

Abstract
Chromosome translocations involving T cell receptor (TCR) loci have been found in tumors from Ataxia telangiectasia (AT) patients and in mouse Atm−/− thymoma, suggesting the involvement of V(D)J recombination in these malignancies. By introducing a RAG-1 deficiency into Atm−/− mice in the presence of a TCR transgene, we show that V(D)J recombination is critical for thymoma development in these mice. Therefore, aberrant V(D)J recombination, normally suppressed by Atm, facilitates tumorigenic events leading to cancer. Because V(D)J recombination is dispensable for lymphomagenesis upon p53 deficiency, this study also indicates that Atm and p53 function by distinct mechanisms in suppressing thymoma.
Keywords: Atm, V(D)J recombination, lymphoma, tumor suppression
Chromosome translocations have been found in many lymphoid malignancies, resulting in activation of proto-oncogenes in those cells (Rabbitts 1994; Nowell 1997). In many cases, the breakpoints of translocations involve immunoglobulin or T cell receptor (TCR) loci, suggesting a predisposition by these loci to the oncogenic events. V(D)J recombination assembles immunoglobulin and TCR gene segments via double-strand break (DSB) intermediates and is active at immunoglobulin and TCR loci during B and T cell development, respectively (Lewis 1994). It has long been hypothesized that errors in this process provide a major source of chromosome translocations in lymphoid malignancies (for review, see Danska and Guidos 1997; Kirsch and Lista 1997). However, the molecules involved in regulating this process to suppress cancer proneness are not understood.
Ataxia telangiectasia (AT), caused by mutations in the ATM gene, is a recessive chromosomal instability disease with pleiotropic phenotypes (Lavin and Shiloh 1997). AT patients have a high risk of cancer, particularly lymphoid malignancies. Chromosome translocations involving the TCR locus occur at a high frequency in AT T-lymphoid tumors (for review, see Taylor et al. 1996; Kirsch 1994). Furthermore, thymic lymphoma also develops in Atm-deficient mice, and evidence for TCR-associated rearrangements within tumors has been demonstrated (Barlow et al. 1996; Elson et al. 1996; Xu et al. 1996). These observations suggest that the tumor suppressor function of ATM acts through a mechanism involving V(D)J recombination. To test this hypothesis, we have analyzed the effect on tumorigenesis of inactivating V(D)J recombination in Atm-deficient mice. Here, by introducing a recombination activating gene (RAG)-1 deficiency into Atm−/− mice, we show that V(D)J recombination is a critical step in thymic lymphoma development, thus identifying a critical role for Atm in suppressing such tumorigenic events in the T-cell lineage.
Results
Recombination-activating genes (RAG), Rag-1 and Rag-2 are required for the first step of V(D)J recombination, that is, the initiation of DNA cleavage at recombination sites (McBlane et al. 1995). Inactivation of either gene by gene targeting results in complete inactivation of V(D)J recombination in lymphocytes (Mombaerts et al. 1992; Shinkai et al. 1992). If V(D)J recombination is involved in the induction of thymoma in Atm-deficient mice, blocking this process by Rag-1 deficiency should inhibit tumorigenesis. Thus, Atm−/− mice were crossed onto a Rag-1−/− background to inactivate V(D)J recombination, and thymic lymphoma development was assessed. As reported previously (Barlow et al. 1996), control Atm−/− mice (n = 8) all died of thymic lymphoma with a mean survival time of 4.8 months (Fig. 1). Similarly, all Atm−/−Rag-1+/− mice (n = 5) died of thymic lymphoma with a mean survival time of 4.7 months. Strikingly, none of the Atm−/−Rag-1−/− mice (n = 9) developed thymic lymphoma, nor were other tumors observed. Three of the nine Atm−/−Rag-1−/− mice died of pneumonia at 5–6 months of age, likely because of impaired immune function in Rag-1−/− mice, as similar infections were observed in Atm+/+Rag-1−/− and Atm+/−Rag-1−/− littermates. Not only was overt thymoma absent in Atm−/−Rag-1−/− mice, thymocyte numbers averaged <106 cells in these mice (not shown), further confirming the absence of lymphoma. Because Atm−/−Rag-1−/− and Atm−/−Rag-1+/− were littermates (Materials and Methods), mouse background differences other than indicated deficiencies did not contribute to the inhibition of thymoma. Thus, prevention of V(D)J recombination by Rag-1 deficiency blocked tumorigenesis in Atm-deficient mice.
