

Abstract
The adenomatous polyposis coli (APC) gene is considered as the true gatekeeper of colonic epithelial proliferation: It is mutated in the majority of colorectal tumors, and mutations occur at early stages of tumor development in mouse and man. These mutant proteins lack most of the seven 20-amino-acid repeats and all SAMP motifs that have been associated with down-regulation of intracellular β-catenin levels. In addition, they lack the carboxy-terminal domains that bind to DLG, EB1, and microtubulin. APC also appears to be essential in development because homozygosity for mouse Apc mutations invariably results in early embryonic lethality. Here, we describe the generation of a mouse model carrying a targeted mutation at codon 1638 of the mouse Apc gene, Apc1638T, resulting in a truncated Apc protein encompassing three of the seven 20 amino acid repeats and one SAMP motif, but missing all of the carboxy-terminal domains thought to be associated with tumorigenesis. Surprisingly, homozygosity for the Apc1638T mutation is compatible with postnatal life. However, homozygous mutant animals are characterized by growth retardation, a reduced postnatal viability on the B6 genetic background, the absence of preputial glands, and the formation of nipple-associated cysts. Most importantly, Apc1638T/1638T animals that survive to adulthood are tumor free. Although the full complement of Apc1638T is sufficient for proper β-catenin signaling, dosage reductions of the truncated protein result in increasingly severe defects in β-catenin regulation. The SAMP motif retained in Apc1638T also appears to be important for this function as shown by analysis of the Apc1572T protein in which its targeted deletion results in a further reduction in the ability of properly controlling β-catenin/Tcf signaling. These results indicate that the association with DLG, EB1, and microtubulin is less critical for the maintenance of homeostasis by APC than has been suggested previously, and that proper β-catenin regulation by APC appears to be required for normal embryonic development and tumor suppression.
Keywords: Tumorigenesis, β-catenin, SAMP, Apc, development
Germ-line mutations at the adenomatous polyposis coli (APC) gene lead to familial adenomatous polyposis (FAP), an autosomal dominant condition characterized by the development of hundreds to thousands of colorectal adenomatous polyps in the second to third decade of life (Groden et al. 1991; Nishisho et al. 1991). Virtually all polyps are characterized by a somatic mutation at the wild-type APC allele (Ichii et al. 1993; Levy et al. 1994). Somatic APC mutations have also been observed in >80% of sporadic adenomas and carcinomas (Miyoshi et al. 1992; Powell et al. 1992; Smith et al. 1993). Thus, mutation of the APC gene represents a very early event in the multistep process of colorectal cancer progression. FAP patients are also at increased risk for several extraintestinal manifestations (Talbot 1994). These include desmoids, hepatoblastomas, osteomas, endocrine adenomas, brain tumors, and cutaneous cysts. The development of these lesions underscores the importance of APC in maintaining homeostasis in many different tissue types derived from all three embryonic lineages.
The APC gene encodes a 2843-amino-acid polypeptide shown to participate in a number of cellular processes ranging from cell cycle regulation, to apoptosis, cell adhesion, cell migration, microtubule assembly, and cell fate determination (Munemitsu et al. 1994; Baeg et al. 1995; Rubinfeld et al. 1995; Morin et al. 1996; Näthke et al. 1996). These functions are accomplished by a number of functional domains scattered throughout the APC protein (see also Fig. 1D). At its most amino-terminal end, the APC protein contains a coiled-coil domain involved in dimerization of the protein (Joslyn et al. 1993; Su et al. 1993b), followed by seven Armadillo repeats of yet unknown function (Polakis 1997). The most obvious feature of the central domain of APC is the presence of three 15-amino-acid repeats followed by a 20-amino-acid motif repeated seven times. These repeats have been shown to bind to and down-regulate β-catenin, respectively (Rubinfeld et al. 1993; Su et al. 1993a; Munemitsu et al. 1995). β-Catenin is well known not only for its function in regulating cell–cell adhesion by binding to cadherins, but also for its role as a signaling molecule in the Wnt signal transduction pathway (for review, see Willert and Nusse 1998). β-Catenin associates with members of the lymphoid enhancer factor/T-cell factor (LEF/TCF) family of transcriptional activators and thereby modulates the transcription of Wnt target genes (Behrens et al. 1996; Molenaar et al. 1996; Korinek et al. 1997; Morin et al. 1997). In the absence of a Wnt signal, APC, on phosphorylation by glycogen synthase kinase 3β (GSK3β), promotes the down-regulation of β-catenin, thereby preventing its signaling activity (Rubinfeld et al. 1996). Recently, two additional proteins, conductin and axin, have also been implicated in the regulation of β-catenin. Beside associating with β-catenin and GSK3β, they interact with APC polypeptides containing a Ser-Ala-Met-Pro (SAMP) motif present three times in its central domain (Behrens et al. 1998; Hart et al. 1998; Nakamura et al. 1998). The carboxyl terminus of APC has been shown to bind to several proteins including homologs of the Drosophila discs large tumor suppressor protein (DLG), EB1-like proteins, microtubulin, and possibly to p34cdc2 (Munemitsu et al. 1994; Smith et al. 1994; Su et al. 1995; Matsumine et al. 1996; Makino et al. 1997; Polakis 1997; Renner et al. 1997; Trzepacz et al. 1997).
Figure 1.




(A) Generation of the Apc1638T mutation. Schematic drawing of exon 15 of the mouse Apc gene, the targeting vector, and the resulting mutant allele. The PGK–hygromycin cassette was inserted at the SmaI site corresponding to codon 1638. Some of the restriction sites used for cloning or Southern analysis are shown. For the generation of the Apc1572T mutation, the PGK–hygromycin cassette was inserted between the same SmaI site and the SspI site corresponding to codon 1572. (B) Southern blot analysis of BglII and HindIII-digested DNA isolated from the various Apc-targeted ES clones, hybridized with a 3-kb fragment encompassing the 5′ half of exon 15. Molecular sizes of wild-type and mutant fragments are depicted. (Lane 1) Apc+/+; (lane 2) Apc+/1638T; (lane 3) Apc+/1572T; (lane 4) Apc1638N/1572T. (C) Western blot analysis of the various Apc-targeted ES clones. Molecular sizes of wild-type and mutant fragments are depicted. (Lane 1) Apc+/+; (lane 2) Apc+/1638T; (lane 3) Apc1638T/1638T; (lane 4) Apc1638N/1638T; (lane 5) Apc1638N/1638N; (lane 6) Apc+/1572T; (lane 7) Apc1638N/1572T. (D) Schematic representation of the 2843-amino-acid full-length Apc and the truncated 1638 and 1572-amino-acid polypeptides. The Apc1638T polypeptide lacks the carboxy-terminal domains that bind to tubulin, DLG, EB1-like proteins, and possibly p34cdc2. In addition, four of seven β-catenin down-regulating repeats and two of three conductin/axin-binding motifs are absent. The Apc1572T polypeptide also lacks the last remaining conductin/axin-binding motif.
The vast majority of both germ-line and somatic mutations at the APC gene are clustered in the 5′ half and predict the truncation of the protein product (Miyoshi et al. 1992; Nagase and Nakamura 1993). These truncated proteins have been observed by Western analysis of lymphoblastoid and colorectal cancer cell lines carrying chain-terminating mutations up to codon 1577 (Smith et al. 1993; Gismondi et al. 1998). Mutations beyond codon 1600 are rare, and appear to result in undetectable levels of the corresponding truncated polypeptides (Eccles et al. 1996; van der Luijt et al. 1996). Therefore, the mutation spectrum observed at the APC gene suggests that the carboxy-terminal domains and part of the β-catenin regulatory domains are critical for its tumor-suppressing function. However, the precise function of the carboxy-terminal domains in relation to homeostasis and tumorigenesis is still poorly understood.
Several mouse models have been generated by introducing specific mutations into the murine Apc gene. A severe intestinal phenotype characterized by the development of >100 intestinal tumors is observed in mice heterozygous for the Min (multiple intestinal neoplasia) and ApcΔ716 mutations (Moser et al. 1990; Su et al. 1992; Oshima et al. 1995). Mice heterozygous for the Apc1638N mutation are characterized by a milder intestinal tumor phenotype (5–6 tumors per animal) in addition to a broad spectrum of extraintestinal manifestations including desmoids and cutaneous cysts (Fodde et al. 1994; Smits et al. 1998). Whereas the ApcΔ716 and Min alleles result in stable truncated proteins of 716 and 850 amino acids, respectively, the Apc1638N mutation results in residual amounts of a 182-kD truncated protein not detectable by conventional Western analysis (M. Kielman, R. Smits, C. Breukel, W. Edelmann, R. Kucherlapati, and R. Fodde, in prep.).
Homozygosity for the above Apc mutations invariably results in embryonic lethality before day 8 of gestation, suggesting an essential role for Apc during development (Fodde et al. 1994; Moser et al. 1995; Oshima et al. 1995).
Here we report the generation and phenotypic analysis of the Apc1638T mouse model characterized by a stable 182-kD truncated protein comprising the amino-terminal 1638 amino acids of Apc. In sharp contrast with the above Apc mouse models, homozygosity for the Apc1638T mutation is compatible with postnatal life. Surprisingly, heterozygous as well as homozygous Apc1638T animals are tumor free. Therefore, the Apc1638T model delineates critical regions of the Apc protein involved in tumorigenesis and development, indicating that most of the carboxy-terminal domains of Apc are less essential for its function in homeostasis than has been suggested previously.
