

Abstract
Transcription repression of the galactose operon of Escherichia coli requires (1) the binding of the GalR repressor to tandem operators flanking the promoters, (2) the binding of histone-like protein, HU, to a site between the GalR-binding sites, and (3) negatively supercoiled DNA. Under these conditions, protein–protein interactions mediate the formation of a nucleoprotein complex in the form of a DNA loop, which we have termed a repressosome. To analyze the structure of the repressosome, we have screened and isolated galR mutants in which single amino acid substitutions in GalR lead to defects in loop formation while the protein’s operator-binding activity is retained. The mutant proteins were purified and their properties confirmed in vitro. We verified that in the case of the two stronger mutations, the proteins had secondary structures that were identical to that of wild-type GalR as reflected by circular dichroism spectroscopy. Homology-based modeling of GalR by use of the crystal structures of PurR and LacI has enabled us to place the three sites of mutation in a structural context. They occur in the carboxy-terminal subdomain of the GalR core, are surface exposed, and, therefore, may be involved in protein–protein interactions. On the basis of our model of GalR and its structural alignment with LacI and PurR, we have identified additional residues, the substitution of which leads to a specific defect in repression by looping. The effects of the mutations are the same in the presence of HMG-17, a eukaryotic protein unrelated to HU, which can also mediate GalR-dependent repression of the gal promoter. This observation suggests that the mutations define sites of GalR–GalR interaction rather than HU–GalR interaction in the repressosome.
Keywords: Repressosome, transcription, galR mutants, protein–protein interactions, DNA looping, GalR, repression
DNA looping is a general phenomenon in the control of transcription in which two or more DNA-binding proteins recognize DNA sequences flanking a promoter and, by contacting each other, cause a reversible transition to a nucleoprotein complex in which the intervening DNA is looped out and the promoter activity is altered (Adhya 1989; Martin et al. 1990). The relative simplicity of these complexes in prokaryotes compared with their counterparts in eukaryotes has facilitated the investigation by genetic and physicochemical methods of questions critical to the understanding of regulation by DNA looping. Some of these questions are structural in nature: What are the protein–protein and protein–DNA contacts that stabilize these loops? What are the symmetries, if any, of the protein complexes? Other questions are framed in terms of thermodynamics and seek to understand the origin of the negative-free energy of the looped structure. Critical mechanistic questions include how loop formation modulates transcription activity and by what means the stability of the loop is sensitive to environmental conditions.
We have used the nucleoprotein complex formed at the galactose (gal ) operon of Escherichia coli by the gal operon repressor, GalR, and a bacterial histone-like protein, HU, as a model system for the investigation of the structure and function of a repression loop. GalR, a member of the LacI family of gene regulatory proteins (Weickert and Adhya 1992), binds as a dimer with high and approximately equal affinity to two operator sequences separated by 113 bp, encompassing the two gal promoters (Fig. 1A; Majumdar and Adhya 1984; Brenowitz et al. 1990). It lacks the tetramerization domain found in LacI and forms a tetramer in solution, if at all, with much weaker affinity (Majumdar et al. 1987). The formation of a loop absolutely requires HU, an abundant component of the bacterial nucleoid, which, in the presence of DNA supercoiling and occupancy by GalR of both operators, binds to a specific site, hbs, located between the operators (Aki et al. 1996; Aki and Adhya 1997). We have termed the nucleoprotein complex that represses the gal promoters a repressosome, because its formation is by site-specific binding of a histone-like component of the bacterial nucleoid as well as a repressor (Adhya et al. 1998). In this report we describe a genetic screen to identify residues in GalR that are critical to repressosome formation and that we propose define sites of protein–protein contact between GalR dimers. We characterized the mutant proteins both in vivo and in vitro. Homology modeling of GalR with two highly conserved members of the LacI family for which the X-ray crystal structures have been reported, PurR and LacI, permitted us to place the mutated residues in a structural context and generate additional mutations that specifically disrupt loop formation. The usefulness of our findings in formulating a model for the structure of the repressosome is discussed.
Figure 1.

(A) Map of the gal promoter region showing the location of the P1 and P2 promoters, the tandem gal operators, and the center of the HU-binding site hbs. (B) Schematic drawing of the OE+P2−P1+galE–lacZ and OE+P2+P1−galEOI+–gusA fusions. The latter is present as a lysogen.
Results
Genetic screen for galR mutants defective in DNA looping
To isolate mutations in galR, the gene encoding the repressor protein, that prevent the formation of a DNA loop while not affecting the interaction of GalR with the operator, we devised a genetic screen that made use of the fact that the two gal promoters, P1 and P2, are regulated differently. GalR binding to the upstream gal operator, OE, is sufficient for repression of P1. P2, located 5-bp upstream from P1, is activated under these conditions (Choy et al. 1997). The repression of P2 requires the formation of a loop through simultaneous binding of GalR to both OE and the downstream operator, OI, as well as binding of HU to hbs. As shown schematically in Figure 1B, two chromosomal fusions were constructed in the reporter strain (DM0022). In one of these, the gal promoter was mutated to inactivate P2 transcription while retaining P1 transcription (Bingham et al. 1986) and fused to the lacZ gene such that OI was deleted. This fusion, in which lacZ transcription from P1 is repressed by GalR binding to OE, was used to measure GalR-binding activity. To measure loop formation, the gal promoter in a second fusion was mutated so that transcription occurred only from P2 (Bingham et al. 1986) and joined to the gusA reporter gene such that OI was retained. The efficacy of the promoter mutations in completely inactivating P1 or P2 while retaining transcription from the other promoter was verified by measuring RNA synthesis in vivo by primer extension. When cells containing a wild-type gal promoter were induced in the absence of CRP binding, the level of the expected 64- and 69-base products of reverse transcription from a primer complementary to galE was equivalent, indicating that the levels of P1 and P2 transcripts are equal (Fig. 2, lanes 3 and 4). The G-to-A transition at position −14 completely eliminated transcription from P1 (Fig. 2, lane 8), whereas a G-to-T transversion at position −19 abolished transcription from P2 (Fig. 2, lane 6). Galactose-sensitive repression is unaffected by both mutations (Fig. 2, lanes 5 and 7).
Figure 2.

Effect of gal promoter mutations on in vivo transcription at P1 and P2 promoters. Primer extension of gal-specific DNA was performed in strain DM0013 containing the wild-type gal promoter (lanes 3,4), DM0012 in which the gal promoter contains a G-to-T transversion at position −19 (lanes 5,6), or DM0011 containing a G-to-A transition at −14 (lanes 7,8). Cells were grown in the absence (lanes 3,5,7) or presence (lanes 4,6,8) of the inducer d-galactose. (Lane 1) Phosphorylated DNA standards; (lane 2) expected 87-base primer-extension product from a control reaction with a specific primer with homogeneous RNA.
Localized mutagenesis of galR was performed by hydroxylamine treatment of a bacteriophage P1 lysate of a strain in which galR was tightly linked to a kanamycin resistance cassette. The mutagenized phage lysate was used to transduce the kan allele to the reporter strain, DM0022. Screening was carried out for transductants that were derepressed for gusA expression, and, therefore, looping deficient but were still repressed for lacZ expression, indicating that GalR retained operator-binding activity. Of 6000 transductants screened, 350 colonies showed increased levels of P2 transcription as measured by their blue color on indicator plates for β-glucuronidase, and these were assayed for expression of the galP1–lacZ fusion. Six of these mutants were found to be repressed for β-galactosidase expression to levels observed in a galR+ strain. Two mutants were identical and coded for an aspartate-to-asparagine change at residue 258 (D258N). Two mutations resulted in redundant codons causing a lysine substitution at glutamate 230 (E230K). The remaining two mutants had a histidine substitution at arginine 325 (R325H), with one having an additional substitution of valine for isoleucine at position 323. The double mutant was not studied further.
In sequencing the mutants, we observed that the strain JT30 used to mutagenize galR contained an insertion of a kan cassette at position 1067 of the gene corresponding to residue 328. This insertion caused a very slight reduction in the efficiency of P2 repression when compared with wild type (data not shown). We subsequently introduced an amber mutation in galR by site-directed mutagenesis so that the protein terminated at residue 328 and found that deletion of the 15 carboxy-terminal amino acids of GalR had little, if any, effect on nonlooping-mediated repression of P1 or looping-mediated repression of P2 (Table 1). The three mutations discussed above were cloned into a wild-type (full-length galR) background by site-directed mutagenesis and transferred to the chromosome. Their phenotypes are reported below.
Table 1.
Effect of GalR mutations on operator interaction and looping-mediated repression
| GalRa
|
β-glucuronidaseb (OE+P2+P1−OI+–gusA)
|
β-galactosidaseb (OE+P2−P1+–lacZ)
|
|---|---|---|
| Wild type | 1.9 | 15 |
| W165R | 2 | 16 |
| L166K | 2.1 | 14 |
| Q173A | 2 | 18 |
| Q174L | 2.5 | 17 |
| F216Q | 3 | 10 |
| G233N | 3.5 | 10 |
| T322R | 50 | 16 |
| N259M | 30 | 13 |
| H327R | 50 | 15 |
| S222E,Q226R | 2.8 | 23 |
| Δ329-343 | 2.8 | 21 |
| E230K | 15 | 19 |
| D258N | 72 | 18 |
| R325H | 64 | 17 |
| galRc | 2.5 | 45 |
| ΔgalRc | 100 | 100 |
GalR expressed from multicopy plasmids.
Enzyme activities are expressed as a percent of that obtained in the ΔgalR strain. The wild-type results are the mean of four separate experiments.
galR or ΔgalR in single chromosomal copy.
