

Abstract
Hereditary pulmonary arterial hypertension (PAH) is usually caused by mutations in BMPR2. Mutations are found throughout the gene, and common molecular consequences of different types of mutation are not known. Knowledge of common molecular consequences would provide insight into the molecular etiology of the disease. The objective of this study was to determine the common molecular consequences across classes of BMPR2 mutation. Increased superoxide and peroxide production and alterations in genes associated with oxidative stress were a common consequence of stable transfection of the vascular smooth muscle cells, with three distinct classes of BMPR2 mutation, in the ligand binding domain, the kinase domain and the cytoplasmic tail domain. Measurement of oxidized lipids in whole lung from transgenic mice expressing a mutation in the BMPR2 cytoplasmic tail showed a 50% increase in isoprostanes and a two-fold increase in isofurans, suggesting increased reactive oxygen species (ROS) of mitochondrial origin. Immunohistochemistry on BMPR2 transgenic mouse lung showed that oxidative stress was vascular-specific. Electron microscopy showed decreased mitochondrial size and variability in the pulmonary vessels from BMPR2-mutant mice. Measurement of oxidized lipids in urine from humans with BMPR2 mutations demonstrated increased ROS, regardless of disease status. Immunohistochemistry on hereditary PAH patient lung confirmed oxidative stress specific to the vasculature. Increased oxidative stress, likely of mitochondrial origin, is a common consequence of BMPR2 mutation across mutation types in cell culture, mice and humans.
Keywords: Born morphogenetic protein receptor II, idiopathic and heritable pulmonary arterial hypertension, signal transduction
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a disease characterized by progressively worsening pulmonary vascular resistance, leading to right heart failure and death. The heritable form is usually caused by mutations in the type 2 receptor for the bone morphogenic protein pathway, BMPR2.[1,2]
BMPR2 is a 1038 amino acid single-pass transmembrane receptor, consisting of an extracellular ligand-binding domain, a short transmembrane domain, a kinase domain and a long, relatively uncharacterized, cytoplasmic tail. In the canonical signaling pathway, dimers of BMPR2 bind dimers of type 1 receptor (there are three possible) in the presence of the BMP ligand. The kinase domain in BMPR2 phosphorylates the type 1 receptor, which then phosphorylates SMAD1, 5 or 8. Phosphorylated SMAD complexes with SMAD4 and enters the nucleus as a transcription factor.[3] The cytoplasmic tail domain binds and signals through LIMK,[4] a regulator of actin cytoskeletal organization, but may have other functions as well.
BMPR2 mutations in PAH patient families are widely distributed throughout the gene and, to date, a common molecular consequence of BMPR2 mutation has not been found. Mutations in the cytoplasmic tail domain do not appear to affect SMAD signaling.[5,6] While it has been suggested that increased p38 or p42/44 MAPK phosphorylation was a common consequence of BMPR2 mutation,[7,8] it seems likely with additional experimentation that BMPR2 mutation derepresses MAPK activation, but does not consistently cause induction.[9] There is thus no known common downstream consequence of different classes of BMPR2 mutation.
Determining the common molecular consequences of the different types of BMPR2 mutation has important implications for PAH. While penetrance and age of onset may vary between mutations,[6,10] all patients develop the same disease regardless of location of their mutation, which implies a common molecular etiology. Understanding the common molecular consequences of different categories of BMPR2 mutation is thus critical to understanding the molecular etiology of PAH.
To address this, we stably transfected vascular smooth muscle cells with three categories of BMPR2 mutation, all derived from hereditary PAH (HPAH) families. Stable transfections of a cell line were used to avoid the problems with interpreting the effects of genetic, environmental, historical and cell origin variation inherent in the study of patient-derived cells. We used a C118W mutation in the ligand binding domain, an R332X mutation in the kinase domain and a 2579–2580delT frameshift in the cytoplasmic tail domain. mRNA from transfected vascular smooth muscle cells was used for gene array analyses, with follow-up measurements in cell culture, transgenic mice and human samples.
MATERIALS AND METHODS
Generation of A7r5 lines
Wild-type human BMPR2 was polymerase chain reaction (PCR) cloned into pCI-Neo and sequence verified. Next, three different mutation types were created using the Stratagene QuickChange Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Mutations were all derived from human patient families, and included T354G (or C118W) in the ligand binding domain, a C994T (or R332X) mutation in the kinase domain and a 2570–2580delT frameshift resulting in a cytoplasmic tail truncation.
Constructs were stably transfected into A7R5 vascular smooth muscle cells using 5 µg/plate Fugene-6 (Promega, Madison, WI, USA) according to the manufacturer's protocol in p100 plates. Initial selection used 1.6 mg/ml G418S for 7 days, followed by maintenance at 800 g/ml.
Western blots
Cultured cells were rinsed twice with cold phosphate-buffered saline (PBS) and then resuspended in RIPA buffer with 2 phosphatase inhibitors (SigmaP 5726, P2850) and a protease inhibitor (Sigma P8340). Lysates were placed on ice for 30 min and then spun at 13,000 rpm for 15 min at 4°C.
Protein was quantified using the Bradford Protein Assay (Bio-Rad, Hercules CA, USA). The gel system was the 8–16% Tris-glycine gel method using Invitrogen and the chemiluminescent detection by ECL Plus Kit (GE Healthcare, Piscataway, NJ, USA, Invitrogen, Carlsbad, CA, USA). The imaging system used was Alpha Innotech's FluorChem HD2. The transfer membrane was PVDF (GE Healthcare). Antibodies were used pSMAD(Cell Sig 9511), pp38(Cell Sig 9211), pp42/44(Cell Sig 4376), pcofilin (Cell Sig 3313) and β-actin (Abcam 8227).
A7r5 gene expression arrays
RNA was isolated from A7R5 cells using a Qiagen RNeasy mini kit (Valencia, CA, USA). RNA quality was confirmed using a Bioanalyzer RNA 6000 kit (Agilent Technologies, Santa Clara, CA, USA). High-quality RNA from three plates was pooled for each array, and two arrays were used for each mutation type (native or no mutation, CD, KD and ED BMPR2 mutations).
First and second strand complementary DNA was synthesized using standard techniques. Biotin-labeled antisense complimentary RNA was produced by an in vitro transcription reaction. Affymetrix rat genome 230 2.0 microarrays were hybridized with 20 μg cRNA. Target hybridization, washing, staining and scanning probe arrays were performed following an Affymetrix GeneChip Expression Analysis Manual.
The array results have been submitted to the NCBI gene expression and hybridization array data repository (GEO, http://www.ncbi.nlm.nih.gov/geo/), as series GSE21583.
