min rhythmic handgrip exercise period. Hypoxic trials consisted of a 3 min baseline, 5 min of steady-state hypoxia at ∼85% arterial oxygen saturation (monitored via pulse oximeter; transition ∼2 min) and 5 min rhythmic handgrip exercise. Each trial was performed under control condition (saline infusion) and with combined NO–PG blockade (l-NMMA and ketorolac, respectively); thus, a total of four trials were performed. Twenty minutes of rest separated each experimental trial. The order of normoxic and hypoxic trials before and after NO–PG blockade was counterbalanced between subjects. Arterial and venous blood samples were taken at the end of rest, normoxic exercise, steady-state hypoxia and hypoxic exercise (Fig. 1).
Figure 1. Timeline and experimental protocol.
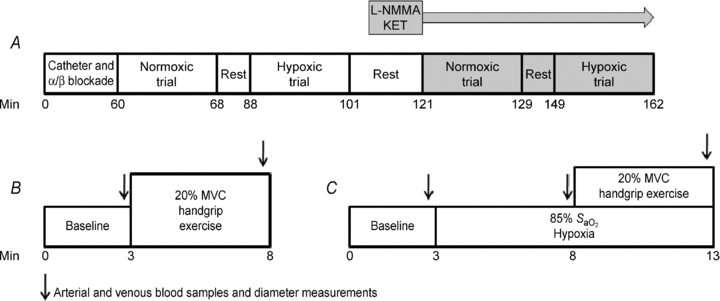
A, overall experimental protocol; subjects' non-dominant arm was instrumented with a brachial catheter and a deep venous catheter. Phentolamine and propranolol were administered intra-arterially to block α- and β-adrenoceptors, respectively, before the experimental trials. Twenty minutes of rest separated each trial. After both normoxic and hypoxic trials were performed in the control (saline) condition, l-NMMA and ketorolac (KET) were administered intra-arterially to block NO and PG synthesis, respectively. Normoxic and hypoxic trials were repeated under the blockade condition (shaded boxes). The order of the normoxic and hypoxic trials was counter-balanced. B, normoxic trial timeline; baseline measurements were made for 3 min, followed by 5 min of 20% MVC rhythmic handgrip exercise. C, hypoxic trial timeline; baseline measurements were made for 3 min, oxygen saturations were then lowered to ∼85% (within first 2 min) and maintained for duration of trial. After 3 min of steady-state hypoxia, subjects performed 5 min of 20% MVC rhythmic handgrip exercise.
Data acquisition and analysis
Data were collected and stored on a computer at 250 Hz and were analysed off-line with signal-processing software (WinDaq, DATAQ Instruments, Akron, OH, USA). MAP was determined from the brachial arterial pressure waveform. FBF, HR, MAP and oxygen saturations (pulse oximetry) represent an average of the last 30 s of the appropriate time period. Minute ventilation and end-tidal CO2 were determined from an average of the data over the last minute of each time period. The sampling timeframe used for averaging was greater for respiratory variables than haemodynamic variables in order to insure an adequate number of sampling points. Blood gas values were determined from blood samples obtained during each condition. From the blood gas data, arteriovenous oxygen difference (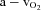 ) was calculated as the difference between arterial and venous oxygen content. Oxygen consumption across the forearm (
) was calculated as the difference between arterial and venous oxygen content. Oxygen consumption across the forearm (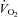 ) was calculated as: (FBF ×
) was calculated as: (FBF ×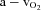 difference) and expressed in ml min−1. Oxygen delivery was calculated as: (FBF × arterial
difference) and expressed in ml min−1. Oxygen delivery was calculated as: (FBF × arterial 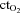 ) and expressed in ml min−1. Oxygen extraction, reported as a per cent, was calculated as: (arterial
) and expressed in ml min−1. Oxygen extraction, reported as a per cent, was calculated as: (arterial 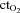 – venous
– venous 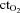 )/arterial
)/arterial 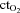 × 100.
× 100.
As an alternative way of expressing the data, we calculated the ‘hypoxic augmentation’ as the difference between the absolute FBF during hypoxic exercise and during normoxic exercise within control (saline) or combined NO–PG blockade (l-NMMA and ketorolac) conditions.
Statistics
Data are presented as mean ± SEM. Differences within and between trials and conditions were determined via 2-way repeated measures analysis of variance (ANOVA). Due to large differences in the magnitude of values of exercise and rest, for comparisons of FBF, FVC, 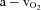 , oxygen extraction, oxygen delivery and oxygen consumption, two ANOVAs were completed, one including both exercise conditions (normoxic and hypoxic) and one including both rest periods and hypoxia. For all other variables collected, all time points were included in the ANOVA analysis. Specific hypothesis testing was performed using 2-tailed Student's t test for paired data and when appropriate, post hoc comparisons were made with Tukey's HSD test. Significance was set a priori at P < 0.05.
, oxygen extraction, oxygen delivery and oxygen consumption, two ANOVAs were completed, one including both exercise conditions (normoxic and hypoxic) and one including both rest periods and hypoxia. For all other variables collected, all time points were included in the ANOVA analysis. Specific hypothesis testing was performed using 2-tailed Student's t test for paired data and when appropriate, post hoc comparisons were made with Tukey's HSD test. Significance was set a priori at P < 0.05.
Results
Effect of regional α- and β-adrenoceptor blockade
Prior to all experimental trials, combined blockade of α- and β-adrenoreceptors significantly increased resting FBF (40 ± 4 vs. 87 ± 10 ml min−1) and FVC (43 ± 5 vs. 92 ± 10 ml min−1 (100 mmHg)−1) by ∼100% (P < 0.05). MAP (93 ± 2 vs. 95 ± 2 mmHg; P = 0.30) and HR (57 ± 2 vs. 56 ± 2 beats min−1; P = 0.24) did not change due to local α- and β-receptor blockade.
Systemic haemodynamic and respiratory responses
Systemic haemodynamic and respiratory responses are presented for each trial and condition in Table 1. MAP was not different during exercise between all trials (P = 0.34–0.92). As expected, hypoxia resulted in significant increases in HR and ventilation (P < 0.05). Ventilation data were not obtained for one subject (technical difficulties); therefore, all ventilation data represent an average of nine subjects. The targeted oxygen saturation of ∼85% was achieved in all hypoxic conditions. Subjects remained isocapnic across all experimental trials, as no significant differences were observed in end-tidal CO2 (P = 0.18 – 0.67).