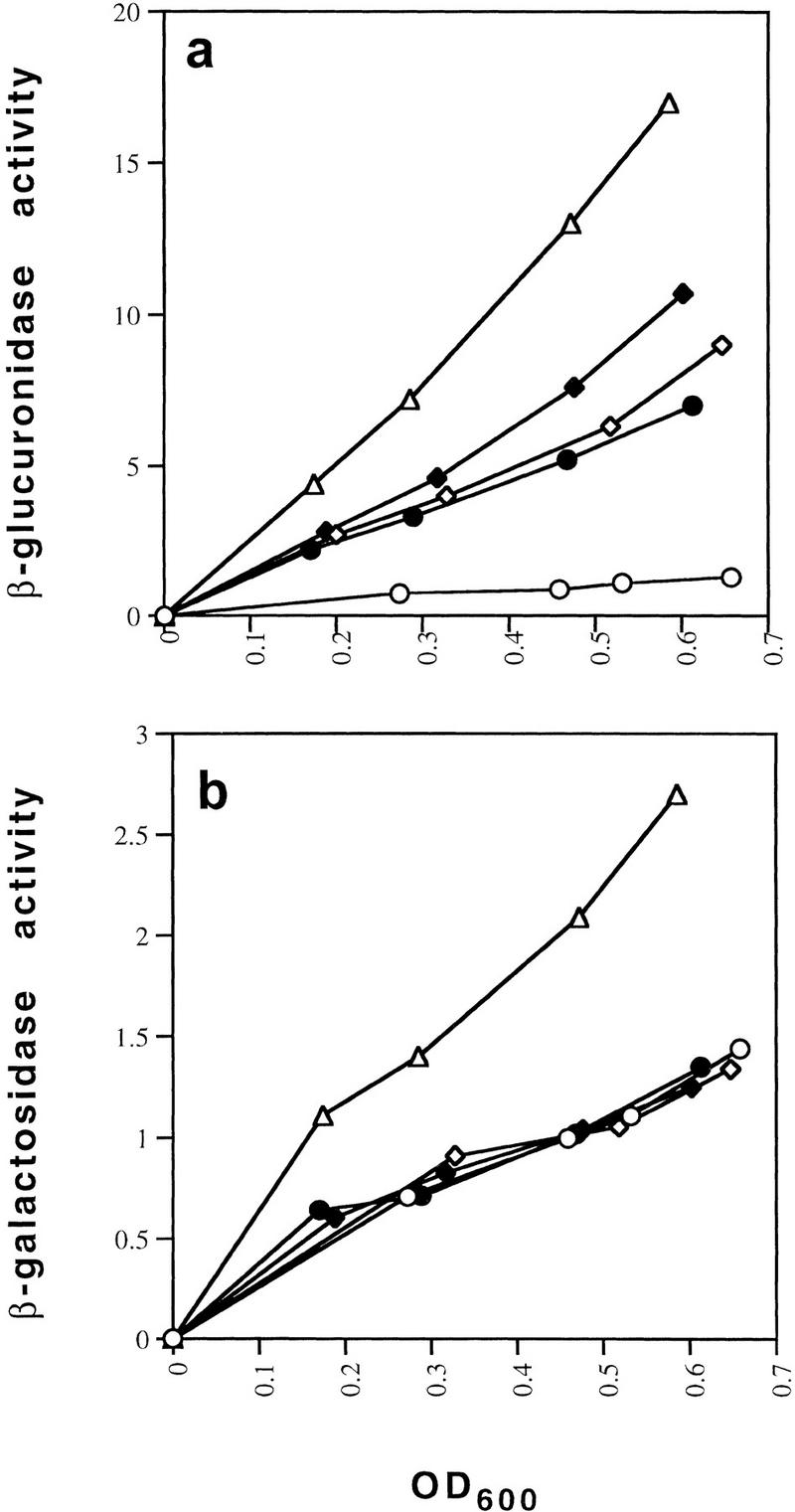
The mutants were assayed under conditions of exponential growth for expression from the P1 and P2 gal fusions. As shown in Figure 3a, the D258N mutant of GalR was nearly completely defective for repression of P2 expression exhibiting a level of β-glucuronidase activity that was comparable with that of the ΔgalR strain. The galR mutants R325H and E230K showed derepression of P2 expression to levels that were 78% and 50%, respectively of that of the ΔgalR strain. In wild-type cells in the absence of the downstream gal operator, OI, the P1 promoter as assayed by β-galactosidase activity was repressed such that expression was 40% of the level in the ΔgalR strain, suggesting that the intracellular concentration of GalR is one that makes the fusion a sensitive reporter of affinity (Fig. 3b). Overexpression of GalR forms a multicopy plasmid in which galR is under the control of an IPTG-inducible Trc promoter-abolished expression of the P1–lacZ fusion (data not shown). The E230K mutant caused a slight increase in P1 expression to 49% of that obtained in the ΔgalR strain. D258N and R325H caused barely measurable increases to levels that were 41% and 44%, respectively, of the expression in the ΔgalR strain (Fig. 3b). When the mutant and the wild-type GalR proteins were expressed from plasmids, all showed identical levels of repression of the P1–lacZ fusion, whereas the three mutants remained defective for repression of P2 (Fig. 3c,d).
Figure 3.
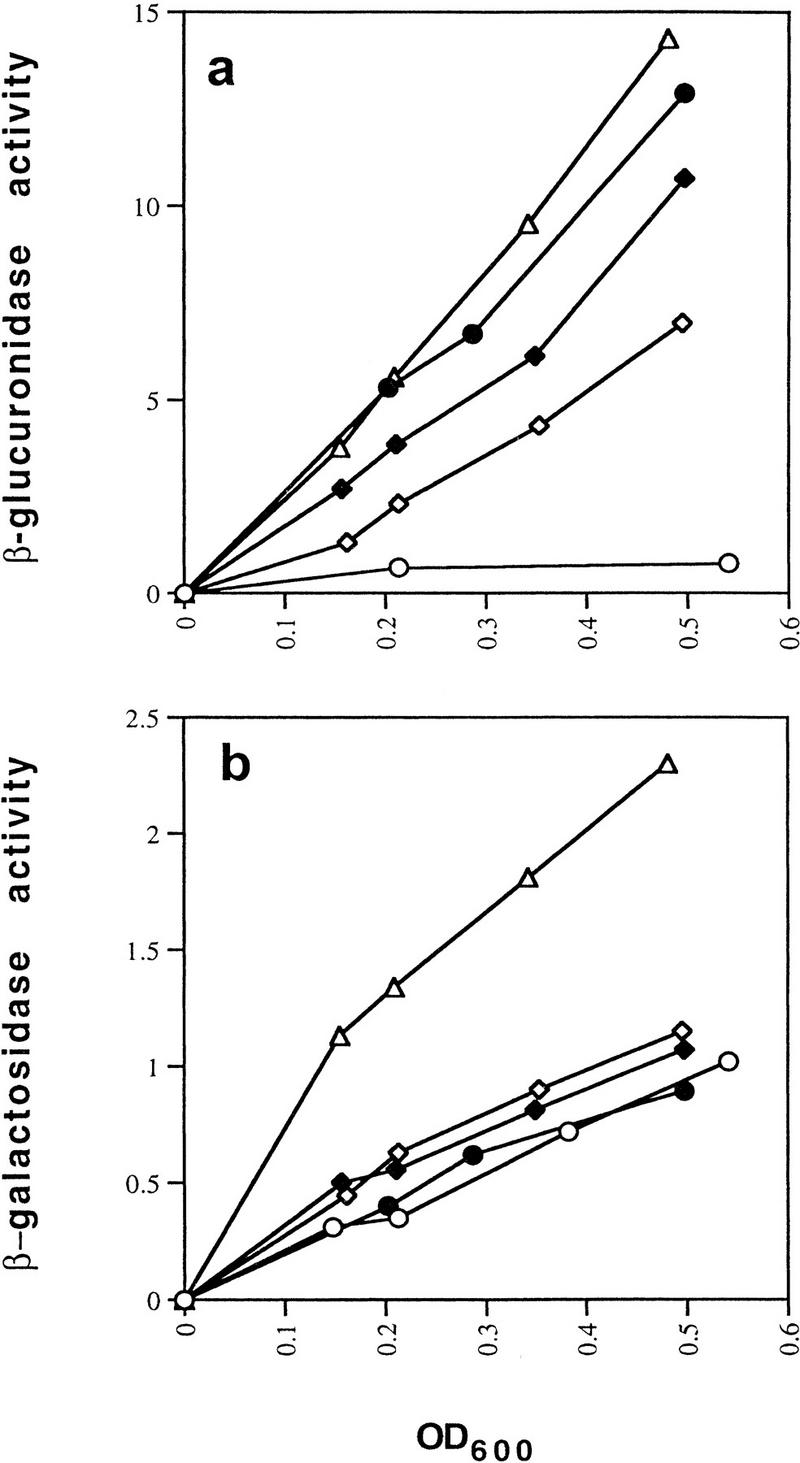
Effect of GalR mutations E230K, D258N, and R325H on looping-dependent repression of galP2 (a) and looping-independent repression of galP1 (b). galR+ (○), galR D258N (●), galR E230K (⋄), galR R325H (♦), and ΔgalR (○) strains were grown in minimal medium and assayed for expression of the P2–gusA fusion containing both operators (a) and the galP1–lacZ fusion containing only OE (b). β-Galactosidase and β-glucuronidase activities are expressed as the change in optical density/min during enzyme assay. (c,d) Expression of the gusA and lacZ reporter genes when the same set of galR alleles were expressed from a multicopy plasmid vector in a host deleted for galR.
In vitro properties of GalR-looping mutants
Wild-type GalR and mutant GalR proteins E230K, D258N, and R325H were purified following overexpression to >95% homogeneity as described in Materials and Methods and assayed as regulators of in vitro transcription with a containing the wild-type gal promoter with both operators present as the template. The products of the transcription reactions after resolution by gel electrophoresis are shown in Figure 4. The results are expressed graphically in Figure 5. In vitro, the binding of GalR to OE is sufficient for repression of transcription of P1 and simultaneous activation of the P2 promoter (Choy and Adhya 1993). At higher concentrations of GalR, transcription termination begins to occur at OI, and, because the resulting short transcripts are not observed in 8% gels, the GalR titration appears biphasic. Looping-mediated repression of the promoters requires HU, which on supercoiled templates in the presence of GalR, binds to the DNA at hbs between the two operators. Figure 5a shows that in the absence of HU, wild-type GalR, E230K, and R325H repressed the P1 promoter with approximately equal efficiency, whereas D258N required about twofold higher concentration to achieve the same level of repression. Wild-type GalR and all of the mutants activated transcription from P2 approximately twofold (Fig. 5b). In the presence of 80 nm HU, the wild-type protein repressed both P1 and P2 efficiently with half-maximal repression of P2 achieved at a slightly higher concentration than that required for P1 (Fig. 5c,d). Under similar conditions, D258N and R325H behaved as they did in the absence of HU as activators of P2 expression, although the level of activation was somewhat less. E230K, which had the smallest effect on P2 repression in vivo, was only partially defective in HU-dependent repression of P2 compared with the wild type.
Figure 4.

Transcription of galP1 and galP2 promoters on the supercoiled template pSA509 in the presence of wild-type GalR and GalR mutants E230K, D258N, and R325H. (Top) Titration with GalR proteins in the absence of HU or HMG-17; (middle, bottom the same reactions in the presence of 80 nm HU dimer or 500 nm HMG-17 (monomer), respectively. The 80-bp RNA 1 transcripts, which do not vary as a function of GalR concentration, served as an internal control between lanes.
Figure 5.