Gene expression array analysis
Affymetrix Cel files were loaded into dChip array analysis software.[11] The overall signal strength from the arrays was normalized to the median array, and the expression levels were determined using the perfect match/mismatch (PM/MM) algorithm. Expression values were base 2 log-transformed for comparisons. Comparisons between mutant lines and native lines used the following criteria: (1) the larger average expression must be at least 7 (128 before log-transformation) in order to avoid values entirely within experimental noise, (2) the sum of the standard deviations of the two groups was subtracted from the absolute value of the difference between the groups, and this value needed to exceed 0.4. This corresponds to roughly a 95% confidence of a minimum 1.3x change, but, on a practical basis, required a much larger average fold change (as only those genes with no error would have an average 1.3x change). This low-fold change requirement is suitable for gene ontology group analyses, which was our goal. A false discovery rate was determined by mixing group identifiers and performing the same analysis.
The resulting lists of genes were imported into a Gene Ontology Tree Machine,[12] where they were sorted into statistically significant Gene Ontology Consortium groups by hypergeometric test internal to the software.
Murine arrays reanalyzed here are part of a recently completed study (companion paper) of actin-organization in Rosa26-BMPR2R899X mice. The arrays referenced here consist of two arrays on control (Rosa26-only) and two in mutant mice (Rosa26-BMPR2R899X) activated for 1 week, with normal right ventricular systolic pressure (RVSP). These arrays have been submitted to the NCBI gene expression and hybridization array data repository (GEO, http://www.ncbi.nlm.nih.gov/geo/), as series GSE21573.
Amplex red assay
Cells were plated in flat bottom white polystyrene 96 well plates (Costar cat #3912) the day prior to the experiment at a concentration of 3×104/well. At the start of the experiment, the media was aspirated and replaced with 100 microliters reaction mixture (50 micromolar Amplex Red reagent [Invitrogen] and 0.1 U/mL HRP in Krebs-Ringer phosphate buffer). Then, the fluorescence was immediately assayed using an Optima fluorescence microplate reader (BMG-Labtech, Offenburg, Germany), with excitation at 544 nm and emission at 590 nm. Fluorescence was read every 10 min for a 1-h period. Values reported are the slope of the resulting line, with the slope of the negative control (empty wells) subtracted.
Luminol assay
Cells were plated at 2.5 × 105cells/plate in 12-well plates (Falcon polystyrene) the day prior to the assay. At the start of the assay, cells were washed once with ice-cold PBS and the media was replaced with Krebs Ringer phosphate buffer (2 ml/well). Luminol was added to a final concentration of 40 micromolar and the plates were immediately placed in the Xenogen IVIS imager and images were captured for 15 s. The luminescence was assayed with Xenogen-Living Image 4.0 software, with background (wells with no cells but otherwise identically treated) subtracted.
Transgenic mouse lines
Rosa26-rtTA2 and TetO7CMV-BMPR2R899X transgenic lines were generated by the University of Cincinnati Trangenic Mouse Core on a FVB/N background, as previously described.[5] Breeding colonies of these lines are maintained independently within the (redacted) Division of Animal Care, and were crossed to produce double transgenic Rosa26-rtTA2 X TetO7CMV-BMPR2R899X mice for these studies.
For gene expression studies, Rosa26-rtTA2 mice or Rosa26-rtTA2 X TetO7CMV-BMPR2R899X mice at 12 weeks of age had transgene expression induced for 1 week by addition of doxycycline at 1 g/kg to chow. For lipid oxidation studies, Rosa26-rtTA2 mice or Rosa26-rtTA2 X TetO7CMV-BMPR2R899X mice at approximately 5 months of age had transgene expression induced for 4 weeks by the addition of doxycycline at 1 g/kg to the chow. Animals used for these studies had normal RVSP, as assessed by right heart catheterization (as previously described).[5]
All studies were approved by the (redacted) Vanderbilt University Animal Care and Use Committee.
Electron micrography
Lungs from Rosa26-rtTA2 (control) mice or Rosa26-rtTA2 X TetO7CMV-BMPR2R899X mice were inflated and fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer and then washed and post-fixed in 1% aqueous osmium tetroxide. Following fixation, the samples were dehydrated through a graded series of ethanols to 100%. The samples were then embedded in Spurr resin. Thin sections of the cured resin were viewed using an FEI CM12 transmission electron microscope operated at 80 KeV. Lung sections containing small vessels were selected (away from broncus and large arteries) for EM. Pulmonary vessels were identified by the presence of blood, which was not flushed for this experiment. Mitochondrial long axis was measured in cells surrounding vessels in three control mice and three BMPR2-mutant mice by a blinded observer.
Immunohistochemical analysis of 15-F2t-isoprostane and isoketals protein adducts in human lung
Immunolocalization of 15-F2t-IsoP was performed on archival paraffin-embedded human lung tissue obtained from control (n=3), HPAH patients (n=3) with BMPR2 mutations. Each of the HPAH cases had a known BMPR2 mutation. Lung sections were deparaffinized, rehydrated and treated with 0.3% hydrogen peroxide for 20 min to block endogenous peroxidase. Sections were incubated with 10% normal rabbit serum in 0.1 M PBS (pH 7.4) for 10 min at room temperature to block non-specific binding of the secondary antibody, followed by incubation with 15-F2t-IsoP antibody (Oxford Biomedical Research, MI; dilution 1:100 of 6 mg/ml protein concentration) overnight at 4°C. The sections were then incubated with biotinylated goat secondary antibody (1:400 dilution; Vector Laboratories Inc., Burlingame, CA, USA) for 1 h at room temperature followed by incubation with HRP-conjugated Streptavidin. Diaminobenzidine (DAB) was used as a substrate for HRP. The sections were dehydrated and mounted in Cytoseal XYL (Richard-Allan Scientific, MI, USA) for the light microscopic examination.
Immunolocalization of isoketal protein adducts was performed on the control and HPAH human lung sections, or on murine lung sections, as mentioned above. Tissue sections were deparaffinized, rehydrated and treated with 0.3% hydrogen peroxide as described previously. Sections were incubated with 0.1 M PBS (pH 7.4) containing 5% normal mouse serum and 5% bovine serum albumin for 30 min at room temperature to block the non-specific binding of the secondary antibody. Sections were incubated with 5 ug/ml D11 ScFv (isoketal antibody) for 2 h at room temperature and then incubated with HRP-labeled anti-E Tag (1:500 dilution, GE Healthcare, Pittsburgh, PA, USA) for 2 h at room temperature. DAB was used as a substrate for HRP. The sections were dehydrated and mounted in Cytoseal XYL (Richard-Allan Scientific) for the light microscopic examination.
