

Abstract
Controversy has arisen as to the role of transforming growth factor-β-induced protein (TGFBI) in the regulation of tumor metastasis. Using lung and breast cancer cell lines (H522 and MCF7, respectively), we established that TGFBI induced cell adhesion to extracellular matrix proteins by activating adhesion-associated signaling and subsequent structure reformation, ultimately leading to cells less motile; whereas TGFBI reduced abilities of colony formation in soft agar, penetration through matrix gel, and activation of matrix metalloproteinases 2 and 9. Furthermore, injection of TGFBI-expressing cells into immuno-deficient mice resulted in a significant reduction in tumor metastasis in vivo. Taken together, these data suggest that TGFBI moderates the metastatic potential of cancer cells.
Keywords: TGFBI, metastasis, extracellular matrix, adhesion-mediated signaling, mouse model
1. Introduction
Tumor metastasis, a complex, multistage process by which primary tumor cells migrate to and colonize distant tissues, is a critical factor in the progression of cancer to lethality [1-3]. Understanding the mechanisms underlying this process is crucial to improve anti-metastatic therapies. One popular misconception is that all cancer cells aggressively colonize many distant tissues and organs and grow relatively unchecked by normal cell proliferative control machinery. Although in some cases this is true, there are, in fact, a range of hurdles a primary tumor cell must overcome in order to metastasize and proliferate outside the progenitor tissue or organ.
Transformed cells must first escape the confines of the primary tumor by breaking down the basement membrane to allow the cancer cells to intravasate into blood vessels. Tumor cells can then be transported to distant tissues, extravasate out of the circulatory system and subsequently colonize remote sites. The capacity of malignant cells to undertake these two stages of metastasis, intravasation and extravasation, is thought to explain the substantial up-regulation of matrix metalloproteinase (MMPs) activity in cancer cells, enzymes that can degrade extracellular matrix (ECM) proteins and permit invasion through the basement membrane [1, 3].
An additional obstacle for metastatic cells is the requirement for adhesion signaling to promote survival. In fully spread cells, adhesion plaques called focal adhesions (FAs) create a link between the ECM outside the cell and the actin cytoskeleton inside the cell. FAs contain a variety of molecules that physically link the cytoskeleton and the ECM, as well as adaptor proteins and kinases that propagate signaling cascades in adherent cells [4]. In normal cells, adhesion molecules on the cell surface must be bound to their ligand to activate intracellular downstream signals, without which cells will cease to proliferate and to undergo apoptosis [4]. However, metastatic cells must, by definition, break away from their progenitor tissue/organ and migrate to distant sites, a process that presumably requires the cell to circumvent normal adhesion-mediated survival signaling. Consequently, a down-regulation of adhesion signaling [5] (although, crucially, not the elimination of adhesion ability [6-8]) is thought to be important for metastatic tumor cells to move through the lymphatic system or blood vessels.
Transforming growth factor-β–induced (TGFBI) protein is detected in most normal human tissues [9], although mRNA was not detected in the brain [10]. TGFBI has been implicated in a number of cellular disease processes including tumorigenesis, angiogenesis, progression and metastasis [11-14]. However, the most extensive studies on TGFBI have been directed toward understanding the role played by the protein in corneal dystrophies, and a number of mutations in the gene have been shown to have a pathological manifestation [11, 15].
In our previous reports, we have focused on the role of TGFBI in tumorigenesis. We have demonstrated that the absence of the protein was associated with increased tumorigenesis in vitro and in vivo [10, 16], and that this down-regulation was associated with hypermethylation of TGFBI gene promoter region [17]. However, since TGFBI is a secreted protein and has been shown as a “linker” participating in the interaction between ECM and integrins [18], we were curious to know whether the absence of the protein affects one of the most important traits of malignant tumors - metastasis. To do this we assessed some of the most prominent in vitro characteristics associated with metastatic tumors by expressing TGFBI in two types of cancer cell lines that had little or no endogenous expression of the protein and measuring invasion ability, matrix metalloproteinase activity and dependence on adhesion for survival signaling. We also assessed this molecule for its ability to modulate tumor metastasis with a mouse model. Very recently, a report suggested that increased TGFBI was associated with a more aggressive metastatic colon cancer type [19]. We wanted to examine if this finding was true for our cells or whether the data were only relevant for the specific metastatic colon cancer cell line used in that study.
2. Materials and methods
2.1. Ectopic expression of TGFBI in lung and breast tumor cells
The MCF-7 and H522 cells were purchased from ATCC (Manassas, VA). MCF-7 or H522 cell lines stably expressing TGFBI and non expression control were created by calcium phosphate transfection of the expression plasmid pRc/CMV2 containing human TGFBI cDNA or vector only. G418-resistant (1 mg/ml) clones were isolated and the cell culture supernatant was screened by Western blot for secreted TGFBI protein. Media were harvested and concentrated 10-fold using SP Sepharose Fast Flow resin (GE Healthcare Bio-Sciences Corp. Piscataway, NJ) before Western blotting. TGFBI antibody was from R&D Systems (Minneapolis, MN). The expression of TGFBI mRNA was determined by quantitative real-time reverse transcription-PCR (RT-PCR) using the ΔΔCt method with GAPDH as a reference as described previously [17]. Briefly, total RNA was isolated by TRIzol Reagent (Invitrogen, Carlsbad, CA) and the single strand of cDNA was synthesized from 2 μg total RNA using SuperScript II First-Strand Synthesis System (Invitrogen). Products were detected using RT2 qPCR Primer Assay, a SYBR Green-based quantitative real-time PCR system on an Applied Biosystems 7300 Real-time PCR System (Applied Biosystems). Primers used to amplify GAPDH and TGFBI mRNA were obtained from SupperArray Biocience Corporation (Frederick, MD). All reactions were done in triplicate. PCR conditions were as follows: 95°C for 15 min followed by 40 cycles at 95 °C for 30 sec, 55 °C for 30 sec, and 72 °C for 30 sec.
2.2. Adhesion to fibronectin
Micro-well plates were coated with fibronectin (FN, 1 μg/ml) in accordance with the manufacturer's instructions (Collaborative Biomedical Products, Bedford, MA) and incubated overnight at 4°C. Plates were washed with deionized H2O to remove unbound FN and were blocked with 0.1% heat-inactivated bovine serum albumin for 1 h and then washed with serum free medium (SFM). Cells were trypsinized, resuspended in SFM and maintained in suspension at 37 °C for 30 to 40 min before plating onto FN coated 96-well plates at 37 °C for the indicated times. The wells were rinsed with PBS, then fixed in 75% ethanol and stained with crystal violet. Bound stain was dissolved with Triton X-100 (0.2% in distilled water) and color density was read at 595nm.
2.3. Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously [20]. Briefly, cells were plated on FN-coated coverslips in DMEM containing 2% bovine serum albumin and incubated at 37°C for the indicated times before fixation (PBS, 4% paraformaldehyde for 10 min) and permeabilization (PBS, 0.5% Triton X-100 for 5 min). Cells were stained with rhodamine phalloidin or antibodies to vinculin (Sigma-Aldrich, St. Louis, MO). Secondary antibodies were from Invitrogen. Images were captured using a laser scan confocal microscope (Nikon, Tokyo, Japan).