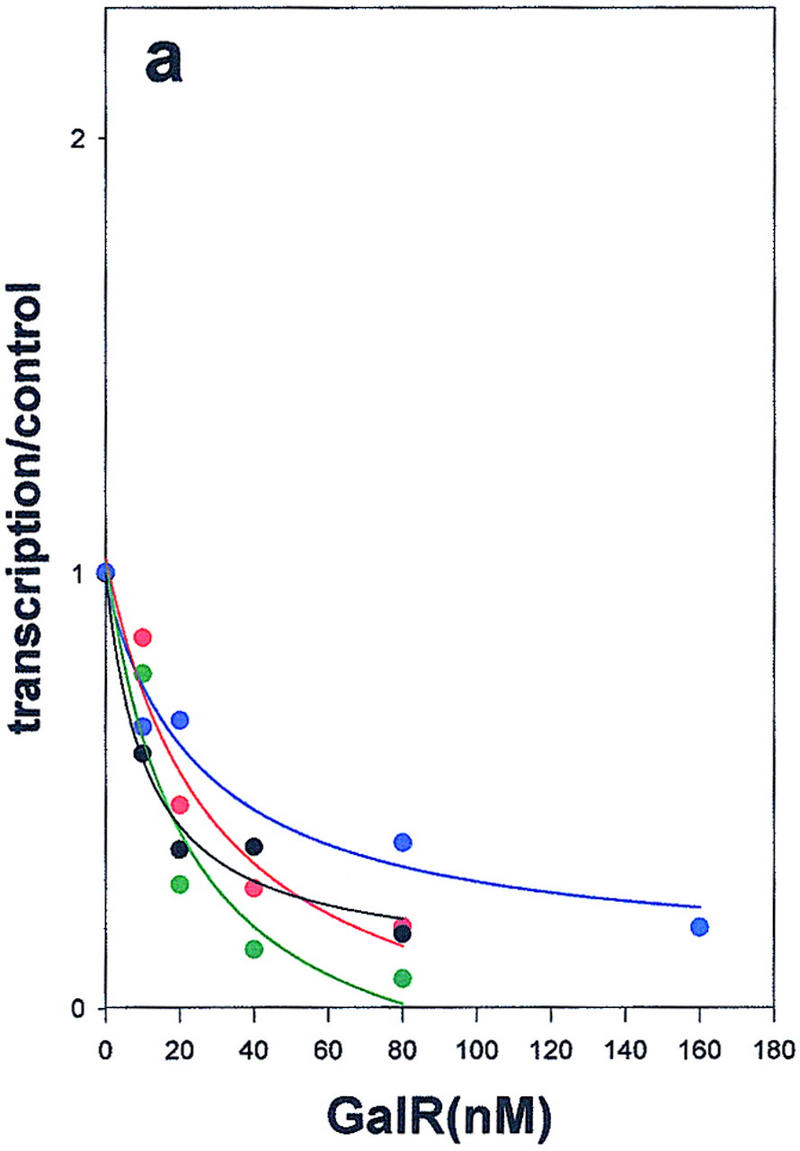
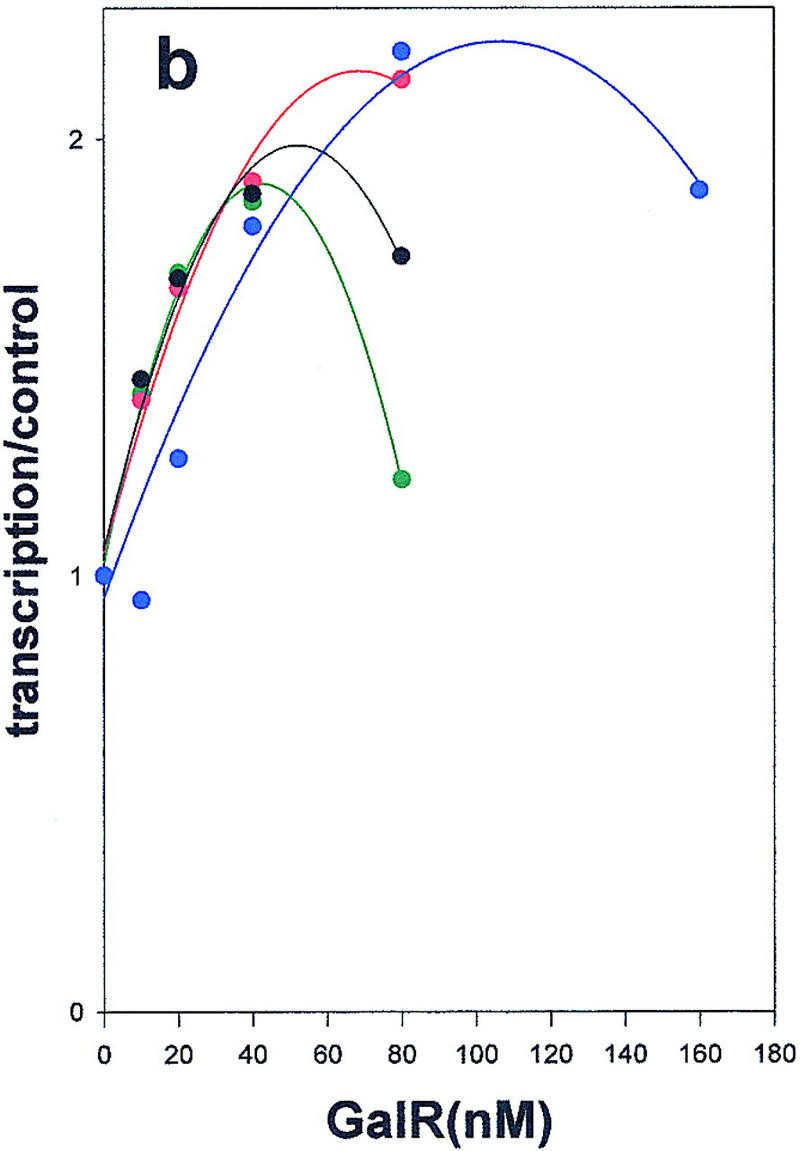
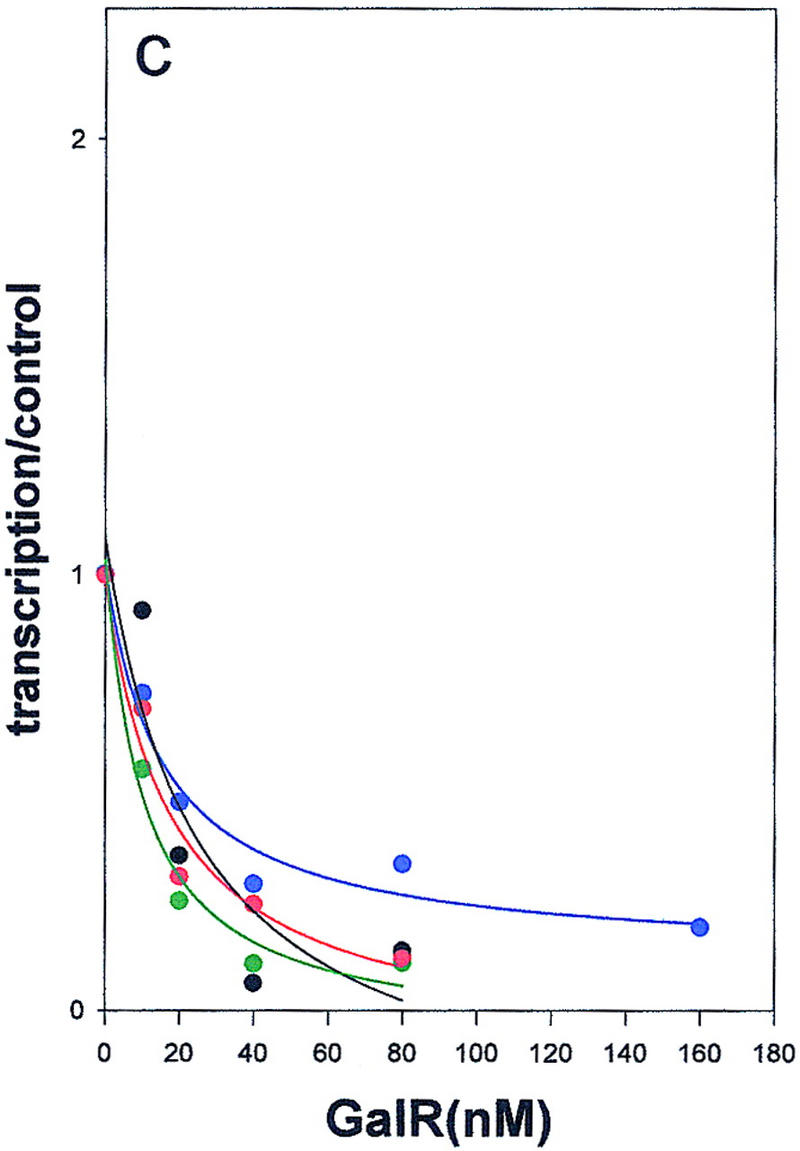
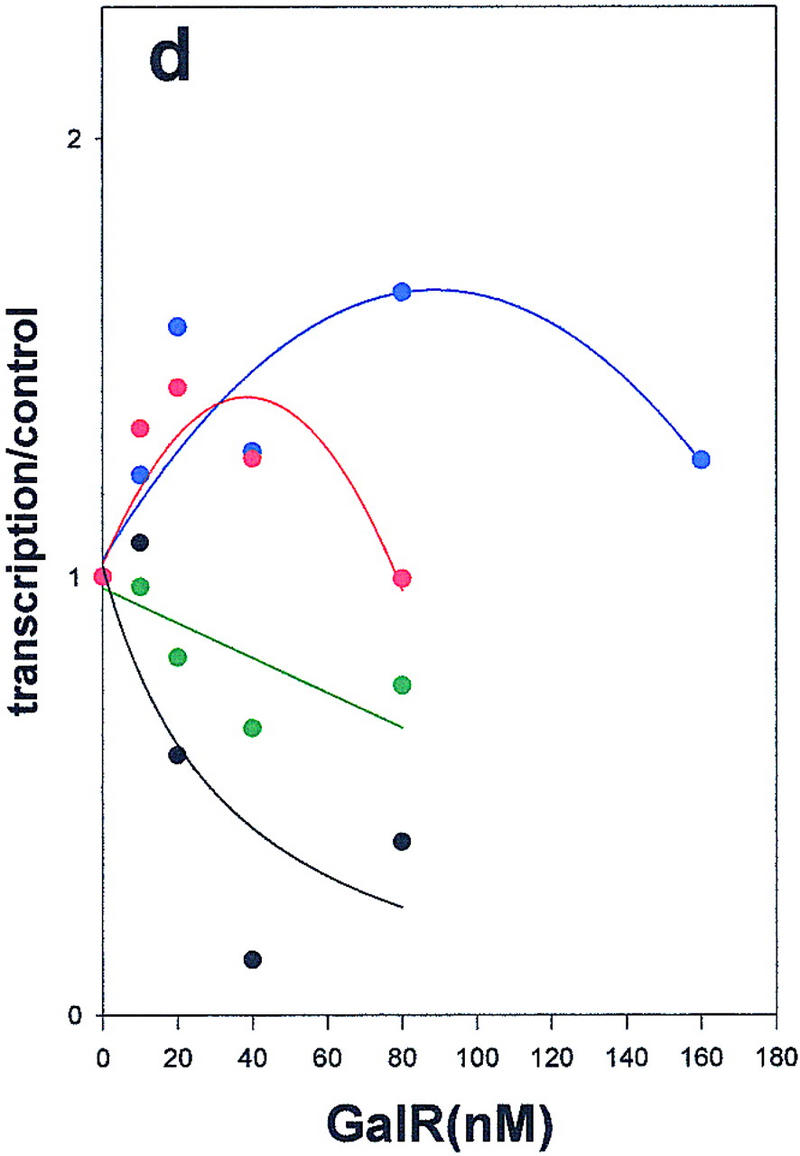
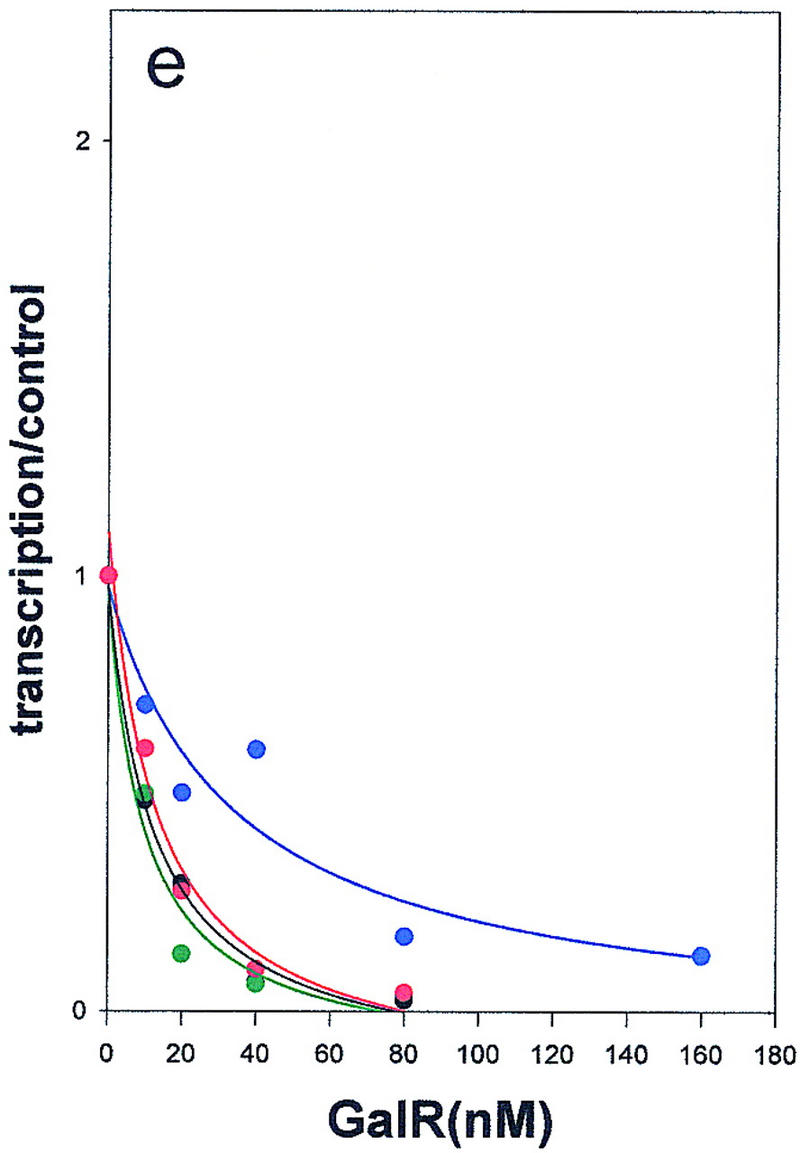
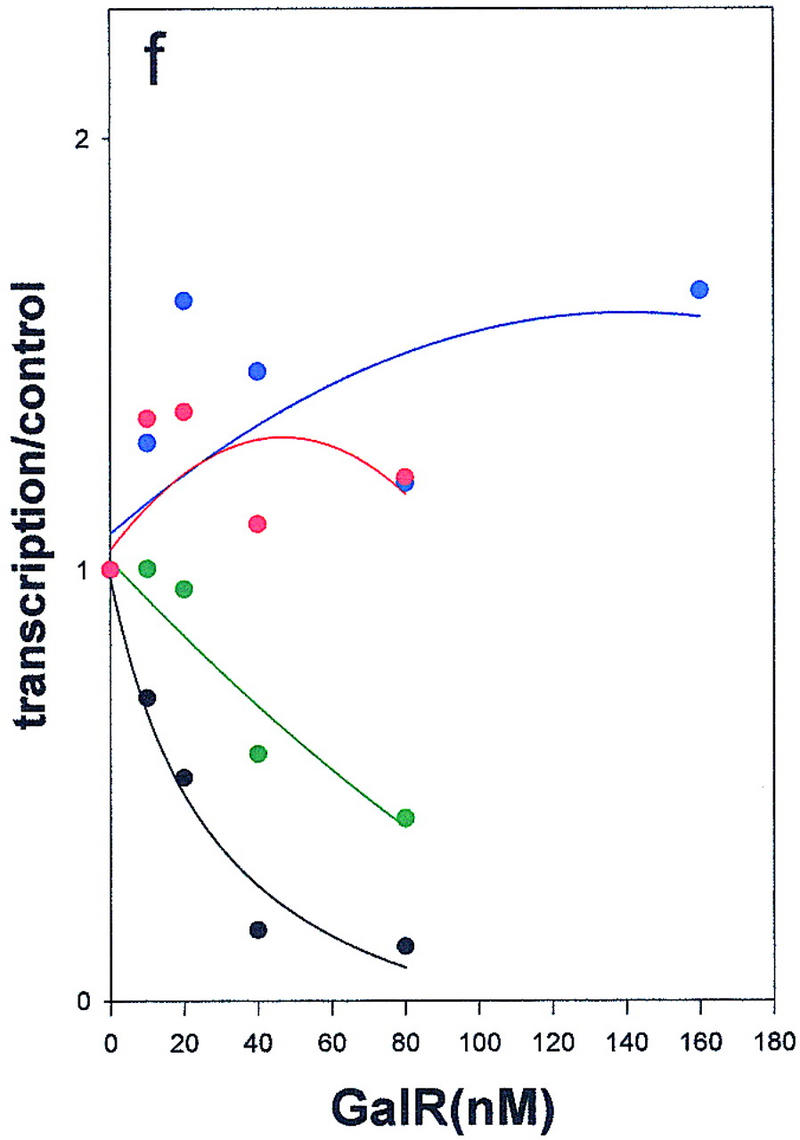
Regulation of the galP1(a,c,e) and galP2 (b,d,f) promoters in the absence of cofactors (a,b) or in the presence of HU (c,d) or HMG-17 (e,f) by wild-type GalR (black), or GalR mutants E230K (green), D258N (blue), or R325H (red). RNA from gels in Fig. 4 was quantified with a PhosphorImager, and transcription is expressed as the fraction of that which was obtained in the absence of the repressor protein. Repression data for P1 and for P2 with wild-type GalR in the presence of HU or HMG-17 were fitted to the function 1/(1 + Kx) in which x is the concentration of the repressor and K is an apparent association constant. All other data were fitted to a second-order polynomial.
We had determined previously that an abundant eukaryotic protein, HMG-17, although not affecting gal transcription by itself, can substitute for HU in mediating GalR-dependent repression of P2 transcription (T. Aki, unpubl.). HMG-17 is an 89-amino acid DNA-binding protein of unknown structure that has been localized to sites at which DNA exits from the nucleosome core (Bustin and Reeves 1996). In spite of their shared