

Abstract
Trialkylborane additives promote reduction of CO2 to formate by bis(diphosphine) Ni(II) and Rh(III) hydride complexes. The late transition metal hydrides, which can be formed from dihydrogen, transfer hydride to CO2 to give a formate-borane adduct. The borane must be of appropriate Lewis acidity: weaker acids do not show significant hydride transfer enhancement, while stronger acids abstract hydride without CO2 reduction. The mechanism likely involves a pre-equilibrium hydride transfer followed by formation of a stabilizing formate-borane adduct.
Introduction
Efficient catalytic conversion of CO2 to fuels and chemicals could be a major contributor towards achieving carbon neutrality.1 We have recently reported (stoichiometric) reactions relevant to conversion of syngas to organic products, making use of a combination of a metal carbonyl complex, a nucleophilic late transition metal hydride, and an intramolecularly appended Lewis acid.2 Because some syngas conversion catalysts are known to convert CO2 as well — for example, the common CuO/ZnO/Al2O3 catalyst can effectively convert a mixture of H2, CO2, and CO to methanol3 — we thought it might be worthwhile to examine the chemistry of our CO-reducing systems with CO2.
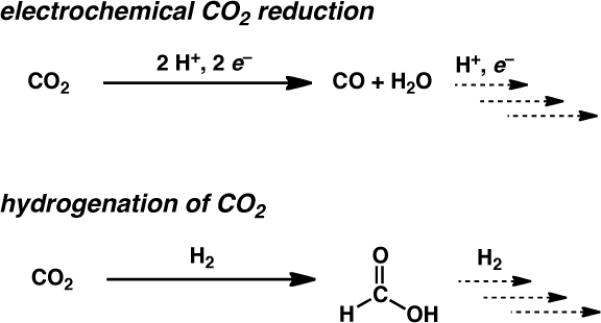
The two main approaches to CO2 reduction currently under consideration are electrocatalytic reduction,1a in which protons and electrons are added sequentially (and/or concertedly), and direct hydrogenation.1b,c Most of the electrocatalytic systems operate at quite reducing potentials to generate CO and H2O, while hydrogenation (under forcing pressures) more commonly produces formic acid (Scheme 1). Lewis (particularly those based on alkali and alkaline earth metals) and Brønsted acids have shown promise in improving electrocatalytic CO2 reduction, notably with low-valent Fe porphyrins.4 Bimetallic catalysts, including the active site of the enzyme [NiFe] CO dehydrogenase, appear to function by having one metal bound to C while the other acts as a Lewis acid to bind O.5 Lewis acids such as A1OX, and TiOx appear to play an important role in heterogeneous catalysts that convert CO2 (and CO) to CH4.6
Scheme 1.
Several examples involving boron compounds have also been reported. The conversion of a diboronyl B–B7 or secondary borane B–H8 bond to a B–O bond helps drive CO2 reduction by homogeneous Cu- and Ni-based catalysts, respectively. Activation of dihydrogen by “frustrated Lewis pairs” can also lead to reduction of CO2; products include methanol (after hydrolysis).9 All of these reactions are stoichiometric in boron; catalysis is precluded by the difficulty of cleaving the strong B–O bonds, unless strong Si–O bonds are formed using silane additives.10
We report here that the late transition metal hydrides, as well as the partially-reduced metal carbonyl components of our CO reductive coupling system, are able to reduce CO2 to formate, with the Lewis acidic trialkylboranes playing a key role. Although no catalysis has yet been achieved, the formate-borane adducts obtained appear to be considerably less strongly bound than in the above-discussed systems, suggesting that this may be a promising new approach to CO2 reduction.
Results and Discussion
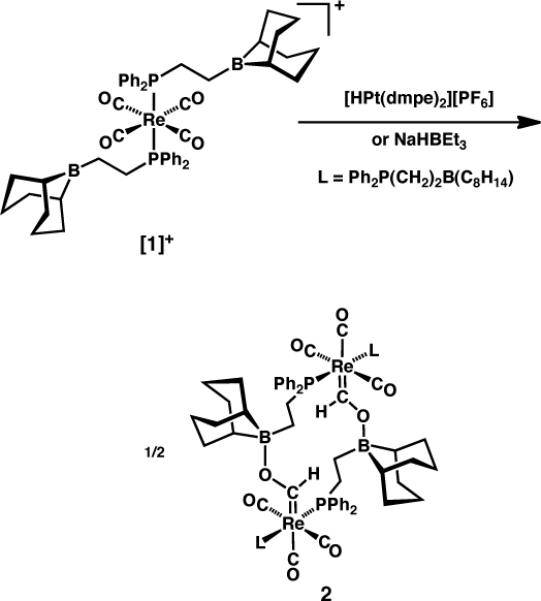
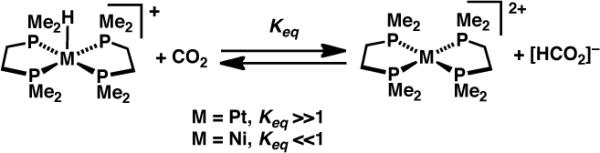
The Re carbonyl cation [(Ph2P(CH2)2B(C8H14))2Re(CO)4][BF4] ([1][BF4]), bearing pendent alkylboranes in the secondary coordination sphere of the metal, reacts with [HPt(dmpe)2][PF6] (dmpe = 1,2-bis(dimethylphosphino)ethane), but not with the analogous Ni hydride, to give boroxycarbene 2 (Scheme 2).2a (Complex 2 can also be prepared by reduction of 1 with NaHBEt3.) Similarly, DuBois has found that CO2 is reduced to [HCO2]− by [HPt(dmpe)2][PF6], whereas [Bu4N][HCO2] transfers hydride to [Ni(dmpe)2]2+ to yield [HNi(dmpe)2]+ (Scheme 3).11 The hydride donor strengths (ΔGH−; smaller values correspond to stronger hydride donors) of [HPt(dmpe)2]+, [HNi(dmpe)2]+ and [HCO2]− have been determined to be 42.5, 50.9 and 44.2 kcal·mol−1 respectively12 (the last value is estimated from the observation that [HCO2]− has ΔGH− similar to that of [HPt(depe)2]+ (depe = 1,2-bis(diethylphosphino)ethane),11 the ΔGH− of which was recalculated from the originally published value12a using DuBois' more recent pKa value12c).