2.4. Determination of anchorage-independent growth
MCF-7 and H522 cells expressing TGFBI were plated in triplicate at a density of 1×103 cells in 1 ml of 0.35% low melting agarose containing 10% FBS DMEM, then overlaid on a 0.7% agar base in a 6-well culture plate. After the agar-cell mixture solidified, 1 ml of 10% FBS DMEM was added on the top. After 3 weeks incubation at 37 °C with 5% CO2, colonies with >30 cells were scored under a light microscope at low magnification.
2.5. Western blot analysis
For plating experiments, cells (∼80 to 90% confluent) were trypsinized, resuspended in SFM and maintained in suspension at 37°C for 30 to 40 min before plating onto FN-coated dishes at 37 °C for the indicated times. Proteins were extracted by lysing cells in extraction buffer (50mM Tris–HCl, pH 8, 150mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate and 1mM phenylmethylsulfonyl fluoride) and the protein concentration was determined by Bio-Rad protein Assay (Bio-Rad, Hercules, CA). Equivalent amounts of protein (30 μg) were fractionated by SDS-PAGE and transferred to PVDF membranes under semi-dry conditions. Antibodies to phospho-Y397-FAK, Phospho-Y402-Pyk2, FAK, Pyk2 and β-actin were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Secondary antibodies were from GE and signals were detected using ECL (Pierce, Rockford, IL). The intensity of protein bands was quantified by ImageJ developed by the National Institutes of Health.
2.6. In vitro invasion assay
Invasion assays were carried out using the 24-well BD Biocoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's recommendations. Briefly, 5×104 cells in 0.5 mL of serum-free DMEM were added to the Matrigel-coated cell culture inserts and control inserts 10% FBS DMEM was added to the lower well. After 24 h at 37 °C, non-invading cells were carefully removed with a cotton swab. The cells on the bottom of inserts were fixed with 70% ethanol and stained with 0.1% crystal violet. Each chamber was transferred to a well containing 0.1% Triton-X100, incubated for 10 min on orbital shaker and OD measured at 595 nm.
2.7. Matrix metalloproteinase activity assay
1×105 cells were grown on 35 mm dishes for 40 h until confluent. The supernatant was collected and cell debris removed by centrifugation. MMP activity in the 50 μL of supernatant was measured using the MMP Gelatinase Activity Assay Kit (CHEMICON International Inc., Millipore Corporate Billerica, MA) as instructed by the manufacturer. Briefly, a biotinylated gelatinase substrate was cleaved by active MMP-2 and MMP-9 enzymes in the sample. The fragments were then added to a biotin-binding 96-well plate and incubated for 30 min at 37°C to allow biotin containing fragments to bind to the plate while digestion continues. The digested but unbound fragments were removed by repeated washing, while undigested biotin-labeled gelatinase that bound to the plate was detected by the addition of a streptavidin-enzyme complex which results in a colored product measured at 450 nm.
2.8. In vivo metastasis: tail vein injection of TGFBI-expressing cells into nude mice
Immuno-deficient Nude mice aged of 3-4 weeks were purchased from Harlan Sprague-Dawley Inc. (Indianapolis, IN) and housed in Columbia University Medical Center animal facility for one week prior to the experiment. 1×105 TGFBI expressing cells as well as controls were resuspended in serum free DMEM and injected via tail vein at one time for each mouse. The mice were monitored bi-weekly for tumor incidence using a Sonix ultrasound scanner (Ultrasonix, Redmond, WA). Mice that had a detectable tumor burden were sacrificed and tumor location and number was recorded.
2.9. Statistical analysis
Statistical analysis was conducted using the t-test or Fisher Exact Test and comparisons between groups with significance at p<0.05.
3. Results
3.1. Ectopic expression of TGFBI in tumor cell lines
This study was designed to assess the effect of TGFBI expression in human cancer cell lines on metastasis. H522 and MCF-7 cell lines, derived from lung and breast cancers, respectively, have only trace amount of endogenous TGFBI. These cells were transfected with human TGFBI, the expression was confirmed by measuring the mRNA levels using Real-Time PCR, and by examining the cell culture medium for secreted TGFBI protein by Western blot (Fig. 1). The intensity of the TGFBI protein band in the Western blot was about 20-fold higher in MCF-7 cells transfected with TGFBI cDNA compared to controls, and more than 5-fold higher in H522 cells compared to controls, indicating the transfected TGFBI was properly expressed in these two tumor cells lines. The large difference in fold increase regarding TGFBI protein in MCF-7 cells might reflect an increased accumulation of the protein compared with H522 cell lines. Numerous clones stably expressing TGFBI were isolated from transfected H522 and MCF-7 cells. Two representative clones of each cell line were chosen to cross check most of the results, which were proved consistent (data not shown). The following data represented results from one clone.
Fig.1.
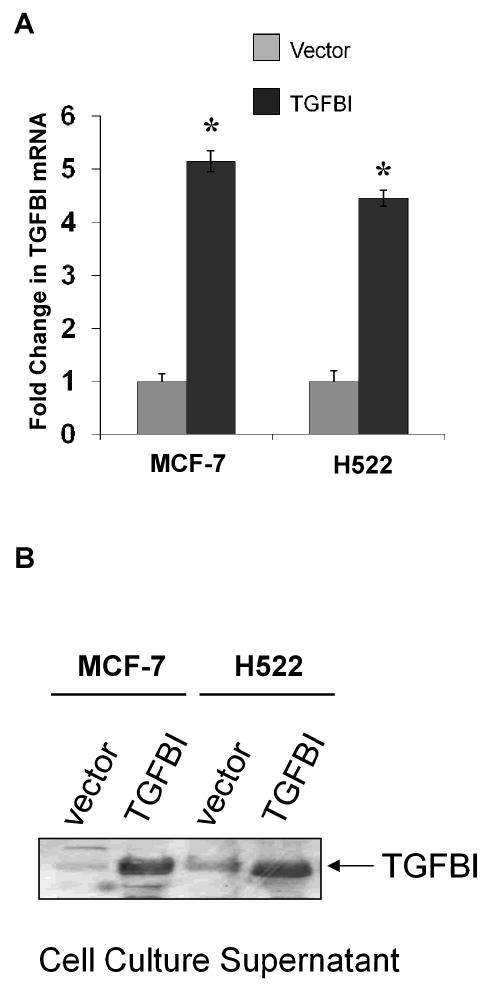
Establishment of cell lines stably expressing TGFBI. MCF-7 and H522 cells were transfected with TGFBI cDNA or empty vector. The expression of exogenous mRNAs and proteins was confirmed by RT-PCR and Western blotting, respectively. (A) Relative quantification of TGFBI mRNA was determined by real-time RT-PCR using the ΔΔCt method with GAPDH as an internal control. Results are mean±SD from three independent experiments. * p<0.05 versus vector controls. (B) Expression of TGFBI in cell supernatant was determined by Western blotting. Experiments were performed three times and a representative immunoblot is shown here.
