

Abstract
The β1D integrin is a recently characterized isoform of the β1 subunit that is specifically expressed in heart and skeletal muscle. In this study we have assessed the function of the β1D integrin splice variant in mice by generating, for the first time, Cre-mediated exon-specific knockout and knockin strains for this splice variant. We show that removal of the exon for β1D leads to a mildly disturbed heart phenotype, whereas replacement of β1A by β1D results in embryonic lethality with a plethora of developmental defects, in part caused by the abnormal migration of neuroepithelial cells. Our data demonstrate that the splice variants A and D are not functionally equivalent. We propose that β1D is less efficient than β1A in mediating the signaling that regulates cell motility and responses of the cells to mechanical stress.
Keywords: Integrin, splice variant, muscle, migration, development, knockout, knockin
Integrins are widely expressed cell-surface receptors that participate in cell–extracellular matrix (ECM) and cell–cell interactions (Hynes 1992; Sonnenberg 1993). Integrins also participate in signal transduction events. In response to ligand binding, they transmit signals that interact with those originating from growth factor receptors to regulate cell growth and differentiation (Schwartz et al. 1995). All integrins are heterodimeric transmembrane proteins, consisting of an α subunit noncovalently associated to a β subunit. At least 16 α and 8 β subunits have been identified. The diversity of the integrin family is further increased by alternative splicing of mRNA. Four isoforms of the β1 integrin subunit with different cytoplasmic domains have now been described. The β1B and the β1C variants are minor isoforms expressed in some human tissues and cells (Altruda et al. 1990; Languino and Ruoslahti 1992), but they are not found in the mouse (Baudoin et al. 1996). The β1D variant is the most prominent β1 isoform in muscle and is highly conserved between species (van der Flier et al. 1995; Zhidkova et al. 1995; Belkin et al. 1996). β1A is the most widely expressed β integrin subunit and it can associate with at least 10 different α subunits. It is involved in a wide range of biological processes. In development, from fertilization (Almeida et al. 1995) and extending through organogenesis, β1A appears to play a role in the migration of many cell types including the parietal endoderm (Sutherland et al. 1993), myotomal myoblasts (Jaffredo et al. 1988), neuroblasts (Galileo et al. 1992), and neural crest cells (Thiery et al. 1985). In organogenesis, β1A also participates in the development of cardiac muscle (Fässler et al. 1996) and facilitates the fusion of myoblasts into myotubes during myogenesis (Yang et al. 1996). In addition, β1A plays a role in the migration of various differentiated cell types postnatally, such as keratinocytes and lymphocytes. Finally, recent findings indicate that β1A-containing integrins are able to transduce a mechanical force into a biochemical signal, a process known as mechanotransduction (Ingber et al. 1994). Mechanotransduction may be a crucial mechanism for integrating different types of events, such as the forces generated by muscle cells and by cytoskeleton stiffening in migrating cells, during tissue remodeling and neurulation in the developing embryo and the mechanical stresses resulting from local tension, compression, or fluid flow (Gordon and Brodland 1987; Ingber et al. 1994).
The β1A and β1D isoforms are very homologous, differing by only 13 amino acids in their cytoplasmic domains, 6 of which are located between the cyto-2 and cyto-3 domains, resulting in the loss of a potential site for phosphorylation and ILK (β1-integrin-linked protein kinase) binding in β1A (Hannigan et al. 1996). In the developing muscle, β1D is expressed in a specific pattern. It is first detectable late in gestation and later in all types of muscle, that is, fast, mixed, and slow fibers and is particularly strongly expressed in the heart, suggesting that it has a crucial function in these tissues (van der Flier et al. 1997). In addition, absence of the integrin α7 subunit, which is the major pairing partner of β1D in heart and skeletal muscles, causes a novel form of muscular dystrophy (Mayer et al. 1997). Because of its specific localization at myotendinous junctions and at intercalated discs, which represent the major force transmission sites in skeletal and heart muscle respectively, it has been proposed that β1D mediates stronger attachment at these sites, thereby providing more resistance to the mechanical forces to which muscles are subjected (Belkin et al. 1996; van der Flier et al. 1995). To increase our understanding of the function of β1D, we have knocked out and knocked in the exon for β1D, thereby generating, for the first time, Cre-mediated exon-specific knockout and knockin animals. Adult β1D knockout mice are viable but display a mild abnormality of cardiac function. In contrast, β1D knockin embryos die during development with multiple defects partly because of abnormal migration of neural cells.
Results
Generation of β1 exon D knockout mice by use of the Cre–loxP system
β1D is an isoform of β1, produced by alternative splicing of the β1 gene. During myogenesis, exon D, specific for the β1D subunit, is spliced into the mature mRNA. To delete exon D in embryonic stem (ES) cells, we have developed a method based on the Cre–loxP system that does not affect the splicing of the remaining exons of the β1 gene. In this method, two steps are involved (Fig. 1A): In the first step, exon D together with the ag/gt splicing consensus sites is replaced, through homologous recombination, by a cassette coding for two selection markers flanked by two loxP sites. In the second step, the Cre enzyme is transiently expressed in the targeted ES cells. Cre catalyzes a loxP site-dependent recombination that efficiently excises the selection marker sequences from the targeted allele, leaving a single loxP site instead of exon D and its splicing consensus sites. The cassette encodes positive and negative selection markers, bacterial neomycin resistance (neor) and herpes simplex thymidine kinase genes (HSV–tk), respectively, linked in tandem and under the control of two separate strong promoters.
Figure 1.
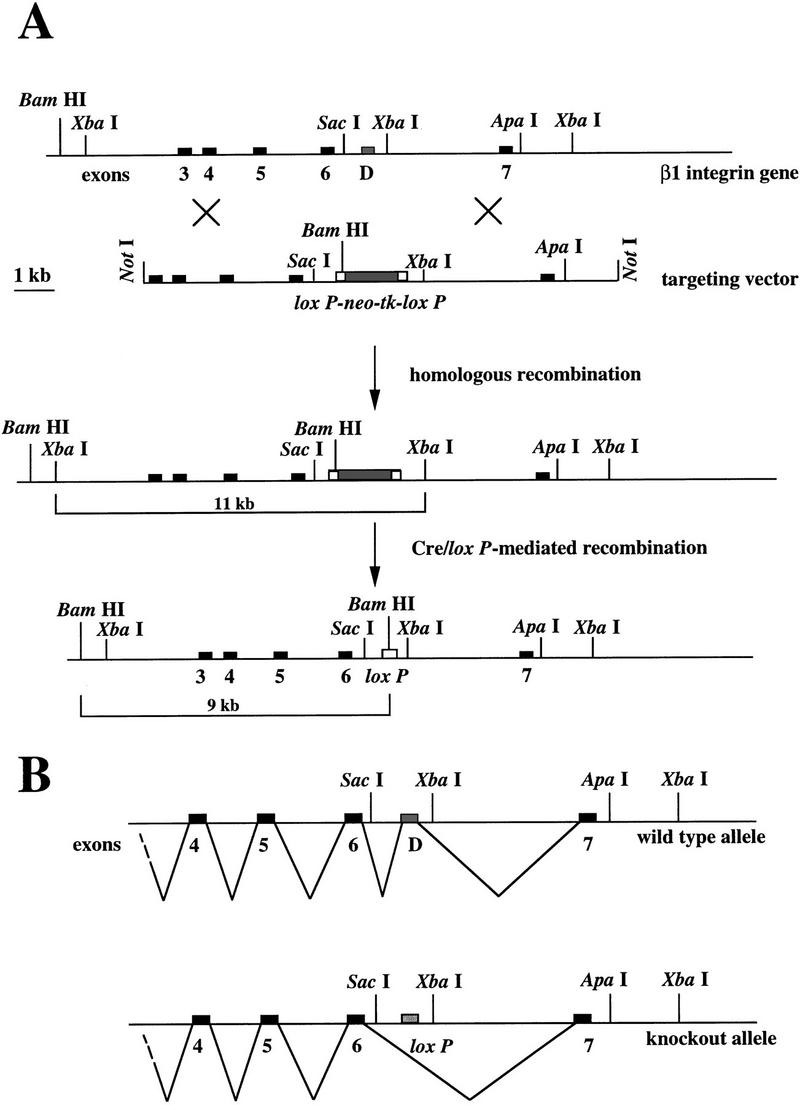
Scheme for exon-specific targeting. (A) Restriction maps for the 3′ portion of the mouse β1 integrin locus, the targeting construct, the homologous recombinant, and the exon D-deleted mutant. In the targeting construct, exon D is replaced by a neo–tk minigene flanked by two loxP sites. After homologous recombination, targeted ES cells are transiently transfected with a Cre-encoding plasmid. After Cre/loxP-mediated recombination, exon D is replaced by a loxP site. Recombination is detected in Southern blot analysis as a new 9-kb BamHI fragment. The probe (331) is a cDNA consisting of exons 3, 4, 5, and 6. (B) In normal muscle cells, the exon D is inserted into the mature β1D mRNA, whereas in the knockout muscle cells, exon 6 and 7 are connected leading to the mature β1A mRNA.
The TC5 targeting construct was designed for homologous recombination (see Materials and Methods). E14 ES cells were transfected with the linearized vector and selected for neomycin resistance. Targeted clones were identified by Southern blot analysis, on the basis of a novel 9-kb band in BamHI-digested genomic DNA, detected with probe 331 (Fig. 2A). Homologous recombination was observed in 60% of cases. The pOG231 plasmid was used to transiently express the Cre enzyme in two independently targeted ES clones. ES cells that had undergone Cre–loxP mediated recombination were selected with gancyclovir. The surviving clones were screened by PCR for the deletion event and the replacement of the exon D by a loxP site. We found that in 100% of the Gancyclovir-resistant colonies the selection marker was deleted, demonstrating the efficiency of the Cre–loxP-mediated recombination. To verify that replacement of exon D by a loxP site did not hamper splicing of the remaining exons (Fig. 1B), PCR and Northern blot analysis were carried out on total RNA extracted from wild-type cells and the mutated ES cell clones. Total RNA was isolated from two independent mutant clones (9D2cre5 and 2G2cre2) and hybridized with a β1 cDNA probe (331). The intensity of the signal in the two clones, as estimated by PhosphorImager analysis, was comparable with that of the parental ES clone (not shown). For PCR analysis, total cDNA from 9D2cre5 and 2G2cre2 clones were amplified with primers coding for exon 6 and 7. No PCR products of abnormal size were detected (not shown). These data demonstrate that there was no aberration in the splicing of the β1 gene. The 9D2cre5 and 2G2cre2 ES clones were injected into C57BL/6 blastocysts and chimeras were obtained. These chimeras successfully allowed germ-line transmission for the two clones and were crossed with FVB or 129/OLA females. Heterozygous offspring was then interbred to produce homozygous mutants, and offspring was tested for exon D deletion by Southern blot and PCR analysis (Fig. 2B).
Figure 2.