Scheme 2.
Scheme 3.
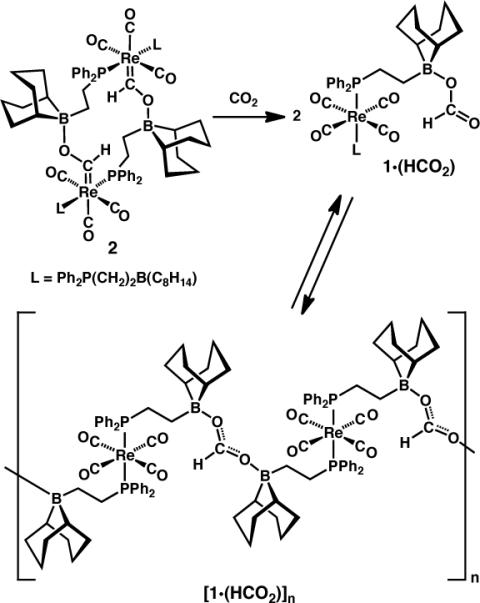
Boroxycarbene 2 can serve as a hydride source, releasing H2 upon treatment with water or weak acids,2d but the above results show that it is intermediate in hydride strength between [HNi(dmpe)2][PF6] and [HPt(dmpe)2][PF6]; is it strong enough to reduce CO2? Admission of 1 atm CO2 to a thoroughly degassed solution of 2 (formed in situ from [1][BF4] and NaHBEt3 in C6D5Cl) led after a few minutes to a new 1H NMR resonance at δ 8.44. 31P NMR showed complete consumption of 2 and formation of a new species with a single signal whose chemical shift was similar to that for [1]+; IR spectroscopy indicated a [trans-L2Re(CO)4]+ substructure (a single strong stretch at 1993 cm−1) as well as a reduced C=O functionality (1620 cm−1). These data are consistent with hydride transfer from 2 to CO2 giving 1·(HCO2) (Scheme 4); the same species could be obtained by addition of [Bu4N][HCO2] to [1][BF4] in C6D5Cl. The apparent symmetry exhibited by the NMR spectra suggests the operation of a fluxional process that exchanges formate between boranes, likely via reversible oligomerization (Scheme 4). After about an hour, large amounts of precipitate formed along with disappearance of the NMR signals assigned to 1·(HCO2); probably this is due to formation of an oligomeric species [1·(HCO2)]n, with –B–OC(H)O–B– linkages. Similar behavior has been observed for some of the CO reduction products studied previously.2d
Scheme 4.
The observation that 2 reacts with CO2 to yield [1·(HCO2)] does not necessarily indicate that 2 is a stronger hydride donor than [HCC2]−, since the B–O interaction in 1·(HCO2) (and the two B–O interactions in [1·(HCO2)]n) presumably afford(s) additional stabilization. Similarly, although transfer of hydride from [HNi(dmpe)2]+ to CO2 should be uphill by ~ 7 kcal·mol−1, addition of CO2 to a mixture of [HNi(dmpe)2][PF6] and [1][BF4] produced significant amounts of 1·(HCO2) over 24 hours at room temperature. This reaction did not proceed to completion, as evidenced by the presence of unreacted [HNi(dmpe)2]+; the chemical shifts for the (single) remaining Re-containing species were intermediate between those for [1]+ and 1·(HCO2), suggesting an equilibrium between these species. (No boroxycarbene 2 was observed.) The precipitate which again formed over time could be partially dissolved by addition of pyridine; the resulting solution showed a slightly shifted 1H NMR signal for the formate group (δ 8.88) and two 31P NMR signals (δ 1.95 d, 2.98 d, JPP = 77 Hz), consistent with formation of the asymmetric pyridine adduct 1·(HCO2)(C5H5N) (Scheme 5).
Scheme 5.
Similar chemistry is promoted by a simple trialkylborane, which could imply that Re may simply be a spectator in the reaction involving [1]+: when [HNi(dmpe)2][PF6] was allowed to react with CO2 (1 atm) in the presence of tBu(CH2)2B(C8H14) in C6D5Cl, over a few hours a new 1H NMR resonance at δ 8.73 grew in, the hydride resonance of [HNi(dmpe)2]+ diminished, and [Ni(dmpe)2][PF6]2 precipitated. The reaction did not go to completion; the ultimate yield of formate (assessed by integration of NMR spectra) was about 50%. As the reaction proceeded, the 1H NMR peak attributed to formate gradually shifted upfield (δ 8.73 at 2 h, 8.66 at 18 h) while that for the tert-butyl group of the borane shifted downfield (δ 0.89 before reaction, 0.95 at 2 h, 0.97 at 18 h). Concurrently the 11B NMR signal shifted upfield (δ 88.2 before reaction, 65 at 2 h, 51 at 18 h) and broadened considerably (Figure 1). Addition of one equivalent of tBu(CH2)2B(C8H14) to [Bu4N][HCO2] in C6D5Cl shifted the 1H NMR resonance for [HCO2]− from δ 9.58 to δ 8.89, consistent with some formate-borane interaction.
Figure 1.
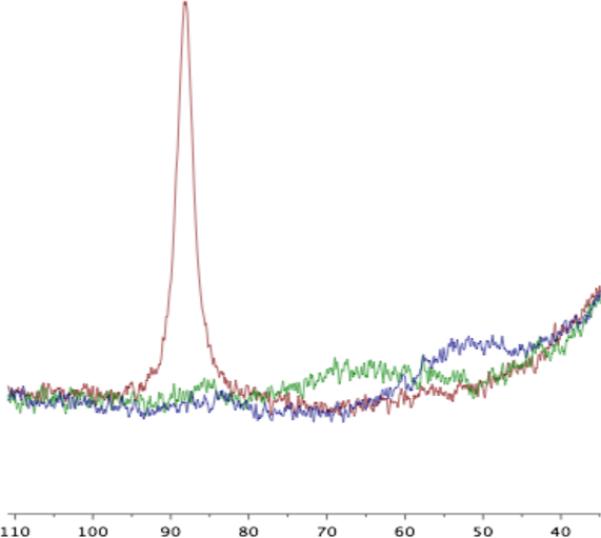
11B NMR spectra of a mixture of [HNi(dmpe)2][PF6] and tBu(CH2)2B(C8H14) in C6D5Cl before CO2 addition (red), 2 hours after addition of CO2 (1 atm) (green), and after 18 hours (blue).