Results
Generation of the Apc1638T mouse model
We introduced a specific Apc chain-terminating mutation into the mouse germ line by gene targeting in ES cells. The targeting vector contained a 10.6-kb genomic SphI fragment encompassing exon 15 of the mouse Apc gene. A PGK–hygromycin cassette was inserted in the same transcriptional orientation as Apc into a unique SmaI site corresponding to codon 1638 of the endogenous gene (Fig. 1A). In total, 113 hygromycin-resistant ES cell clones were screened by Southern analysis with a 3.0-kb fragment encompassing the 5′ half of exon 15. Seven clones showed the 5.5-kb BglII and 5.7-kb HindIII fragments indicative of the correct targeting event (Fig. 1B, lane 2). Western analysis showed that this mutation results in a stable truncated protein of 182 kD (Fig. 1C, lane 2). Therefore, we will hereafter refer to this mutation as Apc1638T (T for truncated). The truncated protein lacks the carboxy-terminal domains that bind to tubulin, DLG, EB1-like proteins, and possibly p34cdc2. In addition, only three of the seven 20-amino-acid β-catenin down-regulating repeats and one of the three conductin/axin-binding motifs are retained in the truncated protein (Fig. 1D). Successfully targeted Apc+/1638T ES cells were used to generate Ola129xB6 mice heterozygous for the Apc1638T mutation.
Generation of Apc1638T/1638T, Apc1638N/1638T, and Apc1638N/1638N ES cells
We have described previously the generation of the Apc1638N mouse model and the broad tumor phenotype associated with this mutation (Fodde et al. 1994; Smits et al. 1998). The PGK–neomycin selectable marker used for the generation of the Apc1638N mutation was inserted at the same codon 1638 of the endogenous mouse Apc gene but in the transcriptional orientation opposite to that of Apc. Western blot analysis of the targeted ES cells and of several tissues derived from Apc+/1638N mice failed to demonstrate the predicted truncated protein (Fodde et al. 1994). This result is in contrast with the Apc1638T mutation, in which the insertion of the hygromycin gene in the same transcriptional orientation as Apc results in the stable expression of the 182-kD truncated protein. The main differences between the two targeting constructs used are the type of selectable marker and the transcriptional orientation of the marker relative to that of the Apc gene. Subsequent investigations conducted to differentiate between these two possibilities revealed that the presence of the truncated protein was dependent solely on the transcriptional orientation of the selection cassette used. Only when either the PGK–neomycin or the PGK–hygromycin gene is inserted in the same transcriptional orientation as Apc is the truncated Apc protein detectable. On the other hand, when either selectable marker is inserted in a transcriptional orientation opposite to that of Apc no truncated protein is detectable by Western analysis (Fodde et al. 1996). Presumably, the latter orientation results in the generation of antisense RNA by the selectable marker complementary to the targeted Apc1638N transcript. By using two successive rounds of targeting with both markers in either the antisense or the sense transcriptional orientation as Apc or a combination of both orientations, we successfully obtained Apc1638T/1638T, Apc1638N/1638T, and Apc1638N/1638N ES cells (Fig. 1C, lanes 3–5).
Mice homozygous for the Apc1638T mutation are viable
Surprisingly, when Ola129xB6 mice heterozygous for the Apc1638T mutation were intercrossed, homozygous mice were obtained at the expected Mendelian frequencies (21 Apc+/+, 52 Apc+/1638T, 20 Apc1638T/1638T). As all Apc mouse models to date are characterized by an early embryonic lethality of homozygous mutant animals, the Apc1638T mutation represents the first example of an Apc truncated protein that retains the essential functions necessary for normal embryonic development.
To validate the correct expression of the 182-kD truncated protein, Western blot analyses were performed on protein lysates derived from brain, liver, and duodenum of all genotypes. A polyclonal antibody raised against amino acids 8–312 of the APC protein (AFPN) detected both the full-length 312-kD and the truncated 182-kD proteins as expected. Probing of the same lysates with an affinity-purified polyclonal antibody raised against amino acids 2260–2843 (AFPC3), revealed the wild-type Apc protein in tissues derived from Apc+/+ and Apc+/1638T, whereas no protein band was detected in lysates of Apc1638T/1638T mice (Fig. 2A).
Figure 2.

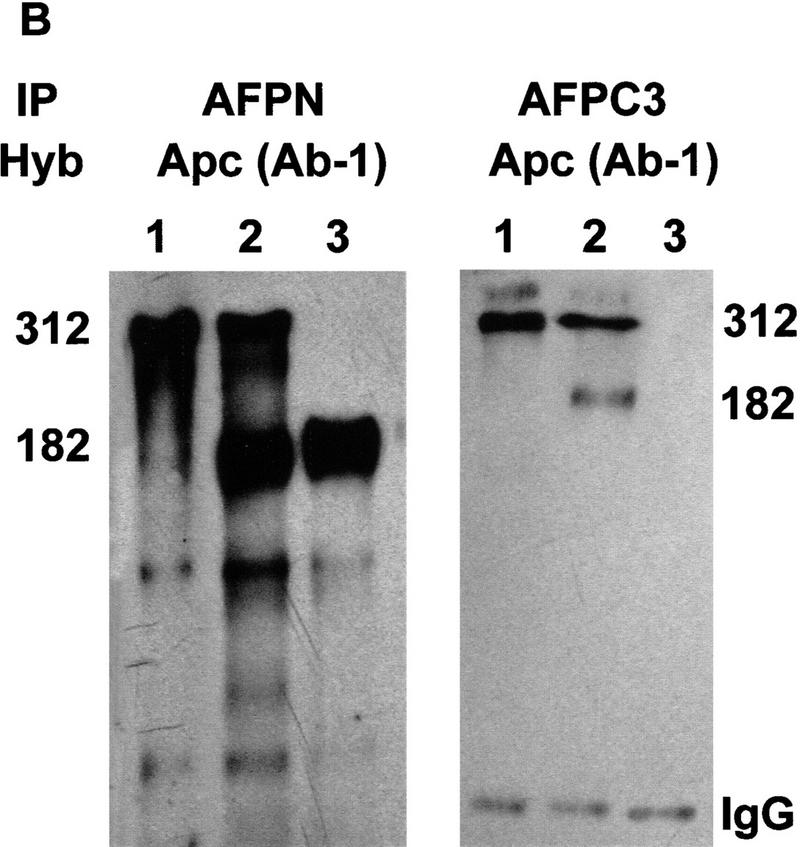
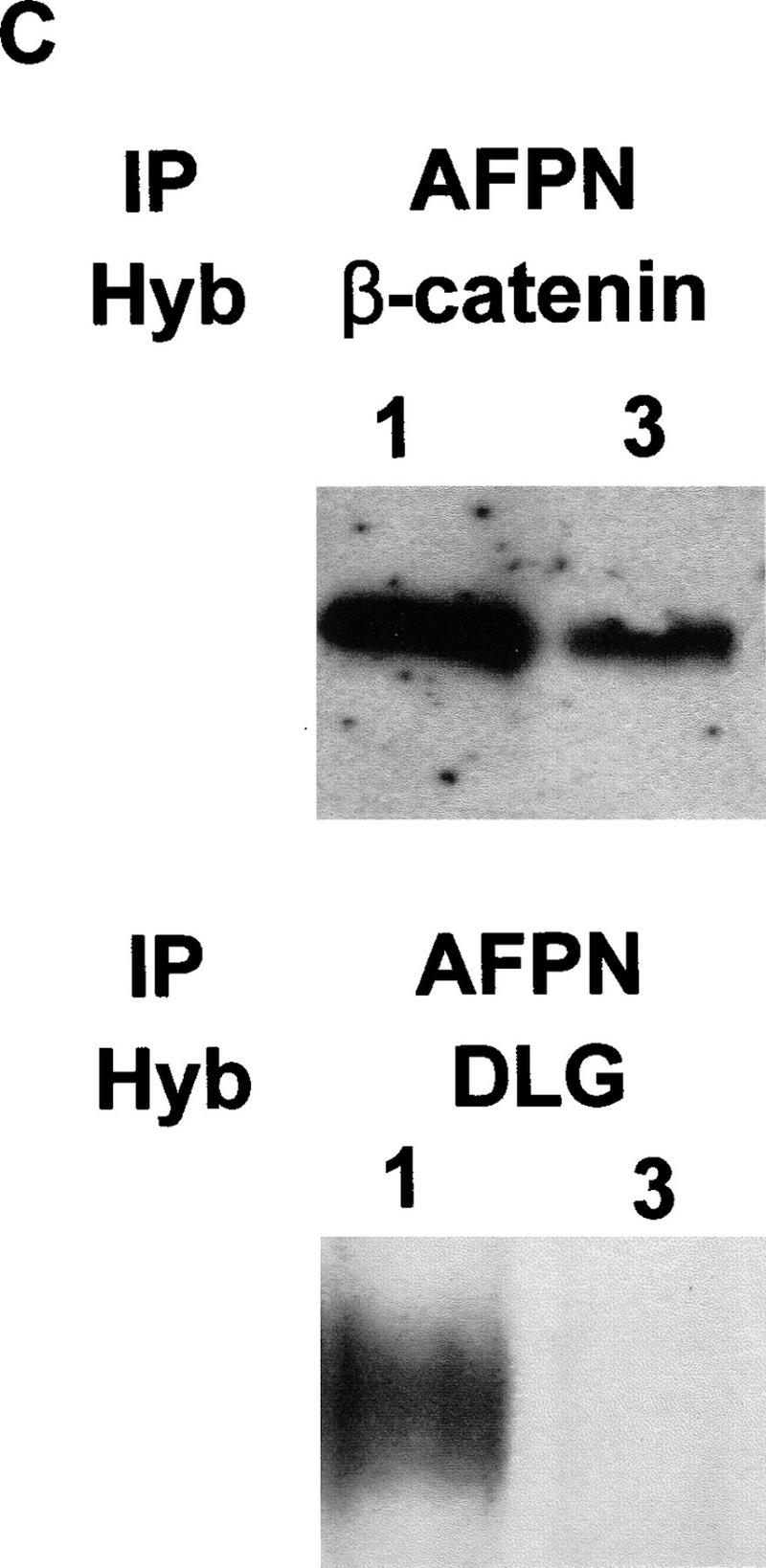
Protein analysis of the Apc1638T mutation. (A) Total protein lysates derived from brains of Apc+/+ (lane 1), Apc+/1638T (lane 2), and Apc1638T/1638T (lane 3) animals were either hybridized with AFPN (left) or AFPC3 (right). Similar results were obtained for liver and duodenum. (B) Apc+/+, Apc+/1638T, and Apc1638T/1638T ES cells were immunoprecipitated with either AFPN or AFPC3, and subsequently hybridized with APC (Ab-1) recognizing an amino-terminal epitope. In Apc+/1638T cells, immunoprecipitation of the full-length Apc with the AFPC3 antibody results in coprecipitation of the Apc1638T protein because of dimerization through the amino-terminal end of Apc. (C) The AFPN immunoprecipitations performed on the ES cells were hybridized with β-catenin and DLG monoclonals, showing that the Apc1638T protein is still capable of binding β-catenin, but does not associate with DLG.