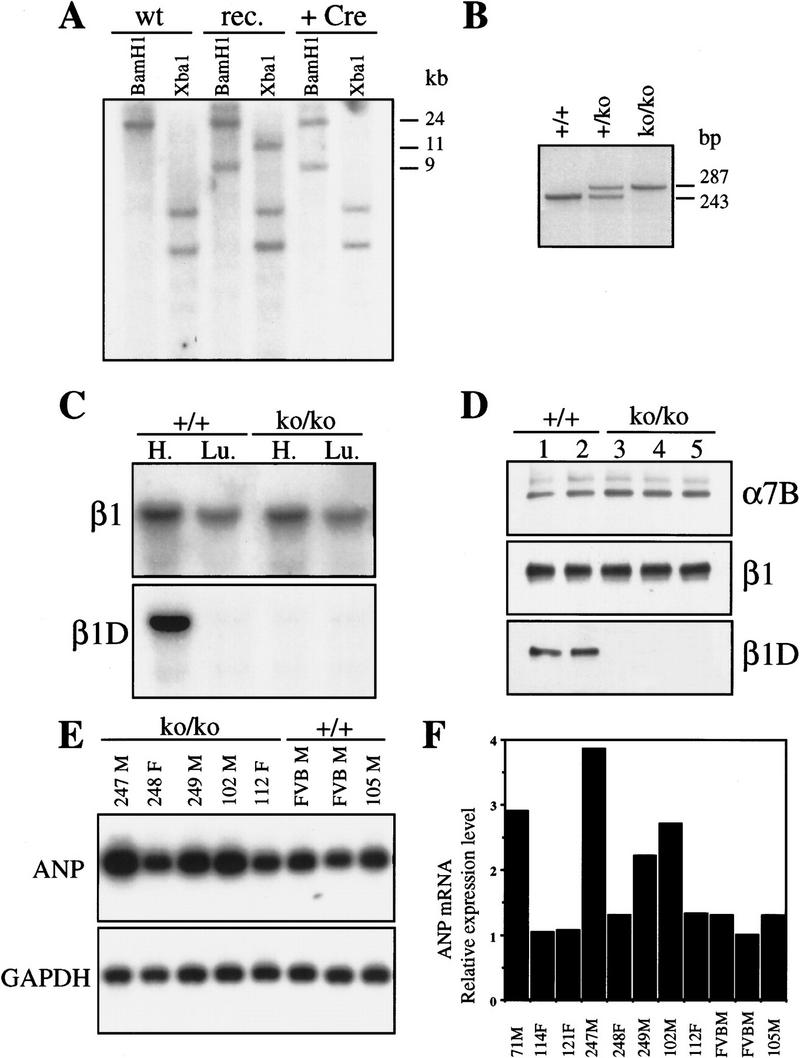
(A) Identification of homologous and Cre/loxP recombination by Southern blot analysis of DNA from targeted ES cell lines. A 24-kb fragment is present in BamHI digested DNA from wild-type ES cells (wt), a 9-kb mutated fragment is also present in DNA from homologous-targeted ES cell before (rec) and after (+Cre) the Cre/loxP mediated recombination. In XbaI-digested DNA, an 11-kb fragment is present in the DNA from homologously targeted ES cells, whereas the fragment disappeared after transfection with the Cre-encoding plasmid, showing that the Cre–loxP recombination had been successful. (B) PCR analysis of tail genomic DNA from +/+, +/ko, and ko/ko β1D mice. Primers flank exon D; the lower band corresponds to exon D and its consensus splicing sites, the upper band corresponds to the loxP site. (C) Northern blot analysis of mRNA derived from heart and lungs of +/+ and β1D ko/ko mice. The blot is successively probed with an antisense oligonucleotide encoding exon D (β1D) and exon 7 (β1). (D) Immunoblot analysis of proteins extracted from the heart of +/+ and ko/ko mice. The blot is successively probed with anti-β1D, β1, and α7B antibodies. (E) Northern blot analysis of RNA extracted from ventricles of +/+ and ko/ko mice. The blot is probed successively with an ANP and GAPDH cDNA probes. (F) Quantification by PhosphorImager analysis of the relative expression level of ANP mRNA in heart ventricles of +/+ and ko/ko mice. (F) Female; (M) male.
The muscles of the β1D knockout mice do not have detectable β1D mRNA or β1D protein
Northern blot analysis, performed with a β1 probe that detects both β1A and β1D transcripts revealed a single transcript of 3.5 kb in the heart and the lungs of the wild-type (+/+) and knockout/knockout (ko/ko) mice (Fig. 2C). PhosphorImager analysis indicated that the levels of β1 mRNA in ko/ko mice were comparable with those in +/+ mice. Subsequent hybridization with a β1D-specific oligonucleotide showed that the 3.5-kb β1D mRNA was present only in our +/+ mice (Fig. 2C). To verify that β1D protein was absent from these animals, we performed immunoblot analysis using the monoclonal antibody, 2B1, which is specific for β1D (van der Flier et al. 1997). As shown in Figure 2D, the antibody recognizes a band of 120 kD in the heart of the +/+ but not the ko/ko animals. Probing with a total anti-β1 antibody revealed that the amounts of β1 protein were similar in the +/+ and the ko/ko mice, suggesting that there was an increase in the expression of β1A in the heart of ko/ko mice (Fig. 2D), which was confirmed by probing with a β1A antibody (not shown).
There are no histological or ultrastructural alterations in muscles of β1D knockout mice
Despite a highly specific expression pattern of β1D in developing muscle (van der Flier et al. 1997), mice homozygous for the β1D–knockout allele were viable and fertile. They could not be distinguished from control animals by overall morphology and behavior. Mice were able to run, swim, or hold a weighted grid without any obvious signs of fatigue as compared with littermate control mice. Light microscopy studies on sections of muscles from the diaphragm, soleus, vastus lateralis, gastrocnemius, tibialis anterior, and heart muscles from β1D ko/ko mice up to 1 year of age, did not reveal any obvious morphological abnormalities characteristic for muscular dystrophies. Also, no necrosis or polynuclear infiltration, characteristic of fiber degeneration/regeneration cycles, were detected. In addition, the myocardium did not show any signs of hypo- or hypertrophy, and no calcium or collagen deposits were present as assessed by von Kossa and Masson trichrome staining. Finally, electron microscopical analysis did not reveal any abnormalities at the sites in which β1D is normally enriched; in skeletal muscles, myotendinous junctions were normally folded with digit-like extensions of the muscle cells into the collagen fibrils. In the heart, myofibrils were correctly organized with a normal lateral alignment, particularly at the intercalated discs (not shown).
To assess whether the distribution of some specific proteins might be affected as a consequence of the loss of β1D, we performed immunofluorescence microscopy on frozen sections of skeletal and cardiac muscle from +/+ and β1D ko/ko mice. Because in cardiac and skeletal muscles the α7A and α7B subunits are the major pairing partners for β1D (Belkin et al. 1996), we first checked for staining for these subunits using isoform specific antibodies on frozen sections of +/+ and β1D ko/ko mice. No differences were observed with respect to the distribution or the expression levels of the α7A and α7B subunits (Fig. 3G–J). Western blot and PhosphorImager analysis revealed that the quantity of α7B protein was the same in the hearts of the +/+ and the ko/ko mice (Fig. 2D), which suggests that there is no preferential association of the α7 subunit with β1A or β1D and that the stability of the α7β1D integrin is not different from that of α7β1A. Staining with anti-α3 and anti-α5 antibodies, which are known to be expressed in the connective tissue and blood vessels present in muscle tissue, also revealed no differences (not shown). Laminin 2 (merosin) is the major laminin isoform found in the extracellular matrix of skeletal and heart muscles and it is a ligand for the α7β1 integrin (Velling et al. 1996; Yao et al. 1996). Staining of muscles with an anti-α2 laminin subunit antibody again revealed no differences with respect to localization or the level of expression (Fig. 3E,F). Finally, we stained sections with an anti-dystrophin antibody to investigate whether the dystrophin–dystroglycan complex may compensate for the loss of β1D. No change in staining was observed in muscles of β1D ko/ko mice. Thus, we conclude that the absence of β1D does not influence the localization or the level of expression of these proteins and that β1D deletion does not result in obvious histological or ultrastructural abnormalities.
Figure 3.
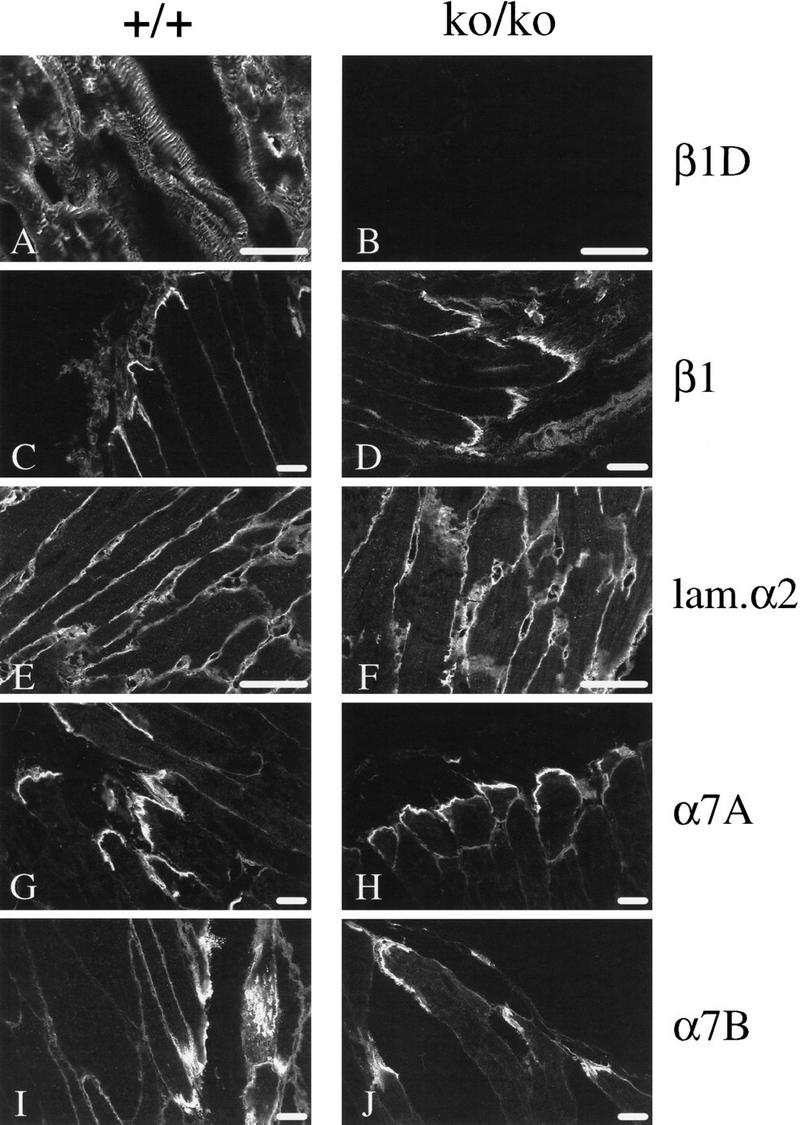
(A–J) Immunostaining of skeletal muscles from β1D +/+ and ko/ko mice. Skeletal muscles of 1-year-old mice were frozen in OCT compound, and sections were prepared and subjected to immunofluorescence histochemistry with various antibodies. The first section of each pair is from +/+ skeletal muscles; the second is from ko/ko skeletal muscles. (A,B) anti-β1D; (C,D) anti-β1; (E,F) anti-α2 laminin subunit; (G,H) anti-α7A integrin; (I,J) anti-α7B integrin. Scale bars, 25 μm.
