

Abstract
Exposure of hematopoietic progenitors to γ-irradiation (IR) induces p53-dependent apoptosis and a p53-independent G2/M cell cycle arrest. These responses to DNA-damage can be inhibited by treatment with cytokine growth factors. Here we report that γ-IR-induced apoptosis and cell cycle arrest are suppressed by specific cytokines (e.g., erythropoietin and interleukin-3) and that activation of the Jak kinase is necessary and sufficient for these effects. Using myleoid cells expressing a series of erythropoietin receptor (EpoR) mutants, we have demonstrated that Jak kinase-dependent signals initiated from the membrane proximal domain of EpoR were sufficient to prevent IR-induced apoptotic cell death, but failed to prevent cell cycle arrest. Cell survival by Epo did not require activation of other known signaling pathways including PI-3 kinase, PLC-γ, Ras or Stats. Signaling targets of Jak kinase pathways included members of the Bcl-2 family of anti-apoptotic proteins, and enforced expression of Bcl-2 or Bcl-xL was as effective as cytokine treatment in blocking IR-induced apoptosis but did not prevent growth arrest. A distinct signal derived from a membrane distal domain of EpoR is required to overcome growth arrest associated with DNA damage. These findings functionally link the Jak signaling pathway to suppression of p53-mediated cell death by cytokines and demonstrate that the apoptotic and growth arrest responses to DNA damage in hematopoietic cells are modulated by distinct, cytokine specific signal transduction pathways.
Keywords: Interleukin-3, erythropoietin, p53, Jak, Bcl-2, Bcl-x
DNA damage in mammalian cells often results in apoptotic cell death or cell cycle arrest. Failure to respond properly to DNA damage could lead to genetic alterations that promote tumor progression. The response to DNA damage varies between different cell types, and whether a cell will undergo apoptosis or cell cycle arrest is dependent on cellular determinants that have not been fully characterized. One established regulator of cell death and cell cycle arrest following DNA damage is the p53 tumor suppressor (Zambetti and Levine 1993). Wild-type p53 is a transcriptional activator that selectively regulates the expression of genes that are involved in growth arrest (e.g., p21/Waf) or apoptosis (e.g., Bax) (el-Deiry et al. 1993; Miyashita and Reed 1995). Treatment of normal cells with DNA-damaging agents, such as ionizing irradiation, results in increased levels of wild-type p53, which in turn leads to the induction of target genes that can mediate apoptosis or cell cycle arrest (Kastan et al. 1992; Kuerbitz et al. 1992; Lowe et al. 1993). Mutation of p53 usually disrupts its DNA binding and transactivation functions and, therefore, mutant p53 is unable to negatively regulate cell growth following DNA damage. Consistent with the importance of p53 in the response to DNA damage, epidemiology studies have revealed that p53 is the most commonly mutated gene detected in human cancer (Hollstein et al. 1996). Interestingly, p53 is not mutated frequently in human leukemias, suggesting that there are mutation-independent mechanisms for functional inactivation of p53 pathways in hematopoietic cells (Canman et al. 1995).
The decision regarding how cells respond to DNA damage is also influenced by the extracellular environment. In particular, specific cytokines that are required growth factors for hematopoietic cells also inhibit or prevent an apoptotic response to DNA damage (Collins et al. 1992; Canman et al. 1995). The cytokines represent several groups of structurally related polypeptide factors including erythropoietin (Epo), interleukin (IL)-3 and IL-2. The receptors for many cytokines comprise a related family that utilize similar mechanisms of signal transduction (Ihle 1995). Whereas members of this receptor family lack intrinsic kinase activity, they are uniquely characterized by their ability to associate with and activate one or more members of the Jak family of protein tyrosine kinases. Jak kinase activation appears to be the initial critical event regulated by cytokine receptors following engagement by their respective ligands. Several other signaling intermediates and pathways have also been shown to be regulated by cytokine receptors, including Ras, Stat transcription factors, PI-3 kinase, PLC-γ and phosphatases (Ihle 1995). Although specific biological responses mediated by these signaling pathways remain largely unresolved, some progress has been made through mutagenic analyses of the cytokine receptors. For the Epo receptor (EpoR), these analyses have demonstrated that a membrane-proximal cytoplasmic domain of the receptor is sufficient to mediate Epo-dependent activation of Jak2 and growth of cells (Miura et al. 1991; Witthuhn et al. 1993). Other known signaling intermediates, such as Ras, PI-3 kinase, and Stats, are regulated through membrane distal domains of EpoR, suggesting that they are dispensable to the mitogenic activity of EpoR (Miura et al. 1994a,c; Damen et al. 1995; Quelle et al. 1996). It has been suggested that signals derived from the membrane distal domain of EpoR may be involved in enhancing cell survival under certain conditions (Nakamura et al. 1992). In contrast, other reports have shown that the membrane proximal domain of EpoR is sufficient to enhance survival of factor-dependent cells under optimal growth conditions (Zhuang et al. 1995)
Previous studies have demonstrated that overexpression of wild-type p53 induces apoptosis in hematopoietic cells and that specific cytokines can override this death signal. For example, p53-null murine myeloid leukemia M1 cells stably expressing a temperature-sensitive mutant p53, undergo rapid apoptosis when shifted to the permissive temperature and treatment of these cells with IL-6 efficiently maintains viability (Yonish-Rouach et al. 1993). Similarly, overexpression of temperature-sensitive mutant p53 in murine erythroleukemia (MEL) cells induces apoptosis at the permissive temperature, and Epo or IL-3 completely suppresses this death program (Lin and Benchimol 1995). The advantage of studying these cell systems is that the apoptotic response is a direct consequence of wild-type p53 activity. However, the levels of p53 expression in these cells are extraordinarily high and not physiologically relevant. Alternatively, Kastan and coworkers have developed a cell culture model for addressing the role of endogenous wild-type p53 in cell cycle arrest and apoptosis (Canman et al. 1995). In these studies, BaF3 murine pro-B cells, which are dependent on IL-3 for cell growth and survival, undergo accelerated apoptotic cell death when exposed to γ-irradiation in the absence of cytokine. In contrast, cells that are irradiated in the presence of IL-3 remain viable. Cell death in response to DNA damage in the absence of cytokine is dependent on p53 as BaF3 cells that are functionally null for p53 are refractory to apoptosis. Therefore, there is a direct role for wild-type p53 in γ-irradiation-induced cell death and cytokines such as IL-3, Epo, and IL-6 are potent suppressors of p53-mediated apoptosis.