Quantification of isofurans and F2-isoprostanes in mouse lung tissue
Lungs were harvested from mice, snap frozen in liquid nitrogen and stored at -80°C until analysis. Tissues were homogenized and lipids were extracted and analyzed as previously described.[13] Briefly, tissues were homogenized in 2:1 chloroform:methanol containing 0.005% butylated hydroxytoluene (BHT). Following quantitative extraction, 0.9% saline was added to precipitate the proteins. Samples were centrifuged at 2000 rpm for 10 min, the upper aqueous phase was removed and the lower organic phase was decanted and evaporated to dryness under nitrogen. Equal parts of methanol containing 0.005% BHT and 15% KOH were added and the samples were incubated at 37°C for 30 min. The pH was adjusted to 3 with 1N HCl, and 1 ng of a [2H4] 15-F2t-IsoP internal standard (Cayman Chemical Company, Ann Arbor, MI, USA) was added. Lipids were partially purified by sequential loading/washing/elution from a C-18 followed by a silica Sep-Pak (Waters Associates, Milford, MA, USA), derivatized to pentafluorobenzyl esters, and further purified by silica thin-layer chromatography. The portion of the thin-layer chromatography plate corresponding to the highest concentration of IsoFs and F2-IsoPs (determined by the relative mobility of a PGF2α methyl ester standard) was scraped free. Lipids were extracted, derivatized to trimethylsilyl ethers and analyzed by gas chromatography/negative ion chemical ionization mass spectrometry with selected ion monitoring. IsoFs were detected at m/z 585, F2-IsoPs at m/z 569 and the [2H4] 15-F2t-IsoP internal standard at m/z 573. Quantification was accomplished by measuring the areas under the portions of the curves for the selected ion currents for IsoFs and F2-IsoPs, respectively, and comparing with the area under the curve for the selected ion current corresponding to the internal standard. Using stable isotope dilution calculations, the amount of IsoFs and F2-IsoPs in each sample was determined and normalized to tissue wet weight.
Quantification of F2-isoprostanes in human urine
The subjects were approached in a clinic or by telephone for enrolment in this study and planning for sample acquisition. In brief, subjects were asked to provide consent prior to study enrolment. They were then given, in person or by subsequent mailing, a box that contained a copy of the consent form already signed by the subject as well as a cold pack for storage and two 50 ml conical tubes. Each tube contained 0.5 ml of 500 mM ascorbic acid for urine preservation per established protocols (1 mg/mL). Each research box came with a set of instructions along with the supplies, with orders to collect the urine sample as a 12-h overnight urine sample. In the morning after collection, the subject filled completely the two 50 ml conical tubes with urine, sealed them and placed the tubes in the package with a cold pack for return by overnight mail to the group's research laboratory (in approved biosample transport packing). The subject called the Research Coordinator upon sending the sample, which was tracked until receipt. All samples were aliquoted into conical tubes and stored at -80°C within 24 h of collection. This study was approved by the (redacted) Human Subjects IRB.
To quantify F2-IsoPs, 0.2 mL urine was added to 2–3 mL deionized water. Internal standard was added, the pH was adjusted to 3 with 1N HCl and analysis was carried out as described above. A separate 1 mL sample from each subject was sent to the core clinical lab for quantification of creatinine.
RESULTS
Different BMPR2 mutations do not cause consistent changes in known downstream pathways
A7r5 rat vascular smooth muscle cells were stably transfected with three categories of human BMPR2 mutation: a cytoplasmic domain (CD) mutation, a kinase domain (KD) mutation, and a ligand binding or extracellular domain (ED) mutation. Quantitative reverse transcriptase PCR was performed to confirm mutant BMPR2 expression comparable with wild-type levels (not shown). We found decreased SMAD1 phosphorylation in KD and ED mutants, but not in CD mutants [Figure 1]. We found increased cofilin phosphorylation,[4] presumably secondary to LIMK dysfunction, in CD, but not in KD and ED mutants. These changes were not dramatic (~40–60%), but were in keeping with our goal of using roughly physiologic stoichiometry between mutant and wild-type mutation.
Figure 1.
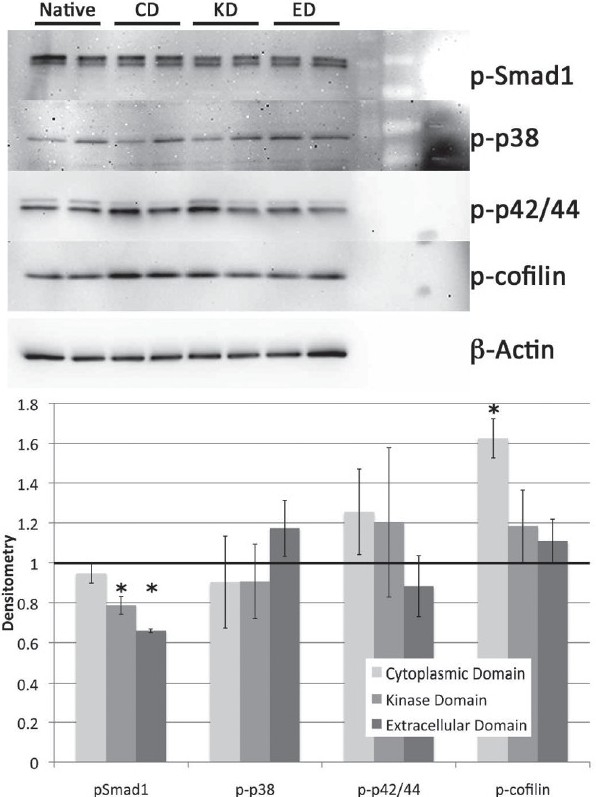
Western blot for phosphorylation of BMPR2 activity targets Smad1, p38, p42/44 and cofilin show no consistent changes across the extracellular domain (ED), kinase domain (KD) and cytoplasmic domain (CD) BMPR2 mutation types. Densitometry (lower panel) indicates a signifi cant decrease in Smad1 phosphorylation in KD and ED mutants, but not CD, and a signifi cant increase in cofi lin phosphorylation in CD, but not KD and ED mutants. Significance is P<0.05 by ANOVA with post hoc t-test
Both p38 and p42/44 phosphorylation showed a great deal of variation from plate to plate in all samples and no consistent changes between mutation types; this was true even after extensive experimentation with timepoints and conditions.