Immunoprecipitation analysis performed with AFPN and AFPC3 on Apc+/+, Apc+/1638T, and Apc1638T/1638T ES cell lines confirmed the results obtained by Western analysis of the tissue lysates (Fig. 2B). Because the Apc1638T protein can still dimerize with full-length Apc, it is coprecipitated in Apc+/1638T cells by the AFPC3 antibody (Fig. 2B, lane 2). Probing both the AFPN and AFPC3 immunoprecipitates with AFPC3 again revealed only the full-length Apc protein in Apc+/+ and Apc+/1638T ES cells, whereas no additional band was detected in homozygous mutant cells (result not shown). Therefore, we conclude that the Apc1638T targeted mutation encodes a 182-kD truncated protein encompassing amino acids 1–1638 of wild-type Apc and that the carboxy-terminal third of the full-length protein is not expressed in Apc1638T/1638T cellular lysates.
Apc1638T and β-catenin regulation
One of the most important functional aspects of Apc is its interaction with β-catenin. The Apc1638T protein still contains all three 15-amino-acid β-catenin binding repeats, three of seven 20-amino-acid β-catenin down-regulating repeats and one of the three conductin/axin-binding motifs. As shown in Figure 2C, β-catenin was coprecipitated with the truncated protein. Moreover, staining of Apc+/+ and Apc1638T/1638T mouse embryonic fibroblasts (MEFs) and ES cells with a β-catenin monoclonal antibody revealed no significant differences in β-catenin levels or subcellular distribution (data not shown).
To investigate whether Apc1638T is still capable of regulating β-catenin/Tcf signaling, transient transfections were performed with a luciferase reporter construct under the control of either optimal (pTOPFLASH) or mutant Tcf-binding motifs (pFOPFLASH). In cells in which this controlling function of Apc is lost, β-catenin can translocate to the nucleus and form a complex with Tcf that specifically enhances transcription of the pTOPFLASH reporter, while leaving the expression of pFOPFLASH unaltered (Korinek et al. 1997). As shown in Figure 3, no increased transcriptional activity of the pTOPFLASH reporter was observed in MEFs of all three genotypes. The same experiment was repeated on Apc+/+, Apc1638T/1638T, Apc1638N/1638T, and Apc1638N/1638N ES cells. Because the Apc1638N allele encodes extremely low levels of the truncated 182-kD protein, it is expected to be defective in β-catenin regulation, thereby leading to enhanced signaling. Accordingly, a 30-fold increase of the transcriptional activity of pTOPFLASH over its mutant pFOPFLASH control was observed in Apc1638N/1638N ES cells (Fig. 3). No specific activation of the pTOPFLASH reporter was observed in wild-type or in Apc1638T/1638T ES cells. In Apc1638N/1638T ES cells haploinsufficient for the Apc1638T protein, a 2.4-fold increase of pTOPFLASH activity was observed. Thus, the Apc1638T protein appears to be sufficient in regulating β-catenin/Tcf signaling only when expressed at wild-type levels, but is slightly impaired in this function when expressed at half this dosage.
Figure 3.
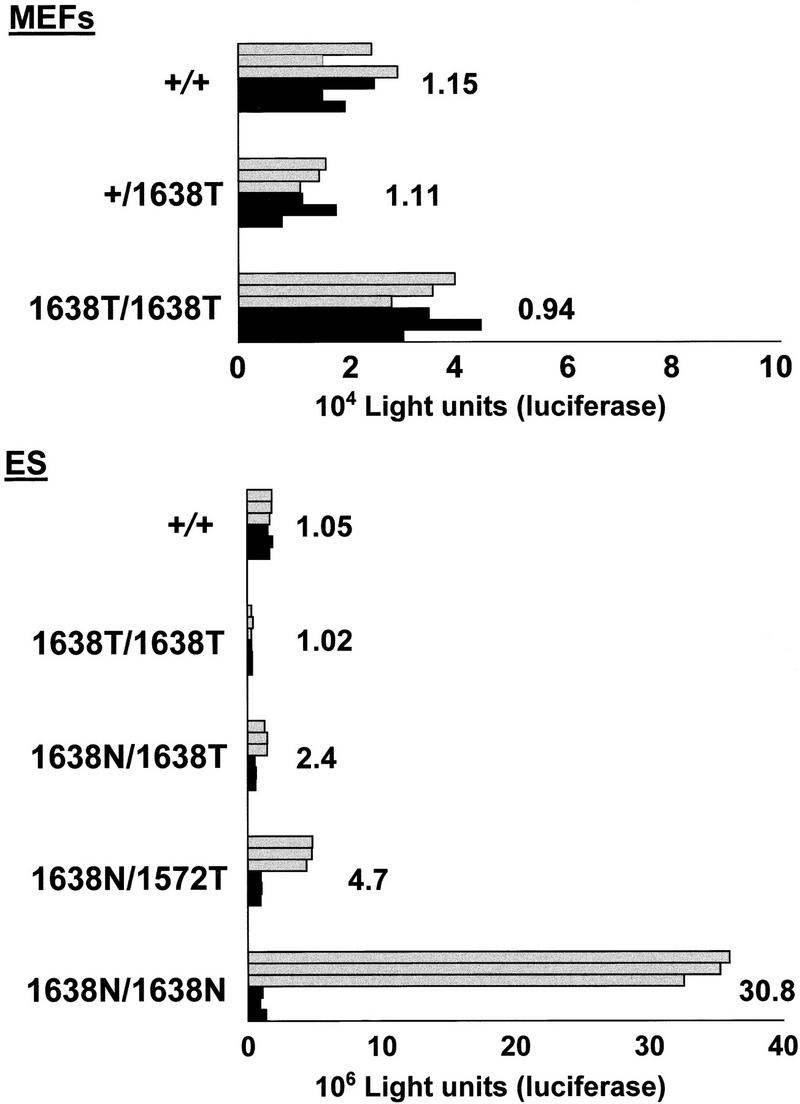
β-Catenin/Tcf reporter assays performed in MEFs and ES cells of the depicted genotypes. Approximately 1 × 105 MEFs or 1 × 106 undifferentiated ES cells were transfected with either 0.75 μg of pTOPFLASH (shaded bars) or pFOPFLASH (solid bars) luciferase reporter construct, and cotransfected with 0.75 μg of CMV-galactosidase serving as an internal control (Korinek et al. 1997). All assays were performed as triplicate transfections as shown. pTOPFLASH/PFOPFLASH ratios are depicted for each cell line. ES cells homozygous for the Apc1638N mutation are clearly defective in β-catenin regulation, as demonstrated by an average 30-fold increase of the transcriptional activity of pTOPFLASH over its mutant pFOPFLASH control. In contrast, the Apc1638T protein appears to be functional both in MEFs and ES cells when expressed at wild-type levels. In Apc1638N/1638T ES cells haploinsufficient for the Apc1638T protein, a twofold increase of pTOPFLASH activity was observed. Apc1572T is more severly impaired in β-catenin down-regulation than Apc1638T, as demonstrated by an average twofold increase of pTOPFLASH activity in the Apc1638N/1572T ES line when compared with the Apc1638N/1638T clone.
The Apc1638T protein differs from the observed truncated proteins in FAP patients and in colorectal tumors, in the presence of the conductin/axin-binding SAMP motif. To investigate the contribution of this motif to the β-catenin down-regulating function of Apc1638T, we generated a novel truncated allele, Apc1572T, by homologous recombination in ES cells. In this case, the targeting construct is identical to Apc1638T with the exception of a 197-bp SspI–SmaI deletion encompassing the first SAMP motif and corresponding to codon 1572 (Fig. 1A,D). In total, 24 of 169 clones showed the 5.3-kb BglII and 5.5-kb HindIII fragments, indicative of a correct targeting event (Fig. 1B, lane 3). Western analysis of the latter revealed the presence of a 175-kD truncated protein in 14 of these lines (Fig. 1C, lane 6). A pool of the Apc+/1572T ES clones was subsequently targeted with the Apc1638N-targeting construct to obtain compound Apc1638N/1572T ES lines, which can be used to determine the β-catenin down-regulating activity of the novel truncated Apc1572T protein. Two clones showing the correct BglII and HindIII restriction fragments and the 175-kD truncated protein were obtained on screening of 230 G418-resistant clones (Fig. 1, B, lanes 4, and C, lane 7). One of these clones was selected for use in the β-catenin/Tcf reporter assay described above. An average twofold increase of pTOPFLASH activity was observed in the Apc1638N/1572T ES line when compared with the Apc1638N/1638T clone, demonstrating that Apc1572T is more severely impaired in β-catenin down-regulation than Apc1638T. Therefore, the unique SAMP motif retained in Apc1638T contributes significantly to the down-regulation of β-catenin.