In the present study, we have demonstrated that activation of Jak signaling pathways is necessary and sufficient for cytokine-mediated rescue of DNA damage-induced apoptosis and growth arrest. Using cell lines that stably express various forms of EpoR, we show that Epo can inhibit both apoptosis and G2 cell cycle arrest following γ-irradiation and that these effects are mediated by two distinct Jak2-dependent signaling pathways. The membrane proximal domain of EpoR is sufficient to prevent apoptosis, whereas the membrane distal domain of EpoR is required to overcome cell cycle arrest following DNA damage. The results presented here uncouple growth arrest from apoptosis and establish a Jak kinase signaling pathway as a key suppressor of p53-mediated apoptosis in hematopoietic cells.
Results
Cytokine suppression of DNA damage-induced apoptosis and growth arrest
IL-3 inhibits apoptosis in responsive BaF-3 hematopoietic cells following γ-irradiation (Collins et al. 1992; Canman et al. 1995). To determine which signaling pathways mediate these effects, we evaluated the effects of γ-irradiation on survival and cell cycle progression of myeloid cell lines responsive to multiple hemopoietins. DA3 cells are a murine myeloid cell line that are dependent on IL-3 for proliferation and die within 2–3 days of factor withdrawal (Ihle and Askew 1989). To test the ability of cytokines to suppress the effects of DNA damage in DA3 cells, asynchronously growing cells were cultured in medium containing 10% serum with or without IL-3 and were compared with cells exposed to 5 Gy of γ-irradiation. At specific intervals following irradiation, the viability of cells was determined by trypan blue dye exclusion and the effects on cell cycle progression determined by FACS analysis of DNA content. As shown in Figure 1A, nonirradiated DA3 cells deprived of IL-3 decline in viability to ∼40% by 46 hr, whereas concomitant exposure of cells to γ-irradiation dramatically accelerated their rate of death, such that by 28 hr no viable cells were detected. In contrast, cells cultured in IL-3 and exposed to 5 Gy showed little reduction in viability. Cell morphology and DNA fragmentation analyses demonstrated that the irradiation-induced cell death in the absence of cytokine was caused by apoptosis (data not shown).
Figure 1.

IL-3 rescue of viability and cell cycle progression following γ-irradiation. Exponentially growing DA3 cells were washed in RPMI 1640 and equivalent aliquots were suspended in medium containing 10% FBS (A) or 0.1% FBS (B). Each aliquot was then split into cultures that were supplemented with IL-3 (squares) or no factor (circles). Cultures then were treated with 5 G of γ-irradiation (solid symbols) or left untreated (open symbols). At various intervals after irradiation, samples of each culture were assayed for viability by trypan blue dye exclusion (left). Alternatively, 24 hr after irradiation, a sample of each culture was assayed for cellular DNA content by propidium iodide staining and FACS (right). The position of 2N and 4N DNA content is indicated for each histogram.
The cell cycle status of DA3 cells grown in the presence of IL-3 exhibited an asynchronous cell cycle distribution, whereas cultures maintained in the absence of IL-3 accumulated in the G1 phase of the cell cycle (Fig. 1A, right panel). Following γ-irradiation, the majority of cells cultured in the absence of IL-3 exhibited a <2N DNA content indicative of DNA fragmentation and apoptotic death. The remaining viable cells were growth arrested in both G1 and G2/M. By comparison, cells irradiated in the presence of IL-3 had only a slight loss of viability and only very modest increases in G1 and G2/M populations, characteristic of partial effects on progression through the G1 and G2/M phases of the cell cycle. Similar results were obtained in evaluating the ability of Epo to rescue DA3 cells expressing Epo receptors, and in IL-3 rescue of myeloid 32D cells or BaF3 pro-B cells (see below).
Contribution of serum to cell cycle progression but not rescue of DNA damage-induced apoptosis
Other factors present in the cell culture medium, particularly serum factors, may contribute to the IL-3-dependent rescue of cell viability following DNA damage. Therefore, we also assessed the ability of IL-3 to rescue viability of irradiated cells cultured in low serum (0.1%). The viability of DA3 cells maintained in 0.1% serum with or without IL-3 was significantly compromised compared with cells maintained in 10% serum (cf. Fig. 1A with B). However, γ-irradiated DA3 cells died at faster rates, as compared with nonirradiated cells, when cultured in medium lacking IL-3 and containing low serum. Addition of IL-3 to cells cultured in low serum abolished the accelerated death following γ-irradiation. Interestingly, γ-irradiation of these cells led to a near complete block in the G1 and G2/M phases of the cell cycle (Fig. 1B, right panel). Thus, although IL-3 is sufficient to rescue myeloid cell viability following γ-irradiation, additional factors present in serum are required to cooperate with cytokine to override cell cycle arrest. To evaluate the maximal effects of cytokine rescue, we therefore performed all subsequent experiments using cells maintained in 10% serum unless otherwise indicated.