β1D knockout mice display mild cardiac functional abnormalities
Because functional abnormalities may precede morphological changes, we analyzed our mice using markers of functional alteration in muscle. We focused on the function of the myocardium because the expression level of β1D is 5- to 10-fold higher in cardiac than in skeletal muscle (van der Flier et al. 1997). The amount of atrial natriuretic peptide (ANP) is a biological marker for the severity of heart dysfunction and predictive for patients’ survival. In ventricles of patients with left ventricular hypertrophy and a failing heart, or in rats with congenital cardiomyopathies, ANP synthesis is increased two- to sixfold as a haemodynamic functional compensatory mechanism (Brooks et al. 1988; Lee et al. 1988; Morita et al. 1990). We hybridized total RNA extracted from the ventricles of the ko/ko and +/+ mice with an ANP cDNA probe. The level of ANP mRNA was significantly increased (two to six times) in 100% (15/15) of the male ko/ko mice compared with controls, whereas no such increase was observed in the female ko/ko mice (0/8) (Fig. 2E–F). In nine wild-type littermates (six males, three females) no differences in the ANP mRNA levels were detected. In addition, we found that in two male β1D ko/ko mice (2/2) the level of the β myosin heavy chain (βMHC), the amount of which is also a marker of heart dysfunction (Izumo et al. 1987), was elevated by 25%, thus confirming a ventricular dysfunction and a compensatory response (Dr. G.S. Buttler-Browne, pers. comm.). As an additional control, the ANP probe was also hybridized with mRNA extracted from 10 α6A ko/ko mice (five males, five females). Although the α6A integrin subunit is expressed in the heart during development, its absence did not result in an increase of the ANP mRNA level (Dr. C. Gimond, pers. comm.). These results show that male β1D ko/ko mice have a mildly affected heart phenotype, as reflected by an increased expression of ANP and βMHC in the ventricles.
Generation of β1 exon D knockin mice: the complementary approach to the β1D knockout
Gene knockin is an approach in which a gene is replaced by another to test whether the product of the two genes are functionally equivalent. To check whether β1D can take over β1A, we replaced part of the gene specifically encoding β1A by a cDNA fragment encoding β1D. To produce β1D knockin/knockin (ki/ki) mice, we designed the TC8 targeting construct for the selective expression of the β1D subunit (Fig. 4A). ES cells were transfected with the targeting construct and selected for neomycin resistance. Homologous recombination was identified by Southern blot analysis of BamHI-digested genomic DNA on hybridization with the probe 331. Recombination can occur via the 5′ and 3′ arms of the construct resulting in a novel 9-kb band (type 1 targeting), or can occur via the 5′ arm and the region located between exon 7 of the neo gene resulting in a novel 14-kb band (type 2 targeting) (Fig. 5A). A high frequency (55%) of homologous recombination was achieved. Two independently targeted clones, 7A1 and 2B1, were injected into C57BL/6 blastocysts and chimeras were obtained. These chimeras successfully gave germ-line transmission for the mutated allele and produced heterozygous offspring when crossed with FVB and 129/OLA females.
Figure 4.
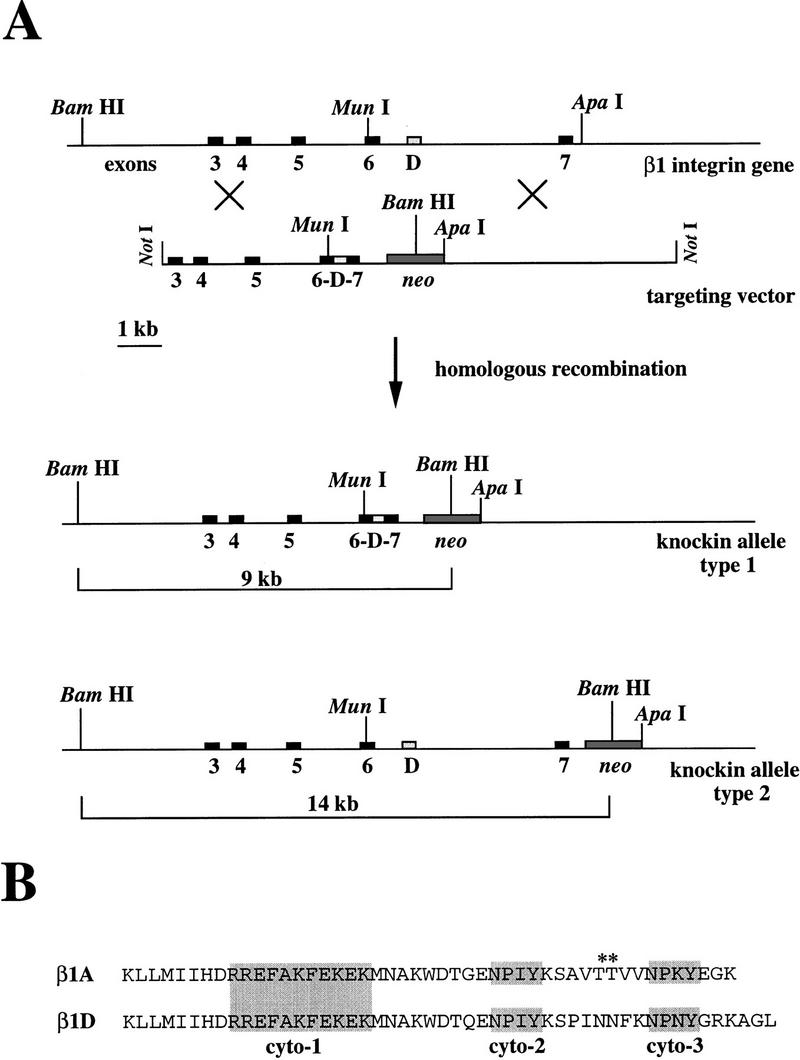
(A) Knockin targeting strategy. Restriction maps for the 3′ portion of the mouse β1 integrin locus, the targeting construct, and the type 1 and 2 homologous recombinants. Homologous recombination occurs between the 5′ and the 3′ end of the construct with the genomic DNA leading to the type 1 recombinant DNA or between the 3′ end and the connecting region between exon 7 and the neo gene with the genomic DNA leading to the type 2 recombinant DNA. After digestion with BamHI, the ES cell DNA is probed with 331, giving a wild-type 24-kb fragment and targeted 14- or 9-kb fragments. (B) The amino acid sequences of the β1A and β1D cytoplasmic domains differ after the KWDT sequence. The conserved cyto-1, cyto-2, and cyto-3, which are important for the localization of integrins in focal contacts, are shaded. Note that the two threonines (*) present in the β1A integrin are absent in the β1D integrin.
Figure 5.
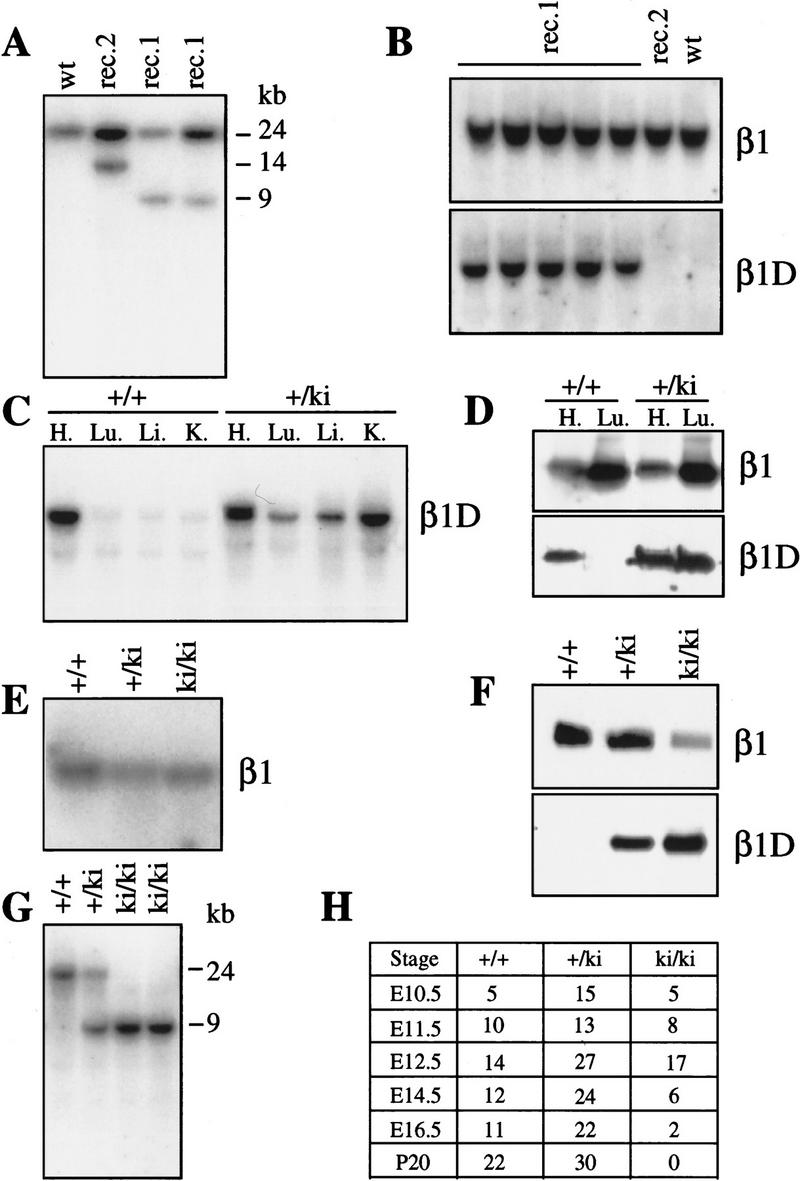
(A) Identification of type 1 and type 2 homologous recombination by Southern blot analysis of DNA from targeted ES cell lines. After digestion with BamHI, DNA is probed with 331. The 24-kb fragment is the wild-type allele (wt), the 9-kb and the 14-kb fragments are the type 1 (rec. 1) and type 2 (rec. 2) mutated alleles, respectively. (B) Northern blot analysis of RNA from total ES cells. The type 1-targeted ES cells (rec. 1) express β1D after homologous recombination. The blot was successively hybridized with an antisense oligonucleotide encoding for exon D (β1D) and exon 7 (β1). (C) Northern blot analysis of RNA from total tissues of heterozygous knockin mice. After knockin recombination, β1D is constitutively expressed in all the tissues analyzed, i.e., heart (H.), lungs (Lu.), liver (Li.), and kidneys (ki.). (D) Western blot analysis of proteins from total tissues of heterozygous knockin mice. Mice expressing β1D at the mRNA level (see Fig. 6C) express β1D at the protein level. (E) Northern blot analysis of total RNAs derived from knockin MEFs in culture. The wild-type +/+, heterozygous +/ki, and homozygous ki/ki β1D knockin MEFs express β1 mRNA at the same level. The blot was hybridized with probe 331, which recognizes all β1 isoforms. (F) Western blot analysis of proteins from a total lysate of MEFs. ki/ki MEFs express β1D. Note the higher expression level of β1 in the +/+ MEFs compared with the ki/ki MEFs. (G) Southern blot analysis of DNA derived from knockin embryos. In ki/ki embryos, only the type 1 mutated allele (9 kb) is present. The blot was hybridized with 331. (H) Genotypes of progeny from intercrosses between heterozygous β1D knockin parents.