Several other boranes, of varying Lewis acidity, were also examined. Addition of CO2 to a mixture of isopropyl pinacol borane and [HNi(dmpe)2]+ did not significantly increase the amount of formate produced relative to the reaction in the absence of any Lewis acid, although the small 1H NMR signal for [HCO2]− was shifted upfield (δ 8.74 vs. 8.84 without borane). Trimesitylborane also failed to effect significant CO2 reduction, but produced a larger upfield shift (δ 8.63), quite similar to that in the trialkylborane reactions. It appears that these formate-borane adducts are considerably less robust, as might be expected for less acidic or more sterically crowded Lewis acids. Triphenylborane did give substantial conversion to formate, but the NMR spectral data indicated formation of unidentified side products as well. The much stronger acid B(C6F5)3 exhibited only hydride transfer from Ni to B and no CO2 reduction.
The results for the trialkylborane, above, clearly indicate formation of a formate-borane adduct (Scheme 6). To further probe this interaction, aliquots of tBu(CH2)2B(C8H14) were added sequentially to a C6D5Cl solution of formate. Upon addition of borane a second [HCO2]− signal appeared upfield of the original signal for free formate; the new signal increased in intensity, while the first decreased correspondingly, and both signals broadened and shifted upfield as more borane was added, up to one equivalent. At and beyond one equivalent of borane, only a single sharp resonance was observed, that shifted still further upfield as more borane was added. Addition of 10 equivalents of pyridine caused the peak to shift back to approximately the same position observed after addition of one equivalent of borane, along with substantial broadening. (Spectra are shown in the SI, Figure S15.) These observations appear most consistent with formation of a strong monoborane adduct, [HCC2BR3]−, which does not exchange with free [HCO2]− on the NMR time scale; and a weaker bis(borane) adduct, [HCO2(BR3)2]−, which does rapidly exchange with the mono adduct; only the latter interaction is cleaved by addition of pyridine. This model does not fully explain the NMR behavior, particularly the changing peak shifts below one equivalent of added borane, but we have observed that other factors (such as counterion speciation and precipitation) can have an effect on NMR shifts (the [Bu4N]+ resonances also shift with added borane in the above experiment), which could account for these details.
Scheme 6.
In the absence of any borane, the reaction of [HNi(dmpe)2][PF6] with 1 atm CO2 gave only about 2% formate product; with borane and the addition of tetraheptylammonium bromide it proceeded to completion, accompanied by precipitation of [Ni(dmpe)2][Br]2. Under the latter conditions the formate 1H NMR signal (δ 8.79) is close to the shift observed for the stoichiometric combination of formate and borane (δ 8.89).
In an attempt to eliminate some of these complications we investigated CQ2 reduction in acetonitrile, in which all of the constituents are soluble (although there was a concern that MeCN would bind the borane too tightly to permit any acid assistance). Reaction of [HNi(dmpe)2][PF6] with CO2 and no added borane led to ~ 5% conversion to [Ni(dmpe)2]2+ and [HCO2]− (δ 8.40; c.f. [Bu4N][HCO2] δ 8.68), while addition of a single equivalent of tBu(CH2)2B(C8H14) gave 60% conversion to [HCO2(BR3)n]− (δ 8.29), and 10 equiv borane effected essentially complete conversion (δ 8.22). Addition of one equivalent of [Bu4N][HCO2] at the end of the last reaction resulted in approximately doubling the peak intensity with a negligible change in position; when the reaction was carried out with one equiv 13CO2, the 1H NMR signal at δ 8.21 appeared as a doublet 1(JCH = 202 Hz), and a new intense 13C{1H} NMR signal appeared at δ 174.16. These findings establish conclusively that the signal around δ 8.2 is indeed due to formate generated by reduction of CO2. The 11B NMR resonance of the free borane in CD3CN is far upfield from that in C6D5Cl, consistent with formation of a CD3CN adduct; that must be weaker than the formate-borane adduct, however, since the 11B NMR resonance shifts further upfield (by ~ 3 ppm) as formate is produced.
In principle reduction of CO2 by H2 could be made catalytic in nickel, by employing a suitable base in concert with [Ni(dmpe)2]2+ to cleave H2,12c followed by hydride delivery to CO2 (Scheme 7). The base must be of sufficient steric bulk to inhibit strong adduct formation with the borane and/or nucleophilic attack at CO2. When a mixture of [Ni(dmpe)2]2+, 5 equiv NEt3, and 5 equiv tBu(CH2)2B(C8H14) in CD3CN was exposed to H2/CO2 (1 atm of a 1:1 mixture), slow conversion to small amounts of formate (~ 8% relative to Ni) was observed over 3 days. The slow reaction is due (at least in part) to slow formation of [HNi(dmpe)2]+ under these conditions: a solution of [Ni(dmpe)2][PF6]2 and NEt3 (4 equiv) in CD3CN under 1 atm H2 was only 30% converted to [HNi(dmpe)2][PF6] after 3 days.
Scheme 7.
In contrast, the reaction of [Ni(dmpe)2][BArF4]2 (BArF4 = tetrakis(3,5-trifluoromethylphenyl)borate) and NEt3 with H2 proceeded far faster in C6D5Cl, with high conversion to [HNi(dmpe)2]+ in 3 hours, and full conversion after 18 hours. We ascribe the difference to coordination of solvent (Scheme 8): the adduct [Ni(dmpe)2(MeCN)]2+ has been structurally characterized, and its formation constant estimated by electrochemical techniques as Keq = 0.3, whereas in chlorobenzene [Ni(dmpe)2]2+ would be present as the 4-coordinate square planar complex (whose structure has also been determined).12a Counteranion effects appear to be unimportant: [Ni(dmpe)2][BArF4]2 reacts at roughly the same rate as [Ni(dmpe)2][PF6]2 with NEt3 under H2 in CD3CN.
Scheme 8.