Different BMPR2 mutations cause dysregulation of energy metabolism or reactive oxygen species (ROS)-related gene expression
To determine the common molecular consequences of different classes of BMPR2 mutation, Affymetrix gene expression array analysis was performed on native A7r5 cells, or cells stably transfected with CD, KD or ED mutations. RNA from three plates was pooled for each array, and two arrays were used per condition. Using requirements for a minimum expression threshold and minimum change in statistically relevant levels of expression (described in Methods), we found 296 probe sets altered between CD and native arrays, representing 249 named genes, with a false discovery rate of 3.7% (Lane and West, unpublished data). With the same conditions, we found 415 probe sets representing 390 genes altered between the ED and the native arrays (Lane and West, unpublished data). Forty-five percent of the alterations on the CD arrays were in common with alterations in the ED arrays. The KD mutation was a strong outlier, with 8444 probe sets altered using the same criteria, with a 0% false discovery rate. This probably represents a wholesale change in the cell differentiation state. When we previously expressed a very similar mutation in the smooth muscle of live mice, we also had a broad dedifferentiation of smooth muscle.[14]
Most of the genes with common altered regulation across mutation types could be grouped into cell cycle or apoptosis, migration or actin organization and energy metabolism or ROS-related genes. Cell-cycle changes related to BMPR2 mutation have been extensively studied.[15] We recently completed a study on the physiologic relevance of dysregulation of actin organization by BMPR2,[16] and thus were most interested in the indicators of altered energy metabolism and increased ROS suggested by the arrays.
Indicators of altered energy metabolism and increased ROS included altered glycolysis genes, decreased mitochondrial ribosomal genes, increased expression of ROS-responsive genes and suggestions of a shift to glutaminolysis, including increased sterol activity and alterations in citrate synthase and glutamine-related genes [Figure 2]. Depicted in Figure 2 are alterations common to all three mutations; each mutation included alterations in additional energy metabolism genes with similar consequences.
Figure 2.
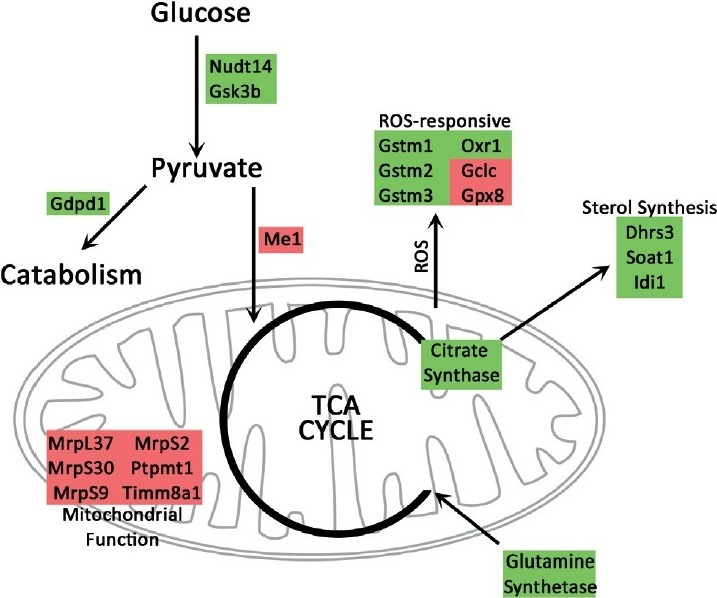
Gene expression changes common to three classes of BMPR2 mutation are consistent with a shift to catabolism and glutaminolysis. Glutaminolysis is normally accompanied by increased glutamine uptake and citrate synthase activity, resulting in increased sterol synthesis and reactive oxygen species. Catabolism is associated with decreased importance of mitochondria in energy production, consistent with decreased mitochondrial functional genes seen in the arrays. Green indicates upregulated genes; red indicates downregulated genes
Different BMPR2 mutations cause increased peroxide and superoxide in culture
Gene expression arrays are an excellent tool for generating a broad overview of the pathways altered in an experiment but, by their nature, are almost useless for determining the functional consequences of alterations in those pathways. To determine whether BMPR2 mutations actually resulted in alterations in ROS production, we directly assayed production of ROS in A7r5 culture by standard methods. The Amplex red assay was used to determine the levels of peroxide;[17] the luminol assay was used to determine the levels of the more labile superoxide.[18] Both assays indicated about a two-fold increase in ROS production across mutation types [Figure 3], although the increase in superoxide was less robust in the CD mutation.
Figure 3.
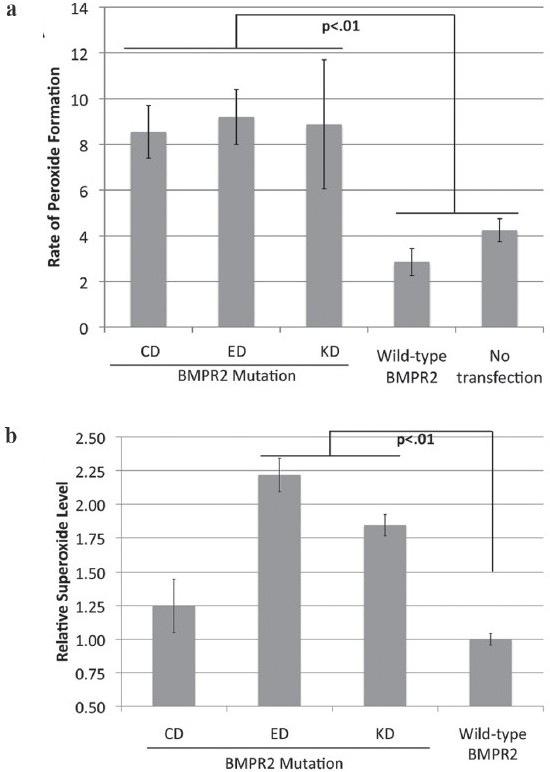
Multiple BMPR2 mutation types show increased reactive oxygen species formation. (a) Cytoplasmic domain (CD), extracellular domain (ED) and kinase domain (KD) mutations all show increased peroxide formation by the Amplex Red assay when stably transfected into A7r5 vascular smooth muscle cells compared with cells stably transfected with wild-type BMPR2, or untransfected cells. (b) Assay of superoxide levels by luminol shows increased levels in ED and KD mutations as compared with wild-type BMPR2. Superoxide levels in CD-mutant A7R5 cells were too noisy across experiments to distinguish from wild-type BMPR2
Cytoplasmic domain BMPR2 mutation alters gene expression in transgenic mice in a manner suggesting increased ROS
Because BMPR2 mutation produces increased ROS in cell culture, we were interested in whether this would be true in vivo as well. The CD mutation was used because it had the weakest effect in culture, and would be the most stringent test of in vivo effect. We recently crossed the BMPR2R899X mutation, previously published with smooth muscle expression,[5] into a universal, Rosa26-driven promoter.[16] This has the advantage that molecular phenotype is far easier to discern because this Rosa26-BMPR2R899X model does not have the dilution-effect problems of smooth muscle-only models.