3.2. Cell adhesion and cytoskeleton formation were enhanced in TGFBI-expressing cells
Many different types of carcinoma cells exhibit invariably decreased levels of intercellular adhesiveness, enhancing their capabilities to overcome cell-cell adhesion, which might underline the characteristics associated with metastasis in vivo [21]. Having established two tumor cell lines that stably express TGFBI, we therefore examined whether these cells exhibited any changes in adhesion characteristics. First we performed adhesion assays to assess the ability of infected cell lines with and without TGFBI expression to bind to fibronectin. Substantial levels of adhesion were obtained in all cell lines after ∼80 min. However, at this time point, for each of the two cell lines expressing TGFBI, adhesion to fibronectin was more than 2-fold higher than for the corresponding control cells. Importantly, even at earlier time points when overall adhesion was significantly below the maximum, cells expressing TGFBI displayed higher levels of adhesion compared to controls (Fig. 2).
Fig.2.
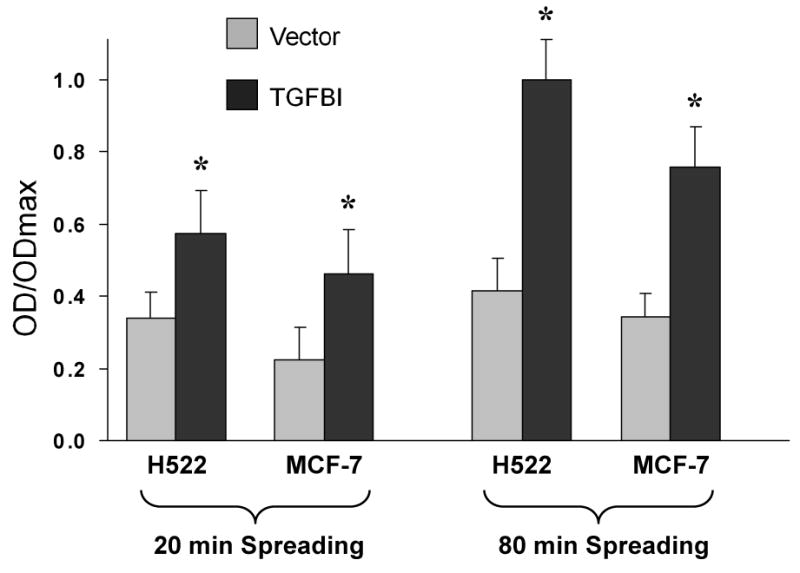
Adhesion of cells expressing TGFBI to fibronectin. 1×104 cells were maintained in suspension for 40 min and then allowed to adhere to fibronectin-coated plates for the indicated times. Bound cells were fixed and stained with crystal violet before optical density (OD) was measured at 595 nm. Adhesion to fibronectin was significantly enhanced after expression of TGFBI in H522 and MCF-7 cells. Data are normalized to the maximum OD detected, and expressed as mean±SD from three independent experiments. *, p<0.05 versus vector controls.
In fully spread cells, adhesion plaques form at the periphery of the cell are called focal adhesions (FAs), which are composed of transmembrane molecules (integrins) that bind to the ECM, as well as a range of cytoplasmic components, adaptor proteins such as vinculin, paxillin and Cas, and signaling molecules such as kinases [4, 22]. Having established that TGFBI-expressing cells gained an increase in adhesion ability, next we determined whether there were phenotypic changes to adhesion structures in these cells when spread on fibronectin. To do this we stained the FAs protein vinculin and F-actin in H522 and MCF7 cells that had been allowed to spread on fibronectin-coated coverslips (Fig. 3A). Images captured of these cells clearly showed that the cells expressing TGFBI were well spread and had pronounced FA and actin stress fiber formation, even after only 30 min of spreading. In contrast, control cells, although adherent, were poorly spread, had few if any FAs, and, even after 60 min of spreading, had a substantially less well-developed actin cytoskeleton. These results were clearly consistent with the data obtained from adhesion assays, indicating that TGFBI-expressing cells had a much greater adhesion ability compared to controls.
Fig.3.
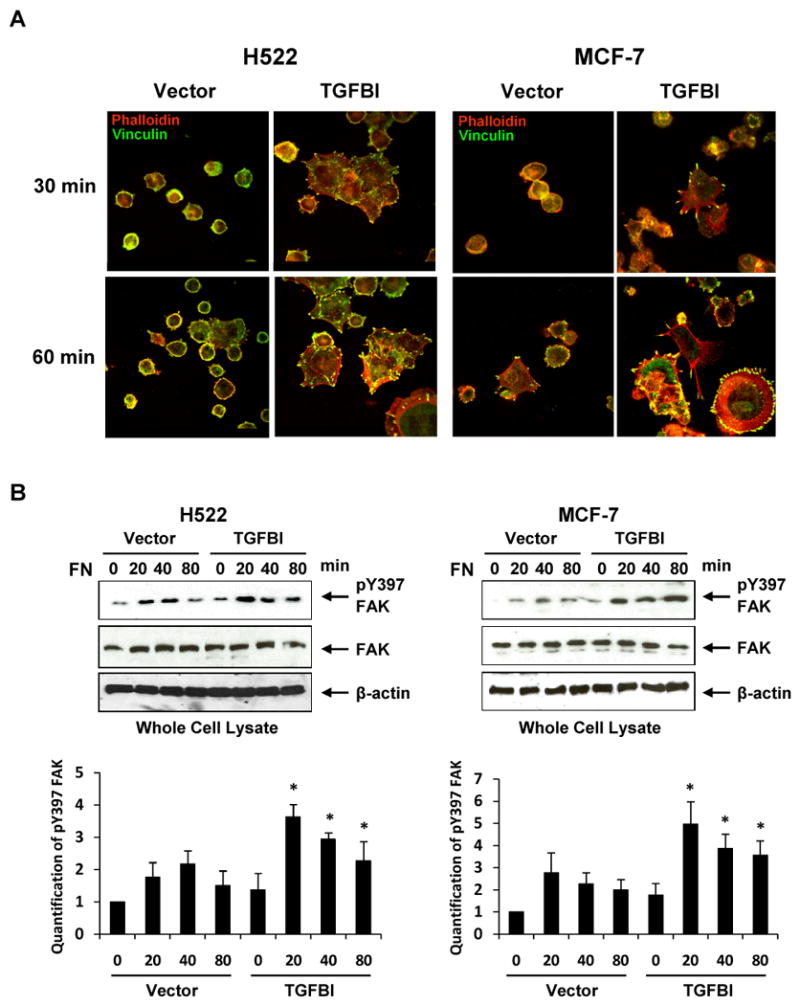
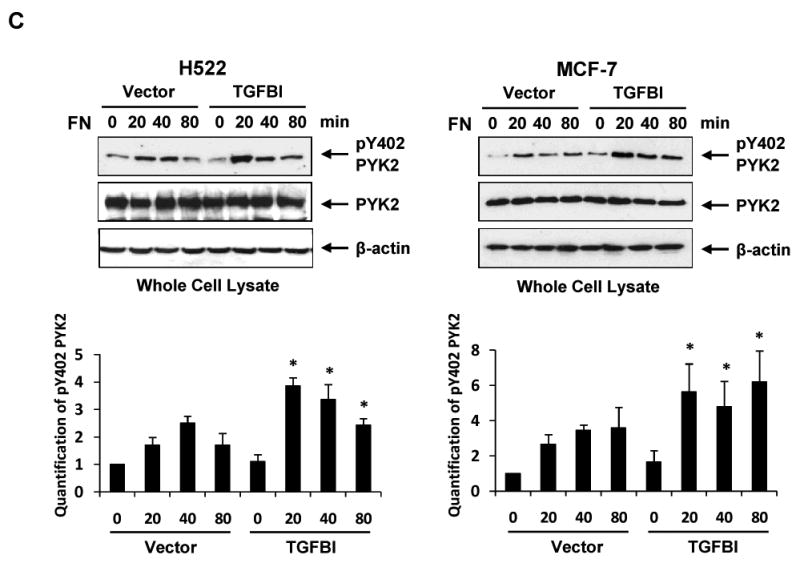
Alteration of focal adhesion and actin stress fiber formation with associated signaling activation in H522 and MCF-7 cells expressing TGFBI. (A) Representative images display focal adhesion and stress fiber formation when spread on fibronectin, which are dramatically increased in cells expressing TGFBI. Cells were plated on fibronectin-coated coverslips in DMEM containing 2% bovine serum albumin and incubated at 37°C for indicated time points. The adherent cells were then fixed and stained with rhodamine phalloidin (red) for stress fiber or antibody to vinculin (green) for focal adhesion (magnification 100×). Activation of (B) FAK and (C) Pyk2 after plating on fibronectin was greatly enhanced in the presence of TGFBI. To detect activated FAK and Pyk2, immunoblot membranes were probed with antibodies to phospho-Y397-FAK and phospho-Y402-Pyk2. Membranes were also reprobed with anti-FAK, anti-Pyk2 and anti-β-actin antibodies to demonstrate equal protein loading. The intensity of protein bands was quantified by ImageJ developed by the National Institutes of Health and plotted using MS-EXCEL. *, p<0.05 versus corresponding time point of vector controls. The figures represent one of the three independent experiments.