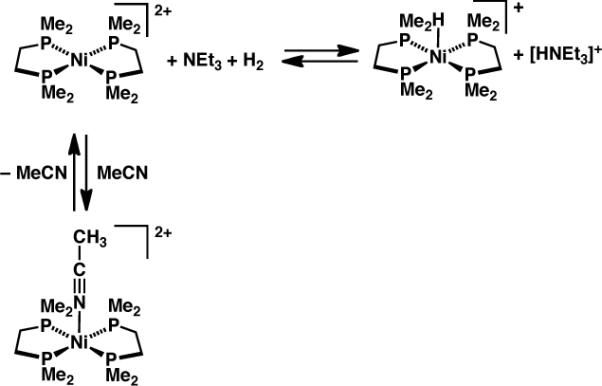
Disappointingly, a solution of [Ni(dmpe)2][BArF4]2, t Bu(CH2)2B(C8H14) and NEt3 under H2/CO2 in C6D5Cl did not produce any detectable formate, despite the steady growth of [HNi(dmpe)2]+ over a few days. It is unclear why no formate is generated. Reactions in CD2Cl2 led to formation of CD2HCl, indicating a hydride transfer side reaction.
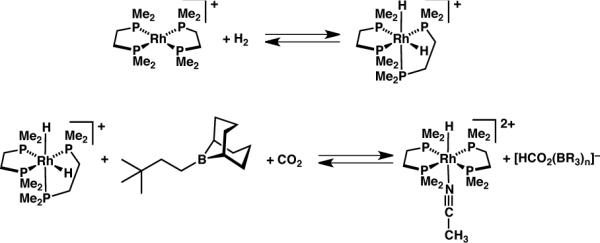
Reduction of CO2 with H2 could, however, be achieved with the rhodium cation [Rh(dmpe)2][OTf] — without the need for an added base, since its H2 oxidative addition product, [H2Rh(dmpe)2][OTf], is a hydride donor of strength (ΔGH– = 50.4 kcal mol−)13 similar to that of [HNi(dmpe)2]+. Treatment of [Rh(dmpe)2][OTf] with H2/CO2 (1 atm of a 1:1 mixture) in CD3CN led to formation of some [H2Rh(dmpe)2][OTf] but no formate over 4 days; however, inclusion of tBu(CH2)2B(C8H14) in the reaction led to the generation of formate (1H NMR signal at δ 8.24) along with the Rh product of hydride transfer, [HRh(dmpe)2(MeCN)]2+, over a few hours (Scheme 9). With one equivalent of trialkylborane, the yield of formate was ~ 25% (by NMR integration). Some [Rh(dmpe)2]+ remained unconverted during the course of the reaction, consistent with the unfavorable thermodynamics of H2 oxidative addition (uphill by 0.44 kcal mol−1)13 and the low (~ 0.5 atm) H2 pressure; 1 atm H2 is required for complete conversion.
Scheme 9.
Conclusions
The late transition metal bis(diphosphine) hydride complexes [HNi(dmpe)2]+ and [H2Rh(dmpe)2]+ (as well as a rhenium-boroxycarbene complex) are able to reduce carbon dioxide to formate, with the assistance of a trialkylborane that shifts the otherwise unfavorable equilibrium by forming a formate-borane adduct. Furthermore, H2 can be used directly in these systems to reduce CO2. As judged by NMR spectroscopy, the formate-borane adduct is relatively weak (in comparison to those generated in the CO reductions studied previously) and hence might be broken sufficiently easily to close a catalytic cycle. While overall catalysis has so far eluded us, the prospects for developing catalytic systems based on this chemistry appear promising.
Experimental Section
General Considerations
All air- and moisture-sensitive compounds were manipulated using standard vacuum line or Schlenk techniques, or in a glovebox under a nitrogen atmosphere. All reactions were carried out in an inert atmosphere, unless otherwise noted. Under standard glovebox conditions, petroleum ether, diethyl ether, benzene, toluene, and tetrahydrofuran were used without purging, such that traces of those solvents were in the atmosphere, and could be found intermixed in the solvent bottles. The solvents for air- and moisture-sensitive reactions were dried over sodium benzophenone ketyl, calcium hydride, or by the method of Grubbs.14 All NMR solvents were purchased from Cambridge Isotopes Laboratories, Inc. Chlorobenzene-d5 (C6D5Cl) and dichloromethane-d2 (CD2Cl2) were freeze-pump-thaw degassed three times before being run through a small column of activated alumina. Tetrahydrofuran-d8 (THF-d8) was purchased in a sealed ampoule, and dried by passage through activated alumina. Unless noted, other materials were used as received. [1][BF4],2a2, [Pt(dmpe)2][PF6]2,15 [HPt(dmpe)2][PF6],15 [Ni(dmpe)2][BF4]2,12a [HNi(dmpe)2][PF6],15tBu(CH2)2B(C8H14),16 [Bu4N][HCO2],17 [Ni(dmpe)2][BArF4]2,2c and [Rh(dmpe)2][OTf]13,18 were synthesized by literature methods. All other materials were readily commercially available, and used as received. 1H and 13C NMR spectra were recorded on Varian Mercury 300 MHz, 400-MR 400 MHz, INOVA 500 MHz, or INOVA 600 MHz spectrometers at room temperature, unless indicated otherwise. Chemical shifts are reported with respect to residual internal protio solvent for 1H and 13C{1H} spectra. Other nuclei were referenced to an external standard: H3PO4 (31P), 15% BF3·Et2O/CDCl3(11B), CFCl3 (19F), all at 0 ppm.