As a first step, we performed a retrospective analysis of the existing arrays from these animals to determine whether there were signs of altered ROS. We compared gene expression in Rosa26-BMPR2R899X mice with transgene activated for 1 week (long before RVSP increases) to control mice also fed doxycycline, by Affymetrix array. Gene expression data from Rosa26-BMPR2R899X mice suggest elevated ROS of mitochondrial origin. The mice showed an increase in the oxidative stress response genes, including glutathione peroxidase,[19] two lysyl oxidases[20] and hydroxyprostaglandin dehydrogenase.[21] However, many NADPH complex genes were downregulated, with approximately two-fold downregulation of Nox2, Nox4, Ncf1 (p47phox) and Ncf4 (p40phox). More broadly, of the 832 genes with at least 3x change in regulation, 386 or 46% fell into the cellular metabolic process gene ontology[22] group, which was thus statistically overrepresented at P=0.005 by hypergeometric test. This suggests that BMPR2 mutations promote widespread alteration in energy metabolism.
Mice with a cytoplasmic domain BMPR2 mutation have increased levels of oxidized lipids in whole lung
While expression arrays suggested broad defects in energy metabolism in vivo, and increased ROS specifically, we wished to directly test the hypothesis that ROS were increased in vivo. Because of their volatility, direct measurement of ROS from animal tissues is extremely error-prone.[18] However, presence of oxidized lipids is an indicator of increased ROS over time. Unlike ROS, lipid oxidation is quite stable, and is the most accurate measure of in vivo oxidant stress.[23]
We thus measured the levels of oxidized lipids in whole lung from either control (transactivator-only) or BMPR2R899X mice, both fed doxycycline for 6 weeks, and all with normal RVSP. Free radical-mediated lipid peroxidation was assessed by measurement of F2-isoprostanes (IsoPs) and isofurans (IsoFs) by GC/MS. Elevated cellular oxygen concentration, which occurs as a result of mitochondrial dysfunction, favors the formation of IsoFs and suppresses the formation of IsoPs.[24] We found approximately a 50% increase in the levels of IsoPs and a 100% increase in the levels of IsoFs in BMPR2-mutant mice compared with controls [Figure 4a and b]. The ratio of IsoF to IsoP increased from 2.3 to 3.2, P=0.02, suggesting that much of the increase was due to mitochondrial dysfunction.
Figure 4.
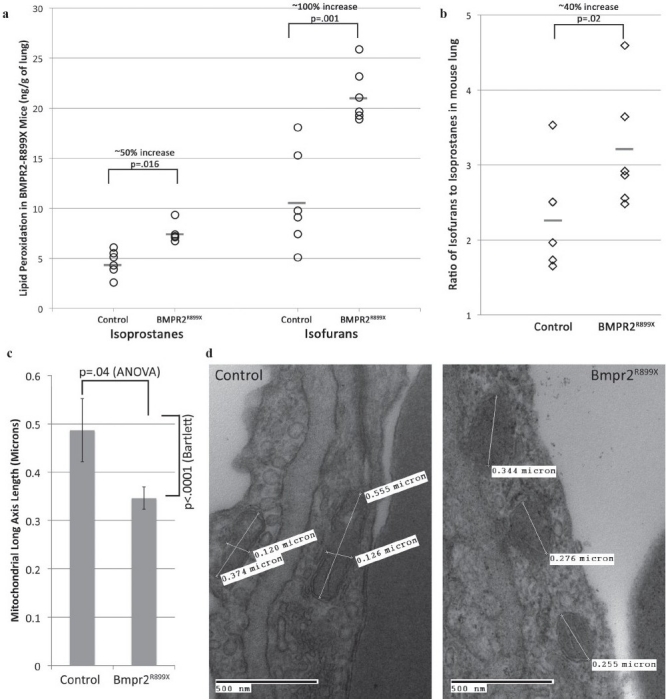
Mice with cytoplasmic domain mutation show increased oxidized lipids, including (a) increases in isoprostane and isofuran levels (signifi cant at indicated levels by unpaired t-test) and (b) an increase in the ratio of isofuran:isoprostane levels (P=0.02 by rank-sum test, appropriate for data not normally distributed). n=6 per group (each symbol corresponds to an individual mouse). (c) Mitochondria in BMPR2-mutant mice are smaller, with less variability in size, than those in controls. (d) Example electron micrography fi elds from BMPR2-mutant mice showing smaller mitochondria
Mitochondria in BMPR2-mutant mice are smaller, with less variable size
Many defects in mitochondrial energy metabolism result in alterations in the mitochondria that are visible by electron microscopy.[25,26] We therefore performed electron microscopy on the lung from Rosa26-BMPR2R899X mice. A blinded observer measured lengths of the long-axis of mitochondria in pulmonary vascular cells, and found that mitochondria from mutant mice were roughly 40% smaller on average, and with three-fold less variability in size than mitochondria from pulmonary vascular cells in controls [Figure 4c and d].
Oxidized lipids in BMPR2-mutant mice are localized to the vasculature
Both gene array and lipid peroxidation data from BMPR2R899X mice were from whole lung. Because expression of the mutant transgene is universal, it was possible that increase in lipid oxidation was also universal. To determine localization, we used antibodies to isoketals[27] for immunohistochemistry. We found that in control mouse lung, isoketal staining was primarily found in likely alveolar macrophages [Figure 5a]. In Rosa26-BMPR2R899X-mutant mice with transgene activated for 4 weeks, but in which RVSP is still normal, there is an increase in vascular but not alveolar isoketal staining [Figure 5b and c]. After an increase in the RVSP, staining is found throughout the area of the lesions [Figure 5d].
Figure 5.