Subcellular localization of Apc-interacting proteins
As a number of interactions between Apc and other proteins have been mapped to the carboxy-terminal third of Apc downstream of codon 1638, we performed immunohistochemical analysis of Apc+/+ and Apc1638T/1638T MEFs with monoclonal antibodies recognizing β-tubulin, EB-1, and discslarge (DLG). This analysis revealed no obvious differences in the subcellular distribution of the above proteins between the two cell lines (Fig. 4). The localization of tubulin throughout the cytoplasm and within the cellular protrusions in which it has been shown to colocalize with Apc (Näthke et al. 1996; Barth et al. 1997; Morrison et al. 1997; Pollack et al. 1997) was unaltered in Apc1638T/1638T MEFs. EB-1 was localized to the microtubule network in a similar fashion in both cell lines, showing that EB-1 localization is not dependent on association with Apc, which is in agreement with previous reports (Berrueta et al. 1998; Morrison et al. 1998). Staining for DLG showed a more intense staining at the cytoplasmic membrane and an occasional perinuclear staining but with no clear differences between the two lines. Probing of the Apc+/+ Apc1638T/1638T immunoprecipitates of Figure 2B with DLG-specific antibodies confirmed the expected inability of Apc1638T binding to DLG (Fig. 2C). A similar experiment with a commercially available antibody raised against EB1 was inconclusive as no interaction with EB1 was detected in either cell line. Similarly, attempts to localize the endogenous levels of Apc1638T protein with various amino-terminal antibodies were not successful.
Figure 4.
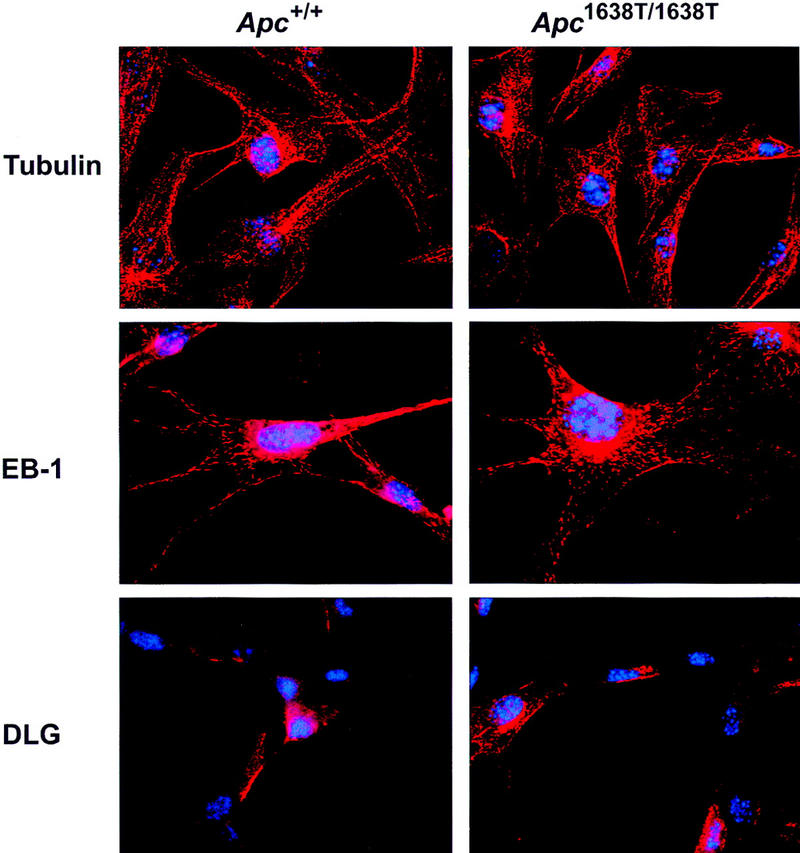
Immunolocalization of β-tubulin, EB-1, and DLG in Apc+/+ and Apc1638T/1638T MEFs. Nuclei are counterstained with DAPI. Original magnification, 630x.
Nuclear localization of the Apc1638T protein
It has been shown that the full-length APC protein can localize to the nucleus. Two putative nuclear localization signals present at amino acids 1773 and 2054 were originally implicated for this nuclear transport (Neufeld and White 1997). However, mutant APC proteins expressed in SW480 and HT29 that do not encompass these nuclear localization signals, have also been detected in the nucleus (Efstathiou et al. 1998; K. Neufeld, pers. comm.). The subcellular localization of the Apc1638T protein was investigated by cell fractionation of homozygous mutant MEFs. As shown in Figure 5, the mutant protein was detectable in the nuclear as well as the membrane/cytoskeletal fraction, but not in the cytoplasmic sample. This observation represents further evidence that the two putative nuclear localization signals present at amino acids 1773 and 2054 are not essential for translocation of APC to the nucleus.
Figure 5.

The Apc1638T protein localizes to both membrane/cytoskeletal and nuclear cell fractions in Apc1638T/1638T MEFs. Following lysis and fractionation, equal amounts of total protein from the different fractions were analyzed by SDS-PAGE and immunoblotting with APC (Ab-1) antibody. The Apc1638T protein is present in the total sample (T), the membrane/cytoskeleton (M), and the nucleus (N) but not in the cytoplasm (C).
Apc1638T mice are tumor free
The most consistent feature of all Apc mouse models described to date is a fully penetrant predisposition to intestinal tumors. The tumor susceptibility of Apc1638T was investigated in 25 Apc1638T/1638T, 22 Apc+/1638T, and 25 Apc+/+ littermates of mixed Ola129/B6 genetic background, with all mice being older than 12 months and with equal contribution of both sexes. Mean age of analysis of all three genotypes was between 15 and 17 months of age. In addition, 21 Apc+/1638T animals were investigated on a B6 genetic background (N9–N12) between 9 and 12.5 months of age with a mean age of analysis of 10.5 months. Animals were subjected to a complete gross necroscopy with special attention to lesions of the intestinal mucosa. Gross lesions were sampled and examined microscopically. Only 4 of 25 homozygous mutant animals had developed a single tumor diagnosed as an osteosarcoma, a follicular center-cell lymphoma, and a hepatocellular adenoma and carcinoma, respectively. Of the 22 Ola129/B6-Apc+/1638T animals, 3 mice showed a single tumor diagnosed as a follicular center cell lymphoma, a broncho-alveolar carcinoma and a hepatocellular adenoma. In addition, a single intestinal adenoma and follicular center cell lymphoma were observed in a 29-month-old heterozygous female. In the 25 wild-type control animals, similar tumor numbers and types were observed. These included two follicular center-cell lymphomas, a hepatocellular adenoma, and a squamous cell papilloma of the lip. Of the 21 B6-Apc+/1638T animals, a single female of 12.5 months of age had developed two 1.5-mm intestinal adenomas, whereas no abnormalities were observed in the other animals. Both the number and type of lesions observed in the Apc1638T animals do not differ significantly from those expected in aging mice (Zurcher et al. 1982; Smith et al. 1973). Therefore, these results indicate that the Apc1638T mutation does not confer a significantly increased predisposition to tumor development.
Absence of preputial glands in Apc1638T/1638T animals
Although Apc1638T mice are not characterized by an increased tumor susceptibility, homozygous mutant animals are affected by a limited number of developmental abnormalities. During a detailed search for pathological changes, it was noted that, in all Apc1638T/1638T males studied, the preputial glands were absent. These accessory-modified sebaceous glands are located between the skin and the abdominal muscles on either side of the genital bulb. Their function consists mainly in the production of sebum used in marking and sexual behavior. These glands arise at day 14 of gestation near the preputial fold as two primary epithelial down-growths into the dermis, thereby giving rise to a hair follicle. Next, the epithelial down-growth forms other secondary outgrowths. At first, no difference is apparent between the anlagen of both sexes. Later on, the male gland develops faster under the influence of testosterone. Postnatally, the female secondary outgrowth remains small and develops into the clitoral gland, whereas the male counterpart differentiates into the larger preputial gland (Cunha 1975). To our knowledge, three mouse models have been described with developmental abnormalities of the preputial gland, namely the X-linked Tabby (Ta) and testicular feminization (Tfm) mutations (Drews 1975), and a Hoxd13 (Spdh) mutant (Johnson et al. 1998). In Spdh mice of both sexes and in Ta males, the preputial gland anlage is completely absent, whereas in Tfm males, the glands are formed but are unable to respond to testosterone and therefore remain rudimentary as in the female.
Serial transversal sections were obtained from the lower portion of Apc1638T/1638T male and female embryos at 18.5 days of gestation, and from their wild-type littermates. As shown in Figure 6, a preputial gland anlagen is clearly present near the genital tubercle in the wild-type mice, whereas it is completely absent in the homozygous mutant mice. This observation suggests that, as in Ta and Spdh, the initial primary downgrowth is not formed in Apc1638T mutant mice or cannot be distinguished from the normally occurring primitive hair follicles. However, other characteristic features of Ta, such as the hair growth abnormalities and the absence of sweat glands in the footpads, or the malformations of the feet in Spdh mice, are not observed in Apc1638T (data not shown).
Figure 6.