3.3. Adhesion-mediated signaling was elevated in TGFBI expressing cells
One prominent signaling molecule which is localized to FAs is focal adhesion kinase (FAK). Activation of FAK, initially by autophosphorylation at tyrosine 397, is crucial for normal cell adhesion and migration [23]. Having identified an increase in overall adhesion, and an improvement in FAs and actin formation in TGFBI-expressing cancer cells, we wondered whether there was a concomitant rise in adhesion signaling. To do this we assessed FAK's phosphorylation state at Y397 at various time points after cellular spreading began. As with other adhesion indicators, FAK Y397 phosphorylation substantially increased in cells expressing TGFBI compared to controls (Fig. 3B). When normalized to the loading control, the intensity of the FAK phospho-Y397 band in both cells lines was remarkably greater in the TGFBI-expressing cells than in the controls.
Adhesion-derived signaling through FAK and other FAs molecules stimulates a range of downstream events that are important in regulating a variety of cellular processes, among them cytoskeletal structure and cell motility. Protein tyrosine kinase 2 (Pyk2), a homolog of FAK [24], was found abundantly expressed in neuron and has been demonstrated to regulate synaptic plasticity [25]. It has been shown that Pyk2 is also involved in regulating the adhesive ability in different types of cell lines [26, 27]. Pyk2 was found expressed in these two cells lines. Therefore, it would be of interest to see if the expression of TGFBI has any effect on Pyk2 activity as well. As shown in Fig. 3C, Pyk2 phosphorylation at tyrosine 402, which is an indicator of Pyk2 activity, was up-regulated in TGFBI-expressing cells upon adhesion to fibronectin-coated surface compared to controls. These results suggest a possible regulatory role of TGFBI in the adhesion-associated signaling pathways involving FAK and Pyk2, etc.
3.4. Anchorage-independent growth was decreased in TGFBI expressing cells
For cells to undergo metastasis, they must detach from the progenitor tissues and overcome adhesion-mediated survival signaling in order to avoid being induced anoikis [28]. Our results indicated that TGFBI-expressing cells had increased adhesion ability and concomitant signaling. We wondered whether the increased adhesion signaling in TGFBI-expressing cells made the cells more sensitive to adhesion-mediated survival signals. To test this we measured the cell's ability to grow in soft agar and we found that the lung and breast cancer cells expressing TGFBI formed fewer colonies in soft agar compared to controls (Fig. 4A and 4B). For both cell lines, the number of colonies formed in soft agar was more than 5-fold lower in TGFBI-expressing cells, showing a correlation between adhesion-mediated survival signal sensitivity and TGFBI expression.
Fig.4.
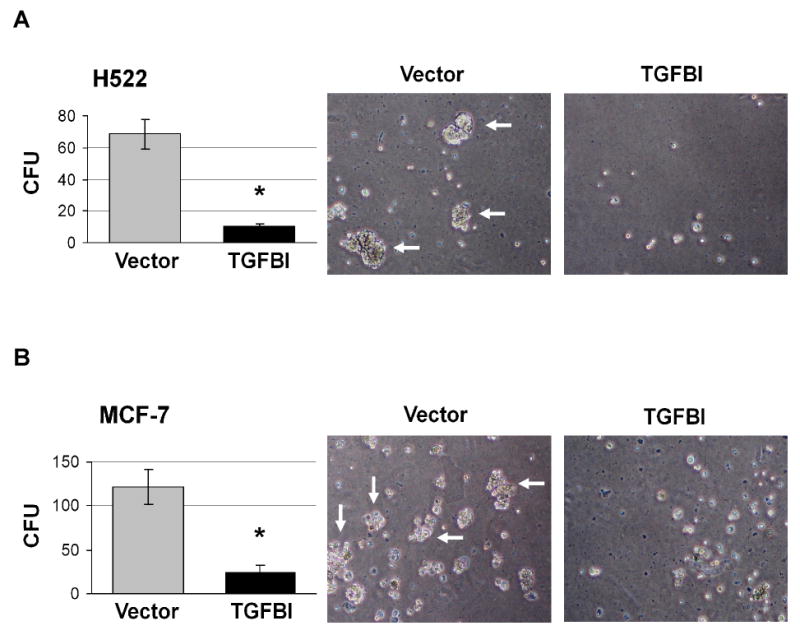
Anchorage-independent growth of cells expressing TGFBI. TGFBI- and empty vector-transfected (A) H522 and (B) MCF-7 cells were cultured in soft agar and colony formation was scored three weeks later. The cells transfected with TGFBI formed more than 5-fold fewer colonies in soft agar than those transfected with vector controls. Graphs (left panel) are mean±SD of colony forming units (CFU) per 1,000 cells from three independent experiments. *, p<0.05 versus vector controls. Images of colonies that formed in soft agar are represented on the right panel (magnification 40×). Arrows indicate colonies with >30 cells.