Experimental Procedures
CO2 reduction involving rhenium carbonyl complexes
Reaction of 2 with CO2
A 10 mL vial was charged with 26.4 mg (0.0251 mmol) [1][BF4] and ~0.6 mL C6D5Cl. With stirring, 25.1 μL (0.0251 mmol) NaHBEt3 (1.0 M in toluene) was added dropwise to provide a pale yellow solution. The reaction mixture was transferred to a J-Young NMR tube and initial spectroscopic measurements showed clean conversion to boroxycarbene 2. The tube was placed under vacuum for 1 minute with gentle shaking to degas, and one atmosphere of CO2 was then admitted to the tube. NMR spectroscopy after 5 minutes showed complete conversion to 1·(HCO2), most of which precipitated overnight. 1H NMR (C6D5Cl, 500 MHz): toluene and BEt3 are omitted, but overlap with some aliphatic peaks, preventing good integration. δ 0.69 (br s, 4H), 1.59 (br s, overlapping), 1.84 (br, overlapping), 2.05 (br m, overlapping), 2.73 (br, 4H), 7.41 (br, 12H), 8.45 (s, 1H, HCO2−). 31P{1H} NMR (C6D5Cl, 121 MHz): δ 2.2. IR (C6D5Cl): νco 1993, 1620 cm−1. 13C NMR spectra could not be acquired because of the low solubility of the product.
Reaction of [1][BF4] with [Bu4N][HCO2]
A J-Young NMR tube was charged with 29.1 mg (0.0276 mmol) [1][BF4], 7.9 mg (0.0276 mmol) [Bu4N][HCO2], and ~0.6 mL C6D5Cl. After 15 minutes, 1H and 31P{1H} NMR spectroscopy showed essentially complete conversion to 1·(HCO2). After about 30 minutes some precipitates were observed, and precipitation of white solids continued over the next 3 hours. At this time, the solution was decanted, and IR spectroscopy showed resonances that matched the preparation of 1·(HCO2) from CO2; an additional peak at 1660 cm−1 is unidentified, however. 1H NMR (C6D5Cl, 400 MHz): [Bu4N]+ peaks omitted. δ 0.68 (br, 4H), 0.95 (br, 4H), 1.58 (br, 4H), 1.83 (br, 16H), 2.03 (br, 4H), 2.75 (br, 4H), 7.1–7.2 (m, 12H), 7.4 (m, 8H), 8.49 (s, 1H, [HCO2]−). 31P{1H} NMR (C6D5Cl, 162 MHz): δ 2.4. IR (C6D5Cl): νco 1993, 1660, 1621 cm−1.
Reaction of [1][BF4] and [HNi(dmpe)2][PF6] with CO2 in C6D5Cl
A J-Young NMR tube was charged with 24.3 mg (0.0231 mmol) [1][BF4], 11.6 mg (0.0231 mmol) [HNi(dmpe)2][PF6], and ~0.6 mL C6D5Cl. The tube was sealed and initial NMR spectroscopic measurements showed no reaction. 1 atm CO2 was added; after ~20 minutes some formate was observed, which grew in over a few hours. During this time precipitates formed, and after about 12 hours there was essentially no formate-containing product in solution. The solids were collected, washed with CD3CN (to extract [Ni(dmpe)2][PF6]2) and C6D5Cl, and treated with C6D5Cl containing 3.8 μL (0.0462 mmol, 2 equiv) pyridine, which dissolved most of the solids. An asymmetric product was observed by NMR spectroscopy which was assigned as 1·(HCO2)(pyridine). 1H NMR (C6D5Cl, 300 MHz): δ 0.44 (br, 2H, Ph2PCH2CH2BR2), 0.68 (br, 2H, Ph2PCH2CH2BR2), 0.95 (2H, Ph2PCH2CH2BR2), 1.09 (2H, Ph2PCH2CH2BR2), 1.34 (6H, Ph2PCH2CH2BR2), 1.5–2.0 (m, 18H, Ph2PCH2CH2BR2), 2.1 (br, 4H, Ph2PCH2CH2BR2), 2.47 (br, 2H, Ph2PCH2CH2BR2), 2.93 (m, 2H, Ph2PCH2CH2BR2), 6.5–7.5 (m, mixture of Ph2PR, free and bound C6H5N), 8.41 (m, 2H, C6H5N), 8.88 (1H, HCO2). 31P{1H} NMR (C6D5Cl, 121 MHz): δ 1.95 (d, JPP = 77 Hz), 2.98 (d, JPP = 76 Hz).
CO2 reduction by nickel and boranes in chlorobenzene
Reaction of [HNi(dmpe)2][PF6] with CO2
A J-Young NMR tube was charged with 12.2 mg (0.0242 mmol) [HNi(dmpe)2][PF6] and ~0.6 mL C6D5Cl. The tube was degased by exposure to vacuum briefly with gentle shaking, and 1 atm CO2 was admitted to the tube. The tube was sealed, shaken well, and monitored by NMR spectroscopy. A small resonance assigned to formate was observed at δ 8.84, integrating 1–2% relative to the Ni hydride. No increase in the formate resonance was observed over 48 hours.
Reaction of [HNi(dmpe)2][PF6] with tBu(CH2)2B(C8H14) and CO2
A solution of 6.7 mg (0.0327 mmol) tBu(CH2)2B(C8H14) in ~0.6 mL C6D5Cl was added to 16.5 mg (0.0327 mmol) solid [HNi(dmpe)2][PF6]. The reaction mixture was transferred to a J-Young NMR tube, and initial spectroscopic measurements were made. After two freeze–pump–thaw cycles, 1 atm CO2 was admitted to the tube, and the reaction was monitored by NMR spectroscopy. After 24 hours the reaction had reached partial conversion (with the formate resonance shifting from δ 8.73 to 8.66), which did not change over 4 days. Addition of ~16.0 mg (0.0327 mmol, 1 equiv nominal; about 2 equiv by NMR integration) [hept4N][Br] led to significant further growth of [hept4N][HCO2(BR3)n], which (along with excess [hept4N][Br]) was the only soluble product (all hydride was consumed). The spectra agreed well with independently synthesized [Bu4N][HCO2(BR3)n] (see below). 1H NMR (C6D5Cl, 400 MHz): δ 0.78 (m, 2H, tBuCH2CH2B(C8H14)), 0.88 (t, J = 6.8 Hz, [hept4N]+), 1.04 (s, 9H, tBuCH2CH2B(C8H14)), 1.1 (br, 2H, tBuCH2CH2B(C8H14)), 1.2–1.3 (br m, [hept4N]+), 1.39 (m, 2H, tBuCH2CH2B(C8H14)), 1.5 (br, [hept4N]+), 1.76 (br, 2H, tBuCH2CH2B(C8H14)), 1.94 (br, 4H, tBuCH2CH2B(C8H14)), 2.2 (br, 6H, tBuCH2CH2B(C8H14)), 3.11 (m, [hept4N]+), 8.83 (1H, [HCO2(BR3)n]−). No discernible signals were observed by 11B NMR spectroscopy, presumably due to broadening and overlapping with the borosilicate glass in the probe construction.