function, HMG-17 and HU have limited sequence homology (see below). As shown in Figure 5e, HMG-17 mediates complete repression of P2 transcription in the presence of wild-type GalR. The defects in the mutant GalR proteins in repressing transcription from P2 in the presence of HMG-17 were indistinguishable from those observed in the presence of HU; E230K was only moderately defective for P2 repression, whereas D258N and R325H remained activators of P2 transcription (Fig 5f). The significance of this finding with respect to the structure of the repressosome is discussed below.
Secondary structure of D258N and R325H
The mutations described above could indicate sites of protein–protein contact as long as they did not lead to large structural alterations. We used circular dichroism spectroscopy, which is sensitive to the secondary structure of proteins, to determine whether such alterations had occurred in the two mutants with the strongest derepression phenotype, D258N and R325H. As shown in Figure 6, the far-UV CD spectra of wild-type, R325H, and D258N proteins are identical, suggesting that these mutations did not detectably alter the GalR secondary structure.
Figure 6.
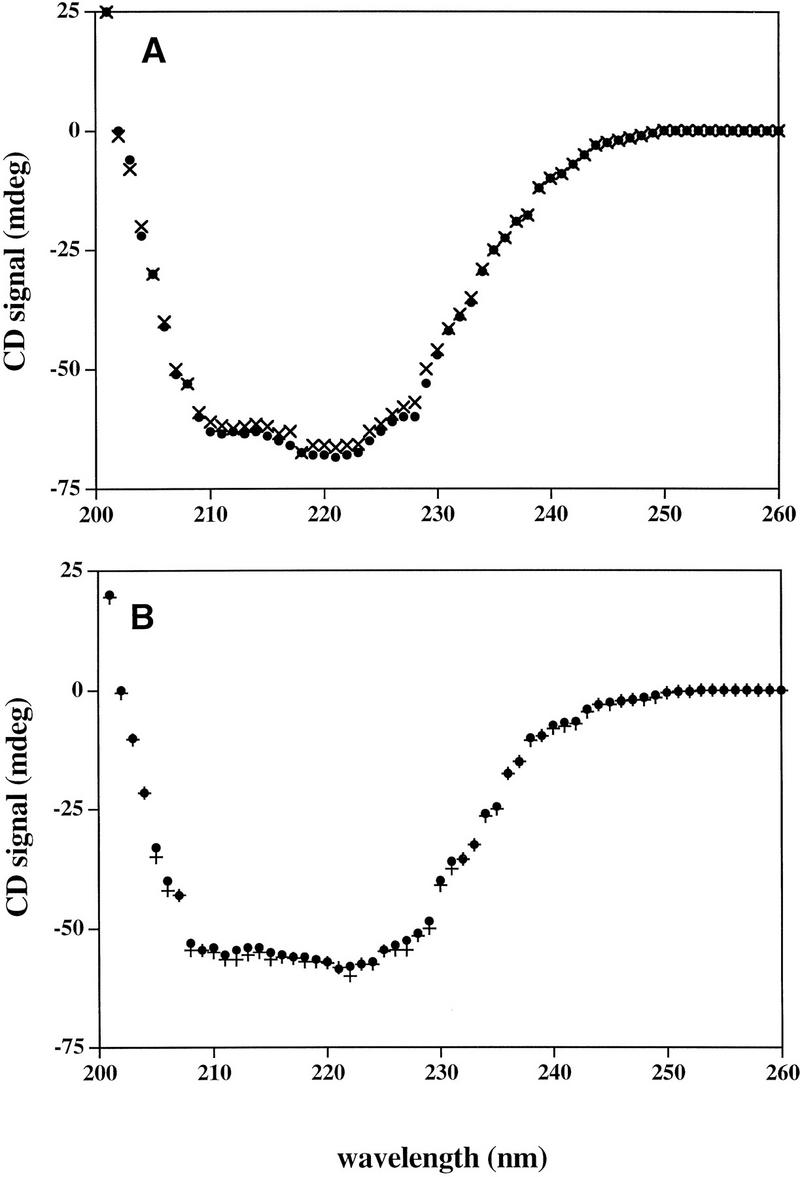
Far-UV CD spectra of GalR wild type (●) and GalR mutant D258N (×) (A), and GalR wild type (●) and GalR mutant R325H (+) (B).