3.5. TGFBI-expressing cells had decreased activity of MMP-2 and 9 and were impaired in vitro tumor cell invasion
One of the most important characteristics of a metastatic cell is the ability to degrade matrix proteins in vivo as this enables the cell to invade through the basement membrane. Having closely examined the adhesion characteristics of the TGFBI expressing cells, we wanted to investigate another aspect of a typical metastatic cell's behavior. To do this we first measured the activity of MMP-2 and MMP-9 using an in vitro matrix degradation activity assay. Members of the matrix metalloproteinase (MMP) family have been demonstrated to play a pivotal role in tumor cell invasion and metastasis [29]. MMP-2 and MMP-9, which are the gelatinase types of the matrix metalloproteinases and mainly degrade type IV and V collagens, have been shown to correlate with the invasiveness of various types of cell lines [30-33]. Interestingly, we found that the ability of TGFBI-expressing cells to degrade collagen was significantly reduced, by almost 2-fold compared to MCF-7 control, and more than 4-fold compared to H522 control (Fig. 5A). Next we wanted to determine if the reduction in MMP activity affected the ability of the cells to invade into a collagen matrix by measuring the penetration of cells through a Matrigel matrix. As expected, TGFBI-expressing cells had a reduced ability to infiltrate the matrix, further indicating that these cells had a reduction in invasion ability in vitro (Fig. 5B).
Fig.5.
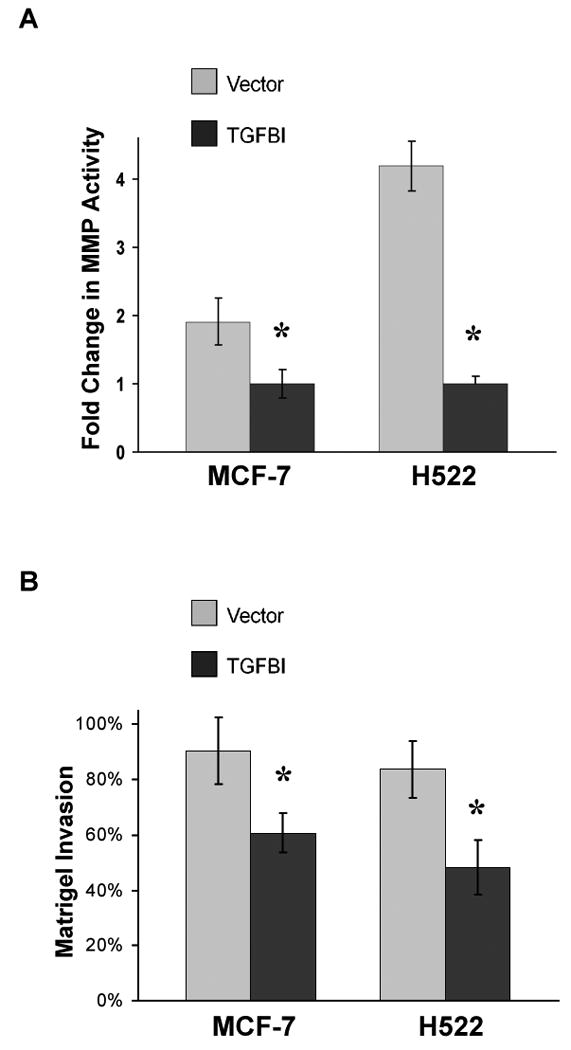
In vitro matrix metalloproteinase activity and invasion ability in cells expressing TGFBI. (A) Activity of MMP-2 and 9 in cell culture supernatant was measured using a MMP-2 and 9 gelatinase activity assay kit. The biotinylated gelatinase substrates were cleaved by active MMPs in the samples and the fragments were added to a biotin-binding plate. The digested but unbound fragments were removed by washing; whereas the bound undigested biotin-labeled gelatinase substrates were detected with streptavidin-enzyme complex producing a colored product measured at 450 nm. In vitro MMP-2 and 9 activities were significantly decreased in TGFBI-expressing cells. Data are mean±SD from three independent experiments and are presented as the fold change compared with vector control. *, p<0.05, compared to vector controls. (B) Invasion ability was measured using a matrigel invasion chamber. Cells in serum-free DMEM were seeded in an invasion chamber and serum-containing DMEM was added to the lower well. After incubation for 24 h, non-invading cells were removed from the upper surface and the cells at the bottom were fixed, stained with crystal violet and OD was read at 595 nm. In vitro invasion activities were significantly impaired with the expression of TGFBI in MCF-7 and H522 cells. Data are mean±SD from three independent experiments with values for cells invading through Matrigel insert membrane given as a percentage of the cell migration through control insert membranes. *, p<0.05, compared to vector controls.
3.6. TGFBI decreased tumor metastasis in vivo
Having established that a number of in vitro characteristics typical of metastatic cells were altered in TGFBI-expressing cells, we next wanted to determine whether these cells had decreased metastasis in vivo. To achieve this we injected pooled clones from culture dishes of both TGFBI-expressing cells or controls into the tail vein of immuno-deficient nude mice and monitored the mice for tumor incidence using ultrasound imaging (Fig. 6A). Tumors were detectable in various organs after 8 weeks in mice injected with both H522 and MCF-7 control cells (Fig. 6A and 6B). In contrast, in TGFBI-expressing groups, tumors were detected 1 to 2 weeks later with a much lower incidence (Fig. 6B). After 10 weeks, when the experiment was terminated and all the remaining animals were sacrificed, 100% (10/10) of the mice injected with control MCF-7 cells and 90% (9/10) of the mice injected with control H522 contained tumors. In contrast, only 20% or 30% of animals injected with either of the two TGFBI-expressing cells (MCF-7=2/10 and H522=3/10) contained tumors (Fig. 6B, p<0.05). Finally, not only was tumor onset observed at later time points in mice injected with TGFBI-expressing cells, but the number of tumors in these animals was also fewer than in controls. In animals injected with MCF-7 control cells, a total of 15 tumors were detected in 10 mice with 4 animals possessing more than one tumor. Similarly, in mice injected with control H522 cells, a total of 13 tumors were detected in 9 mice with 3 animals possessing more than one tumor. In contrast, in the mice that developed tumors when injected with either of the TGFBI-expressing cells, each animal developed only one tumor (Table 1). Overall, lung, gastrointestinal tracts (G.I.), and liver were the three organs that had the highest tumor incidence, likely due to the relatively high blood flow in these organs. It should be noted that the animals were sacrificed once tumors were detected by ultrasound.
Fig.6.