Reaction of [Bu4N][HCO2] with tBu(CH2)2B(C8H14) in C6D5Cl
A J-Young NMR tube was charged with 15.8 mg (0.0549 mmol) [Bu4N][HCO2], 11.3 mg (0.0549 mmol) tBu(CH2)2B(C8H14), and ~0.6 mL C6D5Cl. NMR spectroscopy revealed a formate resonance at δ 8.89, well upfield of [Bu4N][HCO2] in the absence of borane (δ 9.58). 1H NMR (C6D5Cl, 500 MHz): δ 0.76 (m, tBuCH2CH2B(C8H14), 2H), 0.86 (t, J = 7.3 Hz, N(CH2CH2CH2CH3)4, 12H), 1.04 (s, tBuCH2CH2B(C8H14), 9H), 1.25 (q, J = 7.2 Hz, N(CH2CH2CH2CH3)4, 8H), 1.4 (m, 10H, N(CH2CH2CH2CH3)4 and tBuCH2CH2B(C8H14) overlapping), 1.83 (br, tBuCH2CH2B(C8H14), 2H), 1.97 (br, tBuCH2CH2B(C8H14), 4H), 2.28 (br, tBuCH2CH2B(C8H14), 6H), 3.00 (m, N(CH2CH2CH2CH3)4), 8.89 (s, [HCO2(BR3)n]−, 1H). 13C{1H} NMR (C6D5Cl, 126 MHz): δ 13.66, 19.87, 23.91, 26.80, 30.05, 31.35, 33.0 (br), 40.75, 58.66, 168.48. 11B NMR (C6D5Cl, 160 MHz): δ 2.7.
Titration of [Bu4N][HCO2] with tBu(CH2)2B(C8H14) in C6D5Cl
A J-Young NMR tube was charged with 22.1 mg (0.0769 mmol) [Bu4N][HCO2] and 0.600 mL C6D5Cl. Initial NMR spectra were collected. A 1.0 M solution of tBu(CH2)2B(C8H14) in C6D5Cl was prepared, and ~19 μL (~0.25 equiv) was added by syringe, followed by collection of NMR spectra. Integration showed that 0.20 equiv was added. This procedure was repeated so that spectra with 0, 0.2, 0.75, 1.5, 2.4, 3.4, 5.1, and 7.0 equiv borane (by NMR integration) were obtained. Spectra are shown in the SI as Figure S15.
General procedure for reactions of [HNi(dmpe)2] [PF6] with CO2 and other boranes
A solution of borane (~0.03 mmol) in ~0.6 mL C6D5Cl was added to an equimolar amount of solid [HNi(dmpe)2][PF6]. The reaction mixture was transferred to a J-Young NMR tube, and initial spectroscopic measurements were made. After two freeze–pump–thaw cycles, 1 atm CO2 was admitted to the tube, and the reaction was monitored by NMR spectroscopy.
Isopropyl pinacol borate
The reaction proceeded essentially the same as that with no borane added. The formate resonance appears between δ 8.70 and 8.74 during the reaction.
Triphenylborane
Complete disappearance of [HNi(dmpe)2]+ was observed, along with appearance of a presumed formate resonance at δ 8.8 and some unidentified peaks (δ 5–6).
Trimesitylborane
No change in conversion was observed as compared to the reaction without borane. A minor formate resonance was observed at δ 8.63.
B(C6F5)3
Shortly after mixing B(C6F5)3 and [HNi(dmpe)2][PF6], a mixture of [Ni(dmpe)2]2+ and [HNi(dmpe)2]+ was observed by 1H NMR spectroscopy. 19F and 11B NMR showed complete consumption of B(C6F5)3 and were consistent with formation of a 4-coordinate borate. The observed 11B NMR resonance is slightly downfield from the expected chemical shift of [HE(C6F5)3]− and the resonance is broadened beyond being able to observe coupling (probably due to exchange with some free borane). Even after CO2 addition, no formate was observed after ~12 hours at room temperature. 1H NMR (C6D5Cl, 500 MHz): δ −14.40 ([HNi(dmpe)2]+), 1.0 (m, [Ni(Me2PCH2CH2PMe2)2]2+), 1.18 (m, [HNi(Me2PCH2CH2PMe2)2]+), 1.28 (m, [Ni(Me2PCH2CH2PMe2)2]2+), 1.55 (m, [HNi(Me2PCH2CH2PMe2)2]+). 31P{1H} NMR (C6D5Cl, 121 MHz): δ 24.3 ([HNi(dmpe)2]+), 43.0 ([Ni(dmpe)2]2+). 19F NMR (C6D5Cl, 282 MHz): δ −165.35 (m, 6F), −160.38 (t, JFF = 20.6 Hz, 3F), −134.64 (m, 6F). 11B NMR (C6D5Cl, 160 MHz): δ −0.48 (br).
CO2 reductions by nickel and boranes in acetonitrile
General procedure for reactions of [HNi(dmpe)2] [PF6] in acetonitrile
A J-Young NMR tube was charged with [HNi(dmpe)2][PF6] (~0.03 mmol), the appropriate amount of tBu(CH2)2B(C8H14) (0, 1, 10 equiv), and ~0.6 mL CD3CN. The tube was sealed, initial NMR measurements were taken, and the atmosphere replaced with 1 atm CO2 (after freeze–pump–thaw degassing twice). The reactions were monitored periodically by multinuclear NMR spectroscopy until equilibrium was reached (under 24 hours), with conversion at equilibrium estimated by integration.
CO2
In the absence of trialkylborane, only about 5% hydride transfer was observed. The only two species observed in solution were unreacted [HNi(dmpe)2]+ and small amounts of [Ni(dmpe)2]2+.
1 equiv tBu(CH2)2B(C8H14) and CO2
When one equivalent of trialkylborane was added, about 65% hydride transfer was observed.
10 equiv tBu(CH2)2B(C8H14) and CO2
When excess trialkylborane was added, nearly quantitative hydride transfer was observed. Only a trace of unreacted [HNi(dmpe)2]+ was observed.