Table 1.
Systemic haemodynamic and ventilatory responses
| Rest | Normoxic exercise | Rest | Steady-state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| MAP (mmHg) | 95 ± 2 | 98 ± 3 | 94 ± 2 | 98 ± 4 | 100 ± 3† |
| HR (beats min−1) | 56 ± 2 | 58 ± 2 | 57 ± 3 | 70 ± 3† | 72 ± 3†§ |
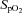 (%) (%) |
97 ± 0 | 97 ± 0 | 98 ± 0 | 84 ± 1† | 84 ± 1†§ |
| Minute vent. (l min−1; BTPS) | 9.0 ± 1.2 | 9.6 ± 0.9 | 9.7 ± 1.3 | 15.0 ± 2.2† | 18.1 ± 2.5†§ |
| End-tidal CO2 (mmHg) | 36.6 ± 1.5 | 38.1 ± 0.9 | 36.9 ± 1.4 | 37.5 ± 1.1 | 38.6 ± 1.2 |
| NO–PG block | |||||
| MAP (mmHg) | 97 ± 2 | 99 ± 2 | 97 ± 2 | 100 ± 3 | 101 ± 3 |
| HR (beats min−1) | 55 ± 2 | 58 ± 2 | 53 ± 2* | 64 ± 3*† | 70 ± 3†§ |
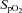 (%) (%) |
97 ± 0 | 97 ± 0 | 98 ± 0 | 84 ± 1† | 84 ± 1†§ |
| Minute vent. (l min−1; BTPS) | 11.0 ± 1.2 | 11.0 ± 0.5 | 10.9 ± 1.1 | 20.1 ± 3.2*† | 21.2 ± 2.0*†§ |
| End-tidal CO2 (mmHg) | 36.2 ± 1.3 | 37.4 ± 0.9 | 36.0 ± 1.3 | 37.5 ± 1.1 | 38.4 ± 1.1 |
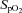 : oxygen saturation via pulse oximetry
: oxygen saturation via pulse oximetry
Forearm blood flow and vascular conductance responses
Absolute values of FBF and FVC for each trial are presented in Fig. 2. No difference was observed in resting FBF or FVC between normoxic and hypoxic trials (P = n.s.). Resting FBF and FVC was ∼38% lower following NO–PG block for both the normoxic and hypoxic trials (P < 0.05). Steady-state hypoxia increased both FBF and FVC in the control condition (P < 0.05), but FBF and FVC were significantly lower than control with NO–PG block (P < 0.05) and no longer different than resting values (both n.s.; Fig. 2).
Figure 2. Forearm haemodynamics at rest, during normoxic exercise, hypoxia and hypoxic exercise.
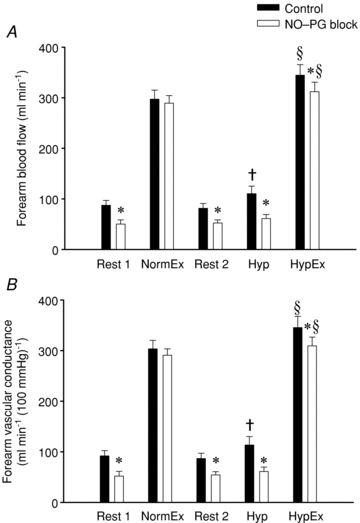
Forearm blood flow (A) and forearm vascular conductance (B) across all trials. Combined inhibition of NO–PG significantly reduced FBF and FVC at all time points except for normoxic exercise (NormEx). In control conditions, hypoxic exercise (HypEx) FBF and FVC was significantly greater than NormEx FBF and FVC. In NO–PG block conditions, this augmentation remained. Hypoxic rest (Hyp) was significantly greater than Rest in control conditions, but not with NO–PG inhibition. *P < 0.05 vs. control condition; †P < 0.05 vs. Rest 2; §P < 0.05 vs. normoxic exercise (within condition).
In the control condition, both FBF and FVC were significantly greater during hypoxic exercise than normoxic exercise (Fig. 2; P < 0.05). Combined blockade of NO and PGs did not significantly reduce FBF (P = 0.33) nor FVC (P = 0.18) during normoxic exercise. In contrast, during hypoxic exercise in the NO–PG block condition, both FBF and FVC were significantly reduced from control hypoxic exercise (P < 0.05). Combined blockade of NO–PG reduced the hypoxic augmentation of exercise FBF by ∼50% (Fig. 3; ΔFBF: control = 48 ± 6 vs. NO–PG block = 23 ± 9 ml min−1; P < 0.05). Despite this reduction, hypoxic exercise FBF and FVC were still significantly greater than normoxic exercise FBF and FVC in the NO–PG block condition (Fig. 2; P < 0.05).
Figure 3. Effect of NO–PG blockade on exercise hyperaemia.
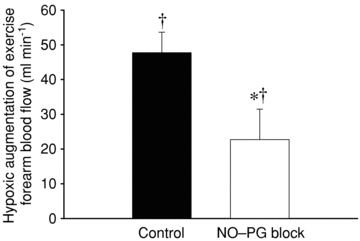
Hypoxic augmentation was calculated for each condition as the difference in absolute forearm blood flow between hypoxic exercise and normoxic exercise. Inhibition of NO and PG synthesis (l-NMMA and ketorolac, respectively) significantly reduced (∼50%) the hypoxic augmentation of exercise hyperaemia. *P < 0.05 vs. control condition; †P < 0.05 vs. zero.
Blood gases and forearm oxygen delivery, extraction and consumption
Blood gas data are presented in Table 2 As anticipated, hypoxia reduced arterial 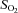 (
(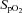 ), arterial
), arterial 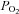 (
(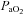 ) and arterial
) and arterial 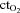 (
(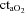 ) as compared to normoxia (P < 0.05). There was no difference in arterial
) as compared to normoxia (P < 0.05). There was no difference in arterial 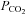 between normoxic exercise and hypoxic exercise within control or NO–PG conditions (P = n.s.). Exercise, both in normoxia and hypoxia, significantly reduced venous pH (pHv), venous
between normoxic exercise and hypoxic exercise within control or NO–PG conditions (P = n.s.). Exercise, both in normoxia and hypoxia, significantly reduced venous pH (pHv), venous 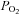 (
(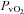 ) and venous (
) and venous (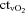 ) (P < 0.05).