Activation of the Jak kinase pathway is required for Epo rescue of DNA damage-induced apoptosis
To assess the signaling pathways required for cytokine-dependent suppression of γ-irradiation-induced apoptosis and cell cycle arrest, we utilized DA3 and 32D myeloid cells engineered to express wild-type or mutant forms of the EpoR. These EpoR mutants have been characterized previously in detail for their ability to mediate (1) Epo-induced proliferation (Miura et al. 1991, 1993), (2) activation of the Jak2 tyrosine kinase (Witthuhn et al. 1993; Miura et al. 1994b), (3) phosphorylation and activation of Stat5 (Quelle et al. 1996), and (4) activation of the Ras, PI-3 kinase, and PLC-γ pathways (Miura et al. 1994a,c; Damen et al. 1995) (Fig. 2A). To assess the effects of γ-irradiation, DA3 or 32D cells expressing EpoR mutants were cultured in Epo, IL-3, or no factor and exposed to 5 Gy of γ-irradiation (Fig. 2B,C). DA3 and 32D cells expressing wild-type EpoR were resistant to γ-irradiation when cultured in Epo or IL-3. Similarly, DA3 cells expressing EpoR mutants deficient in activation of the PI-3 kinase and Ras pathways (EpoR-H), and in Stat5 activation (EpoR-H/Y343F) were resistant to γ-irradiation-induced apoptosis when cultured in Epo (Fig. 2B,C). Epo rescue of DNA damage-induced apoptosis mediated by the EpoR-H/Y343FF mutant in DA3 cells was less effective than that observed for EpoR-H, but is likely the result of less effective activation of Jak2 kinase in these cells (Quelle et al. 1996). Consistent with this interpretation, Epo suppressed γ-irradiation-induced apoptosis efficiently in 32D cells overexpressing the EpoR-S mutant, which is deficient in Ras, PI-3 kinase, and Stat5 activation (Fig. 2C). Therefore, in both DA3 and 32D cells, EpoR-mediated activation of the PI-3 kinase, Ras, and Stat pathways is dispensable for suppression of DNA damaged-induced apoptosis. Notably, Jak2 activation by EpoR is strictly required for Epo to rescue irradiation-induced death as DA3 and 32D cells expressing a point mutant defective in Jak2 activation (EpoR-W282R) died at rates comparable with parental cells deprived of cytokine following γ-irradiation (Fig. 2B,C).
Figure 2.
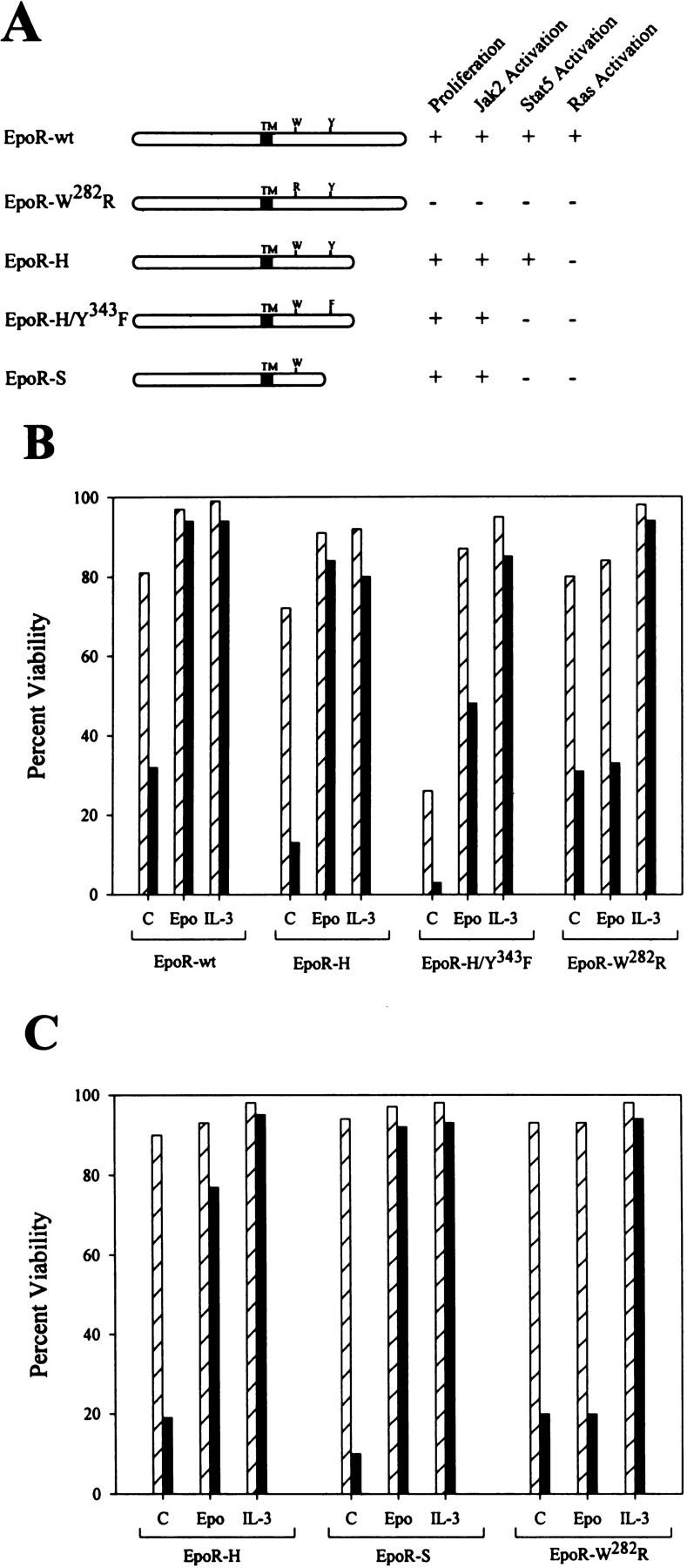
A membrane proximal domain of EpoR is sufficient for Epo-dependent rescue of irradiation-induced cell death, but requires Jak2 activity. (A) Wild-type (EpoR-wt) and various mutant forms of the EpoR are diagrammed. The transmembrane domain (TM), a tryptophane residue at amino acid position 282 (W), and a tyrosine at position 343 (Y) are indicated. Also indicated are the previously reported activities of each mutant receptor with respect to its ability to regulate proliferation and Jak2, Stat5, and Ras activation. DA3 cells (B) or 32D cells (C) expressing various EpoR constructs were washed in RPMI 1640 and suspended in medium plus 10% FBS supplemented with no factor (C), Epo, or IL-3. Cultures were then treated with 5 Gs of γ-irradiation (solid bars) or left untreated (hatched bars). Twenty-five hours after treatment, cells were harvested and assayed for viability by trypan blue exclusion.
Activation of Jak2 kinase activity is sufficient to suppress DNA damage-induced apoptosis