β1D is ubiquitously expressed in targeted ES cells and in heterozygous β1D knockin mice
To assess whether the type 1 homologous recombination resulted in the expression of β1D mRNA, we performed Northern blot analysis of total RNA extracted from targeted ES cells and tissues from heterozygous knockin mice. RNA samples were probed with a radiolabeled antisense oligonucleotide specific for β1D. The 3.5-kb β1D mRNA was detected in the type 1 targeted ES cell clones (Fig. 5B) and in different tissues from heterozygous β1D knockin mice (Fig. 5C). In contrast, no signal was detected with mRNA from wild-type and type 2 targeted clones (Fig. 5B). To quantify the total amount of β1 mRNA, an antisense oligonucleotide for β1 (exon 7) was hybridized with the different blots (Fig. 5B). The level of the β1D mRNA, in type 1 targeted ES cells and in β1D +/ki mice, was 50% of that of β1 mRNA, because β1D, in type 1 targeted ES cells and β1D +/ki mice is expressed by only one allele. Finally, we performed Western blot analysis using the monoclonal antibody 2B1, specific for β1D, to investigate whether the β1D protein is expressed in β1D +/ki mice. As shown in Figure 5D, a 120-kD protein is detected in both heart and lung of heterozygous β1D knockin mice, demonstrating that the homologous recombination has led to the constitutive expression of β1D.
β1D homozygous knockin is embryonic lethal
Next, we investigated whether the exclusive expression of β1D had morphological consequences in heterozygous knockin adult mice. Anatomical and light microscope analysis of tissue sections of β1D +/ki mice showed no obvious signs of degeneration suggesting that β1D does not act as a dominant negative with regard to β1A function.
Strikingly, crossing heterozygous +/ki mice did not result in any live-born homozygous β1D knockin ki/ki pups. Therefore, we determined the developmental age at which the embryos were lost. Twenty percent of the embryos recovered at 10.5 dpc were genotyped as ki/ki, whereas at 16.5 dpc this was only 6% (Fig. 5H). Embryos were collected at various ages between 10.5 and 16.5 dpc, genotyped, analyzed for gross anatomical changes, and for histological and immunohistochemical analysis sections were prepared. At 10.5 dpc, ki/ki embryos were morphologically indistinguishable from +/+ and +/ki embryos (data not shown). At 11.5 dpc, however, about one-third of the ki/ki embryos were obviously abnormal, as shown in Figure 6 (B–I), compared with +/+ or +/ki (Fig. 6A) at the same age. The neural tube was open, and abnormally waved both rostrally and caudally (Fig. 6F,G), some embryos lacked part of the hindbrain (Fig. 6E) and in most embryos, the first branchial arch was shortened (Fig. 6, cf. D and E with control A). In several embryos, this left the tongue exposed (Fig. 6C), where it would normally have been enclosed by developing jaws. At 12.5 dpc, this pattern of abnormalities was maintained (data not shown). Conventional histology on transverse sections at 12.5 dpc showed that the open anterior neural tube in ki/ki embryos was accompanied by a gross expansion of the neuroepithelium (Fig. 7B), in a pattern different from that observed in +/ki embryos in which the neural tube was closed at that level (Fig. 7A). Because the overall phenotype bears resemblance to that described for fibronectin (FN) ko/ko embryos and because the migratory behavior of embryonic fibroblasts isolated from ki/ki embryos at this stage on FN substrates was altered (see below), we stained adjacent sections for FN (Fig. 7C,D) and its major receptor subunit, the α5 integrin subunit (Fig. 6I,J). In the +/+ (not shown) and +/ki embryos (Fig. 7C), FN antibodies stained a thin (cellular) layer surrounding the neural tube, the endothelial cells of blood vessels, a layer around the somites and weakly, mesenchymal cells under the neural tube. The neuroepithelium and somites themselves were negative. In contrast, the β1D ki/ki embryos lacked the thin layer of staining around the open neural tube (Fig. 7D), whereas more caudally in the same embryo, in which the tube was closed, staining appeared to be normal (data not shown). In the mesenchyme under the open neural tube, staining of FN was abnormally strong, whereas staining of FN was normal in the walls of the aorta, but in smaller vessels, levels appeared to be reduced (not shown). This may have resulted in weaker vessel walls and extravasation of red blood cells (RBCs) was evident in various tissues of the ki/ki embryos (Fig. 7H), whereas in contrast, in +/ki embryos, RBCs were only contained within vessels (Fig. 6K). RBCs were also found in the pericardial cavity of the ki/ki embryos (Fig. 7H), but not in the +/ki embryos (Fig. 7G) or +/+ embryos (data not shown). Where extravasation had occurred just under the skin, patches of RBCs were evident immediately on dissection of the ki/ki embryos from the uterus (Fig. 6B), whereas they were not seen in the +/ki (Fig. 7A) or control +/+ embryos (data not shown). The expression and distribution of the FN receptor, α5β1, was unaltered in the ki/ki embryos. The thin layer of α5 around the neural tube seemed to be completely intact (Fig. 7I,J). Other tissues of the ki/ki embryos at 12.5 dpc and compared with +/ki and +/+ tested in serial transverse and sagittal sections included the trigenimal ganglion (V), choroid plexus, optic nerve (II), diencephalon, fore- and hindbrain, Jacobson’s organ, tongue, jaws, ventricles and atria, pericardial cavity, septum and outflow tract, body wall, liver, gut, notochord, urogenital tract, spinal cord, dorsal root ganglia, mesonephros, metanephros, gonads, lung, vagus nerve (X), cartilage, somites, stomach, eye, facial ganglion (VII), and main veins and arteries. Of these, only the choroid plexus was markedly aberrant. Only a rudimentary choroid plexus was present in some sections of a complete series, cut saggitally through the head from the roof of the fourth ventricle (Fig. 7E,F). Its virtual absence correlates with the presence of an abnormally smooth head and small brain cavities, features that were also evident at later stages of development (16.5 dpc) in ki/ki embryos (Fig. 6H). At these late stages, only two embryos were recovered, both of which were abnormal (Fig. 6H, I). A striking feature was the lack of a lower jaw and a dysmorphic lower face, most likely caused by an underdevelopment of the first branchial arch. The heart of the embryos shown in Figure 6H was no longer beating when collected at 16.5 dpc, but development of the ear shows that it was viable until at least 15.5 dpc. Finally, in one embryo, digit formation was delayed in all limbs (Fig. 6I), compared with +/+ littermates (Fig. 6H; data not shown). These results clearly demonstrate that β1D cannot replace the function of β1A during development.
Figure 6.
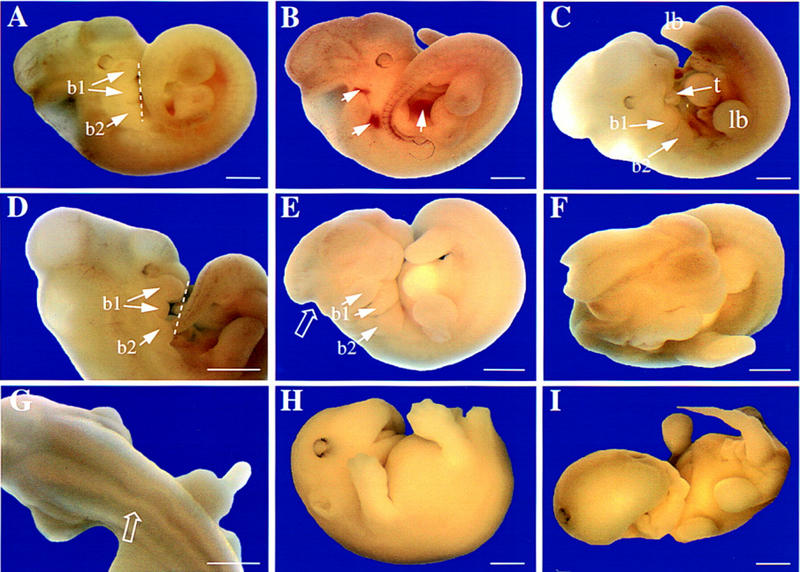
Phenotype of β1D ki/ki embryos between 11.5 and 16.5 dpc. Gross morphology is shown at 11.5 (A–G) and 16.5 dpc (H,I). (A) Control +/ki embryo at 11.5 dpc. The neural tube is closed rostrally and caudally; brain cavities are evident as convolutions in the head; the tail bends to the right, with developing limbs symmetrically positioned on either side. The broken line joins ventral extremities of first and second branchial arches (b1, b2). In b1, the maxillary (top) and mandibular (bottom) components are already separating. (B) β1D ki/ki embryo at 11.5 dpc showing extravasation of RBCs (arrows). (C) β1D ki/ki embryo at 11.5 dpc showing that the maxillary component of the first branchial arch (b1) is missing, leaving the tongue (t) exposed. The limbs (lb) are excentrically positioned with respect to the tail. (D) β1D ki/ki embryo at 11.5 dpc showing abnormal development of the first branchial arch (b1) with the mandibular component missing. The broken line joins ventral extremities of b1 and b2. (E) β1D ki/ki embryo at 11.5 dpc showing lack of part of the hindbrain (open arrow). The body is also abnormally twisted with lower limbs excentrically placed relative to the tail and branchial arches b1 and b2 are underdeveloped. (F) Anterior view of embryo in E showing open neural tube. (G) Dorsal view of a β1D ki/ki embryo at 11.5 dpc showing a kinked neural tube (open arrow). (H) β1D ki/ki embryo at 16.5 dpc. The shortened branchial arch has resulted in an underdeveloped lower jaw, so that the upper palate is visible. The head is abnormally smooth. (F) β1D ki/ki embryo at 16.5 dpc. Both jaws and the lower part of the face are underdeveloped and misformed. One eye has been displaced to the top of the head, probably by edema (arrow). All limbs have retarded digit development. Scale bars, 1.2 mm.
Figure 7.