) (P < 0.05).
Table 2.
Blood gases
| Rest | Normoxic exercise | Rest | Steady-state hypoxia | Hypoxic exercise | |
|---|---|---|---|---|---|
| Control | |||||
| pHa | 7.428 ± 0.010 | 7.410 ± 0.007 | 7.415 ± 0.011 | 7.428 ± 0.009 | 7.408 ± 0.009 |
| pHv | 7.399 ± 0.011 | 7.314 ± 0.008† | 7.390 ± 0.009 | 7.403 ± 0.009 | 7.324 ± 0.010† |
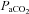 (mmHg) (mmHg) |
35.8 ± 1.4 | 36.0 ± 1.1 | 36.7 ± 1.5 | 35.5 ± 1.2 | 36.5 ± 1.0 |
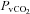 (mmHg) (mmHg) |
39.8 ± 1.7 | 55.0 ± 1.4† | 42.0 ± 1.9 | 38.2 ± 1.4 | 52.1 ± 1.8† |
| [Hb]a (g dl−1) | 15.2 ± 0.5 | 15.1 ± 0.5 | 15.2 ± 0.5 | 15.0 ± 0.5 | 15.0 ± 0.5 |
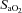 (%) (%) |
96.4 ± 0.2 | 96.3 ± 0.2 | 95.9 ± 0.4 | 85.1 ± 1.0† | 85.7 ± 1.0†§ |
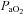 (mmHg) (mmHg) |
84.2 ± 1.8 | 84.0 ± 1.3 | 81.0 ± 2.1 | 49.5 ± 1.5† | 51.7 ± 1.6†§ |
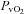 (mmHg) (mmHg) |
44.3 ± 2.8 | 26.3 ± 1.4† | 43.4 ± 2.3 | 37.5 ± 2.0† | 24.3 ± 0.7† |
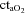 (ml l−1) (ml l−1) |
203 ± 6 | 201 ± 7 | 202 ± 6 | 177 ± 6† | 178 ± 6†§ |
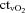 (ml l−1) (ml l−1) |
161 ± 8 | 82 ± 8† | 159 ± 9 | 141 ± 10 | 74 ± 5† |
| Extraction (%) | 21 ± 3 | 60 ± 4 | 22 ± 3 | 21 ± 4 | 59 ± 3 |
| NO–PG block | |||||
| pHa | 7.425 ± 0.012 | 7.419 ± 0.011 | 7.429 ± 0.011 | 7.426 ± 0.008 | 7.411 ± 0.007 |
| pHv | 7.384 ± 0.008 | 7.317 ± 0.008† | 7.388 ± 0.009 | 7.401 ± 0.006 | 7.320 ± 0.006† |
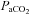 (mmHg) (mmHg) |
34.1 ± 1.3 | 33.8 ± 1.2 | 33.0 ± 1.2* | 32.1 ± 1.1* | 32.8 ± 1.8* |
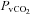 (mmHg) (mmHg) |
40.0 ± 1.3 | 51.9 ± 1.5† | 39.5 ± 1.9 | 36.5 ± 1.5 | 50.1 ± 1.9† |
| [Hb]a (g dl−1) | 14.8 ± 0.5* | 14.9 ± 0.4 | 14.9 ± 0.4* | 14.8 ± 0.4 | 14.9 ± 0.4 |
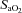 (%) (%) |
96.3 ± 0.3 | 96.5 ± 0.2 | 96.3 ± 0.3 | 85.6 ± 1.0† | 84.7 ± 1.0*†§ |
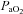 (mmHg) (mmHg) |
84.4 ± 1.5 | 85.2 ± 1.0 | 85.7 ± 2.0* | 52.5 ± 1.6*† | 51.0 ± 1.5†§ |
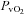 (mmHg) (mmHg) |
33.9 ± 2.0* | 25.0 ± 0.9† | 38.9 ± 3.3* | 36.1 ± 2.2 | 24.8 ± 1.8† |
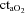 (ml l−1) (ml l−1) |
199 ± 6 | 199 ± 0.6 | 199 ± 5 | 176 ± 5† | 181 ± 8†§ |
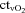 (ml l−1) (ml l−1) |
118 ± 9* | 74 ± 0.3† | 131 ± 10* | 126 ± 9* | 64 ± 4† |
| Extraction (%) | 41 ± 4* | 63 ± 2 | 34 ± 5*‡ | 28 ± 5*† | 64 ± 3* |
Oxygen delivery, presented in Fig. 4A, represents the product of arterial 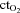 and FBF. In the control condition prior to exercise, oxygen delivery was increased during steady-state hypoxia, as compared to rest (199 ± 33 vs. 167 ± 23 ml min−1; P < 0.05). In the NO–PG block condition, oxygen delivery was no longer greater during hypoxia than at rest (109 ± 17 vs. 106 ± 14 ml min−1; P = n.s.). Oxygen delivery was similar during normoxic and hypoxic exercise prior to NO–PG inhibition. During normoxic exercise, there was no significant reduction in oxygen delivery between control and NO–PG block conditions (599 ± 43 vs. 581 ± 43 ml min−1; P = n.s.). In contrast, for hypoxic exercise, oxygen delivery was significantly lower following NO–PG block (617 ± 47 vs. 561 ± 35 ml min−1; P < 0.05).
and FBF. In the control condition prior to exercise, oxygen delivery was increased during steady-state hypoxia, as compared to rest (199 ± 33 vs. 167 ± 23 ml min−1; P < 0.05). In the NO–PG block condition, oxygen delivery was no longer greater during hypoxia than at rest (109 ± 17 vs. 106 ± 14 ml min−1; P = n.s.). Oxygen delivery was similar during normoxic and hypoxic exercise prior to NO–PG inhibition. During normoxic exercise, there was no significant reduction in oxygen delivery between control and NO–PG block conditions (599 ± 43 vs. 581 ± 43 ml min−1; P = n.s.). In contrast, for hypoxic exercise, oxygen delivery was significantly lower following NO–PG block (617 ± 47 vs. 561 ± 35 ml min−1; P < 0.05).
Figure 4. Forearm oxygen delivery, extraction and consumption for control and NO–PG block conditions.