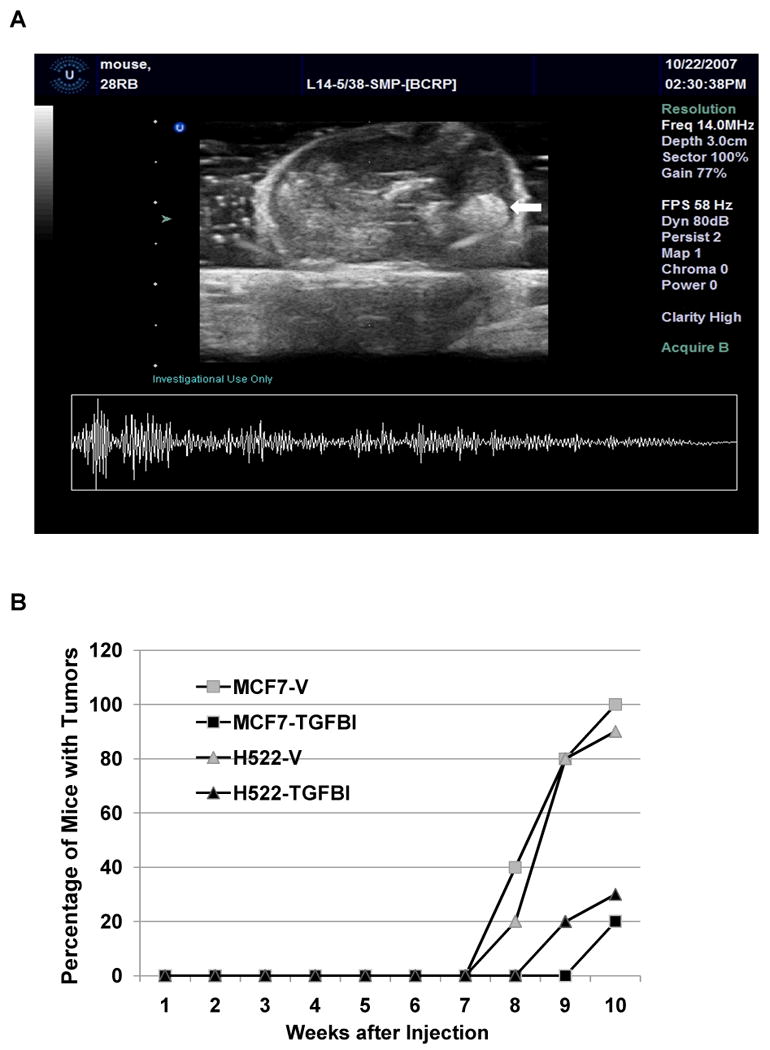
In vivo tumor cell metastasis of TGFBI-expressing cancer cells. The time of tumor formation was monitored in immuno-deficient nude mice injected with cancer cells with or without TGFBI expression. 1×105 cells were injected into nude mice via tail vein, 10 mice for each group. The animals with a tumor burden detected with ultrasound imaging were sacrificed and dissected. (A) A representative ultrasound image demonstrates a tumor. In this image, the mass pointed by arrow was proved a tumor from G.I. in MCF-7/vector group. (B) Tumor cell metastasis was reduced in TGFBI expressing cancer cells in vivo. Fisher Exact Test was used to determine the significant difference of tumor number between groups with or without TGFBI expression. p<0.05, compared to vector controls.
Table 1.
Location, number and average size (in parentheses, cm in diameter) of tumors in nude mice injected with cancer cells expressing TGFBI or vector controls via the tail vein.
| Number of Tumors | Total | |||||||
|---|---|---|---|---|---|---|---|---|
| Liver | Kidney | G.I. | Bone | Lung | Neck | |||
| Vector | 3(0.5) | 1(0.3) | 5(0.5) | 1(0.45) | 5(0.3) | - | 15 | |
| MCF-7 | ||||||||
| TGFBI | - | - | 1(0.45) | - | 1(0.4) | - | 2 | |
| Vector | 3(0.5) | 1(0.4) | 3(0.5) | 2(0.55) | 4(0.45) | - | 13 | |
| H522 | ||||||||
| TGFBI | 1(0.4) | - | 1(0.4) | - | - | 1(0.35) | 3 | |
4. Discussion
TGFBI, otherwise known as Betaig-h3, was first reported to be involved in tumorigenesis more than a decade ago when CHO cells expressing the protein had a reduced ability to form tumors in nude mice [34]. More recently, FAS1 domains of TGFBI have been reported to inhibit tumor angiogenesis and tumor growth, as well as promoting apoptosis, which is consistent with a cancer suppressor role for TGFBI [35]. In addition, our group has shown that: a) many tumor cell lines have decreased expression of TGFBI and b) tumorigenic cells lines exogenously expressing TGFBI have decreased tumorigenesis in vitro and in vivo [10, 16, 35, 36]. Consequently, there is evidence from a number of groups suggesting that TGFBI is a possible tumor suppressor. However, the mechanism by which TGFBI is involved in decreasing tumorigenesis is not well understood.
There has also been considerable interest in the ability of TGFBI to modulate cell adhesion and cytoskeletal formation since it has been considered to be a component of the extracellular matrix (ECM) [18]. Interestingly, a recent study showed that TGFBI as an ECM protein, can sensitize an ovarian cancer cell line to anti-cancer drug paclitaxel through the regulation of microtubule stabilization [34]. It is suggested that TGFBI promotes microtubule stabilization through integrin-mediated FAK and Rho signaling pathways. Our findings of increased actin organization and FAK activation in adherent lung and breast cancer cells expressing TGFBI is in agreement with the data on microtubule stabilization, as FAK has been shown to be required for stable microtubules [37]. These data further contribute to the body of evidence indicating that expression of this protein can positively regulate adhesion and cytoskeletal formation [18].
As a secreted ECM protein, it has also been proposed that TGFBI may be an important regulator of metastasis [19]. The critical steps in the metastasis cascade include loss of cellular adhesion (this presumably requires the cells to overcome, though not completely), the requirement for adhesion-mediated survival signaling, enhanced motility and invasiveness, entry into the circulatory system and colonization at distant tissues [6-8]. In order to see if TGFBI is involved in the regulation of metastasis, we generated two stable cell lines by expressing exogenous TGFBI and tested them for their ability to alter a range of in vitro characteristics that are thought to mimic the phenotype of metastatic cancer cells. In our study, TGFBI-expressing cancer cells exhibited increased adhesion ability and enhanced activation of associated signaling on the extracellular matrix protein fibronectin (these two types of cells exhibited weak adhesion affinity towards Collagen I), detected through phosphorylation of FAK-Y397 and Pyk2-Y402. Furthermore, we observed decreased levels of anchorage-independent growth compared to controls, which was consistent with our previous observations in tumorigenic cells expressing exogenous TGFBI [10, 35, 36]. Down-regulation of FAK signaling has been shown in EGF-induced tumor cell invasion [5]. In vitro, any adherent cell that is detached from its substrate will eventually undergo anoikis [24], and one of the most common attributes associated with in vitro transformation is the anchorage-independent growth capability [38]. Therefore, these two lines of evidences we obtained strongly suggest that expression of TGFBI made the cells more sensitive to adhesion-dependent survival signals and potentially more likely to undergo anoikis during metastasis, which is consistent with a proposed role for TGFBI in suppressing tumor metastasis.