Molecular modeling of GalR
The structure of GalR has not been determined experimentally. To situate the amino acid changes described above in a structure, we modeled GalR by homologous extension using the X-ray crystal structures of two repressors, PurR and LacI (Schumaker et al. 1994; Lewis et al. 1996), which have a significant homology with GalR [31% and 27% sequence identity, respectively (Weickert and Adhya 1992)]. We confined our model to the core of the protein in which all of the mutations occurred. In members of the LacI family, the core is comprised of two globular subdomains (residues 52–343 in GalR) in which inducer binding and dimerization occur. It is separated from the amino-terminal DNA-binding domain by trypsin cleavage. The cores of LacI and PurR have a high degree of structural similarity allowing a close structural alignment [root mean square deviation (RMSD) 1.1 Å]. This structural alignment can improve the sequence alignment of LacI and PurR, and, therefore, a more reliable sequence alignment of these two with GalR can be obtained.
The method used for modeling of GalR is that used for the modeling of the T-cell receptor from the structures of immunoglobulins (Vasmatzis et al. 1996) and consists of the following steps. First, the LacI and PurR amino acid sequences were aligned by the BESTFIT program of the GCG Wisconsin Sequence Analysis Package. This tactic permitted an initial structural alignment in which the backbone atoms of all of the aligned positions of LacI were superimposed on the backbone atoms of PurR. This operation was performed by minimizing the RMSD of the backbone atoms at the aligned positions with a rotation/translation algorithm (Cornette et al. 1993). This initial alignment was not optimal because of the presence of mismatches in the sequence alignment, and therefore, using the aligned structures, we constructed a new sequence alignment that made use of the structural alignment. A second structural alignment was then performed on the basis of the aligned positions in the new sequence alignment. The structures of the cores of LacI and PurR were found to be very similar (1.1 Å RMSD) except for four short segments in domain 2 where the ligands bind and for the carboxyl terminus of the proteins where the unique tetramerization domain of LacI is found. In all other domains, the proteins are highly similar, and we expect that the predicted model of GalR will be more accurate. The PurR backbone atoms corresponding to regions in which the backbone deviated <1.5 Å after superposition between PurR and LacI (the template structure) were used to form a portion of the GalR backbone. To fill in the gaps, we picked the corresponding segments from either PurR or LacI that have the same length as the GalR segments and pasted them on the modeled GalR backbone with priority given to the segments of PurR. If there was no appropriate segment from LacI or PurR, we filled the gap with the GalR segment using a loop-closure algorithm (Vasmatzis et al. 1996). The side chains of the final structure were then mutated to the corresponding side chains of GalR.
The GalR dimer was also modeled on the basis of the PurR and LacI structures. Both PurR and LacI dimerize in a similar manner with the two monomers in a symmetrical topology with an axis of symmetry that runs between the molecules along the larger dimension of the core. The PurR and LacI dimers superimpose with ∼4 Å RMSD with each other. We assumed that the GalR dimer forms in a manner similar to the PurR dimer. Figure 6 shows the structure of the modeled GalR core with the mutated residues described above highlighted in red. The three residues are surface exposed and reside on the carboxy-terminal subdomain of the core.
Site-directed mutagenesis of GalR
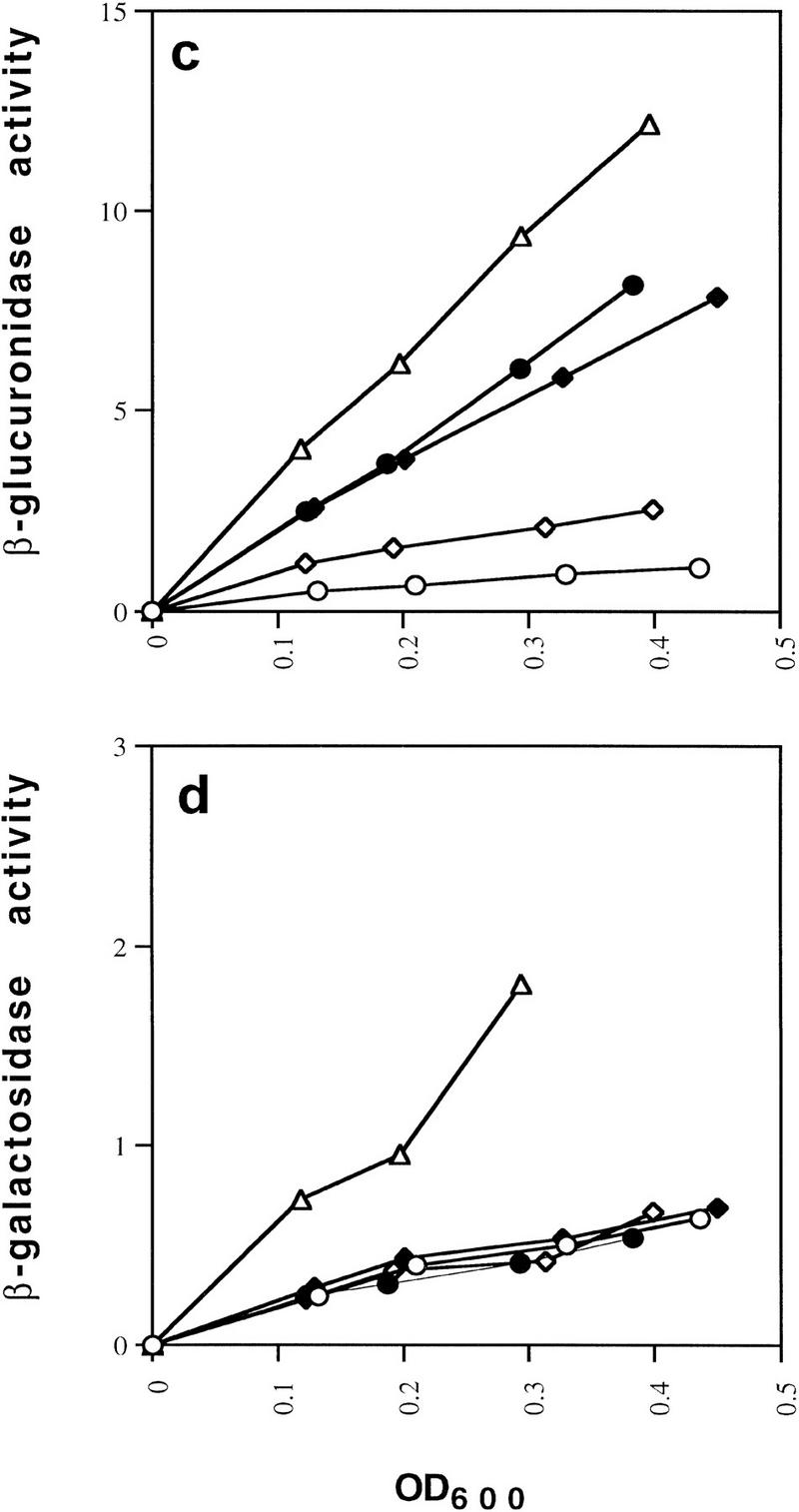
The mutations we identified suggested that domains in the carboxy-terminal subdomain of the protein core made GalR–GalR and/or HU–GalR contacts in the repressosome. To obtain a clearer picture of these domains, we performed site-directed mutagenesis of amino acids, which roughly spanned a region defined by the initial three mutations. We attempted to introduce amino acids that would alter the surface of the GalR dimer but not disrupt the structure of the protein. Utilizing the structural alignment of the PurR and LacI crystal structures and the GalR model described above, we mutated the surface residues in GalR to only those amino acids that were found at corresponding positions (i.e., they were superimposed) in PurR or LacI after structural alignment. For example, the nonconservative substitution of arginine for threonine at position 322 in GalR might disrupt any protein–protein contact in which this residue was involved; however, because the structural alignment we performed indicates that arginine occurs at the corresponding structural position in LacI, its substitution for threonine in GalR would be unlikely to disrupt the overall structure. Ten such substitutions carried on a plasmid vector containing the galR gene and its promoter were assayed in the double-reporter strain deleted for galR (Table 1). In one case, a double mutation was made so that a close interaction between glutamate 223 and arginine 227 of PurR was preserved. All of the mutants were indistinguishable from wild-type GalR for repression of the P1–lacZ fusion, suggesting that the mutations did not destabilize the protein or alter the operator-binding activity to an appreciable extent. Three of the site-directed mutants, N259M, T322R , and H327R derepressed expression from the P2 promoter and are shown in orange in Figure 7. Mutations that had no discernible affect on repression are shown in green. As indicated in Figure 7, residues 259 and 327 are immediately adjacent to 258 and 325, respectively, according to the model of GalR. T322 is located on the same face of the carboxy-terminal subdomain as the other mutations, but somewhat farther from the DNA distal edge of this surface. Because the expression of the P1–lacZ fusion appears to be a more sensitive assay of GalR-binding activity when galR is present in a single chromosomal copy (Fig. 3a,c), we tested the looping-defective mutants, N259M, T322R, and H327R after transfer to the chromosome. All three mutants were indistinguishable from the wild type for P1 repression while showing pronounced defects in P2 repression (Fig. 8).
Figure 7.
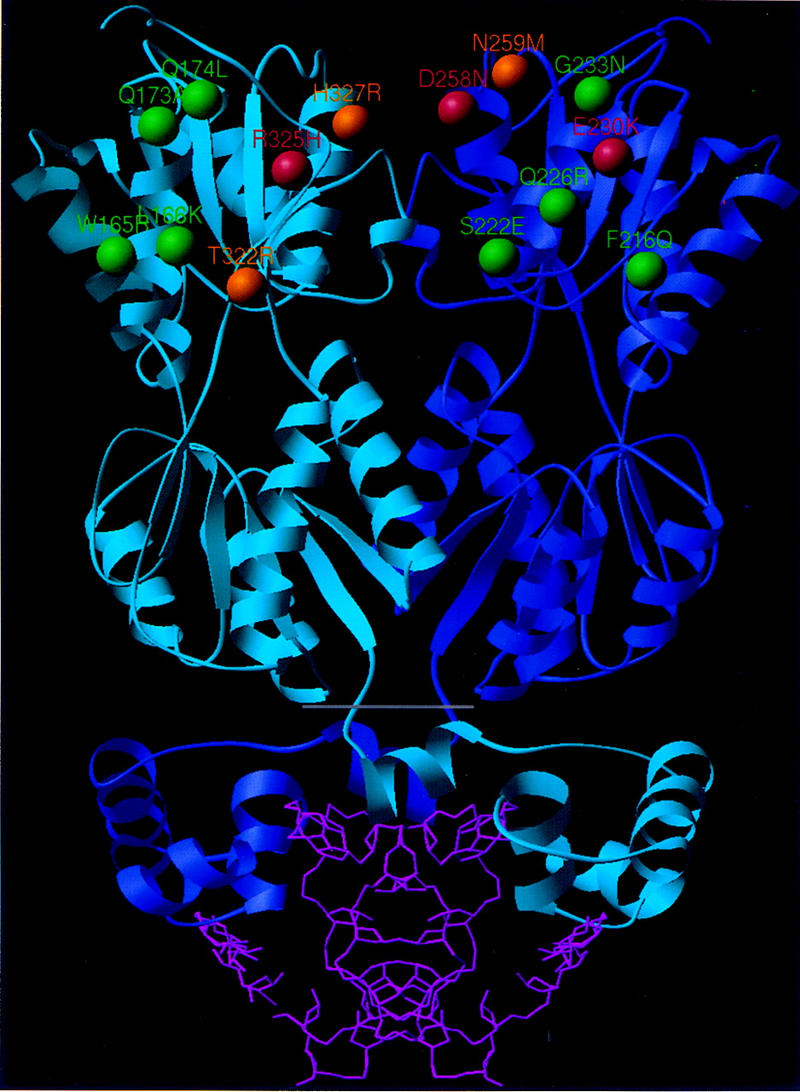
Model of the GalR dimer showing the location of mutations affecting repressosome formation in GalR. The dimeric of the GalR core (above the gray line) was modeled by homologous extension with the PurR and LacI structures. The DNA-binding domain and DNA from the PurR X-ray structure (Schumacher et al. 1994) are shown (below gray line) for reference. The β-carbon positions of the amino acids identified in our screening are shown in red. The location of the site-directed mutations, which specifically disrupted looping-mediated repression, are shown in orange. Shown in green are the α carbons of the nonconservative site-directed substitutions that had no effect on looping-mediated repression. Note that because the structure has C2 symmetry, the same arrangement of residues (not highlighted) occurs on the opposite face of the molecule. Carboxy-terminal residues 329–343, which could not be modeled by homologous extension and whose deletion did not have a marked effect on repression, are not shown.