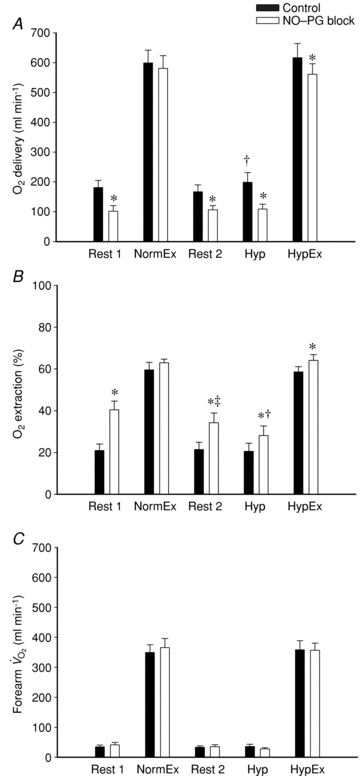
Blockade of NO–PG resulted in a significant attenuation in oxygen delivery (A) at rest, hypoxia (Hyp) and hypoxic exercise (HypEx). Subsequently, oxygen extraction (B) was significantly increased after NO–PG blockade for Rest, Hyp and HypEx. NO–PG blockade had no effect on oxygen delivery or extraction for normoxic exercise (NormEx; P = n.s.). Oxygen consumption (C) was not different during NormEx and HypEx for control or NO–PG block conditions. All values for NormEx and HypEx in both conditions are significantly greater than Rest and Hyp (P < 0.05 within condition). *P < 0.05 vs. control condition; †P < 0.05 vs. rest (within trial and condition); ‡P < 0.05 vs. normoxic rest (within condition).
Oxygen extraction for all trials and conditions is presented in Fig. 4B. At rest and in hypoxia, oxygen extraction was greater after NO–PG block compared to control (Rest 1: 21 ± 3 vs. 41 ± 4%, P < 0.05; Rest 2: 22 ± 3 vs. 34 ± 5%, Hyp: 21 ± 4 vs. 28 ± 5%; P < 0.05). Similarly, for hypoxic exercise, oxygen extraction was greater in the NO–PG block condition than control (59 ± 3 vs. 64 ± 3%; P < 0.05). There was no change in normoxic exercise oxygen extraction as a result of NO–PG blockade (60 ± 4 vs. 63 ± 2%; P = 0.20).
Forearm oxygen consumption (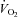 ) is presented in Fig. 4C for all trials and conditions. There were no differences in oxygen consumption at rest or during hypoxia within or between conditions (P = 0.15–0.94). Similarly, there were no differences in oxygen consumption during exercise as a result of hypoxia or NO–PG blockade (P = 0.64–0.99).
) is presented in Fig. 4C for all trials and conditions. There were no differences in oxygen consumption at rest or during hypoxia within or between conditions (P = 0.15–0.94). Similarly, there were no differences in oxygen consumption during exercise as a result of hypoxia or NO–PG blockade (P = 0.64–0.99).
Discussion
The purpose of the present study was to investigate the combined role of NO and PGs to hypoxic exercise hyperaemia, as well as whether combined inhibition reduced skeletal muscle oxygen consumption in resting and exercising muscle during hypoxia. The primary novel findings from this study are as follows. First, under conditions of forearm sympathoadrenal blockade to isolate local vasodilatory mechanisms, acute combined inhibition of NO and PGs reduced exercise hyperaemia in hypoxia but not in normoxia (Fig. 2). As such, the magnitude of augmentation of the hyperaemic response to hypoxic exercise, as compared to normoxic exercise, was significantly attenuated by NO–PG blockade (∼50%; Fig. 3). However, muscle blood flow during hypoxic exercise was still greater than that during normoxic exercise, indicating that other substances/mechanisms play a role in this local augmented hyperaemic response. Second, following combined NO–PG inhibition, despite a decline in hypoxic exercise blood flow and therefore oxygen delivery, a significant increase in oxygen extraction was observed, and thus oxygen consumption was maintained at the same level as in control conditions, and as compared to normoxic exercise (Fig. 4). Finally, data from the present study support previous findings from our laboratory (Markwald et al. 2011) that combined blockade of NO and PGs abolishes the hyperaemic response in skeletal muscle observed during systemic hypoxia at rest in healthy humans (Fig. 2). Similar to exercising conditions, oxygen extraction was greater during systemic hypoxia following combined NO–PG blockade and thus oxygen consumption of the resting forearm was maintained (Fig. 4).
Skeletal muscle blood flow responses to hypoxic exercise
Many investigators have reported that during hypoxic exercise, muscle blood flow is augmented for a given exercise intensity compared with that in normoxia in order to ensure appropriate oxygen delivery to the contracting muscle (Hartley et al. 1973; Rowell et al. 1986; Koskolou et al. 1997; Wilkins et al. 2006, 2008; Casey et al. 2009, 2010). What factors contribute to this augmented exercise hyperaemia during hypoxia has been a topic of recent interest. One idea, tested by Wilkins and colleagues (2006), was that mild-to-moderate-intensity hypoxic exercise would blunt post-junctional α-adrenoceptor-mediated vasoconstriction to a greater extent than during normoxia and thus facilitate greater blood flow to the contracting muscle. However, the vasoconstrictor response to endogenously released noradrenaline (via tyramine) was similar in normoxic and hypoxic exercise conditions, a finding consistent with our previous observations in resting muscle (Dinenno et al. 2003). The finding of no difference in the ability to blunt α-adrenoceptor-mediated vasoconstriction indicates that an enhanced vasodilator signal accounts for the augmented muscle blood flow observed during hypoxic exercise. In a follow-up study, this same group (Wilkins et al. 2008) demonstrated that β-adrenoceptor-mediated dilatation (presumably via circulating adrenaline) was playing a role in the hyperaemic response to hypoxic exercise at mild intensity (10% MVC) but not higher intensity (20% MVC) forearm exercise. Thus, at moderate-intensity hypoxic exercise, the mechanisms and/or substances involved in evoking this enhanced vasodilatation have not been fully elucidated.