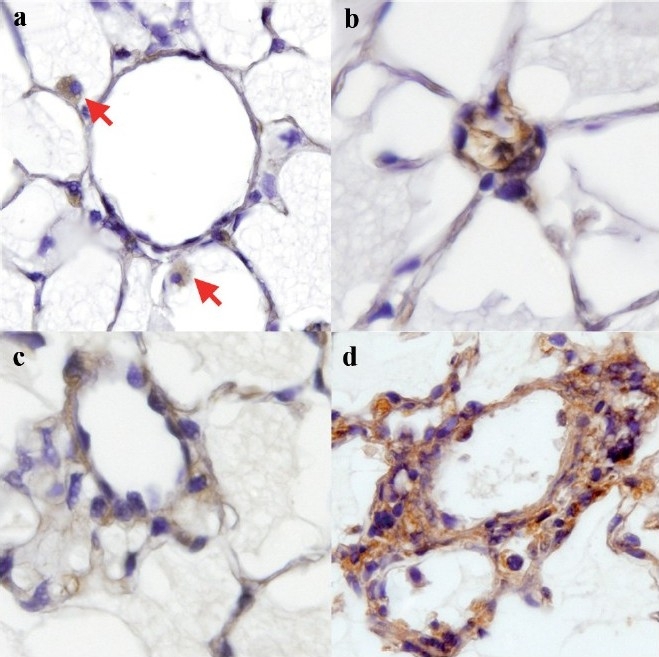
BMPR2-mutant mice have elevated pulmonary vascular isoketals. (a) The control mouse has positive staining limited to macrophages (red arrows). (b, c) Increased isoketal levels are limited to the vasculature in lungs of BMPR2R899X mice that do not yet have elevated RVSP. (d) BMPRR899X mouse with elevated RVSP and adventitial remodeling shows increased isoketal staining throughout the remodeled area
Human BMPR2 mutation carriers have increased oxidized lipids, regardless of disease status
To determine whether increased oxidative stress was present in human BMPR2 mutation carriers, we collected urine from five controls and 17 individuals with BMPR2 mutations, including nine with PAH and eight that were currently asymptomatic, representing six different mutations. We found that HPAH patients had roughly two-fold higher urine isoprostane levels than controls, in agreement with previous studies of urine isoprostanes in idiopathic (IPAH) patients[28] [Figure 6]. However, we discovered that unaffected BMPR2 mutation carriers (UMC) also have increased urine isoprostanes, to a degree statistically indistinguishable from those affected with disease. There was no relationship between isoprostane levels and mutation type, age or treatment.
Figure 6.
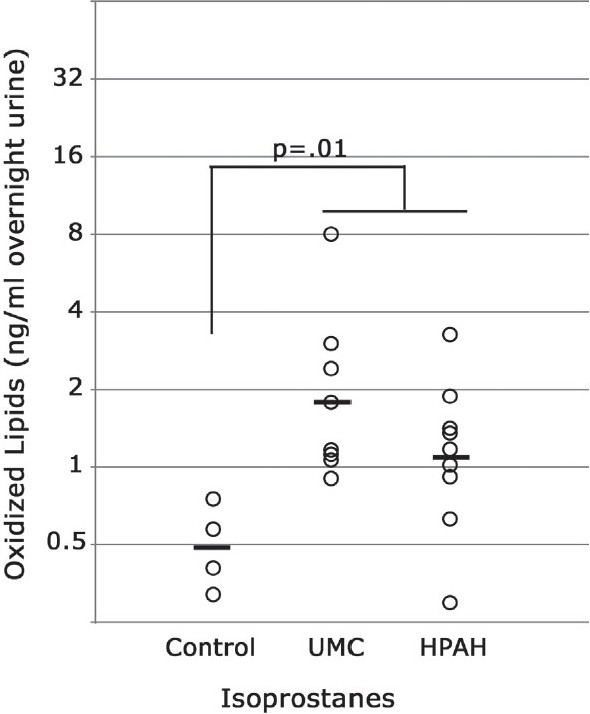
(a) Humans with BMPR2 mutations have increased isoprostane oxidized lipids in urine, regardless of disease status. UMC, unaffected BMPR2 mutation carriers; HPAH, hereditary pulmonary arterial hypertension (also BMPR2 mutation+). Comparisons are by ANOVA with post hoc t-test
As all BMPR2 mutation carriers have abnormal exercise tests,[29] we hypothesize that all have lung pathology. For reasons that are not yet clear in either mice or humans, BMPR2 mutation does not produce sufficient lung pathology for some to develop clinical disease. The increase in ROS seen in asymptomatic BMPR2 mutation carriers may thus be associated with developing disease, but because of the difficulty in obtaining lung samples from asymptomatic individuals, this cannot be directly tested.
Oxidized lipids in human BMPR2-related PAH are localized to the vasculature
In control lung, immunohistochemical analysis revealed the presence of IsoFs [Figure 7a and b] and 15-F2t-IsoP [Figure 7e and f] in a few endothelial and smooth muscle cells of the large and small arteries, respectively. 15-F2t-IsoP was also present in the elastin surrounding these vessels. In HPAH lung, the intensity and number of endothelial and smooth muscle cells exhibiting immunoreactivity to IsoFs [Figure 7c and d] and 15-F2t-IsoP [Figure 7g and h] were increased in the large and small arteries as compared with controls. Immunoreactivity to 15-F2t-IsoP antibody was diffused as compared with IsoFs antibody.
Figure 7.
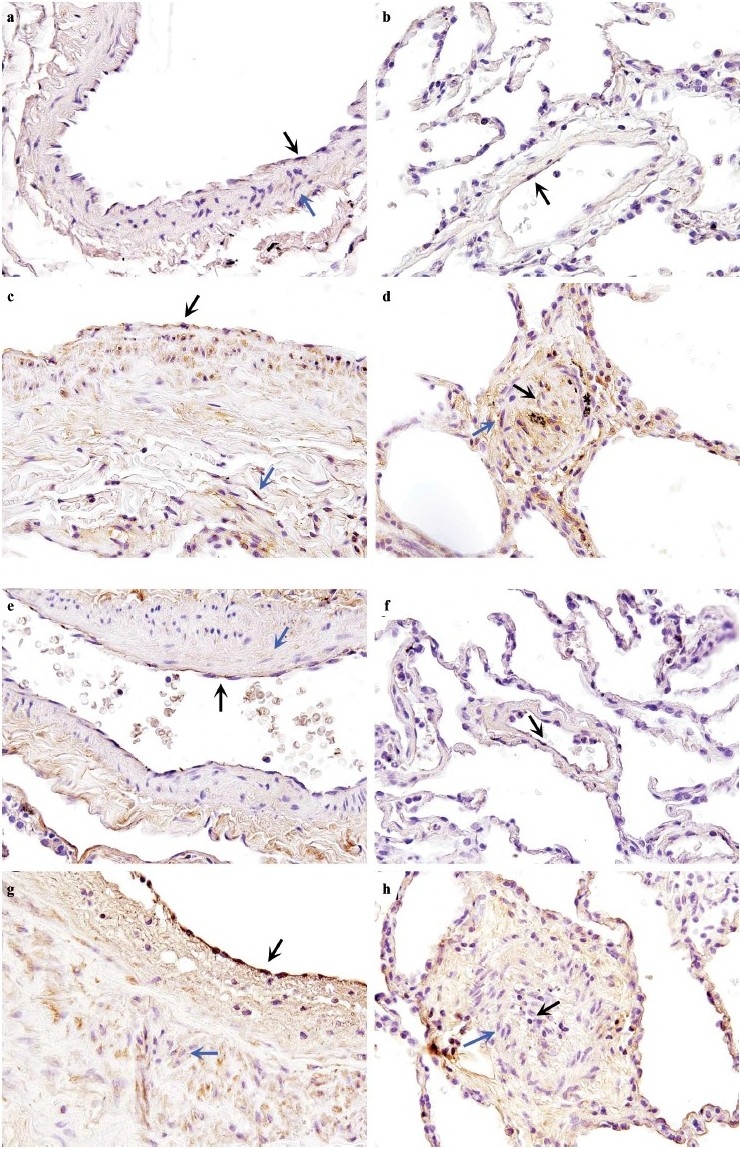
(a–d) Localization of isoketals in human lung from control and hereditary pulmonary arterial hypertension (HPAH) with BMPR2 mutation. In (a) large and (b) small vessels from control lung, localization of isoketals was observed in few endothelial cells (ECs) (black arrows) and smooth muscle cells (SMCs)(blue arrows). In (c) large and (d) small vessels from HPAH lung, the intensity of immunoreactivity and localization of isoketals was signifi cantly increased in ECs and SMCs. (e–h) Localization of 8-isoprostane (8-IP) in human lung from control and HPAH with BMPR2 mutation. In (e) large and (f) small vessels from control lung localization of 8-IP was observed in ECs (black arrows) and SMCs (blue arrows). Immunoreactivity of 8-IP was also observed in elastin surrounding the vessels. In (g) large and (h) small vessels from HPAH lung, the intensity of immunoreactivity and localization of isoketals was significantly increased. Magnification ×600
DISCUSSION
In this study, we show for the first time that increased production of ROS, likely of mitochondrial origin, is a common consequence of BMPR2 mutation in cultured smooth muscle cells, mice and humans. We demonstrate increased ROS directly in cell culture [Figure 3] and through increased lipid oxidation in mice [Figure 4] and humans [Figure 6]. We confirm that the increase in ROS is vascular-specific in both mice and humans [Figures 5 and 7]. Our data suggest that the increased ROS is of mitochondrial origin (gene array data on cell culture and mice; ratio of IsoF to IsoP in mice; electron microscopy in mice). We further show that the increased ROS precedes the development of elevated RVSP in both mice and humans, indicating that it is likely important to pathogenesis, not just a response to high pressure.