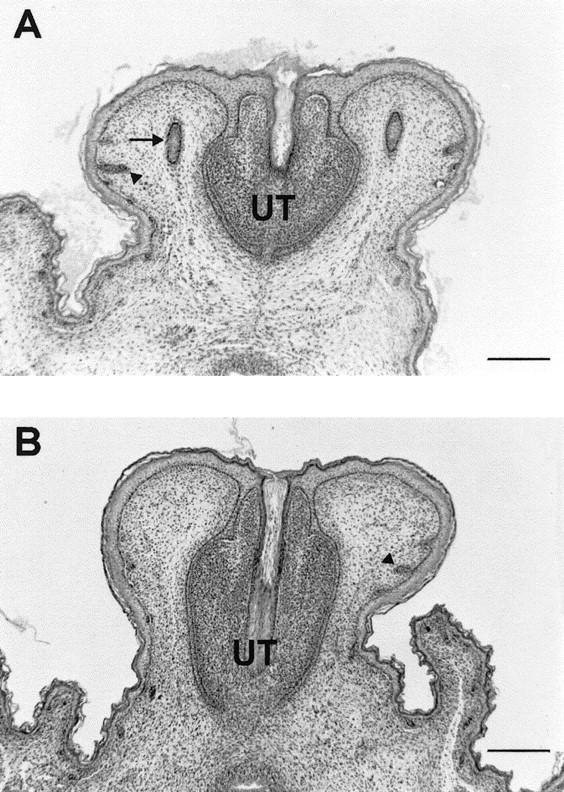
Hematoxylin and eosin-stained cross sections of the genital tubercle of a 18.5-day-old wild-type male embryo (A) and a homozygous Apc1638T male littermate (B). Two preputial gland anlagen (arrow) are clearly present in the mesenchyme adjacent to the urethra (UT) of the wild-type embryo, whereas they are completely absent at the corresponding position in the homozygous mutant embryo. In both embryos, primitive hair follicles (arrowheads) have formed with no apparent histological difference (Bar, 250 μm).
Nipple-associated cutaneous cysts in Apc1638T mutant animals
The vast majority of Apc1638T/1638T males develops one to three cutaneous cysts ranging in size from 1 to 3 mm, which become more prominent in older animals. This phenotypic abnormality is less clear among females in which a cyst smaller than 1 mm has been observed only twice among 11 Apc1638T/1638T females older than 1 year. These cysts were not present in wild-type or heterozygous animals, with the exception of a single 0.5-mm cyst detected in a heterozygous female (n = 12). A remarkable and invariant feature of the Apc1638T cysts is that they are always present as a solitary structure in the proximity of a nipple. No cysts were observed in other parts of the skin, as it has been described for the Apc1638N mouse model (Smits et al. 1998). They represent hair follicle-related lesions, histologically defined as hybrid infundibular/matrical cysts lined by orthokeratinizing squamous epithelium and stretches of matrix cells, although the infundibular differentiation was predominant. In two cases, a connection with the skin surface was evident, a type of cyst often referred to as a dilated pore (Fig. 7). The histological analysis shows that all Apc1638T cysts appear to originate from the pilosebaceous unit, as it has been concluded for the Apc1638N model.
Figure 7.
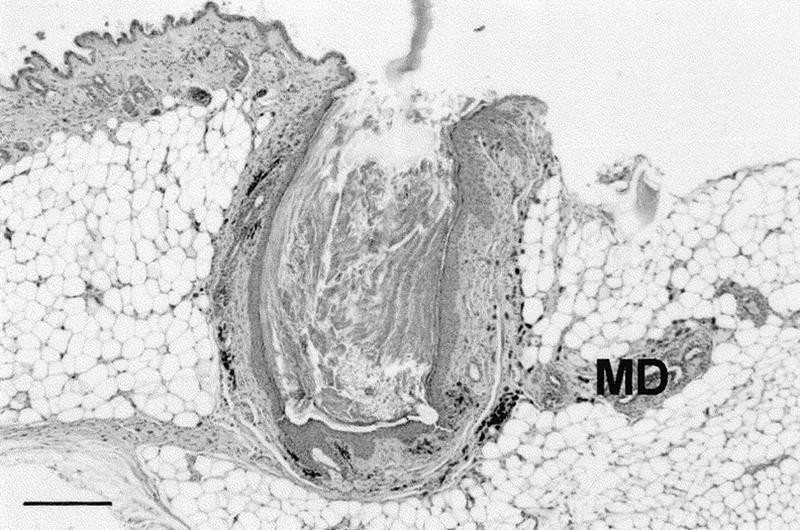
Histological appearance of a cutaneous cyst observed in a Apc1638T/1638T female. In this case, the predominantly infundibular epithelium of the cyst is in open connection with the skin surface. All Apc1638T cysts were present in the proximity of a nipple. Accordingly, some mammary ducts (MD) can be observed in the near vicinity of this cyst. Most of the other Apc1638T cysts were located deep in the dermis or in the subcutaneous fat layer above the panniculus carnosus, as it has been described for the Apc1638N mouse model (Smits et al. 1998). Section was stained with hematoxylin and eosin (Bar, 320 μm).
Postnatal growth retardation of homozygous Apc1638T animals
One of the most apparent developmental abnormalities of homozygous mutant Apc1638T animals is a postnatal growth retardation. Litters generated by matings of Apc+/1638T heterozygotes of mixed Ola129/B6 backgrounds were weighed on a weekly basis from ∼10 days of age up to 8 weeks. In total, 154 mice were analyzed: 31 Apc+/+ (9 males, 22 females), 88 Apc+/1638T (46 males, 42 females), and 35 Apc1638T/1638T (18 males, 17 females). A significant 20% growth retardation was already apparent in homozygous mutant animals of both sexes at 10 days of age (P = 0.001). This growth retardation became more pronounced in older mice. In contrast, heterozygous mice have body weights similar to those of wild-type mice. A growth curve relative to mice between 2 and 8 weeks of age of all three genotypes is shown in Figure 8.
Figure 8.

Growth curve of 35 Apc1638T/1638T mice (█) and their wild-type, (31, □) and heterozygous (88, ●) littermates. All mice were weighed on a weekly basis between 10 days and 8 weeks of age. Standard deviations are indicated as vertical bars. The average weights shown for each time point and genotype represent averages from both sexes. However, similar curves are obtained if the data are analyzed for each sex separately. Therefore, both male and female Apc1638T/1638T animals are characterized by a growth retardation.
To investigate whether the growth retardation of homozygous mutant animals is already present in utero, B6-Apc+/1638T animals were bred and the litters analyzed at 18.5 days of gestation. In total, 46 embryos derived from 6 litters were analyzed, 10 of which were Apc+/+, 23 Apc+/1638T, and 13 Apc1638T/1638T, with equal distribution of both sexes. The average weight was not significantly different among the three genotypes (+/+ 1.15 gram; +/− 1.11 gram; −/− 1.07 gram; P = 0.22). As observed with the mixed Ola129/B6 genetic background, B6-Apc1638T/1638T animals that survive to adulthood (see below) are clearly smaller than their heterozygous and wild-type littermates. Therefore, the growth retardation of Apc1638T/1638T animals becomes apparent postnatally.
Genetic background-dependent postnatal mortality of Apc1638T/1638T mice
As described above, intercrosses of heterozygous animals of mixed Ola129/B6 background resulted in the expected numbers of homozygous mutant mice at weaning. However, when the Apc1638T mutation was backcrossed to B6, it became apparent that with increasing genetic contribution of this inbred strain, a relative lower number of mutant animals survived to adulthood. Of a total of 17 litters derived from intercrosses of N5–N12 backcross generations, only 5 homozygous mutant animals were alive at weaning, compared with 63 heterozygous and 26 wild-type mice. Analysis of 1-day-old litters from B6-Apc+/1638T intercrosses revealed that 12 of 20 succumbed newborns were of the Apc1638T/1638T genotype. This result strongly suggests that homozygosity for the Apc1638T mutation on the B6 genetic background results in a reduced postnatal viability. With the exception of the macroscopic absence of milk in the stomachs of all succumbed animals, no other gross abnormalities were observed. Microscopic examination of five nonautolytic dead newborns and of seven 18.5 dpc embryos did not reveal any lesions or malformations that could explain the high postnatal mortality.
Discussion
The large number of APC mutations identified to date in FAP patients and in colorectal tumors denote a critical region that has to be deleted to trigger the tumorigenic process. In fact, the majority of the above mutations lead to the truncation of the APC protein upstream of the conductin/axin-binding SAMP motifs (Smith et al. 1993; Gismondi et al. 1998). On the basis of these observations, several investigators have concluded that the carboxy-terminal third of APC encompasses important tumor-suppressing functional domains. Of the few mutations located beyond the most amino-terminal SAMP motif, the ones that have been studied by Western analysis were shown not to result in the predicted truncated polypeptides (Eccles et al. 1996; van der Luijt et al. 1996).