Recent studies by Casey and colleagues (2009, 2010, 2011) have sought to further understand the mechanisms involved in the augmented vasodilatation observed during moderate-intensity hypoxic exercise. In similar experimental designs, the role for adenosine was examined via intrabrachial aminophylline infusions (Casey et al. 2009), and the contribution of NO was investigated through acute NOS inhibition (Casey et al. 2010, 2011). The collective data from these studies suggest that the compensatory dilatation that occurs during hypoxic forearm exercise is not mediated by adenosine but seems to have a strong NO component. In fact, their findings suggested that NO derived from endothelial NOS essentially was the sole mechanism underlying the augmented vasodilatation during moderate-intensity (20% MVC) handgrip exercise (Casey et al. 2010). In this study and in contrast to the present study, sympathetic nervous system activity and subsequent α-adrenoceptor-mediated vasoconstriction was intact, and thus the findings regarding muscle blood flow during hypoxic exercise reflect the interaction between NOS inhibition and sympathetic vasoconstriction.
The data from the present study build upon these previous findings in an experimental protocol designed to specifically address the local vasodilator signalling mechanisms involved in hypoxic exercise hyperaemia. In a recent study, we employed pharmacological sympathectomy (via combined α- and β-adrenoceptor blockade) to eliminate sympathoadrenal influences on vascular tone in an effort to understand the local modulators of vascular tone during systemic hypoxia in the resting forearm (Markwald et al. 2011). In this previous study, we found that single inhibition of NO or PGs did not impact the vasodilator response from rest to steady-state systemic hypoxia; however, combined inhibition of NO and PGs abolished the response, indicating an interactive role of these pathways in evoking hypoxic vasodilatation. In the present study, we utilized a similar experimental approach to determine the combined roles of NO and PGs to the augmented exercise hyperaemia during hypoxia. Our data in the resting forearm vasculature are consistent with our previous publication in that combined NO–PG inhibition during systemic hypoxia abolished hypoxic vasodilatation (Fig. 2). With respect to exercise, our data indicate that combined inhibition of NOS and COX does not significantly impact muscle blood flow during moderate-intensity handgrip exercise in humans in normoxia; however, it significantly blunts the compensatory increase in exercise hyperaemia during hypoxia (Fig. 2). In fact, combined NO–PG inhibition reduced the augmented hyperaemia by ∼50% (Fig. 3). However, despite this clear reduction in the response, muscle blood flow during hypoxic exercise was still greater than during normoxia, indicating that other mechanisms contribute to the augmented hyperaemia during hypoxic exercise.
Oxygen delivery, extraction and consumption
In the present study, oxygen consumption did not differ at rest during normoxia and hypoxia, nor did it differ between any of the exercise trials (Fig. 4C). Thus, under conditions in which forearm blood flow and oxygen delivery were reduced at rest or in response to either hypoxia alone or hypoxic exercise via combined NO–PG inhibition, oxygen extraction was augmented and as such, tissue oxygen consumption was maintained. It has been proposed that NOS inhibition can independently reduce oxygen consumption during normoxic exercise (Mortensen et al. 2007, 2009b). Indeed, there is some experimental evidence suggesting that NOS inhibition can reduce oxygen consumption for a given force production; however, these studies were done on isolated muscles subjected to electrical stimulus (King-VanVlack et al. 2002; Baker et al. 2006). Other studies performed in vivo with contracting skeletal muscle have shown no effect of NOS inhibition on oxygen consumption (O'Leary et al. 1994; Frandsen et al. 2001; Casey et al. 2010). Taking this into consideration, we do not believe that l-NMMA infusion had any direct effect on oxygen consumption in the present study. We interpret our findings related to oxygen delivery, extraction and consumption to suggest that at relatively moderate workloads, oxygen extraction can be enhanced to maintain oxygen consumption, if delivery is compromised due to an attenuated hyperaemic response in young healthy humans.
Potential mechanisms for the stimulus of NO and PG synthesis during hypoxic exercise
The underlying mechanism for the augmented stimulus of NO and PGs during hypoxic exercise is not well understood. Given that our experimental trials were performed during α- and β-adrenoceptor blockade, there should not have been any neural (noradrenaline/adrenaline) or adrenal (adrenaline) influences on forearm vascular tone. Thus, the stimulus for NO and PG synthesis probably occurred at the local tissue level. There was no effect of NO and PG block on normoxic exercise hyperaemia, yet there was a significant reduction in the hyperaemic response to both hypoxia at rest and hypoxic exercise. These data suggest that the stimulus for NO and PG synthesis may be linked with the oxygen status of the blood or vessel. In this context, several hypotheses have been proposed for what may function as an ‘oxygen sensor’ to regulate peripheral blood flow. Adenosine has classically been proposed as a modulator of hypoxic vasodilatation at rest and during exercise (Mian & Marshall, 1991; Skinner & Marshall, 1996; Bryan & Marshall, 1999); however, recent work by Casey et al. (2009) indicates that adenosine is not obligatory for hypoxic exercise hyperaemia. More recently, the red blood cell (RBC) has been proposed to regulate vascular tone by release of ATP in response to deoxygenation (Bergfeld & Forrester, 1992; Ellsworth et al. 1995; Gonzalez-Alonso et al. 2001). Some investigators have demonstrated an obligatory role for RBCs in hypoxic vasodilatation (Dietrich et al. 2000), that plasma ATP levels tends to be higher during hypoxic leg extension exercise compared to normoxic exercise (Gonzalez-Alonso et al. 2002), and that combined NOS and COX inhibition modestly reduces ATP-mediated vasodilatation (Mortensen et al. 2009a). Thus, RBC release of ATP and subsequent binding to endothelial P2y receptors could possibly be one stimulus for increased NO and PG production during hypoxic exercise as compared to normoxic exercise. Given previous findings that blood flow during exercise is determined by arterial oxygen content rather than arterial 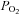 (Roach et al. 1999), the erythrocyte as an ‘oxygen sensor’ is an attractive hypothesis. Additional studies are necessary to determine the specific mechanisms for increased NO and PG synthesis during hypoxic exercise and what other mechanisms may contribute to the local augmented hyperaemic response that remains after NO–PG blockade.
(Roach et al. 1999), the erythrocyte as an ‘oxygen sensor’ is an attractive hypothesis. Additional studies are necessary to determine the specific mechanisms for increased NO and PG synthesis during hypoxic exercise and what other mechanisms may contribute to the local augmented hyperaemic response that remains after NO–PG blockade.