To address whether activation of the Jak2 kinase alone was sufficient to override DNA damage-induced cell death, we utilized 32D cells engineered to express an epidermal growth factor receptor (EGFR)–Jak2 kinase domain chimera receptor. The EGFR/Jak2 chimera contains the extracellular and transmembrane domains of the EGFR fused to the Jak tyrosine kinase domain (796–1129 amino acids) (Nakamura et al. 1996). As a control for Jak2 kinase activity, 32D–EGFR/NW2 cells, expressing an EGFR/Jak2 kinase-inactive chimera (see Materials and Methods), were analyzed in parallel. As shown in Figure 3B, the expression of EGFR/Jak2 receptors on the cell surface is approximately equal between the wild-type and mutant chimera cell lines and EGFR was not detected in the parental cells. Culture of 32D–EGFR/J2wt cells, but not 32D–EGFR/NW2 cells, in medium containing EGF stimulated Jak kinase activity as measured by (1) tyrosine phosphorylation of the chimeric receptor; (2) induction of expression of c-myc mRNA, which is regulated by a Jak kinase dependent pathway; and (3) extended cell viability in the absence of IL-3 (Fig. 3A; data not shown). Treatment of 32D–EGFR/J2wt cells with EGF or IL-3 significantly protected these cells from γ-irradiation-induced cell death (Fig. 3A). In contrast, EGF had no effect on survival of irradiated 32D–EGFR/NW2 cells (Fig. 3A). Therefore, these results demonstrate that activation of the Jak kinase pathway is sufficient to suppress DNA damage-induced death.
Figure 3.

Jak kinase activity is sufficient to inhibit irradiation-induced cell death. (A) 32D–EGFR/J2wt and EGFR/NW2 cells were washed in RPMI 1640, resuspended in medium containing 0.5% FBS, and supplemented with IL-3, EGF, or with no factor. Cultures were treated with 5 G of γ-irradiation (solid bars), or were left untreated (hatched bars). Sixteen hours after treatment, samples of each culture were assayed for viability by trypan blue dye exclusion. Representative data are presented and comparable results have been obtained in three separate experiments. (B) Cell surface expression of the EGFR/Jak chimeric receptors was determined by FACS as described in the Materials and Methods. The relative levels of cell surface receptors are approximately equal between the EGFR/J2wt and EGFR/NW2 cell lines.
bcl-2 and bcl-xL are targets of the Jak kinase pathway and inhibit irradiation-induced apoptosis but not growth arrest
Expression of the anti-apoptotic bcl-2 and bcl-xL genes is transcriptionally up-regulated in response to IL-3 and Epo (Silva et al. 1996). To assess the cytokine signaling pathway(s) that regulate bcl-2 and bcl-xL gene expression, we have analyzed Epo stimulation of 32D cells expressing various EpoR mutants by Northern blot (Fig. 4). Cytokine deprivation of 32D cells results in a low, basal level of bcl-2 and bcl-xL mRNA expression. Erythropoietin stimulation of 32D cells expressing EpoR-S, which is deficient in Ras, PI-3 kinase, and Stat5 activation but competent for activation of Jak2 kinase, dramatically increases bcl-2 and bcl-xL mRNA levels. Similar results are obtained by use of 32DEpoR, 32DEpoR-H cells, and DA3–EpoR cell lines (Fig. 4; data not shown). In contrast, Epo failed to induce bcl-2 and bcl-xL mRNA in 32D cells expressing the EpoR–PB receptor, which does not activate Jak kinase. These results indicate that the Jak kinase pathway regulates bcl-2 and bcl-xL expression.