Regulation of intracellular β-catenin levels is thought to represent one of the most important functions of the APC tumor suppressor protein. Three different motifs in the central domain of APC are responsible for this activity (Fig. 1D). The three 15-amino-acid repeats bind β-catenin, whereas the seven 20-amino-acid repeats both bind and down-regulate β-catenin. More recently, APC polypeptides containing only one of the three SAMP motifs present in the central portion of the full-length protein have been shown to bind to conductin and axin. Although the physical interaction of the SAMP motif has only been formally proven for conductin, on the basis of sequence homology, it is most likely that axin might also bind in a similar fashion. Both conductin and axin can form a multiprotein complex with APC, GSK3β, and β-catenin, thereby promoting the down-regulation of the latter (Behrens et al. 1998; Hart et al. 1998; Nakamura et al. 1998). The Apc1638T truncated protein encompasses three of the seven β-catenin down-regulating repeats and one of the three conductin/axin-binding SAMP motifs. As shown in Figure 3, regulation of intracellular β-catenin appears to be normal in Apc1638T/1638T ES cells and MEFs. Accordingly, heterozygous as well as homozygous mice do not show an increased tumor predisposition when compared with wild-type animals. However, increasingly reduced dosages of the same Apc1638T protein in Apc1638N/1638T and Apc1638N/1638N ES cells result in a 2- and 30-fold increase of pTOPFLASH activity when compared with its Apc1638T/1638T full complement. The targeted deletion of the remaining conductin/axin binding motif results in a further increase in pTOPFLASH activity. In this regard, it is interesting to note that germ-line APC mutations resulting in truncated proteins encompassing three 20-amino-acid repeats do lead to familial polyposis (Nagase et al. 1993; Gismondi et al. 1997). The latter truncated polypeptides, however, do not include any of the SAMP motifs. This suggests that under physiological circumstances, the multiprotein complex of APC, conductin/axin, GSK3β, and β-catenin is necessary for the accurate regulation of intracellular β-catenin levels and that this regulatory activity represents the main tumor suppressing function of APC.
The tumor-free phenotype of Apc1638T mice also indicates that deletion of the carboxy-terminal domains that bind to tubulin, DLG, and EB-1 does not directly predispose to tumor development. A number of investigations have focused on the putative role of APC in mediating microtubular-associated processes that are important for proper cell division and migration. APC fragments containing the microtubule-binding site can induce assembly and bundling of microtubules in vitro (Smith et al. 1994; Munemitsu et al. 1994), in which process EB-1 has also been implicated (Beinhauer et al. 1997; Muhua et al. 1998). Moreover, APC has been shown to cluster in cellular protrusions involved in active cell migration, a localization that was dependent on an intact microtubule cytoskeleton (Näthke et al. 1996; Barth et al. 1997; Morrison et al. 1997; Pollack et al. 1997). Taken together, these data are suggestive for an important role of APC in the assembly and stabilization of microtubules. However, the data presented here suggest that this postulated function of APC is not essential for its tumor-suppressing activity, or at least that the association with the microtubules via its carboxy-terminal domains may not be required in this respect. It remains plausible that, once β-catenin regulation is sufficiently impaired, deletion of the microtubule binding-site or of any other carboxy-terminal domains contributes to APC-driven tumorigenesis.
The early embryonic lethality of the Apc mutant mouse models described to date strongly suggests that Apc is required for normal embryonic development. In the Min, ApcΔ716 and Apc1638N models, embryonic lethality occurs shortly after implantation (Fodde et al. 1994; Moser et al. 1995; Oshima et al. 1995). The severe developmental defects of these Apc mutant mice contrast with the observed viability of Apc1638T homozygotes. Whereas the Min and ApcΔ716 truncated proteins only retain the dimerization domain and the Armadillo repeats, Apc1638T encompasses all three 15-amino-acid β-catenin-binding repeats, three of the seven down-regulating 20-amino-acid repeats and one of three SAMP repeats. These motifs apparently provide sufficient β-catenin regulation to ensure proper embryonic development.
Nevertheless, mice homozygous for the Apc1638T mutation are characterized by several developmental abnormalities such as growth retardation, a reduced postnatal viability on the B6 genetic background, the absence of preputial glands, and the formation of nipple-associated cysts. The causes for these abnormalities could be twofold. They could be the consequence of either the loss of function of the carboxy-terminal domains, or be due to a slightly reduced ability to properly control cytoplasmic β-catenin levels. Although the Apc1638T protein appears to normally regulate β-catenin in ES cells if expressed at the wild-type levels, its β-catenin down-regulating capacity is partly affected when the dosage of protein is reduced. Accordingly, the observation that compound heterozygous Apc1638N/1638T mice are embryonic lethal (M. Kielman, R. Smits, C. Breukel, W. Edelmann, R. Kucherlapati, and R. Fodde, in prep.) supports that dosage of the Apc1638T truncated protein is critical to ensure development. In these animals, the functional dosage of β-catenin regulation provided by Apc1638T is reduced by ∼50%, which is presumably below the critical threshold level necessary for normal development. Likewise, in Apc1638T homozygotes, the level of regulation provided by Apc1638T is below tissue-specific thresholds, thereby resulting in developmental disturbances in a subset of cell types. In the case of the preputial gland that initially arises as other hair follicles, Apc1638T-driven β-catenin regulation may be insufficient to distinguish it from other hair follicles. It should be noted that the cutaneous cysts of both Apc1638T homozygotes as well as Apc1638N heterozygotes are also associated with abnormal hair follicle development (Smits et al. 1998), suggesting that this structure is sensitive to proper regulation by Apc.
A possible explanation for both the growth retardation and the reduced postnatal viability on the B6 genetic background might be represented by a defect in suckling behavior. Accordingly, milk was never found in the stomach of the succumbed newborns and growth retardation becomes apparent only after birth when competition for food may have a dramatic effect on growth. The APC protein is also expressed at high levels in the brain (Bhat et al. 1994; Senda et al. 1998), and it has been shown to bind and colocalize with DLG in a multiprotein complex at synapses of cultured hippocampal neurons (Matsumine et al. 1996; Satoh et al. 1997). These complexes are thought to have a function in the molecular organization of synapses and neuronal cell signaling. Because the Apc1638T protein can no longer associate with DLG, its absence in these complexes may result in a reduced functionality of the central nervous system and possibly result in behavioral abnormalities.
In conclusion, the absence of a tumor phenotype and of major developmental abnormalities in Apc1638T indicates that this mutant protein harbors the majority of the domains required for its normal function. Hence, the regulation of cytoplasmic β-catenin levels by APC must be sufficiently impaired to successfully lead to tumor formation.
Materials and methods
Generation of Apc mutant embryonic stem cells and mice
Four targeting vectors were constructed by inserting a PGK–neomycin or PGK–hygromycin cassette in both possible orientations into a unique SmaI site within a 10.6-kb genomic SphI fragment. This SmaI site corresponds to codon 1638 of the Apc coding sequence. For the generation of the Apc1572T mutation, PGK–hygromycin was inserted in between the unique SmaI site and a SspI site corresponding to codon 1572, obtained after partial digestion of the 10.6-kb SphI fragment. The hygromycin cassette was inserted in the same transcriptional orientation as that of Apc. For all targeting vectors, the introduction of the selectable marker results in a short frameshift and in few additional residues (6–27) at the carboxy terminus of the predicted truncated Apc proteins, differing for each construct. They do not harbor any known destabilizing motifs, possibly explaining the absence of a truncated protein in case PGK–neomycin or PGK–hygromycin are inserted in the transcriptional orientation opposite to that of Apc (see Results).
Approximately 40 μg of each construct was used to transfect 2 × 107 E14 ES cells by electroporation (1000 V/cm, 250 μF). Hygromycin or G418 selection was applied 24 hr after transfection. All candidate ES clones were validated by Southern and Western analysis as described (Fodde et al. 1994). Double-mutant ES cell lines were generated by use of an additional round of targeting with the second available selectable marker.
Correctly modified Apc+/1638T ES clones were injected into C57BL/6J blastocysts, after which chimeric mice with high levels of ES cell contribution were backcrossed to C57BL/6JIco females. To assess germ-line transmission of the mutation, tail DNA was genotyped in a single PCR-reaction with the following primers: Apc-A (5′-TGCCAGCACAGAATAGGCTG-3′) and Apc-C (5′-GTTGTCATCCAGGTCTGGTGTAA-3′) resulting in a 295-bp product indicative of the wild-type allele; PN3 (5′-GCCAGCTCATTCCTCCACTC-3′) in combination with Apc-C resulting in an ∼400-bp product indicative of the Apc1638T mutation. Amplifications were performed in a 25 μl volume containing 10 mm Tris-HCl (pH 8.9), 50 mm KCl, 2.5 mm MgCl2, 10% glycerol, 200 μg/ml BSA, 0.01% gelatin, 0.2 mm of each dNTP, 0.2 units of Taq polymerase, and 10 pmole of each primer. The reactions were heated for 5 min at 94°C followed by 35 PCR cycles at 94°C for 30 sec, 55°C for 60 sec, and 72°C for 90 sec.
Antibodies
The AFPN and AFPC3 polyclonal antibodies were raised in rabbits by injecting a His-tagged fusion protein containing amino acids 8–312 or 2260–2843, respectively, of the human APC protein. AFPC3 was affinity-purified with the His-tagged fragment coupled to CNBr-activated Sepharose (Pharmacia) according to the manufacturer’s instructions. In addition, the following commercially available antibodies were used: APC (Ab-1), a monoclonal antibody recognizing amino acids 1–29 of APC (Oncogene Research Products), clone TUB 2.1, a monoclonal antibody recognizing β-tubulin (Sigma), monoclonal antibodies recognizing β-catenin (clone 14), DLG (clone 12), and EB-1 (clone 5), all from Transduction Laboratories (Lexington, KY).
Immunoprecipitation and immunoblotting
Approximately 2 × 107 ES cells grown without feeder layers were rinsed twice in PBS and lysed for 15 min at 4°C in 800 μl of Triton X-100 immunoprecipitation (IP) buffer (30 mm Tris-Cl at pH 7.4, 250 mm NaCl, 0.1% Triton X-100, 5 mm EDTA, 50 mm NaF) containing a cocktail of protease inhibitors (1 μ/ml aprotinin, 20 μg/ml PMSF, μg/ml leupeptin 10, μg/ml pepstatin 10, 1 μg/ml antipain, and 0.1 mm sodium vanadate). To 400 μl of a cleared lysate obtained after a 10 min centrifugation step at 11,000g, a preformed complex of antibody and protein A–Sepharose beads (Pharmacia, Roosendaal, The Netherlands) was added. After a 1 hr incubation at 4°C with continuous mixing, the beads were washed 3–4 times with 1 ml of IP-buffer. The pellet was resuspended in 50 μl of Laemmli sample buffer (120 mm Tris-Cl at pH 6.8, 20% glycerol, 200 mm DTT, 4% SDS, 0.02% bromophenol blue) and boiled for 5 min.