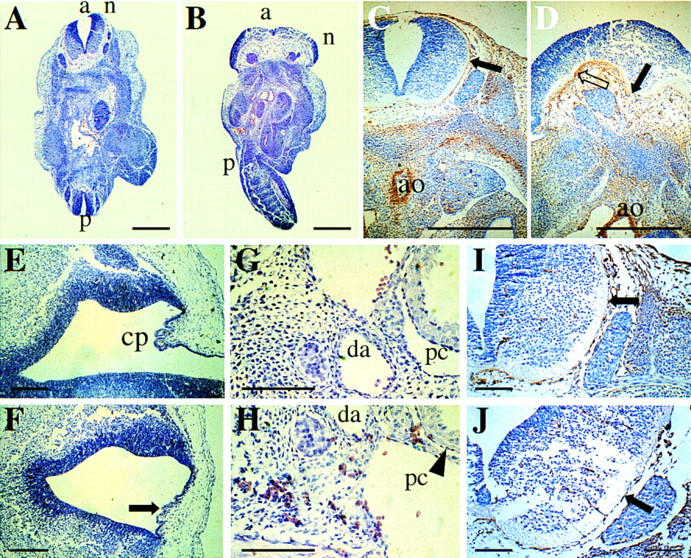
Histological sections of control and β1D ki/ki embryos stained with hematoxilin and eosin. (A) Transverse section of control +/ki embryo (12.5 dpc) at the level of the hind limbs. The embryo is curved so that the anterior (a) and posterior (p) neural tubes (n) are visible and closed. (B) Transverse section of β1D ki/ki embryo (12.5 dpc) at the level of the hind limbs. The neural tube (n) is open and the neuroepithelium overexpanded. Twisting of the body results in an abnormal a–p axis. (C) FN staining of a control +/ki embryo (12.5 dpc) (transverse section through the anterior neural tube). Note the thin layer around the neural tube, the low level of staining in the mesenchyme ventral to the neural tube (solid arrow) and the strong staining around the aorta (ao). (D) FN staining of a β1D ki/ki embryo (12.5 dpc) (transverse section through the anterior neural tube that had failed to close). Note the lack of a thin layer of FN around the neural tube (open arrow), excessive FN in the mesenchyme underlying the neuroepithelium (solid arrow), but the normal amounts of FN around the aorta (a). (E) Sagittal section through the head of a control +/ki embryo at 14.5 dpc showing the choroid plexus (cp) in the roof of the fourth ventricle. (F) Sagittal section through the head of a β1D ki/ki embryo at 14.5 dpc at the position that should contain the choroid plexus (arrow). The head of this embryo is smooth, as in Fig. 6H. (G) Sagittal section through a control +/ki embryo showing RBCs confined within blood vessels [(da) dorsal aorta]. (H) Sagittal section through a β1D ki/ki embryo showing RBCs dispersed throughout the tissue and in the pericardial cavity (pc). (I) α5 integrin staining of a control +/ki embryo at 12.5 dpc. Transverse section adjacent in the anterior neural tube. Note staining of the same thin layer around the neural tube as in C. (J) α5 integrin staining of a β1D ki/ki embryo at 12.5 dpc. Transverse section through the anterior neural tube that had failed to close. The thin layer around the neural tube is stained as in controls in which the tube is closed at the same level. Scale bars, 1 mm in A–D, 150 μm E–J.
Reduced migratory potential of β1D knockin embryonic cells
To assess whether the developmental defects that we observed in the ki/ki embryos might be the result of an altered migratory behavior of the ki/ki embryonic cells, we conducted migration and adhesion assays with cultured 12.5 dpc murine embryonic fibroblasts (MEFs) on different extracellular matrix proteins. The genotype of each 12.5 dpc embryo was tested by PCR and Southern blot analysis (Fig. 5G) and cells were subcultured three times to obtain a homogenous population of fibroblastic cells. First, we assessed the levels of expression at the cell surface of the β1, α1, α3, α5, and α6 integrin subunits and of the αvβ3 and αvβ6 integrins by immunoprecipitation (Fig. 8B) and FACS analysis (not shown). All of these subunits and integrins were expressed at the cell surface. Interestingly, we found that the expression level of β1 was reduced by 50% in the ki/ki MEFs compared to that in the +/+ MEFs (Figs. 8B and 5F) and that, consequently, the expression levels of the α5, α3, and α6 proteins at the cell surface were also reduced by 50%. However, we did not find any difference in the β1 mRNA level in the ki/ki MEFs compared with that in the +/+ MEFs (Fig. 5E). No difference was found for the αvβ3 and αvβ6 integrins.
Figure 8.


(A) Migratory (bars) and adhesive (lines) properties of +/+, +/ki, and ki/ki MEFs on collagen IV, laminin 1, vitronectin, and FN. By use of concentrations of FN that result in the same adhesion of +/+ (1.5 μg/ml) and ki/ki (5 μg/ml) MEFs (bottom right), migration is not restored to normal in the ki/ki MEFs. (Top right) Migration on collagen IV is completely blocked with an anti-β1 antibody. (Bottom left) Migration on FN is blocked completely by an anti-β1 antibody, whereas an anti-α6 antibody has no effect. (B) Immunoprecipitation analysis of various integrin subunits expressed at the surface of the +/+ and ki/ki MEFs. (Cont. Ab.) The secondary antibody used as a control.
On laminin 1, we found that both adhesion and migration of the ki/ki MEFs were greatly reduced (Fig. 8A). In the presence of an anti-α6 antibody (GoH3), migration was completely blocked on laminin 1 (not shown), indicating that in MEFs α6β1 is the only laminin-1 binding integrin. On type IV collagen, we found that migration was reduced, but that adhesion was not altered. An anti-β1 antibody completely blocked migration on collagen IV (Fig. 8A), whereas an anti-α2 antibody had no effect (not shown), which is consistent with α1β1 being the receptor for collagen IV. The level of the α1 integrin subunit was very low but similar on +/+ and ki/ki MEFs, which is in agreement with the fact that both types of MEFs adhered to a similar extent. Adhesion and migration, on FN, were also reduced in ki/ki MEFs (Fig. 8A). To exclude that the reduced adhesion of the ki/ki MEFs, the result of decreased level of β1-containing FN receptors, was responsible for their decreased migration, we performed migration assays using different concentrations of FN. With a concentration of 5 μg/ml, adhesion of ki/ki MEFs was comparable with that of +/+ MEFs at 1.5 μg/ml. In spite of that, migration of the ki/ki MEFs was still strongly reduced. An anti-β1 antibody completely blocked migration on FN, indicating that on MEFs β1 containing integrins are the major receptors for FN (Fig. 8A). Finally, we found that when plated on FN, ki/ki MEFs formed focal contacts that were similar in size and number to those formed by the +/+ MEFs (not shown). These results demonstrate that the ki/ki embryonic cells have a reduced migratory potential, which is probably caused by a reduction in the expression of the β1 containing integrins, in combination with a lower efficiency of β1D, as compared with β1A, in mediating migration.
Discussion
Exon-specific knockout and knockin as complementary tools to assess the function of a splice variant protein
Here, we describe the first exon-specific knockout mice to assess the function of a muscle-specific splice variant of β1, the β1D integrin subunit. Because of the numerous biological functions of β1, the conventional knockout of the β1 gene is embryonically lethal already during gastrulation (Fässler et al. 1995; Stephens et al. 1995), so that the function of the β1D integrin cannot be assessed in these mice. To overcome this problem we have applied a genetic approach on the basis of the Cre–loxP system (Gu et al. 1994; DiSanto et al. 1995; Smith et al. 1995) by which exon D can be specifically deleted. This procedure did not interfere with the germ-line transmission of the mutated gene because chimeric and heterozygous mice were readily obtained. Importantly, the replacement of exon D by a loxP site did not interfere with either the transcription of the β1 gene or the stability or the splicing of the β1 pre-mRNA, as shown by the normal expression of the β1 mRNA in knockout mice (Fig. 2C). Finally, translation of the mature RNA also appears to be normal because the total amount of the β1 proteins in muscles from knockout and control mice were similar (Fig. 2D).
In addition, we have also generated mice that express β1D, instead of β1A, by applying a knockin strategy. This method has been used previously to replace the mouse Engrailed gene En-1 by En-2 (Hanks et al. 1995). Here, we describe, for the first time, the knockin of an exon, replacing the normal mRNA by one encoding one of its splice variants. ES cells were transfected with a construct allowing the constitutive expression of β1D mRNA. Northern blot analysis of RNA demonstrated that the procedure did not affect the transcription of the β1 gene or the stability of the β1D mRNA, that is, the level of β1D in the ki/ki MEFs was the same as that of β1A in the +/+ MEFs (Fig. 5E). Furthermore, the knockin procedure did not interfere with the germ-line transmission of the ki gene because the ratio of heterozygous mice was as expected.
In principle, these two complementary techniques, exon-specific knockout and knockin, should allow the activation and inactivation of any exon in any gene. Thus, it is now possible to study the role of splice variants in a genetically modified animal.
What does the knockout tell us about the function of β1D in muscles?
Considering results of previous studies that indicate that β1 plays an important role in muscle cell differentiation (Fässler et al. 1996), the organization of sarcomeric cytoarchitecture (Volk et al. 1990) and myogenesis (Yang et al. 1996), we were surprised that in our study the lack of β1D did not affect muscle formation and did not cause muscular degeneration or myopathy. A likely explanation for this unexpected finding is that loss of the function of β1D is compensated by β1A. The β1D isoform is very homologous to β1A; the two variants differ only by 13 amino acids of their cytoplasmic tails (van der Flier et al. 1995; Zhidkova et al. 1995; Belkin et al. 1996). Thus, the phenotype observed in the ko/ko mice is only the result of a different function of the β1D-specific amino acids (Fig. 4B), rather than of the total β1 subunit.
In skeletal muscle, β1D is present at the myotendinous junction and at the costameres. These structures are the major junctions through which the skeletal muscle fiber is connected with the extracellular matrix and the tendon. In the heart, β1D is found at the intercalated discs that are the structure units for cell–cell contact of the cardiomyocyte (Belkin et al. 1996). In skeletal muscles, β1D is associated with the α7A and α7B subunits, whereas in heart it is only associated with α7B. Recent work has shown that the cytoplasmic domain of the βPS subunit, which is the β1 integrin-like subunit in Drosophila, is responsible and sufficient for targeting integrins to the termini of muscles in vivo (Martin-Bermudo and Brown 1996). From these observations, it seems that the cytoplasmic domain of β1D plays a role in the localization of integrins at these sites. However, in the β1D ko/ko mice β1A is localized at the same position as β1D in normal muscle, that is, intercalated discs, myotendinous junctions, and costameres, which excludes a specific role for β1D in targeting integrins to these sites. Furthermore, the data suggest that the association of β1A with sarcomeric protein and the actin network is normal and that the role of β1D is not different from that of β1A in the maintenance and the organization of the cytoarchitecture of the mature muscle cells. Finally, β1D ko/ko mice form normal mature muscles, excluding a specific role for β1D in myogenesis. Because β1D is highly enriched at the myotendinous junctions and in intercalated discs, it has been proposed that β1D provides stronger attachment at these sites than β1A (van der Flier et al. 1995; Belkin et al. 1996). Our data do not support this assumption because there are no histological or ultrastructural abnormalities in skeletal and cardiac muscles from ko/ko mice. We also did not observe an increased expression of the dystrophin/dystroglycan complex, which would be a possible compensatory response to a weakening of the attachment of the muscle to the extracellular matrix. Finally, the localization of the α2 laminin subunit appeared to be normal.
What could be the function of the β1D integrin? The production of isoforms by developmentally regulated alternative splicing of pre-mRNAs is a widespread phenomenon that represents an evolutionary strategy aimed at a subtle functional diversification. Consequently, defining the function of a splice variant protein might be more difficult than of a protein in a total gene knockout. Hence, to identify subtle defects, we analyzed our ko/ko mice further for functional alterations of the myocardium, because β1D is 5–10 times more strongly expressed in the heart than in skeletal muscles (van der Flier et al. 1995). In addition, the myocardium is subject to continuous mechanical stress. The amount of ANP is a reliable biological marker for the severity of functional and haemodynamic heart failure. ANP is a 28-amino-acid polypeptide hormone secreted by the heart in response to mechanical and neuroendocrine stimuli (Jarygin et al. 1994; Nakao et al. 1996). In early heart failure, ANP synthesis is increased in the ventricles in which it may play a key role in preserving a compensated state of asymptomatic heart dysfunction (Brandt et al. 1993). In humans and hamsters with congestive heart failure and in rats with aortic valve insufficiency, ANP mRNA levels are increased three- to fivefold, respectively, in the ventricles (Brooks et al. 1988; Morita et al. 1990). In addition, ANP mRNA is also increased sixfold in ventricles of rats with spontaneous biventricular hypertrophy (Lee et al. 1988). The finding that in our ko/ko male mice the ANP mRNA and the βMHC protein levels are increased up to sixfold and by 25%, respectively, is consistent with a ventricular dysfunction despite the lack of any histological abnormalities or hypertrophic responses.