Figure 8.
Effect of the site-directed chromosomal galR mutations N259M (●), T322R (⋄), and H327R (♦) on looping-dependent repression of galP2 (a) and independent repression of galP1 (b) compared with that obtained in wild-type (○) and galR deletion (⋄) strains.
Discussion
Evidence for the existence of a DNA loop encompassing the gal promoter has been obtained by several different means of investigation. In vitro, the repression of galP2 depends on GalR binding to both operators, and HU binding to a specific site with hbs between them (Aki et al. 1996). Deletion of either operator abolishes HU binding and repression, a finding that indicates that tripartite cooperativity among the protein components of the loop leads to the formation of a new structure (Aki and Adhya 1997). The requirement of HU and negative supercoiling for P2 repression in vitro is also observed in vivo. In a ΔhupAΔhupB strain, the P2 promoter is completely derepressed, and addition of the DNA gyrase inhibitor coumermycin, although not affecting the intrinsic activity of the promoter, also leads to complete derepression, suggesting that the structure that forms in the reconstituted system is similar to the intracellular one (Lewis et al. 1999). Atomic force microscopy with a supercoiled minicircle as the DNA template has allowed the direct visualization of a DNA loop comprising HU and GalR (Lyubchenko et al. 1997). The proteins in the loop are visualized as bright blobs at the intersection of the DNA strands, and the size of this loop indicates that the two operators are brought into close proximity where two DNA strands cross over one another.
The genetic screen described above was undertaken to identify amino acid residues in GalR that might make protein–protein contacts in the repressosome. The purified mutant GalR proteins behaved qualitatively differently from the wild type in a way that would be expected of mutants with a specific defect in loop formation. E230K and R325H repressed P1 in the absence of looping with approximately the same concentration dependence as the wild type protein; approximately twofold more of D258N was required to achieve the same level of repression as the wild type. However, even at the highest concentrations, D258N and R325H failed to repress P2. The mutant E230K, which had the weakest effect on P2 repression in vivo, was only slightly defective in P2 repression in vitro. In the case of the two mutants D258N and R325H, which had the strongest P2 derepression phenotypes, we addressed the question as to whether the secondary structure of the proteins was maintained by circular dichroism spectroscopy. This technique has proven useful in determining which among cooperative mutants of bacteriophage λ cI repressor have disrupted structures (Deb et al. 1998). We found that the GalR mutant proteins were unchanged relative to the wild type within the limits of detection of this technique, suggesting that the altered amino acids may correspond to surface-exposed residues making protein–protein contacts in the repressosome.
A molecular model of the GalR core without the DNA-binding domain has been reported on the basis of the structure of the periplasmic glucose/galactose-binding protein (Hsieh et al. 1994). The subsequent reports of the X-ray crystal structures of the PurR and LacI repressors that have significantly higher homology to GalR (31% and 27%, respectively, vs. 21% for the sugar transport protein) led us to construct a new model by homology extension to situate the mutations within a structure with the greatest possible accuracy. The alternative models derived from LacI and PurR or from the sugar-binding protein are very similar; all of the building units (helices, strands, and coils) match structurally. However, there are differences in the alignments proposed by the models, which lead to changes in the orientation of some amino acids within structural domains. According to our model (Fig. 7), the mutant amino acids identified by our screening are exclusively on the surface of the protein. They reside in regions of high structural similarity in LacI and PurR, in which the predicted structure of GalR would be most accurate. All of the amino acids are in the carboxy-terminal subdomain of the protein core—away from the DNA-binding domain, the inducer binding domain, and the dimerization interface. In LacI, tetramerization and therefore, DNA looping, is mediated by a flexible domain that extends from the carboxy-terminal β strand. GalR contains 15 amino acids extending from the this β strand to which we did not assign structure because of the absence of structural conservation in this region in PurR and LacI. The mutations we have described are not in this domain, and deletion of the 15 carboxy-terminal amino acids of GalR by insertion of a stop codon at position 328 did not increase expression of the P2–gusA fusion to a level comparable with that obtained with the looping-defective mutations (Table 1). The results obtained with the mutations created by site-directed mutagenesis support the idea that D258N and R325H are involved in protein–protein contacts. Substitution of N259 immediately adjacent to D258 (Fig. 7) with methionine had a dramatic effect on in vivo P2 repression as did replacement of H327, a residue that is very close to R325, with arginine. Neither of these GalR mutants, when expressed in single copy, affected the operator binding of GalR as reflected by repression of the P1–lacZ fusion (Fig. 8b). As evident in Figure 7, if the core of the dimer is approximated as a hexahedron in the shape of a flat box, all of the mutations that appear to specifically affect repressosome formation reside on the large faces distal to the DNA-binding region. On each face of the dimer, three of the residues that appear to be critical for loop formation are contributed by one of the monomers and three by the other monomer.
We have addressed the question as to whether the mutations cause a defect in a GalR–GalR contact or a GalR–HU contact by creating conditions in which GalR-dependent, looping-mediated repression of P2 occurred in the absence of HU. We observed previously that HMG-17, a eukaryotic DNA-binding protein of unknown structure that associates with the nucleosome core could substitute for HU in permitting GalR-dependent repression of transcription from P2. HMG-17 has very low homology with either subunit of the HU heterodimer. HU-1 (hupB) and HU-2 (hupA), the two homologous subunits of the HU heterodimer, were compared with HMG-17 using GAP of GCG Lite. The two subunits have an identity of 11% and 13%, respectively, with HMG-17. Under conditions of in vitro transcription, GalR mutants did not discriminate between HU and HMG-17; each mutation caused an effect that was of similar magnitude whether HU or HMG-17 was the added cofactor. If one assumes that the mutations define sites of GalR–HU contact, then these results would suggest that HU and HMG-17 contact the same surface of GalR to create interfaces that respond identically to GalR mutations. This scenario seems unlikely given the lack of sequence homology between HU and HMG-17. The most likely explanation for the results is that, in both cases, the mutations are disrupting GalR–GalR contacts. It may be significant that a surface defined by all six residues that were found to specifically affect P2 repression can be brought into close proximity with its counterpart on an opposing GalR dimer. Furthermore, this can be done in a manner that allows the bound DNA strands to crossover at a point the distance of which from the operators is consistent with the size of the repression loop measured by atomic force microscopy. Whether the extremely high affinity of HU for hbs (Aki and Adhya 1997) is explained by specific contacts with GalR remains to be determined.
The gal repressosome appears to be stabilized by relatively weak protein–protein interactions; GalR appears to be a dimer at micromolar concentrations and lacks the tetramerization domain of LacI. The requirement of negative supercoiling and HU for loop formation may allow regulation of the operon by two additional modes beside concentration of the inducer, d-galactose, namely DNA supercoiling and HU concentration. This could be important because the galE gene product, uridinediphosphogalactose-4-epimerase, is required for anabolic functions as well as galactose catabolism. Interestingly, the expression of HU is subject to catabolite repression, whereas its α and β subunits are differentially regulated by the gene-regulatory protein FIS (Claret et al. 1996). Supercoiling in E. coli is also highly dependent on growth conditions (Balke and Gralla 1987). The question as to the structure of a repression complex, the formation of which so completely depends on an abundant component of the bacterial nucleoid as well as DNA topology, warrants continued investigation.
Materials and methods
Bacterial strains, plasmids, and bacteriophage
A list of the bacterial strains, phages and plasmids used in this study is given in Table 2. The OEP2+P1−OI–gusA and OEP2−P1+–lacZ fusions were constructed from plasmid pI24, which contains the gal operon (galETK) as follows: 130 bp EcoR1–BstEII fragments from pAA121(p14A) and pAA121(p19T), the kind gift of Steve Busby, were used to replace the wild-type gal promoters with P2+P1− (pSA805) or P2−P1+ (pSA806) promoters. The gusA gene was amplified from pRAJ275 with PCR primers designed to create a HindIII site followed by a stop codon and then a Shine-Delgarno sequence at the 5′ end and a BspEI site at the 3′ end. It was cloned into pSA805 to create a transcriptional fusion with the first 414 bases of galE under control of the galP2 promoter (pSA809). The lacZ gene minus the first 21 bases was amplified with PCR primers so that the 5′ and 3′ ends of the amplified fragment contained a BstEII site and a BspEI site, respectively. Cloning of the restricted fragment into pSA806 created a translational fusion of lacZ with the first 12 bases of galE in which transcription occurred exclusively from galP1(pSA808). The P2galE–gusA fusion was subcloned to pTS8 (pSA814), which permitted its transfer to a λ phage. Transfer of the fusions to λ phages and to the chromosome was as outlined in Figure 9. Cells into which the galE::lacZ fusion had been transferred by two homologous recombination events between λY3001 and the chromosome were resistant to high temperature and sensitive to chloramphenicol. The P2–gusA fusion was transferred to the cell by site-specific recombination of λB3000 at the λ attachment site. Single lysogens were selected by assaying for low levels of induction by mitomycin. They were also differentiated from multiple lysogens by assaying β-glucuronidase activity, which increased as a linear function of the number of phage that had integrated into the chromosome.
Table 2.