MMPs are a family of proteases that are capable of degrading all kinds of extracellular matrix proteins, among them, MMP-2 and MMP-9, which are the gelatinase types of the MMPs, have been demonstrated to promote tumor cell invasion and metastasis because of their capability to degrade various types of collagens such as IV collagen, etc [39]. In vivo, one of the most important barriers preventing a cancer cell from becoming metastatic is the basement membrane. Once a cancer cell breaks through the basement membrane it is able to enter the circulatory system and move from the primary tumor site, and this is thought to account for the up-regulation of MMPs in many cancers [40]. Consequently, we measured the cell ability to invade through a matrigel, and tested the enzyme activity of secreted MMP-2 and 9. Importantly, in the cancer cell lines we tested, expression of TGFBI resulted in a decreased capacity of MMPs to degrade a gelatin matrix and a reduced ability to penetrate through a matrigel matrix, suggesting a suppressive role of TGFBI in tumor cell invasion ability. Invasive and proliferative phenotypes are fundamental components of cancer cells; tumor metastasis is largely affected by cell proliferation in vivo [41, 42]. In our original characterization of these TGFBI over-expressing cells, the growth of MCF-7 and H522 cells that overexpress TGFBI decreased by 15% and 19%, respectively (data not shown). In the current Matrigel Invasion assay, the reduction among the two lines that over-express TGFBI was 32% and 43%, respectively. While a significantly reduced growth rate over a longer assay time point can certainly altered the invasion assay outcome, the fact that our growth alterations were smaller and the endpoints were determined after 24 hours of plating would suggest that the cell proliferation is unlikely to account for the difference.
Having established that TGFBI expression resulted in a decrease in in vitro metastatic ability, we wanted to determine whether there was an alteration in metastasis in vivo. Animal models of metastasis cannot completely recapitulate metastasis of a primary tumor in humans, for instance, lymphatic infiltration as well as intravasation. However, intravenous injection of cancer cells in immuno-deficient mice does mimic many of the characteristics of malignancy, including extravasation from the circulatory system and colonization of secondary tissues/organs [43, 44]. Significantly, we showed that cancer cells expressed with TGFBI had reduced metastasis in vivo when injected through the tail vein of nude mice, as they produced a lower total number of tumors and, when tumors were evident, they were observed at later time points compared to mice injected with control cancer cells, which is consistent with the previous in vitro evidences. However, the size of tumors derived from each group displays insignificant difference, though slightly decreased in TGFBI expressed groups.
In this study, we have demonstrated by using a lung and breast cancer cell lines that expression of the secreted protein TGFBI ectopically reduces the metastatic ability of these cells in vitro and in vivo, similar results were also obtained in mesothelioma cell lines in a separate report from our group (data not shown). The novelty of this work resides in the results from both in vitro and in vivo, and from well-known cancer cell lines with different origins. The invasion characteristics changed by TGFBI expression were consistent and well explained in the cellular and molecular changes. This is consistent with the findings of our group and others that TGFBI displayed a tumor suppressor function [10, 16, 29, 34, 35]. However, a recent study suggested that TGFBI expression actually increased the metastatic ability of a colon cancer and ovarian cancer cell line [19, 45]. Furthermore, it has been found that TGFBI expression increases in some cancer types [11]. It needs to be addressed whether this is a cell type-specific phenotype, where TGFBI functions differently in a specific scenario; if so, it might be related to the pattern of TGFBI's interaction with different types of integrins [11]. Nevertheless, our findings presented here unequivocally indicate that TGFBI acts as a regulator down-regulating the metastatic process in our in vitro and in vivo systems. These results suggest that TGFBI may be a promising target for the designs of future anti-metastatic therapeutic in certain types of cancers.
Acknowledgments
This work was supported by National Institutes of Health [ES05786, CA49062 and ES09089 to T.K.H.]
Footnotes
Conflicts of Interest Statement: None Declared
References
- 1.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Arribas J. Matrix metalloproteases and tumor invasion. N Engl J Med. 2005;352:2020–2021. doi: 10.1056/NEJMcibr055002. [DOI] [PubMed] [Google Scholar]
- 3.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol. 2001;21:4016–4031. doi: 10.1128/MCB.21.12.4016-4031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphries MJ, Olden K, Yamada KM. A synthetic peptide from fibronectin inhibits experimental metastasis of murine melanoma cells. Science. 1986;233:467–470. doi: 10.1126/science.3726541. [DOI] [PubMed] [Google Scholar]
- 7.Vollmers HP, Imhof BA, Braun S, Waller CA, Schirrmacher V, Birchmeier W. Monoclonal antibodies which prevent experimental lung metastases. Interference with the adhesion of tumour cells to laminin. FEBS Lett. 1984;172:17–20. doi: 10.1016/0014-5793(84)80863-7. [DOI] [PubMed] [Google Scholar]
- 8.Kren A, Baeriswyl V, Lehembre F, Wunderlin C, Strittmatter K, Antoniadis H, Fassler R, Cavallaro U, Christofori G. Increased tumor cell dissemination and cellular senescence in the absence of beta1-integrin function. EMBO J. 2007;26:2832–2842. doi: 10.1038/sj.emboj.7601738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF. cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-β. DNA Cell Biol. 1992;11:511–522. doi: 10.1089/dna.1992.11.511. [DOI] [PubMed] [Google Scholar]
- 10.Zhao YL, Piao CQ, Hei TK. Downregulation of Betaig-h3 gene is causally linked to tumorigenic phenotype in asbestos treated immortalized human bronchial epithelial cells. Oncogene. 2002;21:7471–7477. doi: 10.1038/sj.onc.1205891. [DOI] [PubMed] [Google Scholar]
- 11.Thapa N, Lee BH, Kim IS. TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-β. Int J Biochem Cell Biol. 2007;39:2183–2194. doi: 10.1016/j.biocel.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Wen G, Shao G, Wang C, Lin C, Fang H, Balajee AS, Bhagat G, Hei TK, Zhao Y. TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res. 2009;69:37–44. doi: 10.1158/0008-5472.CAN-08-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nabokikh A, Ilhan A, Bilban M, Gartner W, Vila G, Niederle B, Nielsen JH, Wagner O, Base W, Luger A, Wagner L. Reduced TGF-beta1 expression and its target genes in human insulinomas. Exp Clin Endocrinol Diabetes. 2007;115:674–682. doi: 10.1055/s-2007-984477. [DOI] [PubMed] [Google Scholar]