Why does the absence of β1D lead to an increased synthesis of ANP? In the heart, ANP is secreted in response to stretch induced by blood pressure that is detected by mainly two factors, that is, pressure-sensitive channel proteins (Erdos et al. 1991), and most likely integrins, which may function as mechanotransducers (Nakao et al. 1996; Shyy and Chien 1997). In light of our results, we speculate that β1D acts as a mechanotransducer with a reduced sensitivity to the shear force as compared with β1A. We suggest that on stretching of muscle cells, the signals elicited by β1D and β1A are different. Support for this assumption is derived from the observation that in cultured cells β1A and β1D transmit different signals after crosslinking with antibodies (Belkin et al. 1996). Thus, it is conceivable that in our β1D ko/ko mice, the β1A-mediated reactivity to blood pressure is perturbed, inducing increased synthesis of ANP. This increase in ANP synthesis seems to be sex linked because it was observed in 100% of the male and 0% of the female mice. The reason for this is unknown, but we suspect that male mice have a slightly higher blood pressure than female mice. In addition, studies have reported that female rats are more protected against muscle damage following exercise than male rats. This protective effect is probably the result of female sex hormones, particularly estrogens (Tiidus et al. 1995).
What does the knockin tell us about the functions of β1A in development?
The most striking of the defects that we observed was the shortening of the first branchial arches that resulted in an underdeveloped lower mandible (micrognathia) and an open neural tube. Defective closures, both in the brain (anencephaly) and in the lower spine (myelomeningcele), were evident and, in some cases, were accompanied by an unusual kinking of the back and tail, reminiscent of that described in FN ko/ko and α5 ko/ko embryos at 8.5 dpc (George et al. 1993; Yang et al. 1993). FN, which normally surrounds the neural tube, was absent in the region of spina bifida, which suggests that its absence is related to this defect. Poor elongation of the embryonic axis in the β1D ki/ki embryos accompanied by normal neural tube elongation, however, is a more likely cause of its kinking, as has also been suggested in FN ko/ko embryos. Unexpectedly, the α5 integrin subunit, which we had assumed would bind FN to cells surrounding the neural tube, was normally distributed in the mutant embryos. It is conceivable that α5β1D transmits different signals or interacts with other cystokeletal proteins than α5β1A and, thus, affects the assembly of FN in ki/ki embryos. This hypothesis is supported by the work of Wu et al. (1995) who have shown that integrin postoccupancy events, requiring the cytoplasmic domain of the β chain and an intact cytoskeleton are needed for FN fibrillogenesis. Although other FN-binding integrins may partially compensate for α5β1-induced matrix assembly during development (Yang and Hynes 1996), their ability to do so is less efficient as compared with that of α5β1, as was demonstrated with ES cells (Wennerberg et al. 1996), which explains the altered distribution of FN in our ki/ki embryos. Other striking defects that we observed were an abnormally smooth head with poorly expanded brain cavities, in some cases accompanied by a virtual lack of choroid plexus in the roof of the fourth ventricle. We suspect that an altered migratory potential of neuroepithelial cells may underly these alterations. It is also tempting to speculate that the migratory capacity of the neural crest cells, the major migratory cell population at these mid-gestational stages, is also reduced, because they are an important component of the first branchial arch, in which we observe a defect, and because these cells are thought to be guided to their destination by various extracellular matrix (ECM) molecules, including FN (Morrison-Graham et al. 1992). This hypothesis is supported by the reduced capacity of ki/ki MEFs to migrate on FN. In the mutants, the abnormal distribution of FN may actually represent a barrier to migrating cells. On the other hand, there are other several possible reasons for the observed branchial arch defects. First, there may be a defect in the formation of neural crest cells in the rhombomeres r2 and r3 of the hindbrain where they originate. Second, the paraxial mesoderm of the somites contributes to the outer layer of the first branchial arch and FN normally surrounds developing somites in a thin layer and appears to be essential for their formation (George et al. 1993). Although somites were clearly present at 11.5–16.5 dpc in ki/ki embryos, their rate of formation or their size may have been affected by the altered distribution of FN in ki/ki embryos. Third, there may have been altered apoptosis on pathways followed by migrating neural crest cells, as described for PDGF receptor ko/ko embryos, with altered cell attachment compromising cell survival (Soriano 1997). Cell attachment-induced signaling, via integrins, seems to be a determinant for cell survival and growth (Chen et al. 1997; Frisch and Ruoslathi 1997; Merredith and Schwartz 1997). Fourth, β1D might be less efficient than β1A with regard to signaling induced by mechanical force; neural cell crest migration, which is mediated by cell–ECM interactions requires transmission of mechanical forces (Harris et al. 1980). The severe defects described above are unlikely to be lethal for the ki/ki embryos. Of the 13 ki/ki embryos recovered at 12.5 dpc, 5 were, however, severely anemic. In viable ki/ki embryos recovered at other stages, there were RBCs outside the embryonic vasculature, randomly dispersed throughout various tissues and organs. This again resembles, to some extent, defects in the FN ko/ko mice (Georges et al. 1993), although the vasculature of the yolk sac had a normal appearance in the ki/ki mice. Because FN was only expressed at low levels in the smaller embryonic vessels, it is possible that these vessels may be weak and may rupture easily, which would lead to hemorrhage and death.
What do the knockin and knockout tell us about the function of β1D vs. β1A?
We have demonstrated that the splice variants A and D of β1 are not functionally equivalent. From findings in our knockout mice, it is clear that β1A does not completely compensate for β1D because its replacement by β1A results in mild heart dysfunction, suggesting that β1D has some subtle function that is lacking in β1A. Therefore, it would be particularly interesting to look for genetic mutations in β1D in patients with heart dysfunction of unknown etiology, because a disturbance of the balance between the two isoforms, β1A and β1D, may be causally related to the dysfunction.
In contrast, in nonmuscular tissues, β1D appeared to lack some of the functions of β1A. The lethality of the ki/ki embryos clearly demonstrates that β1A cannot be substituted by β1D. Reduced migratory activity of the ki/ki embryonic cells may be an important factor in contributing to the embryonic phenotype, because fibroblasts isolated from mid-gestation embryos showed striking behavioral changes on different ECM proteins.
What might be the cause of the reduced migratory potential? First, we have observed that ki/ki MEFs have a 50% reduction in the level of the β1 subunit, which is associated with a reduced level of the α3, α5, and α6 subunits. The reason for this reduced level of the β1D protein remains unclear because the amounts of β1A and β1D mRNA are similar in +/+ and ki/ki MEFs. It is possible that, when associated with another α subunit than the muscle-specific α7 subunit, the β1D protein is less stable. Second, β1D may be less efficient than β1A in mediating migration, which is supported by the observation that even when we used concentrations of FN which gave +/+ and ki/ki MEFs the same adhesive capacity, the migratory activity of the ki/ki MEFs was not restored to control levels (Fig. 7). In addition, we found that on collagen IV, the migration rate of the ki/ki MEFs was greatly reduced without a parallel reduction in adhesion. Our observations are supported by the recent demonstration that increasing adhesion proportionally increases the migration rate of cells (Palecek et al. 1997). In conclusion, the reduced migratory potential of the embryonic cells is likely caused by a combination of reduced adhesion and a reduced migrating capacity, although we suspect that the latter is mainly responsible for the embryonic defects that we observed in the ki/ki embryos. This is supported by the observation that despite a 50% reduction in the expression level of the β1, α5, and α6 subunits in the β1 +/ko ES cells, the β1 +/ko embryos developed normally (Fässler et al. 1995). Recently, it has been suggested that β1A acts as a mechanotransducer in cultured cells (Wang et al. 1993) and in tissues (Shyy and Chien 1997). On the basis of our findings and the results of these previous studies, we suggest that β1D acts as a mechanotransducer in muscle but with a lower efficiency than β1A. To some extent, migration may also be considered as a mechanotransduction process (Ingber et al. 1994). In cell migration, a cell spreads and thereby induces stretching and stiffening of the cytoskeleton (Lauffenburger and Horwitz 1996). If β1D is less efficient than β1A in mediating stress-induced signaling, then this might explain the reduced migratory capacity of the ki/ki embryonic cells. In addition to mechanical force-induced signaling, other types of extracellular signals might be less efficiently transduced by β1D in the knockin embryonic cells. The loss of a potential site for phosphorylation and ILK binding in β1D (Hannigan et al. 1996) could explain such a partially defective signaling pathway. Thus, our data suggest that probably because of a difference in their cytoplasmic domains, β1D, generally, transduces signals less efficiently than β1A.
Materials and Methods
Generation of β1D knockout mice
Genomic clones for the generation of the knockout and knockin targeting constructs were isolated from a 129/OLA genomic library (Stratagene), by use of a mouse β1 integrin 3′-end cDNA probe (probe 331). Three clones encoding the most downstream sequences of the β1 gene (λ5, λ7, and λ8) were subcloned and analyzed by restriction enzyme mapping according to standard procedures (Sambrook et al. 1989). To construct the knockout targeting vector, the δ8 clone was used. BamHI sites were introduced by PCR mutagenesis on either side and in the close vicinity of exon D. The DNA was digested with BamHI and exon D with its ag/gt consensus splicing sites removed and replaced by a neo–tk cassette flanked by two loxP sites (a kind gift from Dr. R. Fässler, Max-Planck Institute, Munich, Germany). E14 ES cells were grown on mitotically inactivated MEF and electroporated with 80 μg of linear targeting insert. After 8–10 days in neomycin-supplemented ES cell medium (200 μg/ml G418), colonies were isolated, expanded, and screened by Southern blot. To delete the selection markers, 2 × 106 targeted ES cells were retransfected with 1.5 μg of supercoiled Cre-encoding plasmid DNA by electroporation. The transfected cells were plated on MEFs for 48 hr and then replated at a density of 2 × 103 cells/cm2. On day 3 after replating, 2 μm Gancyclovir was included in the medium, and selection was continued for 5 days. Colonies surviving the selection were picked at days 8–10, and the deletion event was verified by PCR and Southern blot analysis. The following primers were used for the PCR: a 5′ primer 5′-cagtttatagtgtcaatacac-3′ located upstream of the exon D. ES cell clones carrying the exon D deletion were injected into C57BL/6 blastocysts, and the male chimeric mice were crossed with FVB and 129/OLA females to generate heterozygous mutant mice. Mice were genotyped by PCR. DNA from tail biopsies were isolated by proteinase K digestion followed by isopropanol precipitation.