Bacterial strains, phage, and plasmids
|
|
Relevant genotype
|
Source
|
|---|---|---|
| Strains | ||
| MG1655 | F− | S. Adhya |
| CH1105 | MG1655 ΔlacZM15 | S. Adhya |
| CH1200 | CH1105 Δcya854 | S. Adhya |
| MG0002 | CH1105 Δ(manA–uidA) 1298 zdg-232::Tn10 | this study |
| MG0003 | CH1105 zdg-232::Tn10 | this study |
| DM0007 | MG0002 ΔgalE::cat | this study |
| DM0008 | CH1200 Δ(manA–uidA)1298 zdg-232::Tn10 | this study |
| DM0009 | DM0008 ΔgalE::cat | this study |
| DM0011 | DM0009 [λ B3001] | this study |
| DM0012 | DM0009 [λ B3002] | this study |
| DM0013 | DM0009 [λ B3003] | this study |
| DM0018 | DM0009 [λP1galE–lacZΔ(galT)galK] | this study |
| DM0021 | DM0009 P1galE–lacZΔ(galT)galK | this study |
| DM0022 | DM0021 [λ B3001] | this study |
| DM0026 | DM0022 ΔgalR::cat | this study |
| JT7 | galR+[λ ΔgalR::cat] | S. Adhya |
| JT30 | ΔgalR::kan | S. Adhya |
| JT32 | ΔgalR(A–B)::kan | S. Adhya |
| DM0081a | DM0022 ΔgalR(A–B)::kan | this study |
| DM0081b | DM0081agalR–D258N | this study |
| DM0082 | DM0081 agalR–R325H | this study |
| DM0083 | DM0081agalR–E230K | this study |
| DM0108 | DM0081agalR–N259M | this study |
| DM0109 | DM0081agalR–T322R | this study |
| DM0110 | DM0081agalR–H327R | this study |
| Phage | ||
| Y392 | λc1857 gal+int− | this study |
| Y3001 | λP1+P2-galE–lacZ(galT)galK | this study |
| B3000 | λ imm21 galK+Δatt P′ P+ | this study |
| B3001 | B3000 P2+P1−galE–gusA bla | this study |
| B3002 | B3000 P2−P1+galE–gusA bla | this study |
| B3003 | B3000P2+P1+galE–gusA bla | this study |
| W30 | λcI− | NIH collection |
| W248 | λcI−h80 | NIH collection |
| B10 | λimm21 cI− | NIH collection |
| B 482 | λimm21 cI−h80 | NIH collection |
| Plasmids | ||
| pAP1 | pPROEX galR+ | this study |
| pI24 | pBR322Δ(EcoRI–PvuII) gal+ | S. Adhya |
| pRAJ275 | gusA+ | this study |
| pAA121(p14A) | P2+P1−galEK | S. Busby |
| pAA121(p19T) | P2−P1+galEK | S. Busby |
| pSA805 | pI24Δ(EcoRI–BstEII) galP2+P1− | this study |
| pSA806 | pI24Δ(EcoRI–BstEII) galP2−P1+ | this study |
| pSA809 | pSA805(HindIII–BspEI)gusA+ | this study |
| pSA808 | pSA806(BstEII–BspEI)lacZ+ | this study |
| pTS8 | P1−P2−gal ETK P−lacZ λattPOP′int+bla | this study |
| pSA814 | pTS8 Δ (PstI–BspEI) P2+P1−galE–gusA | this study |
| pMT4 | pBR322 Δ (PstI–EcoRI) galR(A–B)::kan | J. Tokeson |
Figure 9.
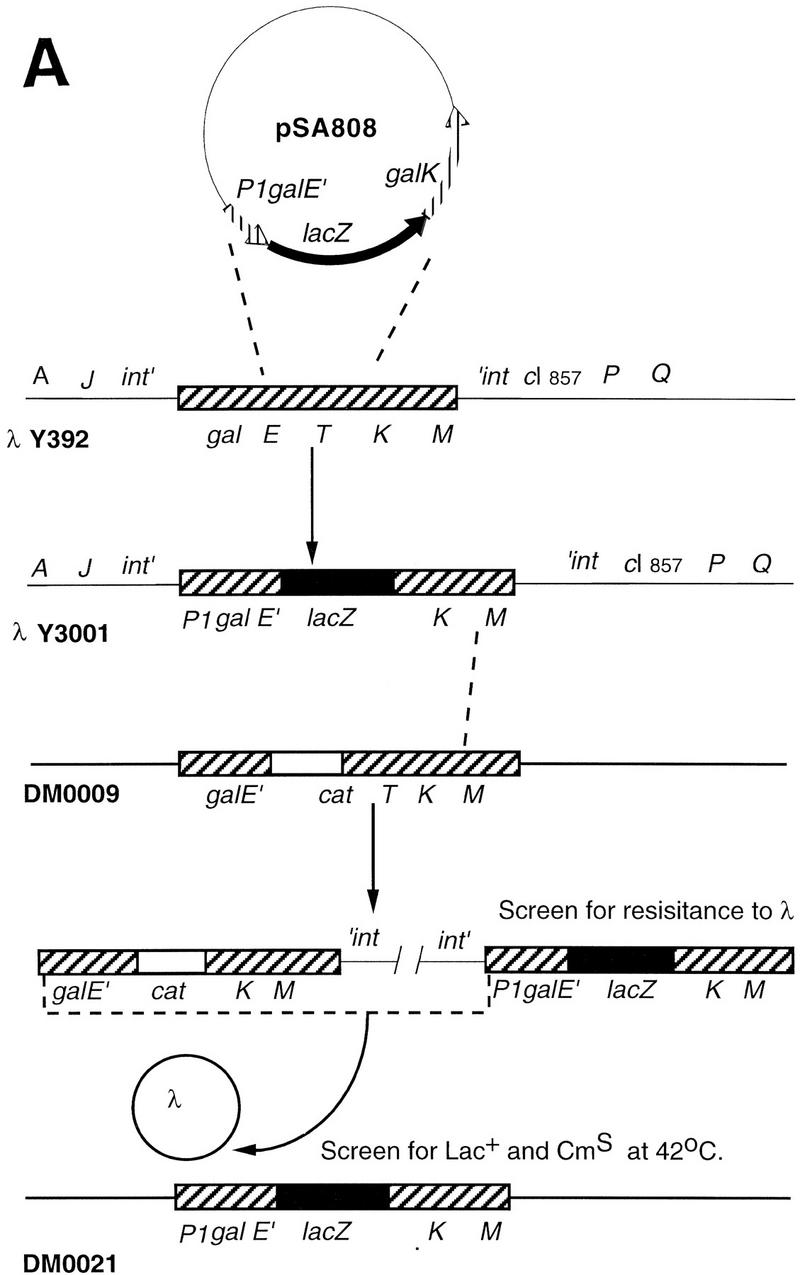
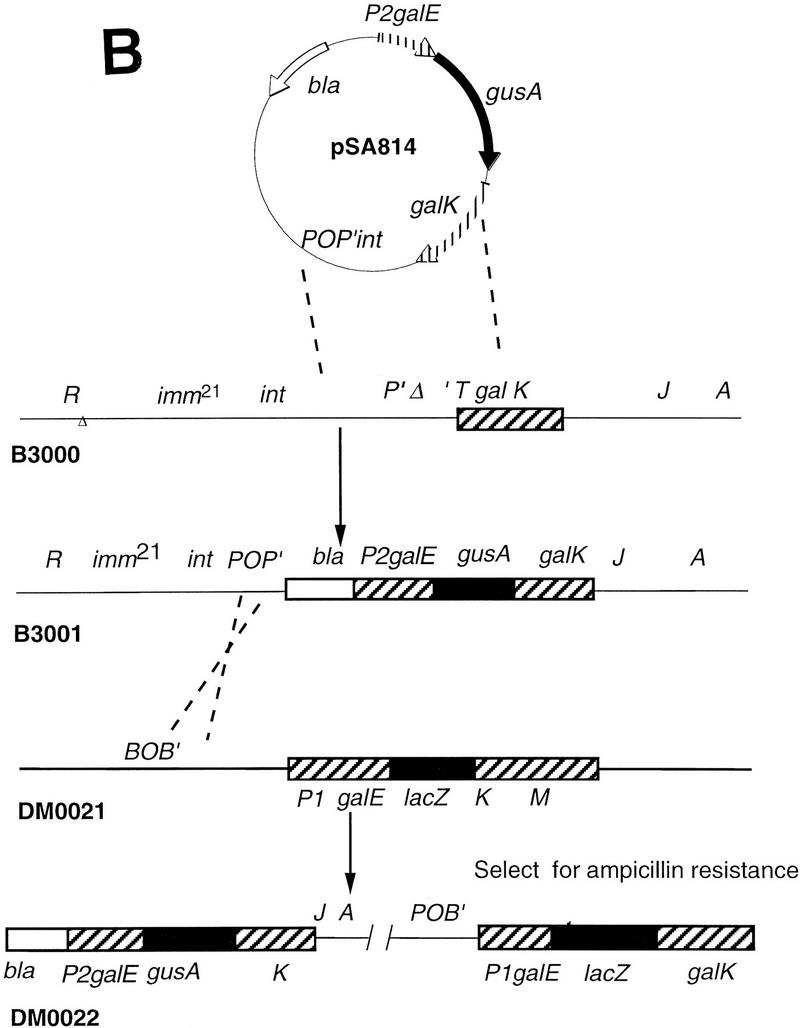
Construction of the dual reporter strain. (A) Transfer of the galP1-lacZ fusion to the chromosome by homologous recombination. (B) Construction of a second reporter fusion by homologous recombination and integration into the bacterial chromosome at the λ att site. Details are given in Materials and Methods.
The three galR mutations that were recloned into a wild-type (full-length galR) background were transferred to the reporter strain, DM0022, by the same method as described for the galP1–lacZ fusion except that the galR mutations were initially transferred into a non-lysogenic strain followed by transduction by bacteriophage P1 of the galR allele into DM0022.
Assay of β-galactosidase and β-glucuronidase activity
Cells were grown overnight in Luria–Bertani medium and diluted 50-fold for further growth in M63 salt solution supplemented with 0.4% (wt/vol) d-fructose, 0.1% (wt/vol) casamino acids and 0.004% (wt/vol) vitamin B1. At various times, aliquots of the cells were washed in the same medium containing 100 μg/ml chloramphenicol and stored on ice (Wilson et al. 1992). β-Glucuronidase or β-galactosidase activities were measured for all cultures simultaneously by the Softmax microplate spectrophotometer system. Aliquots of 100 μl from cell cultures were added to the wells of a microtiter plate followed by solubilization in 50 μl of permeabilization buffer [100 mm Tris-HCl (pH 7.8), 32 mm NaPO4, 5 mm DTT, 5 mm CDTA, 4% (vol/vol) Triton X-100, and 200 μg/ml polymixin B (Schupp et al. 1995)]. After incubation at room temperature for at least 10 min, 50 μl of substrate (4 mm α-p-nitrophenyl β–d–glucuronide for assay of β-glucuronidase or 16 mm o-nitrophenyl β-d-galactoside and 2 mm sodium citrate for assay of β-galactosidase) was added to each well. β-glucuronidase activity was measured at 405 nm and 37°C; β-galactosidase at 420 nm and 28°C.