Experimental limitations and considerations
One potential limitation of the present study is that we did not directly test the efficacy of any of our pharmacological blockers. In our recent study (Markwald et al. 2011), phentolamine and propranolol in the same loading and maintenance doses as used in the present study were shown to be effective throughout an even longer experimental protocol, and thus this should not influence the interpretation of the data. Regarding NOS and COX inhibition, we used similar doses demonstrated to be effective in previous studies (see Methods), and combined inhibition resulted in a significant reduction in resting blood flow which is consistent with effective blockade. Further, based on our findings that NO–PG block did in fact reduce hypoxic exercise hyperaemia, had we not achieved full inhibition of these vasodilator pathways, our results would underestimate the contribution of NO and/or PGs.
Another potential limitation is that our experimental approach of combined inhibition of NO and PGs does not allow us to make conclusions regarding the individual contributions of these vasodilators to the augmented hyperaemic response during hypoxic exercise, or to determine whether any compensatory interactions exist between these endothelium-derived substances. We chose the approach of combined inhibition based on our recent observations that individual blockade of NO or PGs did not influence hypoxic vasodilatation at rest, whereas combined NO–PG inhibition abolished hypoxic vasodilatation in the human forearm (Markwald et al. 2011). It would be expected that this potential interaction would be similar (if not more robust) during an exercise stimulus (elevated tissue metabolic demand) where the compensatory pathways involved in the regulation of muscle blood flow would be predicted to be greater to ensure adequate oxygen delivery. Regardless, subsequent studies would need to be designed to specifically address the effects of independent blockade on hypoxic exercise hyperaemia.
The finding that there was no effect of NO–PG blockade on normoxic exercise hyperaemia (Fig. 2) is in agreement with our previous work that demonstrated that NO and PGs do not play a significant role in normoxic exercise hyperaemia when inhibition occurs prior to forearm exercise onset (Dinenno & Joyner, 2004; Schrage et al. 2004). While this lack of an effect is in contrast to some of the findings within the leg vasculature (Kalliokoski et al. 2006; Mortensen et al. 2007, 2009b), the contributions of NO and PGs to exercise hyperaemia may be intensity dependent (Boushel et al. 2002). The workload utilized in the present study (20% MVC), while higher than that in previous forearm studies (Dinenno & Joyner, 2004; Schrage et al. 2004), may not be great enough to elicit a major contribution of NO and PGs during normoxic exercise hyperaemia. Further, whether our overall findings related to hypoxic exercise blood flow regulation and oxygen consumption would be similar or different in the lower extremities remains unknown.
Another consideration relates to the apparent divergent findings in the present study compared with those presented by Casey and colleagues. Specifically, Casey et al. (2010) demonstrated that NOS inhibition via l-NMMA completely abolished the augmented vasodilatation during moderate-intensity hypoxic handgrip exercise, whereas our data indicate that combined NO–PG inhibition blunts the response by ∼50%. The findings from Casey et al. reflect the interaction between sympathetic α-adrenoceptor vasoconstriction and NO, and as such, it is possible that augmented sympathetic vasoconstrictor activity during hypoxic exercise (Wilkins et al. 2008) masks the potential contribution of other vasodilatory signals involved in this response, as has been proposed in resting muscle tissue (Weisbrod et al. 2001; Markwald et al. 2011). In the present study, we used established pharmacology to block sympathetic vasoconstrictor effects (as well as β-adrenoceptor-mediated vasodilatation) in an attempt to isolate the local vasodilatory mechanisms. Our data clearly indicate that combined blockade of NO and PGs blunts the augmented response, but also highlight that other vasodilatory mechanisms are operative and can have vascular effects when sympathetic vasoconstrictor tone is eliminated.
Perspectives
The present findings indicate that NO and PGs, two endothelium-dependent vasodilators, significantly contribute, in combination, to the augmented hyperaemic response during hypoxic exercise and are necessary to observe local vasodilatation during systemic hypoxia in resting conditions. Given this, we speculate that our findings might have important implications for populations that demonstrate endothelial dysfunction in regards to their ability to regulate blood flow and oxygen delivery during a hypoxic stimulus. For example, chronic pathological conditions such as diabetes, obstructive sleep apnoea, congestive heart failure and even healthy older adults demonstrate impaired endothelial function, which is often due to decreased NO and perhaps prostacyclin bioavailability (Feletou & Vanhoutte, 2006). Along similar lines, the remaining augmentation of blood flow (beyond NO and PGs) during hypoxic exercise could also be a result of the nitrite reductase activity of haemoglobin to produce NO independently of NOS and cause vasodilatation (Gladwin et al. 2004). The potential for this mechanism is significant, given that venous 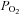 in the present study was close to the P50 of the oxygen dissociation curve of haemoglobin, where this reductase mechanism is optimized (Gladwin, 2008). Importantly, populations that exhibit endothelial dysfunction tend to have decreased nitrite levels, subsequent to generalized decreased NO bioavailability (Kleinbongard et al. 2006). Thus, the potential additional mechanism of vasodilatation through nitrite reduction to NO might be impaired during hypoxic exercise in these groups. Future studies will be needed to address both what accounts for the remaining augmented blood flow during hypoxic exercise in young healthy humans, as well as how populations that demonstrate endothelial function are able to regulate blood flow and oxygen delivery during hypoxic conditions.
in the present study was close to the P50 of the oxygen dissociation curve of haemoglobin, where this reductase mechanism is optimized (Gladwin, 2008). Importantly, populations that exhibit endothelial dysfunction tend to have decreased nitrite levels, subsequent to generalized decreased NO bioavailability (Kleinbongard et al. 2006). Thus, the potential additional mechanism of vasodilatation through nitrite reduction to NO might be impaired during hypoxic exercise in these groups. Future studies will be needed to address both what accounts for the remaining augmented blood flow during hypoxic exercise in young healthy humans, as well as how populations that demonstrate endothelial function are able to regulate blood flow and oxygen delivery during hypoxic conditions.