Generation of β1D knockin mice
To construct the TC8 knockin targeting vector, we used the genomic clone δ8 (see above). A β1D-encoding cDNA fragment was generated by PCR by use of a β1D cDNA clone as a template (Baudoin et al. 1996). This fragment encompasses exons 6, D, and 7 of the β1D cDNA (Tominaga 1988) and contains unique MunI and ApaI restriction sites located, respectively, in exons 6 and 7 (Baudoin et al. 1996). The δ8 clone DNA was digested with MunI and ApaI. After digestion, the genomic region, containing exons 6, D, 7, and their intronic sequences, was removed and the δ8 DNA ligated to the MunI–ApaI β1D-encoding cDNA fragment. This replacement results in a constitutive expression for the β1D subunit. For positive selection, a neomycin cassette was introduced into the construct by use of an ApaI site located between the two polyadenylation signals of the β1 integrin gene. Electroporation, selection with G418, screening of the neomycin-resistant ES cell colonies, and blastocysts injection were done with the same protocol followed for the knockout experiment.
Southern blot, Northern blot, and Western blot analysis
For Southern blot analysis, ES cells and mouse DNA were digested with the BamHI or XbaI enzymes and the blots were hybridized with the 331 probe according to standard procedures (Sambrook et al. 1989). For Northern blot analysis of β1D expression, ES cells and mouse total RNA (10 μg) were hybridized to an 32P-labeled antisense oligonucleotide (5′-gagaccagctttacgtccatagttgggattcttgaaatt-3′) specific for exon D. The blot was hybridized and washed by standard conditions and autoradiographed overnight. For Northern blot analysis of ANP expression, a cDNA probe was generated by RT–PCR amplification by use of the following primers deduced from the ANP cDNA sequence (Seidman et al. 1984): upstream primer 5′-ccaggccatattggagcaaa-3′ and downstream primer 5′-gaagctgttgcagcctagtc-3′. For Western blot analysis, various adult mouse tissues were homogenized in RIPA buffer and spun down. Proteins from the supernatant (40 μg) were electrophoresed on 8% SDS-PAGE (Laemmli 1970) and transferred onto Immobilon membranes. Blots were blocked with 2% milk powder in TBST for 1 hr. Blots were then incubated with primary antibodies and secondary peroxydase-conjugated antibodies, washed and developed by use of ECL reagents (Amersham).
Immunohistochemistry and histological analysis
For histology, mouse tissues or embryos were fixed in formalin, paraffin embedded, sectioned, and stained with hematoxylin and eosin according to standard procedures (Smith and Bruton 1978). For immunohistochemistry of mouse adult tissues, 5-μm cryosections were cut at −20°C and collected on silane-coated slides and air dried. Immunohistochemistry was carried out by standard protocols. FN and α5 integrin were stained on fixed paraffin sections. Primary antibodies used were rat anti-mouse-β1 (MB1.2) (von Ballestrem et al. 1996), rat anti-β1 (9EG7) (Lenter et al. 1993), rabbit anti-β1Acyto and rabbit anti-α5 (DeFilippi et al. 1991), rabbit anti-αv (Hirsch et al. 1994), mouse anti-α3A (29A3), mouse anti-α3B (54B3) (De Melker et al. 1997), rat anti-mouse α7 (CA5) (Yao et al. 1996), rabbit anti-α7Acyto (a7CDA2), rabbit anti-α7Bcyto (a7CDB1) (Martin et al. 1996), mouse anti-β1D (2B1) (van der Flier et al. 1997), mouse anti-dystrophin (Mandra 1, Sigma, ref. D-8043), and goat anti-fibronectin (Sigma, ref. F-1509).
Analysis of knockin embryos
Embryos were dissected as described previously (Cockroft 1990). Embryos were fixed in 4% paraformaldehyde for 4–6 hr at 4°C, dehydrated with ethanol, cleared in butanol, and embedded in paraffin. Sections (10 μm) were stained with hematoxylin and eosin, examined, and photographed. Only defects found in at least five embryos were described. Genomic DNA was extracted from the yolk sacs and genotyped by PCR or Southern blot analysis.
Culture of knockin MEFs
To culture embryonic cells, the whole E12.5 embryo was washed twice in DMEM and dissociated with sterile tweezers. The minced tissues from single embryos were trypsinized for 5 min at 37°C and the suspension of each embryo plated in a T75 flask with 25 ml of DMEM supplemented with 10% FCS. Cells were subcultured three times to obtain a homogenous population of fibroblastic cells. The genotype of each embryonic cell culture was tested by PCR and Southern blot analysis.
Adhesion and chemotactic migration assays
Chemotactic cell migration was performed as described (Grotendorst 1984) in a Boyden chamber-type with polycarbonate filters (8-μm pores; Transwell, Costar) coated on both sides with different ECM proteins. MEFs were trypsinized, washed once with DMEM containing 0.1% BSA, resuspended in the same medium at the density of 5 × 105 cells/ml, and introduced in the upper compartment of the chamber (100 μl). The lower compartment contained 500 μl of DMEM supplemented with 25 ng/ml PDGF. After 1, 2, or 3 hr at 37°C cells that had migrated on the lower side were fixed, stained with crystal violet, and eight microscopic fields per filter counted. For adhesion assays, the attachment of MEFs to ECM proteins followed procedures described previously (Aumailley et al. 1989). Tissue culture 96-well plastic plates were coated with ligands at 4°C overnight. After blocking with 1% BSA, 5 × 104 MEFs resuspended in 100 μl of DMEM was added to each well and incubated for 20–40 min, depending on the ligand, at 37°C. Wells were washed with PBS, and the attached cells were fixed with 1% glutaraldehyde, stained with 0.1% crystal violet for 30 min, and washed extensively with water. Cell-bound stain was measured at 550 nm in an ELISA reader after solubilization with 0.2% Triton X-100. ECM proteins were human vitronectin (Preissner et al. 1985), pepsinized human collagen type IV (Vandenberg et al. 1991), mouse laminin 1 (Paulsson et al. 1985), and human plasma FN (Sigma Chemical).
Acknowledgments
We thank R. van der Valk, M. van Rooijen, and J. Korving for histological analysis of the mice; L. Oomen for confocal laser scanning microscopy; D. Kramer for FACS analysis; D. Prins for PCR analysis of mice DNA; F. Veerbeek with embryo illustrations; and A. van der Flier for generating and providing us with the 2B1 antibody. We also thank S.J. Kaufman for the α7A and α7B antisera; G. Tarone for the β1A, α5, and αv antisera; B.M.C. Chan and D. Westerber for the rat monoclonal antibodies to murine β1, MB1.2, and 9EG7, respectively; R. Kramer for the rat monoclonal antibody to murine α7; K. Preissner for human vitronectin; K. Kühn for human collagen IV; and R. Timpl for mouse laminin. We are grateful to J.L. Samuel for her help and advice; R. Fässler for providing us with the neo–tk cassette and many useful suggestions; and L. Borradori, R. van der Neut, E. Roos, C.P.E. Engelfriet, and the several colleagues in our laboratory for critical comments on the manuscript. Finally, special thanks go to C. Gimond for her contribution to the adhesion assays. All of the ES cell work was carried out in the ES cell laboratory of the Netherlands Cancer Institute (Prof. A. Berns). This work was supported by a Training and Mobility of Researchers Marie Curie Research training grant from the European Commission (ERBFMBICT961823 to C.B.) and a grant (NHS 96.006) from the Netherlands Heart Foundation (to A.S.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL asonn@nki.nl; FAX (31) 20 512 1944.
References
- Almeida EAC, Huovila APJ, Sutherland AE, Stephens LE, Calarco PG, Shaw LM, Mercurio AM, Sonnenberg A, Primakoff P, Myles DG, White JM. Mouse egg integrin α6β1 functions as a sperm receptor. Cell. 1995;81:1095–1104. doi: 10.1016/s0092-8674(05)80014-5. [DOI] [PubMed] [Google Scholar]
- Altruda F, Cervella P, Tarone G, Botta C, Balzac F, Stefanuto G, Silengo L. A human integrin β1 subunit with a unique cytoplasmic domain generated by alternative mRNA processing. Gene. 1990;95:261–266. doi: 10.1016/0378-1119(90)90369-3. [DOI] [PubMed] [Google Scholar]
- Aumailley M, Mann K, von der Mark H, Timpl R. Cell attachment properties of collagen type IV and Arg-Gly-Asp dependent binding to its α2(VI) and α3(VI) chains. Exp Cell Res. 1989;181:463–474. doi: 10.1016/0014-4827(89)90103-1. [DOI] [PubMed] [Google Scholar]
- Baudoin C, Van der Flier A, Borradori L, Sonnenberg A. Genomic organization of the mouse β1 gene: Conservation of the β1D but not of the β1B and β1C integrin splice variants. Cell Adhes Commun. 1996;4:1–11. doi: 10.3109/15419069609010759. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Zhidkova NI, Balzac F, Altruda F, Tomatis D, Maier A, Tarone G, Koteliansky VE, Burridge K. β1D integrin displaces the β1A isoform in striated muscles: Localization at junctional structures and signaling potential in nonmuscle cells. J Cell Biol. 1996;132:211–226. doi: 10.1083/jcb.132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt RR, Wright RS, Redfield MM, Burnett JC. Atrial natriuretic peptide in heart failure. J Am Coll Cardiol. 1993;22:86A–92A. doi: 10.1016/0735-1097(93)90468-g. [DOI] [PubMed] [Google Scholar]
- Brooks SE, Douglas MA, Lee ME, Reeder GS, Wold LE, Burnett JC. Identification of atrial natriuretic factor within ventricular tissue in hamsters and humans with congestive heart failure. J Clin Invest. 1988;81:82–86. doi: 10.1172/JCI113314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Cockroft DL. Dissection and culture of postimplantation embryos. In: Copp AJ, Cockroft DL, editors. Postimplantation mammalian embryos, a practical approach. Oxford, UK: Oxford University Press; 1990. pp. 15–40. [Google Scholar]
- DeFilippi P, Van Hinsbergh V, Bertolotto A, Rossino P, Silengo L, Tarone G. Differential distribution and modulation of expression of α1β1 integrin on human endothelial cells. J Cell Biol. 1991;114:855–863. doi: 10.1083/jcb.114.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Melker AA, Sterk LMT, Delwel GO, Fles DLA, Daams H, Weening JJ, Sonnenberg A. The A and B variants of the α3 integrin subunit: Tissue distribution and functional characterization. Lab Invest. 1997;76:547–563. [PubMed] [Google Scholar]
- DiSanto JP, Muller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with targeted deletion of the interleukin 2 receptor γ chain. Proc Natl Acad Sci. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdos T, Butler-Browne G, Rappaport L. Mechanogenetic regulation of transcription. Biochimie. 1991;73:1219–1231. doi: 10.1016/0300-9084(91)90007-n. [DOI] [PubMed] [Google Scholar]
- Fässler R, Pfaff M, Murphy J, Noegel AA, Johansson S, Timpl R, Albrecht R. Lack of β1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J Cell Biol. 1995;128:979–988. doi: 10.1083/jcb.128.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fässler R, Rohwedel J, Maltsev V, Bloch W, Lentini S, Guan K, Gullberg D, Hescheler J, Addicks K, Wobus AM. Differentiation and integrity of cardiac muscle cells are impaired in the absence of β1 integrin. J Cell Sci. 1996;109:2989–2999. doi: 10.1242/jcs.109.13.2989. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Ruoslathi E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Galileo DS, Majors J, Horwitz AF, Sanes JR. Retrovirally introduced antisense integrin RNA inhibits neuroblast migration in vivo. Neuron. 1992;9:1117–1131. doi: 10.1016/0896-6273(92)90070-t. [DOI] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Gordon R, Brodland GW. The cytoskeletal mechanics of brain morphogenesis: Cell state splitters cause primary neural induction. Cell Biophys. 1987;11:177–238. doi: 10.1007/BF02797122. [DOI] [PubMed] [Google Scholar]