Reaction of [Ni(dmpe)2][PF6]2 with [Bu4N][HCO2]
A small vial was charged with 19.7 mg (0.0359 mmol) [Ni(dmpe)2][PF6]2 and 10.3 mg (0.0359 mmol), and ~0.6 mL CD3CN. As the mixture was stirred a color change from yellow to dark orange was observed. The mixture was transferred to a J-Young NMR tube, and monitored. The color faded to a lighter orange over a few minutes. After 15 minutes, almost complete conversion (~95%) to [HNi(dmpe)2][PF6] was observed, although a slight excess of [HCO2]− (δ 8.58) was still present.
Addition of [Bu4N][HCO2] after reduction
A 1:10 mixture of [HNi(dmpe)2][PF6] and tBu(CH2)2B(C8H14) was treated with CO2 according to the general procedure. After the reaction was complete the tube was brought into a glovebox and 1 equiv [Bu4N][HCO2] was added to the tube. The resonance at δ 8.210 roughly doubled in intensity (and shifted by only 0.003 ppm), thereby confirming that formate is the product of CO2 reduction.
Reaction of [HNi(dmpe)2]+ and tBu(CH2)2B(C8H14) with 13CO2
A J-Young NMR tube was charged with 18.9 mg (0.0374 mmol) [HNi(dmpe)2][PF6], 38.6 mg (0.187 mmol, 5 equiv) tBu(CH2)2B(C8H14), and ~0.6 mL CD3CN. After initial NMR spectroscopic measurements, the tube was subjected to two freeze–pump–thaw cycles, and 0.0187 mmol (0.5 equiv) 13CO2 was condensed from a 2.87 mL calibrated gas bulb (11.8 mmHg). About 5% conversion to [Ni(dmpe)2][PF6]2 had occurred after 30 minutes, and a small doublet could be seen by 1H NMR. After 12 hours, a large doublet at δ 8.21 (202 Hz) was visible, consistent with formation of [H13CO2(BR3)n]−.
Attempted catalytic reactions
Attempted catalysis in acetonitrile
A J-Young NMR tube was charged with 5.0 mg (0.0102 mmol, 1 equiv) [Ni(dmpe)2][BF4]2, 7.1 μL (0.0511 mmol, 5 equiv) NEt3, 10.5 mg (0.0511 mmol, 5 equiv) tBu(CH2)2B(C8H14), and ~0.6 mL CD3CN. The tube was subjected to two freeze–pump–thaw cycles, and 1 atm of a 1:1 H2:CO2 mixture (mixed in a large round bulb on a vacuum line) was admitted. The reaction was monitored by NMR spectroscopy, and a small formate peak gradually grew in over 3 days, along with another product, δ ~6.8. Heating the tube to 60 °C for ~24 hours generated visible amounts of [HNi(dmpe)2]+, but also several unidentified products.
Attempted catalysis in chlorobenzene
A J-Young NMR tube was charged with 35.5 mg (0.0174 mmol, 1 equiv) [Ni(dmpe)2][BArF4]2, 10.0 μL (0.0695 mmol, 4 equiv) NEt3, 3.6 mg (0.0174, 1 equiv) tBu(CH2)2B(C8H14), and ~0.6 mL C6D5Cl. The Ni2+ formed a yellow oil at the bottom of the tube if it was allowed to settle. The tube was subjected to two freeze–pump–thaw cycles, and 1 atm of a 1:1 mixture of H2:CO2 (mixed in a large round bulb on a vacuum line) was admitted to the tube. NMR spectroscopic monitoring showed steady generation of [HNi(dmpe)2][BArF4], but no evidence of formate was observed.
Hydrogen cleavage reactions
Hydrogen cleavage in acetonitrile
A J-Young NMR tube was charged with 32.6 mg (0.0159 mmol) [Ni(dmpe)2][BArF4]2, 8.9 μL (0.0639 mmol, 4 equiv) NEt3, and ~0.6 mL CD3CN. The tube was subjected to two freeze–pump–thaw cycles and 1 atm of H2 was admitted. The reaction was monitored by NMR spectroscopy, revealing about 30% conversion to [HNi(dmpe)2]+ over 3 days.
Hydrogen cleavage in chlorobenzene
A J-Young NMR tube was charged with 29.0 mg (0.0140 mmol) [Ni(dmpe)2][BArF4]2, 7.9 μL (0.0568 mmol, 4 equiv) NEt3, and ~0.6 mL C6D5Cl. The Ni dication was insoluble, and formed an oil at the bottom of the tube. Initial NMR measurements detected no Ni species. The tube was subjected to two freeze–pump–thaw cycles and 1 atm H2 was admitted. The tube was sealed and monitored by NMR, which revealed relatively rapid growth of [HNi(dmpe)2][BArF4]. After 18 hours, no insolubles were visible, and a large amount of Ni hydride was present. The two phase reaction prevented precise yields or equilibrium measurements.
CO2 reduction by rhodium and boranes in acetonitrile
Reaction of [Rh(dmpe)2][OTf] with H2/CO2
A J-Young NMR tube was charged with 19 mg (0.0344 mmol) [Rh(dmpe)2][OTf] and ~0.6 mL CD3CN. The tube was placed under 1 atm of a 1:1 mixture of H2 and CO2, mixed by diffusion in a large round bulb on a vacuum manifold. The reaction was monitored by multinuclear NMR, which showed formation of [H2Rh(dmpe)2][OTf], but no more than a trace of formate was observed.
Reaction of [Rh(dmpe)2][OTf] with tBu(CH2)2B(C8H14) and H2/CO2
A J-Young NMR tube was charged with 15.9 mg (0.0288 mmol) [Rh(dmpe)2[OTf], 5.9 mg (0.0288 mmol) tBu(CH2)2B(C8H14), and ~0.6 mL CD3CN. The tube was placed under 1 atm of a 1:1 mixture of H2 and CO2, mixed by diffusion in a large round bulb on a vacuum manifold. The reaction was monitored by NMR spectroscopy, which showed growth of signals attributable to [H2Rh(dmpe)2]+ (δ −10.25, br d, JPH = 146.4), and eventually [HCO2(BR3)n]− (δ 8.25) and [HRh(dmpe)2(MeCN)]2+ (δ −18.57, m).