Conclusions
The results from the present investigation demonstrate that during local sympathoadrenal blockade, acute combined inhibition of NOS and COX abolishes the local hyperaemic response at rest and significantly reduces it during moderate-intensity handgrip exercise with systemic isocapnic hypoxia. Inhibition of NO and PG synthesis does not affect normoxic exercise hyperaemia. Oxygen consumption at rest, during hypoxia and during hypoxic exercise was maintained after NO–PG block (despite a reduction in blood flow in all conditions), due to a compensatory increase in oxygen extraction. After combined NO–PG blockade, ∼50% of the augmented hyperaemia during hypoxic exercise remains, suggesting that other local factors (e.g. hyperpolarizing stimuli, nitrite reduction) also play a role in mediating this response. Given that NO and PGs are endothelial-derived vasodilators, our novel findings on the important contribution of these substances to the local regulation of muscle blood flow during hypoxic exercise may have potential implications for populations that demonstrate endothelial dysfunction and deficiencies in these vasodilator pathways.
Acknowledgments
We would like to thank the subjects who volunteered for this study as well as Julia A. Davis for her administrative assistance. This research was supported by the National Institutes of Health awards HL087952 and HL095573 (F.A.D.).
Glossary
Abbreviations
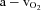
arteriovenous oxygen difference
- COX
cyclooxygenase
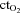
oxygen content
- FBF
forearm blood flow
- FVC
forearm vascular conductance
- Hb
haemoglobin
- HR
heart rate
- l-NMMA
NG-monomethyl-l-arginine
- MAP
mean arterial pressure
- MBV
mean blood velocity
- MVC
maximal voluntary contraction
- NO
nitric oxide
- NOS
nitric oxide synthase
- PG
prostaglandin
- RBC
red blood cell
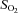
oxygen saturation
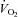
oxygen consumption
Author contributions
A.R.C. contributed to the design of the experiment, collection, analysis and interpretation of the data, and writing of this article. B.S.K. contributed to the design of the experiment, collection and interpretation of the data, and critical revision of this article. W.F.V. contributed to the experimental design, provided invasive methodology for data collection, and critical revision of this article. F.A.D. contributed to the conception and design of the experiment, collection, analysis and interpretation of the data and writing of this article. All authors gave final approval of the article. All experiments were performed in the Human Cardiovascular Physiology Laboratory at Colorado State University.
References
- Baker DJ, Krause DJ, Howlett RA, Hepple RT. Nitric oxide synthase inhibition reduces O2 cost of force development and spares high-energy phosphates following contractions in pump-perfused rat hindlimb muscles. Exp Physiol. 2006;91:581–589. doi: 10.1113/expphysiol.2005.032698. [DOI] [PubMed] [Google Scholar]
- Banzett RB, Garcia RT, Moosavi SH. Simple contrivance “clamps” end-tidal PCO2 and PO2 despite rapid changes in ventilation. J Appl Physiol. 2000;88:1597–1600. doi: 10.1152/jappl.2000.88.5.1597. [DOI] [PubMed] [Google Scholar]
- Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698. doi: 10.1113/jphysiol.2002.021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Curry TB, Wilkins BW, Joyner MJ. Nitric oxide mediated vasodilation becomes independent of β-adrenergic receptor activation with increased intensity of hypoxic exercise. J Appl Physiol. 2011;110:687–694. doi: 10.1152/japplphysiol.00787.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Curry TB, Eisenach JH, Wilkins BW, Joyner MJ. Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J Physiol. 2010;588:373–385. doi: 10.1113/jphysiol.2009.180489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Madery BD, Pike TL, Eisenach JH, Dietz NM, Joyner MJ, Wilkins BW. Adenosine receptor antagonist and augmented vasodilation during hypoxic exercise. J Appl Physiol. 2009;107:1128–1137. doi: 10.1152/japplphysiol.00609.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Nitric oxide, but not vasodilating prostaglandins, contributes to the improvement of exercise hyperemia via ascorbic acid in healthy older adults. Am J Physiol Heart Circ Physiol. 2010;299:H1633–H1641. doi: 10.1152/ajpheart.00614.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol Heart Circ Physiol. 2000;278:H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- Dietz NM, Halliwill JR, Spielmann JM, Lawler LA, Papouchado BG, Eickhoff TJ, Joyner MJ. Sympathetic withdrawal and forearm vasodilation during vasovagal syncope in humans. J Appl Physiol. 1997;82:1785–1793. doi: 10.1152/jappl.1997.82.6.1785. [DOI] [PubMed] [Google Scholar]
- Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinenno FA, Dietz NM, Joyner MJ. Aging and forearm postjunctional α-adrenergic vasoconstriction in healthy men. Circulation. 2002;106:1349–1354. doi: 10.1161/01.cir.0000028819.64790.be. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ. Combined NO and PG inhibition augments α-adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Heart Circ Physiol. 2004;287:H2576–H2584. doi: 10.1152/ajpheart.00621.2004. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ, Halliwill JR. Failure of systemic hypoxia to blunt α-adrenergic vasoconstriction in the human forearm. J Physiol. 2003;549:985–994. doi: 10.1113/jphysiol.2003.042507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenach JH, Clark ES, Charkoudian N, Dinenno FA, Atkinson JL, Fealey RD, Dietz NM, Joyner MJ. Effects of chronic sympathectomy on vascular function in the human forearm. J Appl Physiol. 2002;92:2019–2025. doi: 10.1152/japplphysiol.01025.2001. [DOI] [PubMed] [Google Scholar]