For the detection of Apc proteins by Western analysis, lysates were resolved on SDS-agarose gels as described (Fodde et al. 1994). Lower molecular weight proteins were resolved on a 10% SDS–polyacrylamide gel and transferred onto PVDF membranes (Immobilon-P, Millipore) by electroblotting. The membranes were blocked for at least 1 hr with 5% nonfat dry milk in TBST (10 mm Tris-HCl at pH 8.0, 150 mm NaCl, 0.05% Tween 20). After a 1-hr incubation with primary antibody, the blot was washed 2–3 times with TBST, and subsequently incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hr. The peroxidase was visualized by enhanced chemiluminescence according to the manufacturer’s instructions (Amersham). Antibodies were used at the following dilutions: Apc (Ab-1) (1:2000); AFPN (1:3000); AFPC3 (1:2000); monoclonal β-catenin (1:5000).
Immunofluorescence
For immunofluorescence analyses, Apc+/+ and Apc1638T/1638T mouse embryonic fibroblasts were seeded on 3-aminopropyltriethoxy-silane coated coverslips to reach 70% to 80% confluency. Cells were washed twice in CB buffer (10 mm Mes at pH 6.1, 150 mm NaCl, 5 mm EGTA, 5 mm MgCl2, 5 mm glucose) and subsequently fixed for 5 min in CB containing 3% paraformaldehyde and 0.3% Triton X-100. After a single wash in CB, the cells were fixed for an additional 15 min in CB containing 3% paraformaldehyde, followed by two washes in CB. For EB-1 staining, cells were washed in PBS and fixed in methanol at −20°C for 5 min, followed by three washes in PBS. After fixation, the cells were incubated in TBS (20 mm Tris-HCl at pH 7.5, 150 mm NaCl, 2 mm EGTA, 2 mm MgCl2) containing 5% nonfat dry milk for 30 min. Primary antibodies were incubated for 1 hr at room temperature in TBS/5% nonfat dry milk at the following dilutions: β-tubulin (1:200); EB-1 (1:50); DLG (1:25); β-catenin (1:1000). After three washes in TBS containing 0.2% Tween-20, the cells were incubated for 30 min in a 1:500 dilution of Alexa 594 goat anti-mouse IgG secondary antibody (Molecular Probes), diluted in TBS/5% nonfat dry milk containing 0.5 μg/ml DAPI for DNA counterstaining. Following three washes in TBS containing 0.2% Tween-20, the coverslips were mounted in Gelvatol. Stained cells were examined by a Leitz DM-RBE microscope with a 63× objective and mounted with a Photometrics Series 200, KAF1400 CCD camera.
β-catenin/Tcf reporter assays
Approximately 1 × 105 MEFs or 1 × 106 undifferentiated ES cells were transfected with 0.75 μg of the luciferase reporter construct pTOPFLASH or pFOPFLASH kindly provided by Dr. H. Clevers (Korinek et al. 1997) using the Fugene lipofection reagent (Boehringer Mannheim). Both constructs contain a luciferase gene driven by a minimal TK promotor downstream of an optimal TCF-binding motif (pTOPFLASH) or a mutant motif (pFOPFLASH). Cells were cotransfected with 0.75 μg of pcDNA3.1/Myc-His/lacZ (Invitrogen) containing a β-galactosidase gene under the control of a CMV promoter, serving as an internal control. After 40 hr, the cells were lysed in cell culture lysis reagent provided by the Luciferase Assay System (Promega) and luciferase activity was determined on a Berthold Lumat LB9501 scintillation counter. β-Galactosidase activity was determined as described (Sambrook et al. 1989). All assays were performed as triplicate transfections.
Cell fractionation
Cellular fractionations were performed as described previously (Neufeld and White 1997). Following fractionation, 70 μg of total protein from each fraction was resolved by SDS-PAGE and transferred to nitrocellulose for Western blot analysis.
Histological processing of tissues
Tissues were fixed overnight at 4°C in Notox (Earth Safe Industries, Inc., Belle Mead, NJ) or 10% buffered formalin and embedded in paraffin according to standard procedures. Sections were stained with hematoxylin and eosin.
For the analysis of the preputial gland from 18.5-dpc C57BL/6J (N10–N12) embryos, serial transversal sections of the lower half of the body were made, with a maximum interval between two successive sections of 100 μm.
Intact, nonautolytic Apc1638T/1638T mice were found dead shortly after birth, and 18.5-dpc embryos were fixed in Notox or 10% buffered formalin, cut longitudinally in 1-mm-thick sections, paraffin embedded, and processed to hematoxifin- and eosin-stained sections. Microscopic examination allowed screening for lesions or malformations in most organs or tissues.
Growth measurements of animals
Heterozygous animals of mixed Ola129/B6 backgrounds (N1–N3 backcrosses to B6) were crossed to generate mice of all Apc1638T genotypes for the growth curve determination. At ∼10 days of age, the mice were tagged, weighed, and genotyped. Subsequent weight measurements were performed weekly up to 8 weeks of age. For growth determination of 18.5-day embryos, heterozygous B6-Apc+/1638T animals were mated. All extraembryonic tissues were removed, followed by the weight measurement. Tail DNA was isolated for genotyping and sex determination. In a single PCR-reaction with the same conditions as described for the genotyping, primers for the Y-chromosome specific Sry gene, that is, Sry-1 (5′- GAGAGCATGGAGGGCCAT-3′) and Sry-2 (5′-CCACTCCTCTGTGACACT-3′) resulting in a 265-bp product, were combined with X-chromosomal primers, that is, D79F1 (5′-AATAAATGTTTTACAACTCCTGATTCC-3′) and D79R4 (5′-TGCATAGACGTGTAAAACCTGC-3′) resulting in a 194 bp-product.
To statistically evaluate the body weight data of mice 10 days to 8 weeks of age, a two-way ANCOVA was performed with gender and genotype as factors and littersize as covariable. A one-way ANOVA with genotype as factor was performed to analyze the data of the 18.5-dpc embryos.
Generation of mouse embryonic fibroblasts
Embryonic fibroblasts were isolated essentially as described by Hogan et al. (1994). Embryos aged 14.5–16.5-dpc were obtained from matings of Apc+/1638T animals. After removal of the internal organs, limbs, and head, followed by several washes in PBS, embryos were minced and treated for 15 min at 37°C with 10 ml of 0.3% trypsin/PBS. DMEM supplemented with 10% fetal calf serum was added to inactivate the trypsin, after which the cells were seeded onto culture dishes.
Acknowledgments
We thank Dr. Peter de Knijff for his assistance with the statistical analyses. This project was supported by grants of the Dutch Cancer Society (94-817, R.S. and C.B.; 95-1102, N.H. and S.J-C), the Dutch Research Council Netherlands Organisatie voor Wetenschappelijk Onderzoek (901-01-166, M.K.), the Dutch Royal Academy of Sciences (R.F.), the National Institutes of Health (CA76329-01, W.E.; CA67944, R.K.; CA09602, K.N.), the Huntsman Cancer Institute (R.W. and K.N.), and the American Cancer Society (R.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL fodde@ruly46.medfac.Leidenuniv.nl; FAX 31-71-5276075.