Figure 4.

The Jak kinase signaling pathway regulates expression of bcl-2 and bcl-xL. 32D cells expressing the EpoR-H (defective in activation of PI-3 kinase and Ras, but activates Stat5 and Jak2), Epo-S (activates Jak2) or Epo-PB (defective in Jak activation) mutant receptors were deprived of IL-3 for 15 hr (0 time point). Cells were then stimulated with Epo for the indicated times. Total RNA was prepared and analyzed for expression of bcl-2 (top) and bcl-x (bottom) by Northern blot, as described in Materials and Methods.
To examine whether expression of these proteins could substitute for cytokine treatment following DNA damage, 32D cells stably expressing human Bcl-2 or murine Bcl-xL cDNA constructs were assayed. Bcl-2 and Bcl-xL overexpression was confirmed by Western blot analysis (data not shown). As shown in Figure 5A, 32D cells expressing Bcl-xL or Bcl-2 maintained viability in the absence of cytokine with or without exposure to γ-irradiation. However, these cells were factor-dependent on the basis of FACS data (Fig. 5A) and cell proliferation assays (not shown). In contrast, parental 32D cells required IL-3 to preserve cell viability and to inhibit DNA damage-induced apoptosis. Interestingly, γ-irradiation of 32D–Bcl-2 and 32D–Bcl-xL in the absence of IL-3 resulted in a near complete G1 and G2/M cell cycle arrest (Fig. 5B). Comparison of the percentage of G2 phase cells in γ-irradiated cultures versus nonirradiated cultures demonstrated that irradiation of 32D–Bcl-xL or 32D–Bcl-2 cells in the absence of IL-3 was associated with a substantial increase in the G2/M population (7.3- and 5.9-fold, respectively) (Fig. 6C). In contrast, parental 32D, 32D–Bcl-xL, or 32D–Bcl-2 cells cultured in IL-3 showed only a modest increase in the G2/M population (1.5- to 1.9-fold) following γ-irradiation. Thus, consistent with data presented in Figures 1 and 2, IL-3 substantially attenuated the γ-irradiation-induced cell cycle block, and this was not suppressed by overexpression of either Bcl-xL or Bcl-2.
Figure 5.

Expression of antiapoptotic proteins in 32D cells prevents irradiation-induced cell death but does not prevent cell cycle arrest. Parental 32D cells, or 32D cells stably expressing cDNAs encoding Bcl-xL (32D–Bcl-xL) or Bcl-2 (32D–Bcl-2), were washed in RPMI 1640 and suspended in medium plus 10% FBS, supplemented with IL-3 (+IL-3), or with no factor (−IL-3). Cultures then were treated with 5 G of γ-irradiation (solid bars), or were left untreated (hatched bars). Eighteen hours after treatment, samples of each culture were assayed for viability by trypan blue exclusion (A). Alternatively, samples of each culture were harvested at 20 hr after irradiation, stained with propidium iodide, and analyzed by FACS (B). The position of 2N and 4N DNA content is indicated for each histogram. The percentage of G2/M cells (4N DNA content) was calculated from the FACS data. A ratio of the percentage of G2 cells in irradiated vs. nonirradiated cultures (G2+/γ/G2+γ) is presented in C for cells treated with IL-3 (solid bar) or with no factor (hatched bars). The results shown are representative of two independent experiments.
Figure 6.

The membrane distal domain of EpoR is required for Epo-dependent inhibition of irradiation-induced cell cycle arrest. (A) Twenty-four hours after irradiation, samples of the EpoR-wt, EpoR-H, and EpoR-H/Y343F cultures analyzed in Fig. 5 were stained with propidium iodide and analyzed by FACS. The percentage of G2/M cells (4N DNA content) was calculated from the FACS data. A ratio of the percentage of G2 cells in irradiated vs. nonirradiated cultures (G2+/γ/G2+γ) is presented in B for cells treated with IL-3 (hatched bars) or cells treated with Epo (solid bars). Comparable results were obtained in three separate experiments by use of EpoR-H-expressing cells.
Distinct domains of the Epo receptor are required for rescue of DNA damage-induced apoptosis and growth arrest
To define the domains of EpoR required to override irradiation-induced G1 and G2 arrest, we assessed the effects of γ-irradiation on cell cycle progression for DA3 cells expressing EpoR mutants. Figure 6A shows representative cell cycle profiles of γ-irradiated versus nonirradiated cells, whereas Figure 6B compares the ratio of G2 phase cells in γ-irradiated cultures versus nonirradiated cultures. Rescue of cell death by the wild-type EpoR or by IL-3 was associated with only a slight increase in the G2/M population (1.2- to 1.8-fold) with cells remaining largely asynchronous. In contrast, Epo rescue of cells expressing either the EpoR-H or EpoR-H/Y343F mutant receptor was associated with a substantial increase in the G2/M population (fourfold) compared with nonirradiated Epo-stimulated cells. Thus, two distinct EpoR domains appear to function in attenuation of the effects of DNA damage in myeloid cells. The membrane proximal domain is required for the maintenance of cell viability, whereas the membrane distal domain is required to overcome DNA damage-induced cell cycle arrest. Both of these activities are dependent on the ability of the receptor to activate Jak2.