There has been an explosion of interest in the relationship between ROS and pulmonary hypertension recently, with 64 papers on the topic from 2003 to 2010. The majority of these are in the context of persistent pulmonary hypertension of the neonate or hypoxia-induced PAH animal models, in which the increased ROS appears to be at least in part through NOS uncoupling.[30] Recently, Xu et al. found that IPAH patients had increased glucose uptake, decreased mitochondrial numbers and an apparent shift to “Warburg effect” aerobic glycolysis metabolism in their lungs.[31,32] While the authors hypothesized that their findings were secondary to dysregulation of NO, these findings are highly supportive of our current results.
The data from this earlier study combined with our own results suggests that a common consequence of BMPR2 mutation, and indeed of any PAH, is a shift in cellular metabolism away from production of energy and toward the production of proteins, lipids, etc., necessary for proliferation, similar to the shift in energy metabolism found in cancers.[31] This mechanism is thus very similar to the likely mechanism for spontaneous PAH in fawn-hooded rats.[33] Under this hypothesis, increased ROS is a marker of a pathologic shift in energy metabolism, rather than being directly pathologic itself.
On the other hand, there are multiple mechanisms by which the increased ROS could be directly pathologic. There is a strong crosstalk between mitochondrial ROS and NADPH,[34] and thus NO metabolism through BH4 depletion.[35] High levels of ROS can cause DNA damage, potentially leading to genetic instability.[36] Increased ROS could cause cross-linking of lipids or collagen, directly leading to increased vascular stiffness.[37,38] ROS can also more directly drive inflammatory processes,[39] including through induction of NF-kappaB.
While this study does not directly address the protein-protein interactions by which BMPR2 mutation results in the observed mitochondrial defects, a strong hypothesis can be formed by combining the data from this study with our previous studies and the literature. BMPR2 has multiple points of direct interaction with cytoskeletal regulatory elements, including binding of LIMK, SRC and TCTEX, among others.[40] We have recently shown in mice that BMPR2 mutation causes broad-based defects in f-actin-related pathways and cytoskeletal trafficking;[16] this is supported by the arrays in this study (Lane and West, unpublished data). These are the central pathways regulating mitochondrial fission and fusion;[41] defects in mitochondrial fission and fusion have already been implicated in pulmonary hypertension and oxidant stress.[42]
Limitations of our study include use of urine, the only available substrate from asymptomatic BMPR2 mutation carriers, to assess lipid oxidation in human patients. Additional limitations to our study include the use of systemic vascular smooth muscle cells for cell culture, although confirmation of cell culture results in both animal and human experiments somewhat ameliorates this. Further, we were only able to examine ROS in one mutation type because of the time and expense of animal studies. Finally, while the direct mechanism linking BMPR2 to mitochondrial defects seems very likely to be through altered f-actin dynamics, we have not directly shown that yet.
Our central finding is that increased oxidative injury is a common consequence of BMPR2 mutations, probably because of a shift in mitochondrial metabolism. Identification of disease genes carries with it the promise of understanding molecular etiology and thus identification of important targets for intervention. The functional significance to this study is that mechanisms for attacking the Warburg effect in cancer patients could be addressed to PAH patients. Use of dichloroacetate,[43] blood glucose control[44,45] or other small-molecule drugs targeted at aerobic glycolysis[46] provide potential treatment options entirely distinct from the current therapies. Interventions against mitochondrial dysfunction and increased ROS may allow resolution of the vascular injury that is the hallmark of PAH.
Footnotes
Source of Support: NIH grant (P01 HL 72058).
Conflict of Interest: None declared.