Supplementary Material
Acknowledgment
This research was generously funded by BP through the Methane Conversion Cooperative (MC2) Program and by the Moore Foundation. The Varian 400-MR spectrometer was purchased through NIH grant RR027690.
Footnotes
Supporting Information Available. Accompanying NMR spectra for reactions described in the Experimental Section. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Benson EE, Kubiak CP, Sathrum AJ, Smieja JM. Chem. Soc. Rev. 2009;38:89. doi: 10.1039/b804323j. [DOI] [PubMed] [Google Scholar]; (b) Leitner W. Angew. Chem. Int. Ed. 1995;34:2207. [Google Scholar]; (c) Jessop PG, Joó F, Tai C. Coord. Chem. Rev. 2004;248:2425. [Google Scholar]
- 2.(a) Miller AJM, Labinger JA, Bercaw JE. J. Am. Chem. Soc. 2008;130:11874. doi: 10.1021/ja805108z. [DOI] [PubMed] [Google Scholar]; (b) Elowe PR, West NM, Labinger JA, Bercaw JE. Organometallics. 200928:6218. [Google Scholar]; (c) Miller AJM, Labinger JA, Bercaw JE. J. Am. Chem. Soc. 2010;132:3301. doi: 10.1021/ja100574n. [DOI] [PubMed] [Google Scholar]; (d) Miller AJM, Labinger JA, Bercaw JE. Organometallics. 2010;29:4499. [Google Scholar]; (e) West NM, Miller A,JM, Labinger JA, Bercaw JE. Coord. Chem. Rev. 2011;255:881. [Google Scholar]; (f) West NM, Labinger JA, Bercaw JE. Organometallics. 2011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rozovskii AY, Lin GI. Top. Catal. 2003;22:137. [Google Scholar]
- 4.(a) Hammouche M, Lexa D, Momenteau M, Saveant JM. J. Am. Chem. Soc. 1991;113:8455. [Google Scholar]; (b) Bhugun I, Lexa D, Saveant J-M. J. Am. Chem. Soc. 1994;116:5015. [Google Scholar]; (c) Bhugun I, Lexa D, Saveant J-M. J. Phys. Chem. 1996;100:19981. [Google Scholar]; (d) Bhugun I, Lexa D, Saveant J-M. J. Am. Chem. Soc. 1996;118:1769. [Google Scholar]; (e) Gennaro A, Isse AA, Severin M-G, Vianello E, Bhugun I, Saveant J-M. J. Chem. Soc, Faraday Trans. 1996;92:3963. [Google Scholar]; (f) Saveant J-M. Chem. Rev. 2008;108:2348. doi: 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]; (g) Wong K-Y, Chung W-H, Lau C-P. J. Electroanal. Chem. 1998;453:161. [Google Scholar]
- 5.(a) Steffey BD, Curtis CJ, DuBois DL. Organometallics. 1995;14:4937. [Google Scholar]; (b) Dubois MR, Dubois DL. Ace. Chem. Res. 2009;42:1974. doi: 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]; (c) Jeoung J-H, Dobbek H. Science. 2007;318:1461. doi: 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]
- 6.Boffa A, Lin C, Bell AT, Somorjai GA. J. Catal. 1994;149:149. [Google Scholar]
- 7.Laitar DS, Muller P, Sadighi JP. J. Am. Chem. Soc. 2005;127:17196. doi: 10.1021/ja0566679. [DOI] [PubMed] [Google Scholar]
- 8.Chakraborty S, Zhang J, Krause JA, Guan HR. J. Am. Chem. Soc. 2010;132:8872. doi: 10.1021/ja103982t. [DOI] [PubMed] [Google Scholar]
- 9.(a) Stephan DW, Erker G. Angew. Chem. Int. Ed. 2010;49:46. doi: 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]; (b) Ashley AE, Thompson AL, O'Hare D. Angew. Chem. Int. Ed. 2009;48:9839. doi: 10.1002/anie.200905466. [DOI] [PubMed] [Google Scholar]; (c) Tran SD, Tronic TA, Kaminsky W, Heinekey DM, Mayer JM. Inorg. Chim. Acta. 2011;369:126. [Google Scholar]
- 10.(a) Matsuo T, Kawaguchi H. J. Am. Chem. Soc. 2006;128:12362. doi: 10.1021/ja0647250. [DOI] [PubMed] [Google Scholar]; (b) Berkefeld A, Piers WE, Parvez M. J. Am. Chem. Soc. 2010;132:10660. doi: 10.1021/ja105320c. [DOI] [PubMed] [Google Scholar]
- 11.DuBois DL, Berning DE. Appl. Organomet. Chem. 2000;14:860. [Google Scholar]
- 12.(a) Berning DE, Noll BC, DuBois DL. J. Am. Chem. Soc. 1999;121:11432. [Google Scholar]; (b) Berning DE, Miedaner A, Curtis CJ, Noll BC, Rakowski DuBois MC, DuBois DL. Organometallics. 2001;20:1832. [Google Scholar]; (c) Curtis CJ, Miedaner A, Ellis WW, DuBois DL. J. Am. Chem. Soc. 2002;124:1918. doi: 10.1021/ja0116829. [DOI] [PubMed] [Google Scholar]
- 13.Wilson AD, Miller AJM, DuBois DL, Labinger JA, Bercaw JE. Inorg. Chem. 2010;49:3918. doi: 10.1021/ic100117y. [DOI] [PubMed] [Google Scholar]
- 14.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 15.Miedaner A, DuBois DL, Curtis CJ, Haltiwanger RC. Organometallics. 1993;12:299. [Google Scholar]
- 16.Hirano K, Yorimitsu H, Oshima K. Org. Lett. 2005;7:4689. doi: 10.1021/ol051917b. [DOI] [PubMed] [Google Scholar]
- 17.Silavwe ND, Goldman AS, Ritter R, Tyler DR. Inorg. Chem. 1989;28:1231. [Google Scholar]
- 18.DuBois DL, Blake DM, Miedaner A, Curtis CJ, DuBois MR, Franz JA, Linehan JC. Organometallics. 2006;25:4414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.