Discussion
The occurrence of DNA damage in mammalian cells is associated with the rapid onset of cell cycle arrest and/or apoptosis. Treatment of cells with cytokines such as IL-3 has been reported to rescue cell viability following DNA damage (Collins et al. 1992; Canman et al. 1995). To address the molecular basis for this rescue, we have examined multiple cell systems responsive to various hemopoietins and growth factors. In each cell line, cytokines substantially promoted cell survival following DNA damage. Using mutants of the Epo receptor, we have shown that suppression of γ-irradiation-induced apoptosis localizes to the membrane proximal domain of the receptor and is dependent on Jak kinase activation. Similar requirements have been defined for the proliferation signal activated by Epo through its receptor (Miura et al. 1991, 1993; Quelle et al. 1996), and it is probable that both growth and survival signals are required for balanced mitogenesis. Thus, it is likely that inhibition of irradiation-induced apoptosis is an outcome of the same Jak-dependent signaling cascade. Although the components of this Jak kinase pathway have not been defined, several immediate-early gene targets regulated by the minimally active Epo receptor have been identified and include c-myc, pim-1, bcl-2, and bcl-x (Fig. 4; Quelle et al. 1996). Enforced expression of Bcl-xL or Bcl-2 is just as effective at rescuing 32D cells from irradiation-induced death as is treatment with IL-3 (Fig. 5). These results are consistent with previous studies demonstrating that wild-type p53 stimulates expression of the apoptotic inducer Bax (Miyashita and Reed 1995). However, it remains to be established whether the induced expression of Bcl-x or Bcl-2 is the primary mechanism by which cytokines rescue cell viability following DNA damage.
The domains of EpoR that are dispensable for suppression of irradiation-induced apoptosis activate several known signaling pathways, including the activation of Stat transcription factors, PI-3 kinase, various components of the classic Ras pathway (Shc, Ras, Raf, Map kinases, etc.), and recruitment and activation of phosphatases (Miura et al. 1994c; Damen et al. 1995; Klingmuller et al. 1995; Quelle et al. 1996; Tauchi et al. 1996). These results indicate that activation of the PI-3 kinase pathway, and consequently the regulation of the serine/threonine Akt protein kinase, is dispensable for cytokine-mediated survival of 32D or DA3 cells following DNA damage. Clearly, the PI-3 kinase/Akt signaling pathway is important for survival of other cell types, such as fibroblasts and neuronal cells, that are grown under very different conditions than used here (for review, see Franke et al. 1997). Several other lines of evidence demonstrate that the Ras/Raf pathway is also not required to block DNA damage-induced cell death. Continuous activation of the Ras pathway in 32D cells through the expression of an active form of the Raf-1 kinase (v-Raf) (Cleveland et al. 1994) failed to rescue cell survival after γ-irradiation (data not shown). In addition, hepatocyte growth factor (HGF), which functions through the c-Met tyrosine kinase receptor (Bottaro et al. 1991) to specifically activate Ras (Hartmann et al. 1994; Ridley et al. 1995), is sufficient for mitogenesis of murine myeloid NFS-60 cells, but unable to promote cell survival following DNA damage (data not shown). Although not required, it is possible that activation of signaling pathways such as PI-3 kinase/Akt and/or Ras may contribute to cell survival following DNA damage. Nevertheless, at least in these myeloid cells, rescue from irradiation-induced apoptosis is not a general effect of growth factors but is rather dependent on cytokine receptors that utilize Jak kinases as an integral part of their signal transduction cascades.
In previous studies, cytokine rescue of irradiation-induced apoptosis of an IL-3-dependent pro-B cell line was associated with G1 and G2 blocks in cell cycle progression (Canman et al. 1995). This appears to be the case if cytokine rescue experiments are performed in reduced serum concentrations. However, when experiments were performed under optimal growth conditions with 10% serum, suppression of apoptosis by cytokines was associated with only a modest block in cell cycle progression. These observations suggest that serum factors, which are not in themselves sufficient to rescue cell viability, contribute along with cytokine receptor signals to overcome cell cycle arrest following DNA damage. Cytokine function is clearly required for attenuation of cell cycle arrest on DNA damage, as loss of this activity in EpoR truncation mutants (e.g., EpoR-H) leads to a prolonged delay in G2/M. Thus, the membrane proximal and membrane distal domains of EpoR are uniquely required for Epo-dependent inhibition of apoptosis and Epo-dependent cell cycle progression, respectively, following DNA damage. This differential requirement suggests that these effects are mediated by separable signal transduction pathways, a concept consistent with the observation that enforced expression of Bcl-2 or Bcl-xL is sufficient to rescue cell viability, but fails to overcome cell cycle arrest associated with DNA damage.
The mechanism involved in establishing a G1 block in cell cycle progression following DNA damage is often ascribed to p53. In contrast, the G2 arrest following γ-irradiation is largely mediated by a p53-independent pathway. In myeloid cells, a requirement for p53 in mediating an irradiation-induced G1 block cannot be properly evaluated because withdrawal of cytokine results in a G1 arrest even in the absence of DNA damage. However, degradation of p53 in pro-B BaF3 cells through E6 expression had no effect on the accumulation of G2/M-arrested cells following γ-irradiation (data not shown). This suggests that at least the G2/M block does not require p53. In myeloid cells, the ability to override the DNA damage-induced G2/M arrest requires multiple extracellular signals because the presence of serum factors and a fully active cytokine receptor were required. The membrane distal domain of EpoR appears to mediate one or more of these signals as the removal of this domain prevented release of the G2/M block. Interestingly, the membrane distal domain of EpoR is entirely dispensable to Epo-induced cell cycle progression in the absence of DNA-damage (Fig. 2; Miura et al. 1991; Quelle et al. 1996). Thus, EpoR signaling effectors of cell cycle progression are either distinct in irradiated versus nonirradiated cells or are differentially regulated by irradiation. The membrane distal domain of EpoR is known to interact with several signaling intermediates, and their regulation in irradiated cells should be enlightening.