Figure 1.

Rag-1 is required for thymic lymphoma development in Atm−/− mice. Tumor-free survival of mice is plotted with time. Each data point represents the sacrifice or death of a terminally ill mouse. A drop in percentage reflects the sacrifice or death of a mouse with thymoma. As with Atm−/− mice (□; n = 8), 100% of the Atm−/−Rag-1+/− mice (⋄; n = 5) developed thymic lymphoma. The mean survival time was 4.8 months for Atm−/− mice and 4.7 months for Atm−/−Rag-1+/− mice. All Atm−/−Rag-1−/− mice (○; n = 9) died or were sacrificed with no overt thymic lymphoma or other tumors. The mean survival time was 9 months, comparable to Rag-1−/− mice housed under the same conditions. Three of the nine Atm−/−Rag-1−/− mice died of pneumonia at 5–6 months with no sign of thymic lymphoma. Mouse food containing antibiotics was used to treat one Atm−/−Rag-1−/− mouse of this data set with similar symptoms.
As a result of inactivating V(D)J recombination, Rag deficiency blocks thymocyte development at the CD4/CD8 double-negative (DN) stage, causing a decrease in total thymic cellularity. It is conceivable that this indirect effect on thymocyte number, rather than V(D)J recombination per se, could alter thymoma development in Atm−/− mice. For example, reduction in thymus cellularity could reduce the size of target cell population for secondary tumorigenic events and increase survival time. However, reduced cellularity did not inhibit thymoma development in p53-deficient mice (Liao et al. 1998; Nacht and Jacks 1998). Also, Atm−/−Rag-1−/− mice survived beyond 14 months of age with no sign of tumor development (Fig. 1), indicative of a complete inhibition of tumorigenesis rather than a delay. Another formal possibility, however, is that tumorigenesis was inhibited because of the