- 14.Becker J, Erdlenbruch B, Noskova I, Schramm A, Aumailley M, Schorderet DF, Schweigerer L. Keratoepithelin suppresses the progression of experimental human neuroblastomas. Cancer Res. 2006;66:5314–5321. doi: 10.1158/0008-5472.CAN-05-3049. [DOI] [PubMed] [Google Scholar]
- 15.Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, Schorderet DF. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet. 1997;15:247–251. doi: 10.1038/ng0397-247. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, El-Gabry M, Hei TK. Loss of Betaig-h3 protein is frequent in primary lung carcinoma and related to tumorigenic phenotype in lung cancer cells. Mol Carcinog. 2006;45:84–92. doi: 10.1002/mc.20167. [DOI] [PubMed] [Google Scholar]
- 17.Shao G, Berenguer J, Borczuk AC, Powell CA, Hei TK, Zhao Y. Epigenetic inactivation of Betaig-h3 gene in human cancer cells. Cancer Res. 2006;66:4566–4573. doi: 10.1158/0008-5472.CAN-05-2130. [DOI] [PubMed] [Google Scholar]
- 18.Billings PC, Whitbeck JC, Adams CS, Abrams WR, Cohen AJ, Engelsberg BN, Howard PS, Rosenbloom J. The transforming growth factor-beta-inducible matrix protein (beta)ig-h3 interacts with fibronectin. J Biol Chem. 2002;277:28003–28009. doi: 10.1074/jbc.M106837200. [DOI] [PubMed] [Google Scholar]
- 19.Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, Wang XF. Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev. 2008;22:308–321. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Partridge MA, Marcantonio EE. Initiation of attachment and generation of mature focal adhesions by integrin-containing filopodia in cell spreading. Mol Biol Cell. 2006;17:4237–4248. doi: 10.1091/mbc.E06-06-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavallaro U, Christofori G. Multitasking in tumor progression: signaling functions of cell adhesion molecules. Ann N Y Acad Sci. 2004;1014:58–66. doi: 10.1196/annals.1294.006. [DOI] [PubMed] [Google Scholar]
- 22.Turner CE. Paxillin interactions. J Cell Sci. 2000;113(Pt 23):4139–4140. doi: 10.1242/jcs.113.23.4139. [DOI] [PubMed] [Google Scholar]
- 23.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong WC, Mei L. Roles of FAK family kinases in nervous system. Front Biosci. 2003;8:s676–82. doi: 10.2741/1116. [DOI] [PubMed] [Google Scholar]
- 26.Roelle S, Grosse R, Buech T, Chubanov V, Gudermann T. Essential role of Pyk2 and Src kinase activation in neuropeptide-induced proliferation of small cell lung cancer cells. Oncogene. 2008;27:1737–1748. doi: 10.1038/sj.onc.1210819. [DOI] [PubMed] [Google Scholar]
- 27.Yuan TC, Lin FF, Veeramani S, Chen SJ, Earp HS, 3rd, Lin MF. ErbB-2 via PYK2 upregulates the adhesive ability of androgen receptor-positive human prostate cancer cells. Oncogene. 2007;26:7552–7559. doi: 10.1038/sj.onc.1210570. [DOI] [PubMed] [Google Scholar]
- 28.Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, Lengyel E. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001;7:2396–2404. [PubMed] [Google Scholar]
- 29.Skonier J, Bennett K, Rothwell V, Kosowski S, Plowman G, Wallace P, Edelhoff S, Disteche C, Neubauer M, Marquardt H. beta ig-h3: a transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol. 1994;13:571–584. doi: 10.1089/dna.1994.13.571. [DOI] [PubMed] [Google Scholar]
- 30.Moser TL, Young TN, Rodriguez GC, Pizzo SV, Bast RC, Jr, Stack MS. Secretion of extracellular matrix-degrading proteinases is increased in epithelial ovarian carcinoma. Int J Cancer. 1994;56:552–559. doi: 10.1002/ijc.2910560415. [DOI] [PubMed] [Google Scholar]
- 31.Young TN, Rodriguez GC, Rinehart AR, Bast RC, Jr, Pizzo SV, Stack MS. Characterization of gelatinases linked to extracellular matrix invasion in ovarian adenocarcinoma: purification of matrix metalloproteinase 2. Gynecol Oncol. 1996;62:89–99. doi: 10.1006/gyno.1996.0195. [DOI] [PubMed] [Google Scholar]
- 32.Fishman DA, Bafetti LM, Banionis S, Kearns AS, Chilukuri K, Stack MS. Production of extracellular matrix-degrading proteinases by primary cultures of human epithelial ovarian carcinoma cells. Cancer. 1997;80:1457–1463. doi: 10.1002/(sici)1097-0142(19971015)80:8<1457::aid-cncr13>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Afzal S, Lalani el-N, Foulkes WD, Boyce B, Tickle S, Cardillo MR, Baker T, Pignatelli M, Stamp GW. Matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-2 expression and synthetic matrix metalloproteinase-2 inhibitor binding in ovarian carcinomas and tumor cell lines. Lab Invest. 1996;74:406–421. [PubMed] [Google Scholar]
- 34.Nam JO, Jeong HW, Lee BH, Park RW, Kim IS. Regulation of tumor angiogenesis by fastatin, the fourth FAS1 domain of betaig-h3, via alphavbeta3 integrin. Cancer Res. 2005;65:4153–4161. doi: 10.1158/0008-5472.CAN-04-2705. [DOI] [PubMed] [Google Scholar]
- 35.Zhao YL, Piao CQ, Hei TK. Overexpression of Betaig-h3 gene downregulates integrin alpha5beta1 and suppresses tumorigenicity in radiation-induced tumorigenic human bronchial epithelial cells. Brit J Cancer. 2002;86:1923–1928. doi: 10.1038/sj.bjc.6600304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed AA, Mills AD, Ibrahim AE, Temple J, Blenkiron C, Vias M, Massie CE, Iyer NG, McGeoch A, Crawford R, Nicke B, Downward J, Swanton C, Bell SD, Earl HM, Laskey RA, Caldas C, Brenton JD. The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell. 2007;12:514–527. doi: 10.1016/j.ccr.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 2004;303:836–839. doi: 10.1126/science.1091325. [DOI] [PubMed] [Google Scholar]
- 38.San RH, Laspia MF, Soiefer AI, Maslansky CJ, Rice JM, Williams GM. A survey of growth in soft agar and cell surface properties as markers for transformation in adult rat liver epithelial-like cell cultures. Cancer Res. 1979;39:1026–1034. [PubMed] [Google Scholar]
- 39.Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 40.McCawley LJ, Matrisian LM. Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today. 2000;6:149–156. doi: 10.1016/s1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- 41.Gao CF, Xie Q, Su YL, Koeman J, Khoo SK, Gustafson M, Knudsen BS, Hay R, Shinomiya N, Vande Woude GF. Proliferation and invasion: plasticity in tumor cells. Proc Natl Acad Sci USA. 2005;102:10528–10533. doi: 10.1073/pnas.0504367102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med. 2000;6:100–102. doi: 10.1038/71429. [DOI] [PubMed] [Google Scholar]
- 43.Bradley MO, Kraynak AR, Storer RD, Gibbs JB. Experimental metastasis in nude mice of NIH 3T3 cells containing various ras genes. Proc Natl Acad Sci USA. 1986;83:5277–5281. doi: 10.1073/pnas.83.14.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res. 2006;66:4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. [DOI] [PubMed] [Google Scholar]
- 45.Ween MP, Lokman NA, Hoffmann P, Rodgers RJ, Ricciardelli C, Oehler MK. Transforming growth factor-beta-induced protein secreted by peritoneal cells increases the metastatic potential of ovarian cancer cells. Intl J Cancer. 2011;128:1570–1584. doi: 10.1002/ijc.25494. [DOI] [PubMed] [Google Scholar]