We have demonstrated that at least three signals are required to fully escape negative regulation of cell growth following exposure of myeloid cells to DNA-damaging agents. These signaling pathways include hemopoietin-dependent regulation of cell survival and cell cycle progression, whereas additional signals for cell growth are regulated by serum factors. The components of each of these signaling pathways would represent likely targets for activating mutations allowing escape from negative regulators of cell growth.
Materials and methods
Cell lines and culture conditions
DA3 and 32D.3 cells expressing EpoR and various mutant EpoRs (Miura et al. 1991, 1993; Quelle et al. 1996), 32D-Raf (Cleveland et al. 1994), 32D-Bcl-2 (Nip et al. 1997), and NFS-60 (Mizuno et al. 1993) cell lines have been described previously. The 32D-Bcl-XL cell line was derived by stable transfection of 32D.3 cells with a spleen focus-forming virus (SFFV) long terminal repeat (LTR) vector expressing a murine Bcl-XL cDNA. The 32D–EGFR/J2wt cell line was derived as described previously for the generation of the EG-J2 cell line (Nakamura et al. 1996). 32D–EGFR/NW2 cells were derived by stable transfection of 32D.3 cells with a plasmid expressing an EGF receptor-Jak2 mutant chimera, in which tyrosines 868, 913, 966, 1008, and 1021 in the Jak kinase domain have been changed to phenylalanine by site directed mutagenesis. These tyrosine mutations completely abolish Jak kinase activity. Cell surface staining for EGFR/Jak chimeric receptor expression was performed by use of mouse monoclonal antibody specific for human EGFR (sc-120; Santa Cruz Biotechnology, Santa Cruz, CA) as described previously (Ashmun et al. 1989). All cell lines were maintained in RPMI 1640 plus 10% fetal bovine serum (FBS) and recombinant murine IL-3 (1 ng/ml).
For DNA-damage studies, exponentially growing cells were washed in RPMI 1640 medium. Washed cells were then suspended (6 × 105 cells/ml) in medium containing 10% or 0.1% FBS, as indicated. Aliquots of cells were then mixed with equal volumes of medium plus FBS supplemented with Epo (40 U/ml), IL-3 (4 ng/ml), HGF (50 ng/ml), EGF (50 ng/ml), or no factor. Parallel cultures were exposed to γ-irradiation with a 137Cs source (J.L. Shepherd, San Fernando, CA) at a dose rate of 675 rads/min., or were left untreated. All cultures were then returned to a 37°C, 5% CO2 incubator until they were assayed for viability or cell cycle status. Cell viability was measured as a function of cell membrane integrity as determined by Trypan blue dye exclusion (Life Technologies).
Determination of cell cycle distribution
Cells were collected by centrifugation and resuspended at 1 × 106 cells/ml in 0.1% sodium citrate containing 50 μg/ml propidium iodide and treated with 1 μg/ml RNase at room temperature for 30 min. DNA fluorescence of the stained cells was measured with a FACSCAN flow cytometer (Becton Dickinson, San Jose, CA). The percentages of cells within the G1, S, and G2/M phases of the cell cycle were calculated by use of ModFit software (Verity Software House, Topsham, ME).
Northern blot analysis
Total RNA was isolated from cells by the guanidinium isothiocyanate method followed by centrifugation through cesium chloride as described previously (Cleveland et al. 1994). The RNA (20 μg) was electrophoretically separated on 1% agarose/6% formaldehyde gels, transferred to nitrocellulose filters, and hybridized under stringent conditions with 32P-radiolabeled bcl-2 or bcl-x cDNA probes. The filters were washed extensively and analyzed by autoradiography.
Acknowledgments
We thank Dr. Richard Ashmum and Sam Lucas for cell cycle analyses, and Linda Snyder, Xiaoping He, and Hui Yang for technical assistance. This work was supported in part by National Institutes of Health/National Cancer Institute (NIH/NCI) grant CA63230 (G.P.Z.), NIH/National Institute of Diabetes and Digestive and Kidney Diseases grant DK44158 (J.L.C.), The University of Iowa Bioscience Initiative Funding (F.W.Q.), NIH/NCI Cancer Center Support core grant 5 P30 CA21765, and the American Lebanese Syrian Associated Charities of St. Jude Children’s Research Hospital.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gerard.zambetti@stjude.org; FAX (901) 525-8025.