Primer extension
RNA purification was performed as recommended in the RNeasy Midi Protocol (Qiagen). A 32P-labeled probe complementary to galE mRNA from +48 to +64 was used in reverse transcription as outlined in the Promega Primer System. Products were resolved on 8% DNA polyacrylamide–urea sequencing gels.
GalR purification
Wild-type and mutant galR genes were cloned into the pProEXHta expression plasmid so that the amino terminus of the protein minus the amino-terminal methionine was fused to a cleavable sequence containing six histidines. After overexpression, cells were lysed by French press and the histidine-tagged GalR was bound to Ni-NTA resin and eluted with a buffer containing imidazole followed by desalting as described previously (Polayes 1996). The histidine sequence was removed by incubation of 1 mg of the purified protein in 1.5 ml of buffer [50 mm Tris-HCl (pH 8.0), 0.5 mm EDTA, 1 mm DTT, and 5% glycerol] which contained 300 units of recombinant TEV protease, for 1 hr at 4°C followed by 10 min at 30°C.
In vitro transcription
Transcription reactions were performed as described previously (Geanacopoulos and Adhya 1997). The reaction mixture (50 μl) contained 20 mm Tris acetate (pH 7.8), 10 mm magnesium acetate, 100 mm potassium glutamate, 2 nm DNA template, 1.0 mm ATP, 0.1 mm GTP, 0.1 mM CTP, 0.01 mM UTP, and 10–20 μCi of [α-32P]UTP (3000 Ci/mmole). After incubation of the reactions at 37°C for 5 min, RNA polymerase was added to a concentration of 20 nanomolar. Reactions were terminated after 10 min by addition of an equal volume of RNA loading buffer [80% vol/vol deionized formamide/ TBE (89 mm Tris borate, 2 mm EDTA), 0.025% bromophenol blue, 0.025% xylene cyanol]. After heating at 90°C for 5 min, the samples were loaded onto 8% DNA polyacrylamide–urea sequencing gels. Radioactive transcription products were quantified with a PhosphorImager. RNA bands were normalized for varying background by subtraction of the total radioactivity of an equal area immediately above or below the band of interest. The RNA1 transcript originating from the plasmid, which was not affected by GalR, HU, or HMG-17 addition, served as an internal control between lanes.
Mutagenesis of galR
GalR was mutagenized by treatment of bacteriophage P1 lysate of a strain (JT30) in which galR was closely linked to a kan allele with hydroxylamine as described previously (Hong and Ames 1971). Mutagenesis was allowed to proceed at 37°C until the transducing titer was reduced 100-fold (∼20 hr). Transduction of the kan allele to the reporter strain, DM0022, was carried out by standard methods. Cells were plated on minimal glucose kanamycin plates supplemented with 0.1% casamino acids to which 5-bromo-4-chloro-3-indolyl-N-actyl-β-d-glucuronide had been added to a concentration of 50 μg/ml. Site-directed mutagenesis of galR was performed with the Quick-Change site-directed mutagenesis kit from Stratagene. All mutants were sequenced by standard methods.
Circular dichroism
Circular dichroism spectra were obtained in a JASCO J-600 spectropolarimeter with a 0.1-cm pathlength cuvette. The concentrations of mutant and wild-type GalR were adjusted to ∼5 micromolar as measured by absorbance at 280 nm. Measurements were performed in a buffer containing 0.02 m Tris-HCl (pH 8.5) and 15% glycerol.
Molecular modeling of GalR
For the sequence alignment and similarity calculations, we used the package Bestfit program (http://molbio.info.nih.gov/molbio/gcglite/compare.html; Needleman and Wunsch 1970). Parameters used in the alignment program were as follows: gap weight 3.000, average match, 0.540, length weight 0.1, and average mismatch −0.396. The structural alignment algorithm (Cornette et al. 1993), and both the Monte Carlo algorithm for loop closure and the homologous extension procedure (Vazmatzis et al. 1994) have been described previously.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sadhya@helix.nih.gov; FAX (301) 402-1344.
References
- Adhya S. The galactose operon. In: Neidhardt FC, Ingraham JL, Low KB, Maganasid B, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. Washington, D.C.: American Society for Microbiology; 1987. pp. 1503–1512. [Google Scholar]
- ————— Multipartite genetic control elements: Communication by DNA loop. Annu Rev Genet. 1989;23:227–250. doi: 10.1146/annurev.ge.23.120189.001303. [DOI] [PubMed] [Google Scholar]
- Adhya S, Geanacopoulos M, Lewis DEA, Roy S, Aki T. Transcription regulation by repressosome and by RNA polymerase contact. Cold Spring Harbor Symp Quant Biol. 1998;62:1–10. doi: 10.1101/sqb.1998.63.1. [DOI] [PubMed] [Google Scholar]
- Aki T, Adhya S. Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J. 1997;12:3666–3674. doi: 10.1093/emboj/16.12.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aki T, Choy HE, Adhya S. Histone-like protein HU as a specific transcriptional regulator: Co-factor role in repression of gal transcription by gal repressor. Genes Cells. 1996;2:179–188. doi: 10.1046/j.1365-2443.1996.d01-236.x. [DOI] [PubMed] [Google Scholar]
- Balke V, Gralla J. Changes in the linking number of supercoiled DNA accompany growth transitions in Escherichia coli. J Bacteriol. 1987;169:4499–4506. doi: 10.1128/jb.169.10.4499-4506.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham AHA, Pannambalan S, Chan B, Busby S. Mutations that reduce expression from the P2 promoter of the Escherichia coli. Gene. 1986;41:67–74. doi: 10.1016/0378-1119(86)90268-4. [DOI] [PubMed] [Google Scholar]
- Brenowitz M, Jamison E, Majumdar A, Adhya S. Interaction of the Escherichia coli Gal repressor protein with its DNA operators in vitro. Biochemistry. 1990;29:3374–3383. doi: 10.1021/bi00465a033. [DOI] [PubMed] [Google Scholar]
- Bustin M, Reeves R. High-mobility group chromosomal proteins: Architectural components that facilitate chromatin function. Prog Nucleic Acid Res. 1996;54:35–94. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- Choy HE, Adhya S. RNA polymerase idling and clearance in gal promoters: Use of a supercoiled minicircle DNA template made in vivo. Proc Natl Acad Sci. 1993;90:472–476. doi: 10.1073/pnas.90.2.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy HE, Hanger R, Aki T, Mahoney M, Murakami K, Ishihama A, Adhya S. Repression and activation of promoter-bound RNA polymerase activity by Gal repressor. J Mol Biol. 1997;272:293–300. doi: 10.1006/jmbi.1997.1221. [DOI] [PubMed] [Google Scholar]
- Claret L, Rouviere-Yaniv J. Regulation of HUa and HUb by CRP and FIS in Escherichia coli. J Mol Biol. 1996;263:126–139. doi: 10.1006/jmbi.1996.0564. [DOI] [PubMed] [Google Scholar]
- Cornette JL, King BL, Silverman MD, DeLisi C. Graphical representations of the class I MHC cleft. J Mol Graph. 1993;3:174–179. doi: 10.1016/0263-7855(93)80069-4. [DOI] [PubMed] [Google Scholar]
- Deb S, Bandyopadhyay S, Roy S. Spectroscopic study of Y210 Cl-repressor: Implications for cooperative interaction. Protein Eng. 1998;11:481–487. doi: 10.1093/protein/11.6.481. [DOI] [PubMed] [Google Scholar]
- Geanacopoulos M, Adhya S. Functional characterization of roles of GalR and GalS as regulators of the gal regulon. J Bacteriol. 1997;179:228–234. doi: 10.1128/jb.179.1.228-234.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JS, Ames BS. Localized mutagenesis of any specific small region of the bacterial chromosome. Proc Natl Acad Sci. 1971;68:3158–3162. doi: 10.1073/pnas.68.12.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh M, Hensley P, Brenowitz M, Fetrow JS. A molecular model of the inducer binding domain of the galactose repressor of Escherichia coli. J Biol Chem. 1994;269:13825–13833. [PubMed] [Google Scholar]
- Lewis D, Geanacopoulos M, Adhya S. Roles of HU and DNA supercoiling in transcription repression: Specialized nucleoprotein repression complex at gal promoters in Escherichia coli. Mol Micro. 1999;31:451–461. doi: 10.1046/j.1365-2958.1999.01186.x. [DOI] [PubMed] [Google Scholar]
- Lewis M, Chang G, Horton NC, Kercher MA, Pace HC, Schumacher MA, Brennan RG, Lu P. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996;271:247–254. doi: 10.1126/science.271.5253.1247. [DOI] [PubMed] [Google Scholar]
- Lyubchenko YL, Shlyakhtenko LS, Aki T, Adhya S. Atomic force microscopic demonstration of DNA looping by GalR and HU. Nucleic Acids Res. 1997;25:873–876. doi: 10.1093/nar/25.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Adhya S. Demonstration of two operator elements in gal: in vitro repressor binding studies. Proc Natl Acad Sci. 1984;81:6100–6104. doi: 10.1073/pnas.81.19.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Rudikoff S, Adhya S. Purification and properties of Gal Repressor:pL-galR fusion in pKC31 plasmid vector. J Biol Chem. 1987;262:2326–2331. [PubMed] [Google Scholar]
- Martin KJ, Schleif RJ. The DNA loop model for ara repression: AraC protein occupies the proposed loop sites in vivo and repression-negative mutations lie in these same sites. Proc Natl Acad Sci. 1992;83:3654–3658. doi: 10.1073/pnas.83.11.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Polayes DA. Prokaryotic protein expression with the ProEXHT expression vector. Focus. 1996;18:50–56. [Google Scholar]
- Schumacher MA, Choi KY, Zalkin H, Brennan RG. Crystal structure of LacI member, PurR, bound to DNA: Minor groove binding by alpha helices. Science. 1994;266:763–770. doi: 10.1126/science.7973627. [DOI] [PubMed] [Google Scholar]
- Schupp JM, Travis SE, Price LB, Shand RF, Keim P. Rapid bacterial permeabilization reagent useful for enzyme assay. Biotechniques. 1995;19:18–20. [PubMed] [Google Scholar]
- Vasmatzis G, Brower R, Delisi C. Predicting immunoglobulin-like hypervariable loops. Biopolymers. 1994;34:1669–1680. doi: 10.1002/bip.360341211. [DOI] [PubMed] [Google Scholar]
- Vasmatzis G, Cornette J, Sezerman U, DeLisi C. TcR recognition of the MHC-peptide dimer: structural properties of a ternary complex. J Mol Biol. 1996;261:72–89. doi: 10.1006/jmbi.1996.0442. [DOI] [PubMed] [Google Scholar]
- Weickert M, Adhya S. A family of bacterial regulators homologous to gal and lac repressors. J Biol Chem. 1992;267:15899–15874. [PubMed] [Google Scholar]
- Wilson KJ, Hughes SG, Jefferson RA. The Escherichia coli gus operon: Induction and expression of the gus operon in E.coli and the occurrence and use of GUS in other bacteria. In: Gallagher SR, editor. Gus protocols: Using the gus gene as a reporter of gene expression. New York, NY: Academic Press; 1992. pp. 7–21. [Google Scholar]