REFERENCES
- 1.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 2.Cogan JD, Vnencak-Jones CL, Phillips JA, 3rd, Lane KB, Wheeler LA, Robbins IM, et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med. 2005;7:169–74. doi: 10.1097/01.gim.0000156525.09595.e9. [DOI] [PubMed] [Google Scholar]
- 3.ten Dijke P, Korchynskyi O, Valdimarsdottir G, Goumans MJ. Controlling cell fate by bone morphogenetic protein receptors. Mol Cell Endocrinol. 2003;211:105–13. doi: 10.1016/j.mce.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, et al. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–98. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, et al. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol. 2008;295:L744–55. doi: 10.1152/ajplung.90255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sankelo M, Flanagan JA, Machado R, Harrison R, Rudarakanchana N, Morrell N, et al. BMPR2 mutations have short lifetime expectancy in primary pulmonary hypertension. Hum Mutat. 2005;26:119–24. doi: 10.1002/humu.20200. [DOI] [PubMed] [Google Scholar]
- 7.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–25. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Davies RJ, Southwood M, Long L, Yang X, Sobolewski A, et al. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102:1212–21. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]
- 9.Herpin A, Cunningham C. Cross-talk between the bone morphogenetic protein pathway and other major signaling pathways results in tightly regulated cell-specific outcomes. FEBS J. 2007;274:2977–85. doi: 10.1111/j.1742-4658.2007.05840.x. [DOI] [PubMed] [Google Scholar]
- 10.Austin ED, Phillips JA, Cogan JD, Hamid R, Yu C, Stanton KC, et al. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res. 2009;10:87. doi: 10.1186/1465-9921-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Schmoyer D, Kirov S, Snoddy J. GO Tree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrow JD, Scruggs J, Chen Y, Zackert WE, Roberts LJ., 2nd In vivo Evidence that the E2-isoprostane, 15-E2t-isoprostane (8-iso-prostaglandin E2) is formed. J Lipid Res. 1998;39:1589–93. [PubMed] [Google Scholar]
- 14.Tada Y, Majka S, Carr M, Harral J, Crona D, Kuriyama T, et al. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1556–63. doi: 10.1152/ajplung.00305.2006. [DOI] [PubMed] [Google Scholar]
- 15.Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc. 2006;3:680–6. doi: 10.1513/pats.200605-118SF. [DOI] [PubMed] [Google Scholar]
- 16.Johnson J, Hemnes A, Lane K, Robinson L, Gladson S, West J. New Orleans, Louisiana: 2010. ACE2 Reverses Established Pulmonary Arterial Hypertension in BMPR2R899X Mice. Paper presented at: American Thoracic Society International Conference. [Google Scholar]
- 17.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–8. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 18.Münzel T, Afanas’ev IB, Kleschyov AL, Harrison DG. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol. 2002;22:1761–8. doi: 10.1161/01.atv.0000034022.11764.ec. [DOI] [PubMed] [Google Scholar]
- 19.Förstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–49. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 20.Johnson CD, Balagurunathan Y, Dougherty ER, Afshari CA, He Q, Ramos KS. Insight into redox-regulated gene networks in vascular cells. Bioinformation. 2007;1:379–83. doi: 10.6026/97320630001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12:955–62. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 22.The Gene Ontology in 2010: extensions and refinements. Nucleic Acids Res. 2010;38:D331–5. doi: 10.1093/nar/gkp1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milne GL, Yin H, Brooks JD, Sanchez S, Jackson Roberts L, 2nd, Morrow JD. Quantification of F2-isoprostanes in biological fluids and tissues as a measure of oxidant stress. Methods Enzymol. 2007;433:113–26. doi: 10.1016/S0076-6879(07)33006-1. [DOI] [PubMed] [Google Scholar]
- 24.Fessel JP, Jackson Roberts L. Isofurans: novel products of lipid peroxidation that define the occurrence of oxidant injury in settings of elevated oxygen tension. Antioxid Redox Signal. 2005;7:202–9. doi: 10.1089/ars.2005.7.202. [DOI] [PubMed] [Google Scholar]
- 25.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–85. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–22. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 27.Davies SS, Amarnath V, Roberts LJ., 2nd Isoketals: highly reactive gamma-ketoaldehydes formed from the H2-isoprostane pathway. Chem Phys Lipids. 2004;128:85–99. doi: 10.1016/j.chemphyslip.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Robbins IM, Morrow JD, Christman BW. Oxidant stress but not thromboxane decreases with epoprostenol therapy. Free Radic Biol Med. 2005;38:568–74. doi: 10.1016/j.freeradbiomed.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 29.Grunig E, Weissmann S, Ehlken N, Fijalkowska A, Fischer C, Fourme T, et al. Stress Doppler echocardiography in relatives of patients with idiopathic and familial pulmonary arterial hypertension: results of a multicenter European analysis of pulmonary artery pressure response to exercise and hypoxia. Circulation. 2009;119:1747–57. doi: 10.1161/CIRCULATIONAHA.108.800938. [DOI] [PubMed] [Google Scholar]
- 30.Firth AL, Yuan JX. Bringing down the ROS: a new therapeutic approach for PPHN. Am J Physiol Lung Cell Mol Physiol. 2008;295:L976–8. doi: 10.1152/ajplung.90515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:171–85. doi: 10.1007/978-1-60761-500-2_11. [DOI] [PubMed] [Google Scholar]
- 34.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 35.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–47. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naka K, Muraguchi T, Hoshii T, Hirao A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid Redox Signal. 2008;10:1883–94. doi: 10.1089/ars.2008.2114. [DOI] [PubMed] [Google Scholar]
- 37.Goh SY, Cooper ME. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–52. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- 38.Hunter KS, Albietz JA, Lee PF, Lanning CJ, Lammers SR, Hofmeister SH, et al. In vivo measurement of proximal pulmonary artery elastic modulus in the neonatal calf model of pulmonary hypertension: development and ex-vivo validation. J Appl Physiol. 2010;108:968–75. doi: 10.1152/japplphysiol.01173.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao H, Yang SR, Kode A, Rajendrasozhan S, Caito S, Adenuga D, et al. Redox regulation of lung inflammation: role of NADPH oxidase and NF-kappaB signalling. Biochem Soc Trans. 2007;35:1151–5. doi: 10.1042/BST0351151. [DOI] [PubMed] [Google Scholar]
- 40.West J. Cross talk between Smad, MAPK, and actin in the etiology of pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:265–78. doi: 10.1007/978-1-60761-500-2_17. [DOI] [PubMed] [Google Scholar]
- 41.Bereiter-Hahn J, Vöth M, Mai S, Jendrach M. Structural implications of mitochondrial dynamics. Biotechnol J. 2008;3:765–80. doi: 10.1002/biot.200800024. [DOI] [PubMed] [Google Scholar]
- 42.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–8. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 43.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–94. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shanmugam M, McBrayer SK, Rosen ST. Targeting the Warburg effect in hematological malignancies: from PET to therapy. Curr Opin Oncol. 2009;21:531–6. doi: 10.1097/CCO.0b013e32832f57ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rippe C, Lesniewski L, Connell M, Larocca T, Donato A, Seals D. Short-term Calorie Restriction Reverses Vascular Endothelial Dysfunction in Old Mice by Increasing Nitric Oxide and Reducing Oxidative Stress. Aging Cell. 2010;9:304–12. doi: 10.1111/j.1474-9726.2010.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Granchi C, Bertini S, Macchia M, Minutolo F. Inhibitors of Lactate Dehydrogenase Isoforms and their Therapeutic Potentials. Curr Med Chem. 2010;17:672–97. doi: 10.2174/092986710790416263. [DOI] [PubMed] [Google Scholar]