- Eklund B, Kaijser L. Effect of regional α- and β-adrenergic blockade on blood flow in the resting forearm during contralateral isometric handgrip. J Physiol. 1976;262:39–50. doi: 10.1113/jphysiol.1976.sp011584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol Heart Circ Physiol. 1995;269:H2155–H2161. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–H1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Frandsen U, Bangsbo J, Sander M, Hoffner L, Betak A, Saltin B, Hellsten Y. Exercise-induced hyperaemia and leg oxygen uptake are not altered during effective inhibition of nitric oxide synthase with NG-nitro-l-arginine methyl ester in humans. J Physiol. 2001;531:257–264. doi: 10.1111/j.1469-7793.2001.0257j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin MT. Evidence mounts that nitrite contributes to hypoxic vasodilation in the human circulation. Circulation. 2008;117:594–597. doi: 10.1161/CIRCULATIONAHA.107.753897. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res. 2002;91:1046–1055. doi: 10.1161/01.res.0000044939.73286.e2. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alonso J, Richardson RS, Saltin B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol. 2001;530:331–341. doi: 10.1111/j.1469-7793.2001.0331l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwill JR, Lawler LA, Eickhoff TJ, Dietz NM, Nauss LA, Joyner MJ. Forearm sympathetic withdrawal and vasodilatation during mental stress in humans. J Physiol. 1997;504:211–220. doi: 10.1111/j.1469-7793.1997.211bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada A, Sander M, Gonzalez-Alonso J. Human skeletal muscle sympathetic nerve activity, heart rate and limb haemodynamics with reduced blood oxygenation and exercise. J Physiol. 2003;551:635–647. doi: 10.1113/jphysiol.2003.044024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley LH, Vogel JA, Landowne M. Central, femoral, and brachial circulation during exercise in hypoxia. J Appl Physiol. 1973;34:87–90. doi: 10.1152/jappl.1973.34.1.87. [DOI] [PubMed] [Google Scholar]
- Johnsson G. The effects of intra-arterially administered propranolol and H 56-28 on blood flow in the forearm – a comparative study of two β-adrenergic receptor antagonists. Acta Pharmacol Toxicol (Copenh) 1967;25:63–74. doi: 10.1111/j.1600-0773.1967.tb02997.x. [DOI] [PubMed] [Google Scholar]
- Kalliokoski KK, Langberg H, Ryberg AK, Scheede-Bergdahl C, Doessing S, Kjaer A, Kjaer M, Boushel R. Nitric oxide and prostaglandins influence local skeletal muscle blood flow during exercise in humans: coupling between local substrate uptake and blood flow. Am J Physiol Regul Integr Comp Physiol. 2006;291:R803–R809. doi: 10.1152/ajpregu.00808.2005. [DOI] [PubMed] [Google Scholar]
- King-VanVlack CE, Mewburn JD, Chapler CK, MacDonald PH. Endothelial modulation of skeletal muscle blood flow and VO2 during low- and high-intensity contractions. J Appl Physiol. 2002;92:461–468. doi: 10.1152/japplphysiol.01152.2000. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Voyles WF, Carlson RE, Dinenno FA. Graded sympatholytic effect of exogenous ATP on postjunctional α-adrenergic vasoconstriction in the human forearm: implications for vascular control in contracting muscle. J Physiol. 2008;586:4305–4316. doi: 10.1113/jphysiol.2008.154252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BS, Voyles WF, Simpson CB, Carlson RE, Schrage WG, Dinenno FA. Endothelium-dependent vasodilatation and exercise hyperaemia in ageing humans: impact of acute ascorbic acid administration. J Physiol. 2009;587:1989–2003. doi: 10.1113/jphysiol.2008.167320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinbongard P, Dejam A, Lauer T, Jax T, Kerber S, Gharini P, Balzer J, Zotz RB, Scharf RE, Willers R, Schechter AN, Feelisch M, Kelm M. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med. 2006;40:295–302. doi: 10.1016/j.freeradbiomed.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Koskolou MD, Calbet JA, Radegran G, Roach RC. Hypoxia and the cardiovascular response to dynamic knee-extensor exercise. Am J Physiol Heart Circ Physiol. 1997;272:H2655–H2663. doi: 10.1152/ajpheart.1997.272.6.H2655. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Kirby BS, Crecelius AR, Carlson RE, Voyles WF, Dinenno FA. Combined inhibition of nitric oxide and vasodilating prostaglandins abolishes forearm vasodilatation to systemic hypoxia in healthy humans. J Physiol. 2011;589:1979–1990. doi: 10.1113/jphysiol.2011.205013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM. The Joan Mott Prize Lecture. The integrated response to hypoxia: from circulation to cells. Exp Physiol. 1999;84:449–470. [PubMed] [Google Scholar]
- Mian R, Marshall JM. The role of adenosine in dilator responses induced in arterioles and venules of rat skeletal muscle by systemic hypoxia. J Physiol. 1991;443:499–511. doi: 10.1113/jphysiol.1991.sp018847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Bune L, Saltin B, Pilegaard H, Hellsten Y. ATP induced vasodilatation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins and adenosine. Am J Physiol Regul Integr Comp Physiol. 2009a;296:R1140–R1148. doi: 10.1152/ajpregu.90822.2008. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol. 2007;581:853–861. doi: 10.1113/jphysiol.2006.127423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP, Nyberg M, Thaning P, Saltin B, Hellsten Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension. 2009b;53:993–999. doi: 10.1161/HYPERTENSIONAHA.109.130880. [DOI] [PubMed] [Google Scholar]
- O'Leary DS, Dunlap RC, Glover KW. Role of endothelium-derived relaxing factor in hindlimb reactive and active hyperemia in conscious dogs. Am J Physiol Regul Integr Comp Physiol. 1994;266:R1213–R1219. doi: 10.1152/ajpregu.1994.266.4.R1213. [DOI] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JA, Saltin B. Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol. 1999;276:H438–H445. doi: 10.1152/ajpheart.1999.276.2.H438. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Saltin B, Kiens B, Christensen NJ. Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am J Physiol Heart Circ Physiol. 1986;251:H1038–H1044. doi: 10.1152/ajpheart.1986.251.5.H1038. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Eisenach JH, Joyner MJ. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J Physiol. 2007;579:227–236. doi: 10.1113/jphysiol.2006.124313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol. 2004;557:599–611. doi: 10.1113/jphysiol.2004.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG, Wilkins BW, Johnson CP, Eisenach JH, Limberg JK, Dietz NM, Curry TB, Joyner MJ. Roles of nitric oxide synthase and cyclooxygenase in leg vasodilation and oxygen consumption during prolonged low-intensity exercise in untrained humans. J Appl Physiol. 2010;109:768–777. doi: 10.1152/japplphysiol.00326.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation induced in the rat by acute systemic hypoxia. J Physiol. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Victor RG, Seals DR. Reflex stimulation of sympathetic outflow during rhythmic exercise in humans. Am J Physiol Heart Circ Physiol. 1989;257:H2017–H2024. doi: 10.1152/ajpheart.1989.257.6.H2017. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Pike TL, Martin EA, Curry TB, Ceridon ML, Joyner MJ. Exercise intensity-dependent contribution of β-adrenergic receptor-mediated vasodilatation in hypoxic humans. J Physiol. 2008;586:1195–1205. doi: 10.1113/jphysiol.2007.144113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BW, Schrage WG, Liu Z, Hancock KC, Joyner MJ. Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol. 2006;101:1343–1350. doi: 10.1152/japplphysiol.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]