References
- Baeg GH, Matsumine A, Kuroda T, Bhattacharjee RN, Miyashiro L, Yoyoshima K, Akiyama T. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 1995;14:5618–5624. doi: 10.1002/j.1460-2075.1995.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AIM, Pollack AL, Altschuler Y, Mostov KE, Nelson WJ. NH2-terminal deletion of β-catenin results in stable colocalization of mutant β-catenin with adenomatous polyposis coli protein and altered MDCK cell adhesion. J Cell Biol. 1997;136:693–706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow B-A, Würtele M, Grimm J, Asbrand C, Wirtz R, Kühl M, Wedlich D, Birchmeier W. Functional interaction of an Axin homolog, Conductin, with β-Catenin, APC, and GSK3β. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Beinhauer JD, Hagan IM, Hegemann JH, Fleig U. Mal3, the fission yeast homologue of the human APC-interacting protein EB-1 is required for microtubule integrity and the maintenance of cell form. J Cell Biol. 1997;139:717–728. doi: 10.1083/jcb.139.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrueta L, Kraeft S-K, Tirnauer JS, Schuyler SC, Chen LB, Hill DE, Pellman D, Bierer BE. The adenomatous polyposis coli-binding protein EB1 is associated with cytoplasmic and spindle microtubules. Proc Natl Acad Sci. 1998;95:10596–10601. doi: 10.1073/pnas.95.18.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Baraban JM, Johnson RC, Eipper BA, Mains RE. High levels of expression of the tumor suppressor gene APC during development of the rat central nervous system. J Neurosci. 1994;14:3059–3071. doi: 10.1523/JNEUROSCI.14-05-03059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha GR. Hormonal influences on the morphogenesis of the preputial gland of embryonic mice. Anat Rec. 1975;181:35–54. doi: 10.1002/ar.1091810104. [DOI] [PubMed] [Google Scholar]
- Drews U. Direct and mediated effects of testosterone: The development of intersexes in sex reversed mosaic mice, heterozygous for testicular feminization. Anat Embryol. 1975;146:325–340. doi: 10.1007/BF00302178. [DOI] [PubMed] [Google Scholar]
- Eccles DM, van der Luijt R, Breukel C, Bullman H, Bunyan D, Fisher A, Barber J, duBoulay C, Primrose J, Burn J, Fodde R. Hereditary desmoid disease due to a frameshift mutation at codon 1924 of the APC gene. Am J Hum Genet. 1996;59:1193–1201. [PMC free article] [PubMed] [Google Scholar]
- Efstathiou JA, Noda M, Rowan A, Dixon C, Chinery R, Jawhari A, Hattori T, Wright NA, Bodmer WF, Pignatelli M. Intestinal trefoil factor controls the expression of the adenomatous polyposis coli-catenin and the E-cadherin-catenin complexes in human colon carcinoma cells. Proc Natl Acad Sci. 1998;95:3122–3127. doi: 10.1073/pnas.95.6.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Meera Khan P, Kucherlapati R. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R, Smits R, Breukel C, Hofland N, Edelmann W, Kucherlapati R, Meera Khan P. Hereditary cancer, second international research conference on familial cancer. Basel, Switzerland: Karger; 1996. Genotype-phenotype correlations in intestinal carcinogenesis: Lessons from mouse models; pp. 35–45. [Google Scholar]
- Gismondi V, Bafico A, Biticchi R, Pedemonte S, Molina F, Heouaine A, Sala P, Bertario L, Presciuttini S, Strigini P, Groden J, Varesco L. Characterization of 19 novel and 6 recurring APC mutations in Italian adenomatous polyposis patients, using two different mutation detection techniques. Hum Mut. 1997;9:370–373. doi: 10.1002/(SICI)1098-1004(1997)9:4<370::AID-HUMU14>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Gismondi V, Stagnaro P, Pedemonte S, Biticchi R, Presciuttini S, Grammatico P, Sala P, Bertario L, Groden J, Varesco L. Chain-terminating mutations in the APC gene lead to alterations in APC RNA and protein concentration. Genes Chromosomes Cancer. 1998;22:278–286. [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3β. Curr Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. pp. 260–261. [Google Scholar]
- Ichii S, Takeda S, Horii A, Nakatsuru S, Miyoshi Y, Emi M, Fujiwara Y, Koyama K, Furuyama J, Utsunomiya J, Nakamura Y. Detailed analysis of genetic alterations in colorectal tumors from patients with and without familial adenomatous polyposis (FAP) Oncogene. 1993;8:2399–2405. [PubMed] [Google Scholar]
- Johnson KR, Sweet HO, Donahue LR, Ward-Bailey P, Bronson RT, Davisson MT. A new spontaneous mouse mutation of Hoxd13 with a polyalanine expansion and phenotype similar to human synpolydactyly. Hum Mol Genet. 1998;7:1033–1038. doi: 10.1093/hmg/7.6.1033. [DOI] [PubMed] [Google Scholar]
- Joslyn G, Richardson DS, White R, Alber T. Dimer formation by an N-terminal coiled coil in the APC protein. Proc Natl Acad Sci. 1993;90:11109–11113. doi: 10.1073/pnas.90.23.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin P, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54:5953–5958. [PubMed] [Google Scholar]
- Makino K, Kuwahara H, Masuko N, Nishiyama Y, Morisaki T, Sasaki J-I, Nakao M, Kuwano A, Nakata M, Ushio Y, Saya H. Cloning and characterization of NE-dlg: A novel human homolog of the Drosophila discs large (dlg) tumor suppressor protein interacts with the APC protein. Oncogene. 1997;14:2425–2433. doi: 10.1038/sj.onc.1201087. [DOI] [PubMed] [Google Scholar]
- Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg G-H, Kawahara T, Kobayashi S, Okada M, Toyoshima K, Akiyama T. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science. 1996;272:1020–1023. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Madino J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum Mol Genet. 1992;4:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Vogelstein B, Kinzler KW. Apoptosis and APC in colorectal tumorigenesis. Proc Natl Acad Sci. 1996;93:7950–7954. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Morrison EE, Askham JM, Clissold P, Markham AF, Meredith DM. The cellular distribution of the adenomatous poliposis coli tumour suppressor protein in neuroblastoma cells is regulated by microtubule dynamics. Neuroscience. 1997;81:553–563. doi: 10.1016/s0306-4522(97)00099-7. [DOI] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Moser AR, Shoemaker AR, Connelly CS, Clipson L, Gould KA, Luongo C, Dove WF, Siggers PH, Gardner RL. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Dev Dyn. 1995;203:422–433. doi: 10.1002/aja.1002030405. [DOI] [PubMed] [Google Scholar]
- Muhua L, Adames NR, Murphy MD, Shields CR, Cooper JA. A cytokinesis checkpoint requiring the yeast homologue of an APC-binding protein. Nature. 1998;393:487–491. doi: 10.1038/31014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Souza B, Müller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–3681. [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993;2:425–434. doi: 10.1002/humu.1380020602. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T. Axin, an inhibitor of the Wnt signalling pathway, interacts with β-catenin, GSK-3β and APC and reduces the β-catenin level. Genes Cells. 1998;3:395–402. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- Näthke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld KL, White RL. Nuclear and cytoplasmic localizations of the adenomatous polyposis coli protein. Proc Natl Acad Sci. 1997;94:3034–3039. doi: 10.1073/pnas.94.7.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack AL, Barth AIM, Altschuler Y, Nelson WJ, Mostov KE. Dynamics of β-catenin interactions with APC protein regulate epithelial tubulogenesis. J Cell Biol. 1997;137:1651–1662. doi: 10.1083/jcb.137.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta. 1997;1332:F127–F147. doi: 10.1016/s0304-419x(97)00008-5. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Renner C, Pfitzenmeier J-P, Gerlach K, Held G, Ohnesorge S, Sahin U, Bauer S, Pfreundschuh M. RP1, a new member of the adenomatous polyposis coli-binding EB1-like gene family, is differentially expressed in activated T cells. J Immunol. 1997;159:1276–1283. [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SC, Masiarz F, Munemitsu S, Polakis P. Association of the APC gene product with β-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E-cadherin form similar but independent complexes with α-catenin, β-catenin, and plakoglobin. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-Catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. pp. 16.66–16.67. [Google Scholar]
- Satoh K, Yanai H, Senda T, Kohu K, Nakamura T, Okumura N, Matsumine A, Kabayashi S, Toyoshima K, Akiyama T. DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95. Genes Cells. 1997;2:415–424. doi: 10.1046/j.1365-2443.1997.1310329.x. [DOI] [PubMed] [Google Scholar]
- Senda T, Lino S, Matsushita K, Matsumine A, Kobayashi S, Akiyama T. Localization of the adenomatous polyposis coli tumour suppressor protein in the mouse central nervous system. Neuroscience. 1998;83:857–866. doi: 10.1016/s0306-4522(97)00459-4. [DOI] [PubMed] [Google Scholar]
- Smith GS, Walford RL, Mickey MR. Lifespan and incidence of cancer and other diseases in selected long-lived inbred mice and their F1 hybrids. J Natl Cancer Inst. 1973;50:1195–1213. doi: 10.1093/jnci/50.5.1195. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Johnson KA, Bryan TM, Hill DE, Markowitz S, Willson JKV, Paraskeva C, Petersen GM, Hamilton SR, Vogelstein B, Kinzler KW. The APC gene product in normal and tumor cells. Proc Natl Acad Sci. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Levy DB, Maupin P, Pollard TD, Vogelstein B, Kinzler KW. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–3675. [PubMed] [Google Scholar]
- Smits R, van der Houven van Oordt W, Luz A, Zurcher C, Jagmohan-Changur S, Breukel C, Meera Khan P, Fodde R. Apc1638N: A mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology. 1998;114:275–283. doi: 10.1016/s0016-5085(98)70478-0. [DOI] [PubMed] [Google Scholar]
- Su L-K, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Su L-K, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993a;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- Su L-K, Johnson KA, Smith KJ, Hill DE, Vogelstein B, Kinzler KW. Association between wild type and mutant APC gene products. Cancer Res. 1993b;53:2728–2731. [PubMed] [Google Scholar]
- Su L-K, Burrell M, Hill DE, Gyuris J, Brent R, Wiltshire R, Trent J, Vogelstein B, Kinzler KW. APC binds to the novel protein EB1. Cancer Res. 1995;55:2972–2977. [PubMed] [Google Scholar]
- Talbot IC. Pathology. In: Phillips RKS, Spigelman AD, Thomson JPS, editors. Familial adenomatous polyposis and other polyposis syndromes. London, UK: Edward Arnold; 1994. pp. 15–25. [Google Scholar]
- Trzepacz C, Lowy AM, Kordich JJ, Groden J. Phosphorylation of the tumor suppressor adenomatous polyposis coli (APC) by the cyclin-dependent kinase p34cdc2. JBiol Chem. 1997;272:21681–21684. doi: 10.1074/jbc.272.35.21681. [DOI] [PubMed] [Google Scholar]
- van der Luijt RB, Meera Khan P, Breukel C, Tops CMJ, Vasen HFA, Scott RJ, Fodde RF. Germline mutations in the 3′ part of APC exon 15 do not result in truncated proteins and are associated with Attenuated Adenomatous Polyposis Coli. Hum Genet. 1996;98:727–734. doi: 10.1007/s004390050293. [DOI] [PubMed] [Google Scholar]
- Willert K, Nusse R. β-catenin: A key mediator of Wnt signaling. Curr Opin Genet Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- Zurcher C, van Zwieten MJ, Solleveld HA, Hollander CF. Aging research. In: Foster HL, Small JD, Fox JG, editors. The mouse in biomedical research, Vol. 4. Experimental biology and oncology. New York, NY: Academic Press; 1982. pp. 11–35. [Google Scholar]