blockage of thymocyte development. Although normal thymocytes develop from DN to CD4+CD8+ double-positive (DP), then to mature CD4+CD8− or CD4−CD8+ single-positive (SP) cells, thymocyte development in Atm−/−Rag-1−/− mice was arrested at the DN stage as expected due to the Rag deficiency (Fig. 2A). Although most Atm−/− thymic lymphomas are reported to consist of DP cells (Barlow et al. 1996; Elson et al. 1996; Xu et al. 1996), we have observed a single case of DN Atm−/− thymoma, indicating that these cells can give rise to Atm−/− thymoma (data not shown).
Figure 2.
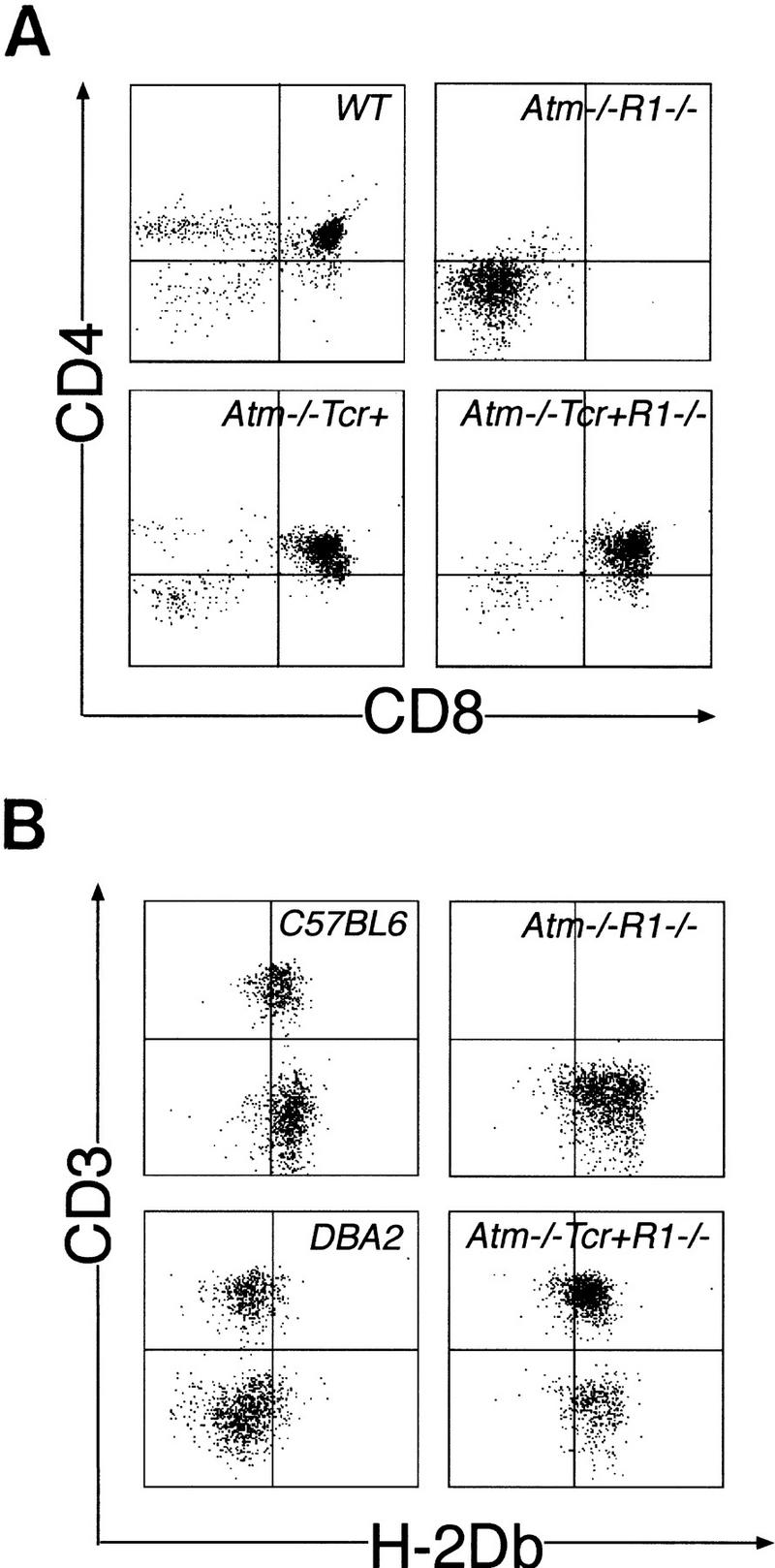
Rag-1−/− thymocyte differentiation is rescued by a TCR transgene. (A) CD4/CD8 expression was analyzed by FACS (Materials and Methods). Atm−/−Rag-1−/− thymocytes (Atm−/−R1−/−) are blocked at the CD4−CD8− DN stage compared to wild-type (WT) thymocytes. Expression of LCMV–TCR transgenes in Atm−/−Rag-1−/− thymocytes (Atm−/−TCR+R1−/−) induced a CD4+CD8+ DP population. The Atm−/−TCR+ thymocyte population is shown as a control. TCR+ thymocytes have the same profile as those from Atm−/−TCR+ mice (not shown). (B) Lack of positive thymic selection is not a factor in thymoma inhibition. Most of the Atm−/−TCR+Rag-1−/− mice are H-2Db positive, and have mature T cells in peripheral blood. Atm−/−TCR+R1−/− mice were screened for the presence of the H-2Db MHC molecule and the TCR component CD3 using PBLs (Materials and Methods). Profiles of PBLs from C57BL6 and DBA2 mice are shown as positive and negative controls for the presence of H-2Db, respectively. One typical profile of the seven Atm−/−TCR+R1−/− PBLs is shown, which confirms H-2Db-positive and CD3-positive (mature) T cells. An Atm−/−R1−/− PBL profile serves as the negative control for CD3 expression of mature T cells.