- Grotendorst GR. Alteration of the chemotactic response of NIH/3T3 cells to PDGF by growth factors, transformation, and tumor promotors. Cell. 1984;36:279–285. doi: 10.1016/0092-8674(84)90221-6. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Hanks M, Wurst W, Anson-Cartwright LA, Auerbach AB, Joyner AL. Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. Science. 1995;269:679–682. doi: 10.1126/science.7624797. [DOI] [PubMed] [Google Scholar]
- Hannigan GE, Leunghagesteijn C, Fitzgibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new β1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Harris AK, Wild P, Stopack D. Silicone rubber substrata: A new wrinkle in the study of cell locomotion. Science. 1980;208:177–180. doi: 10.1126/science.6987736. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Gullberg D, Balzac F, Altruda F, Silengo L, Tarone G. αv integrin subunit is predominantly located in nervous tissue and skeletal muscle during mouse development. Dev Dyn. 1994;201:108–120. doi: 10.1002/aja.1002010203. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ingber DE, Dike L, Hansen L, Karp S, Liley H, Maniotis A, McNamee H, Mooney D, Plopper G, Sims J, Wang N. Cellular tensegrity: Exploring how mechanical changes in the cytoskeleton regulate cell growth, migration, and tissue pattern during morphogenesis. Int Rev Cytol. 1994;150:173–225. doi: 10.1016/s0074-7696(08)61542-9. [DOI] [PubMed] [Google Scholar]
- Izumo S, Lompre AM, Matsuoka R, Koren G, Schwartz K, Nadal-Ginard B, Mahdavi V. Myosin heavy chain mRNA and protein isoform transitions during cardiac hypertrophy. J Clin Invest. 1987;79:970–977. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffredo T, Horwitz AF, Buck CA, Rong PM, Dieterlen-Lievre F. Myoblast migration specially inhibited in the chick embryo by grafted CSAT hybridoma cells secreting an anti-integrin antibody. Development. 1988;103:431–446. doi: 10.1242/dev.103.3.431. [DOI] [PubMed] [Google Scholar]
- Jarygin C, Hanze J, Lang RE. Gene expression of atrial natriuretic peptide in rat papillary muscle. Rapid induction by mechanical loading. FEBS Lett. 1994;364:185–188. doi: 10.1016/0014-5793(94)00464-1. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:23439–23442. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Languino LR, Ruoslahti E. An alternative form of the integrin β1 subunit with a variant cytoplasmic domain. J Biol Chem. 1992;267:7116–7120. [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee RT, Bloch KD, Pfeffer MA, Neer EJ, Seidman CE. Atrial natriuretic factor gene expression in ventricles of rats with spontaneous biventricular hypertrophy. J Clin Invest. 1988;81:431–434. doi: 10.1172/JCI113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenter M, Uhlig H, Hamann A, Jenö P, Imhof B, Vestweber D. A monoclonal antibody against an activation epitope on mouse integrin chain β1 blocks adhesion of lymphocytes to the endothelial integrin α6β1. Proc Natl Acad Sci. 1993;90:9051–9055. doi: 10.1073/pnas.90.19.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Kaufman SJ, Kramer RH, Sanes JR. Synaptic integrins in developing, adult, and mutant muscle: Selective association of α1, α7A, and α7B integrins with the neuromuscular junction. Dev Biol. 1996;174:125–139. doi: 10.1006/dbio.1996.0057. [DOI] [PubMed] [Google Scholar]
- Martin-Bermudo MD, Brown NH. Intracellular signals direct integrin localization to sites of function in embryonic cells. J Cell Biol. 1996;134:217–226. doi: 10.1083/jcb.134.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer U, Saher G, Fässler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Pöschl E, von der Mark K. Absence of integrin α7 causes a novel form of muscular dystrophy. Nature Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- Merredith JE, Jr, Schwartz MA. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997;7:146–150. doi: 10.1016/S0962-8924(97)01002-7. [DOI] [PubMed] [Google Scholar]
- Morita H, Tanaka I, Oda T, Ichimaya A, Yamasaki T, Uematsu T, Nakashima M, Yoshimi T. Atrial natriuretic peptide mRNA and pepide in rats with aortic valve insufficiency. Peptides. 1990;11:843–847. doi: 10.1016/0196-9781(90)90202-g. [DOI] [PubMed] [Google Scholar]
- Morrison-Graham K, Scatteman GC, Bork T, Bowen-Pope DF, Weston JA. A PDGF receptor mutation in the mouse (patch) pertubs the development of non-neural crest-derived cells. Development. 1992;115:133–142. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- Nakao K, Itoh H, Saito Y, Mukoyama M, Ogawa Y. The natriuretic peptide family. Curr Opin Nephrol Hypertens. 1996;5:4–11. doi: 10.1097/00041552-199601000-00003. [DOI] [PubMed] [Google Scholar]
- Palecek SP, Loftus C, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Paulsson MR, Deutzmann R, Timpl R, Dalzoppo R, Odermatt E, Engel J. Evidence for coiled-coil α-helical regions in the long arm of laminin. 1985. EMBO J. 1985;4:309–316. doi: 10.1002/j.1460-2075.1985.tb03630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissner KT, Wassmuth R, Muller-Berghaus G. Physicochemical characterization of human s-protein and its function in the blood coagulation system. Biochem J. 1985;231:349–355. doi: 10.1042/bj2310349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins-emerging paradigms of signal-transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Seidman CE, Bloch KD, Klein KA, Smith JA, Seidman JG. Nucleotide sequences of the human and mouse atrial natriuretic factors genes. Science. 1984;226:1206–1209. doi: 10.1126/science.6542248. [DOI] [PubMed] [Google Scholar]
- Shyy JYJ, Chien S. Role of integrins in cellular responses to mechanical stress and adhesion. Curr Opin Cell Biol. 1997;9:707–713. doi: 10.1016/s0955-0674(97)80125-1. [DOI] [PubMed] [Google Scholar]
- Smith A, Bruton J. A colour atlas of histological staining techniques. Wolfe Medical Atlases, no. 18. London, UK: Wolfe Medical Publications Ltd.; 1978. [Google Scholar]
- Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, Rabbitts P. A site-directed chromosomal translocation induced in embryonic stem cells by Cre–loxP recombination. Nature Genet. 1995;9:376–385. doi: 10.1038/ng0495-376. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A. Integrins and their ligands. Curr Top Microbiol Immunol. 1993;184:7–35. doi: 10.1007/978-3-642-78253-4_2. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGFα receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of β1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes & Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- Sutherland AE, Calarco PG, Damsky CH. Developmental regulation of integrin expression at the time of implantation in the mouse embryo. Development. 1993;119:1175–1186. doi: 10.1242/dev.119.4.1175. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Duband JL, Trucker GC. Cell migration in the vertebrate embryo: Role of cell adhesion and tissue environment in pattern formation. Annu Rev Cell Biol. 1985;1:91–113. doi: 10.1146/annurev.cb.01.110185.000515. [DOI] [PubMed] [Google Scholar]
- Tiidus PM. Can estrogens diminish exercise-induced muscle damage? Can J Appl Physiol. 1995;20:26–38. doi: 10.1139/h95-002. [DOI] [PubMed] [Google Scholar]
- Tominaga S. Murine mRNA for the β-subunit of integrin is increased in Balb/c-3T3 cells entering the G1 phase from the Go phase. FEBS Lett. 1988;238:315–319. doi: 10.1016/0014-5793(88)80503-9. [DOI] [PubMed] [Google Scholar]
- Vandenberg P, Kern A, Ries A, Luckenbill-Edds L, Mann K, Kühn K. Characterization of a type IV collagen major cell binding site with affinity to the α1β1 and α2β1 integrins. J Cell Biol. 1991;113:1475–1483. doi: 10.1083/jcb.113.6.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier A, Kuikman I, Baudoin C, Van der Neut R, Sonnenberg A. A novel β1 integrin isoform produced by alternative splicing: Unique expression in cardiac and skeletal muscles. FEBS Lett. 1995;369:340–344. doi: 10.1016/0014-5793(95)00814-p. [DOI] [PubMed] [Google Scholar]
- van der Flier A, Gaspar AC, Thorsteindóttir S, Baudoin C, Groeneveld E, Mummery CL, Sonnenberg A. Spatial and temporal expression of the β1D integrin during mouse development. Dev Dyn. 1997;210:472–486. doi: 10.1002/(SICI)1097-0177(199712)210:4<472::AID-AJA10>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Velling T, Collo G, Sorokin L, Durbeej M, Zhang H, Gullberg D. Distinct α7Aβ1 and α7Bβ1 integrin expression patterns during mouse development: α7A is restricted to skeletal muscle but α7B is expressed in striated muscle, vasculature, and nervous system. Dev Dyn. 1996;207:355–371. doi: 10.1002/(SICI)1097-0177(199612)207:4<355::AID-AJA1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Volk T, Fessler LI, Fessler JH. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell. 1990;63:525–536. doi: 10.1016/0092-8674(90)90449-o. [DOI] [PubMed] [Google Scholar]
- Von Ballestrem CG, Uniyal S, McCormick JI, Chau T, Singh B, Chan BMC. VLA-β1 integrin subunit-specific monoclonal antibodies MB1.1 and MB1.2: Binding to epitopes is not dependent on thymocyte development or regulated by phorbol ester and divalent cations. Hybridoma. 1996;15:125–132. doi: 10.1089/hyb.1996.15.125. [DOI] [PubMed] [Google Scholar]
- Wang N, Butler J, Ingber D. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fässler R. β1 integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. 1996;132:227–238. doi: 10.1083/jcb.132.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Keivens V, O’Toole TE, McDonald J, Ginsberg MH. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell. 1995;83:715–724. doi: 10.1016/0092-8674(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Yang JT, Hynes RO. Fibronectin receptor functions in embryonic cells deficient in α5β1 integrin can be replaced by αv integrins. Mol Cell Biol. 1996;7:1737–1748. doi: 10.1091/mbc.7.11.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- Yang JT, Rando TA, Mohler WA, Rayburn H, Blau HM, Hynes RO. Genetic analysis of α4 integrin functions in the development of mouse skeletal muscle. J Cell Biol. 1996;135:829–835. doi: 10.1083/jcb.135.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao CC, Ziober BL, Sutherland AE, Mendrick DL, Kramer RH. Laminins promote the locomotion of skeletal myoblasts via the α7 integrin receptor. J Cell Sci. 1996;109:3139–3150. doi: 10.1242/jcs.109.13.3139. [DOI] [PubMed] [Google Scholar]
- Zhidkova NI, Belkin AM, Mayne R. Novel isoform of β1 integrin expressed in skeletal and cardiac muscles. Biochem Biophys Res Commun. 1995;214:279–285. doi: 10.1006/bbrc.1995.2285. [DOI] [PubMed] [Google Scholar]