References
- Ashmun RA, Look AT, Roberts WM, Roussel MF, Seremetis ST, Ohtsuka M, Sherr CJ. Monoclonal antibodies to the human CSF-1 receptor (c-fms proto-oncogene product) detect epitopes on normal mononuclear phagocytes and on human myeloid leukemic blast cells. Blood. 1989;73:827–837. [PubMed] [Google Scholar]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Canman CE, Gilmer TM, Coutts SB, Kastan MB. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes & Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- Cleveland JL, Troppmair J, Packham G, Askew DS, Lloyd P, Gonzalez-Garcia M, Nunez G, Ihle JN, Rapp UR. v-raf suppresses apoptosis and promotes growth of interleukin-3-dependent myeloid cells. Oncogene. 1994;9:2217–2226. [PubMed] [Google Scholar]
- Collins MK, Marvel J, Malde P, Lopez-Rivas A. Interleukin 3 protects murine bone marrow cells from apoptosis induced by DNA damaging agents. J Exp Med. 1992;176:1043–1051. doi: 10.1084/jem.176.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen JE, Cutler RL, Jiao H, Yi T, Krystal G. Phosphorylation of tyrosine 503 in the erythropoietin receptor (EpR) is essential for binding the P85 subunit of phosphatidylinositol (PI) 3-kinase and for EpR-associated PI 3-kinase activity. J Biol Chem. 1995;270:23402–23408. doi: 10.1074/jbc.270.40.23402. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Hartmann G, Weidner KM, Schwarz H, Birchmeier W. The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase met requires intracellular action of Ras. J Biol Chem. 1994;269:21936–21939. [PubMed] [Google Scholar]
- Hollstein M, Shomer B, Greenblatt M, Soussi T, Hovig E, Montesano R, Harris CC. Somatic point mutations in the p53 gene of human tumors and cell lines: Updated compilation. Nucleic Acids Res. 1996;24:141–146. doi: 10.1093/nar/24.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle JN. Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- Ihle JN, Askew D. Origins and properties of hematopoietic growth factor-dependent cell lines. Int J Cell Cloning. 1989;7:68–91. doi: 10.1002/stem.5530070202. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Benchimol S. Cytokines inhibit p53-mediated apoptosis but not p53-mediated G1 arrest. Mol Cell Biol. 1995;15:6045–6054. doi: 10.1128/mcb.15.11.6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- Miura O, D’Andrea A, Kabat D, Ihle JN. Induction of tyrosine phosphorylation by the erythropoietin receptor correlates with mitogenesis. Mol Cell Biol. 1991;11:4895–4902. doi: 10.1128/mcb.11.10.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura O, Cleveland JL, Ihle JN. Inactivation of erythropoietin receptor function by point mutations in a region having homology with other cytokine receptors. Mol Cell Biol. 1993;13:1788–1795. doi: 10.1128/mcb.13.3.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura O, Nakamura N, Ihle JN, Aoki N. Erythropoietin-dependent association of phosphatidylinositol 3-kinase with tyrosine-phosphorylated erythropoietin receptor. J Biol Chem. 1994a;269:614–620. [PubMed] [Google Scholar]
- Miura O, Nakamura N, Quelle FW, Witthuhn BA, Ihle JN, Aoki N. Erythropoietin induces association of the JAK2 protein tyrosine kinase with the erythropoietin receptor in vivo. Blood. 1994b;84:1501–1507. [PubMed] [Google Scholar]
- Miura Y, Miura O, Ihle JN, Aoki N. Activation of the mitogen-activated protein kinase pathway by the erythropoietin receptor. J Biol Chem. 1994c;269:29962–29969. [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Higuchi O, Ihle JN, Nakamura T. Hepatocyte growth factor stimulates growth of hematopoietic progenitor cells. Biochem Biophys Res Comm. 1993;194:178–186. doi: 10.1006/bbrc.1993.1801. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Komatsu N, Nakauchi H. A truncated erythropoietin receptor that fails to prevent programmed cell death of erythroid cells. Science. 1992;257:1138–1141. doi: 10.1126/science.257.5073.1138. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Chin H, Miyasaka N, Miura O. An epidermal growth factor receptor/Jak2 tyrosine kinase domain chimera induces tyrosine phosphorylation of stat5 and transduces a growth signal in hematopoietic cells. J Biol Chem. 1996;271:19483–19488. doi: 10.1074/jbc.271.32.19483. [DOI] [PubMed] [Google Scholar]
- Nip J, Strom DK, Fee BE, Zambetti G, Cleveland JL, Hiebert SW. E2F-1 cooperates with topoisomerase II inhibition and DNA damage to selectively augment p53-independent apoptosis. Mol Cell Biol. 1997;17:1049–1056. doi: 10.1128/mcb.17.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y, Ihle JN. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol Cell Biol. 1996;16:1622–1631. doi: 10.1128/mcb.16.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Comoglio PM, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol. 1995;15:1110–1122. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M, Grillot D, Benito A, Richard C, Nunez G, Fernandez-Luna JL. Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood. 1996;88:1576–1582. [PubMed] [Google Scholar]
- Tauchi T, Damen JE, Toyama K, Feng GS, Broxmeyer HE, Krystal G. Tyrosine 425 within the activated erythropoietin receptor binds Syp, reduces the erythropoietin required for Syp tyrosine phosphorylation, and promotes mitogenesis. Blood. 1996;87:4495–4501. [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- Yonish-Rouach E, Grunwald D, Wilder S, Kimchi A, May E, Lawrence JJ, May P, Oren M. p53-mediated cell death: relationship to cell cycle control. Mol Cell Biol. 1993;13:1415–1423. doi: 10.1128/mcb.13.3.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambetti GP, Levine AJ. A comparison of the biological activities of wild-type and mutant p53. FASEB J. 1993;7:855–865. doi: 10.1096/fasebj.7.10.8344485. [DOI] [PubMed] [Google Scholar]
- Zhuang H, Niu Z, He TC, Patel SV, Wojchowski DM. Erythropoietin-dependent inhibition of apoptosis is supported by carboxyl-truncated receptor forms and blocked by dominant-negative forms of Jak2. J Biol Chem. 1995;270:14500–14504. doi: 10.1074/jbc.270.24.14500. [DOI] [PubMed] [Google Scholar]