To definitively address the possibility that indirect effects of inactivating V(D)J recombination contributed to the inhibition of Atm−/− thymoma, we rescued thymocyte development in Atm−/−Rag-1−/− mice by introducing a TCR transgene [TgN(TcrLCMV) (lymphocyte choriomeningitis virus); Pircher et al. 1989]. Transgenic expression of the prerearranged TCRα and TCRβ genes in Rag-1−/− thymocytes induces thymocyte development in the absence of V(D)J recombination. Similar to the results in previous studies (Pircher et al. 1989; Shinkai et al. 1993), expression of LCMV–TCRα and TCRβ genes in Atm−/−Rag-1−/− thymocytes rescued thymocyte development to DP and SP stages (Fig. 2A) and restored thymus cellularity to the normal level (not shown). To ensure that transgenic TCR-expressing thymocytes were positively selected by matched major histocompatibility complex (MHC) molecules, Atm−/−TCR+Rag-1−/− mice were screened for histocompatibility. Two subtypes of H-2D MHCs, H-2Db and H-2Dd occur in mice in this study due to their mixed genetic background. Transgenic LCMV–TCR-expressing thymocytes survive in the presence of the H-2Db subtype (Pircher et al. 1989). FACS analysis showed that all mice included in this study were H-2Db positive (Fig. 2B). The β-chain segment Vβ8.1 of the transgene also reacts with the Mlsa superantigen, which can lead to the elimination of mature SP but not DP cells (Pircher et al. 1989). Because of the DP origin of Atm−/− tumors, the Mlsa antigen (if present) should not affect the target cell population (DP). Nevertheless, to test whether negative selection had affected the T-cell population in Atm−/−TCR+Rag-1−/− mice, PBLs were analyzed for the presence of a mature CD3+ T cells. All Atm−/−TCR+Rag-1−/− PBL samples contained mature CD3+ T cells, indicating that negative selection did not delete this population and further supporting a matched MHC type. As a control, mature cells were absent from Atm−/−Rag-1−/− mice in which thymocyte development was blocked (Fig. 2B). Therefore, the presence of the TCR transgene did not cause deletion of critical subsets of thymocytes that could be possible targets for tumorigenesis.
Despite the restoration of thymocyte differentiation in Atm−/−TCR+Rag-1−/− mice, however, thymic tumors were not observed in these mice. Control littermate Atm−/−TCR+Rag-1+/− mice (n = 12) all died of thymic lymphoma at a time similar to that of Atm−/− mice (n = 8; mean survival of 5.0 months and 4.8 months, respectively). However, Atm−/−TCR+Rag-1−/− mice (n = 9) did not develop tumors. All mice lived beyond the mean Atm−/− survival time of 5 months with no evidence of thymoma development (Fig. 3). The thymuses of several mice that died of infections (mean survival, 8 months; range, 6–9 months) were normal compared to age-matched controls. Three mice are currently healthy at 8–9 months. Therefore, an intact V(D)J recombination pathway is critical for the induction of thymic lymphoma in Atm-deficient mice. By extension, these results implicate Atm in the regulation or checkpoint control of thymocyte V(D)J recombination.
Figure 3.

V(D)J recombination is required for Atm-deficient thymoma. All Atm−/− mice (□; n = 8) and Atm−/−TCR+Rag-1+/− mice (⋄; n = 12) died of thymic lymphoma with a mean survival time of 4.8 and 5.0 months, respectively. Of the Atm−/−TCR+Rag-1−/− mice shown (○; n = 9), 6 died of infections at 6–9 months (mean survival, 8 months) with no sign of thymic lymphoma. Others are currently alive at 8 months with no sign of lymphoma. Because Atm−/−TCR+Rag-1+/− and Atm−/−TCR+Rag-1−/− mice are from the same cross, genetic background does not contribute to the observed difference.
Discussion
Chromosomal translocations are the most common genetic alterations in human lymphoid malignancies (Rabbitts 1994). The presence of a high frequency of antigen recognition gene translocations suggests that V(D)J recombination is involved in most of these cases (Nowell 1997). Recent in vitro studies have shown that the RAG proteins can mediate DNA transposition-like events, suggesting that RAG proteins may have a role in oncogenic translocations (Agrawal et al. 1998; Hiom et al. 1998). The Atm-deficient mouse model accurately reflects the occurrence of such translocations, which develop in human AT T-cell neoplasms (Taylor et al. 1996). Similar translocations are also observed in non-AT lymphoid tumors (albeit at a lower frequency), indicating a general underlying mechanism. Our study shows that V(D)J recombination can drive tumorigenic events leading to lymphomagenesis. Moreover, these results further link Atm to a tumor suppression mechanism that is tied to V(D)J recombination, as aberrant V(D)J recombinase activity is normally inhibited or eliminated in mouse thymocytes in the presence of Atm. Thus, loss of Atm function predisposes to lymphoid malignancy. These findings represent an inroad to the molecular understanding of the respective V(D)J oncogenic and Atm tumor suppression mechanisms. In addition, because early death of Atm-deficient mice from thymic lymphoma normally hampers prolonged study of any neurological defects of AT in these mice, Atm-deficient mice in which thymomagenesis is inhibited (in a Rag-1-deficient background) will facilitate further study of this disease. Although no overt ataxia was observed in mice of our study, close examination may yield further insight.
Role of ATM in V(D)J recombination fidelity
Several possible explanations exist for Atm’s role in suppressing chromosomal translocations. First, ATM acts in cell cycle checkpoints to signal DSBs at G1, S, and G2/M phases (Kastan et al. 1992; Meyn 1995; Shiloh and Rotman 1996). Premature entry into S phase or mitosis in Atm-deficient cells with unrepaired DSBs could promote recombination by erroneous, nonhomologous end joining leading to translocations (Elledge 1996; Paulovich et al. 1997). Second, Atm has a role in meiotic chromosome synapsis and recombination (Plug et al. 1997). It is possible that Atm participates in the synaptic structures that orchestrate V(D)J recombination and prevent interchromosomal recombination. Failure in this orchestration would lead to translocation. The level of hybrid TCR genes generated by interlocus recombination is 10- to 100-fold higher in human AT lymphocytes compared with normal lymphocytes (Lipkowitz et al. 1990; Kobayashi et al. 1991). Third, Atm deficiency could affect chromatin organization and TCR locus accessibility, resulting in overexuberant recombination at these sites (Kirsch 1994). DSBs observed in AT cells are present more frequently as chromatin breaks suggesting that ATM may participate in managing chromatin structure at some DSBs (Pandita and Hittelman 1992). Fourth, it is possible that Atm functions in detecting and eliminating cells with illegitimate V(D)J recombination products by inducing apoptosis. In mouse CNS development, Atm is required for apoptosis after irradiation-induced DNA damage (Herzog et al. 1998), although some controversy surrounds the issue of whether Atm is involved in a similar response in thymocytes (Xu and Baltimore 1996; Barlow et al. 1997; Westphal et al. 1997). Finally, a DSB repair defect distinct from cell cycle checkpoint defects has been suggested in AT cells (Jeggo et al. 1998). Higher than normal levels of chromosome breaks and rearrangements were observed in noncycling AT cells upon irradiation (IR). Further experiments are required to distinguish whether one or more of these mechanisms has a role in Atm-mediated tumor suppression.
ATM and p53 suppress thymoma by distinct mechanisms
Previous evidence supports the hypothesis that ATM acts upstream of p53 in signaling IR-induced DNA damage in fibroblasts. Human AT cells are deficient in up-regulating p53 (Kastan et al. 1992), and ATM acts as a Ser/Thr protein kinase in phosphorylating and activating p53 at Ser-15 upon irradiation (Banin et al. 1998; Canman et al. 1998). The fact that both Atm- and p53-deficient mice develop thymoma was suggestive that these factors function in the same pathway in thymocytes as well. However, our data indicate different mechanisms for Atm and p53 tumor suppression in mouse thymocytes. We (Liao et al. 1998) and others (Nacht et al. 1998) examined thymoma development previously in p53-deficient mice using a similar strategy as described here for Atm. Thymomas developed in p53-deficient mice in the absence of V(D)J recombination (inhibited by a deficiency of Rag-1 or Rag-2), although longer latencies were observed. We showed further that thymomas developed in a Rag1−/−/TCR+ background without effect on frequency and with no increased latency. Furthermore, the major aberration detected in p53-deficient thymomas was aneuploidy (Liao et al. 1998), whereas chromosome translocations, including those associated with TCR chromosomes, are common in Atm-deficient thymomas (Barlow et al. 1996; Liao et al. 1998). No such translocations were observed in p53-deficient thymomas (Liao et al. 1998).
The observation that Atm and p53 function by distinct mechanisms in thymocytes is consistent with a previous report that p53 and Atm cooperate in thymoma suppression. Thymoma development is accelerated in p53/Atm doubly deficient mice relative to either deficiency alone, indicating that Atm and p53 function in distinct complementary pathways (Westphal et al. 1997). Together, these data suggest that there may be multiple mechanisms for ATM tumor/checkpoint regulation. Although its function in p53 activation is important for cell cycle arrest upon irradiation-induced DNA damage in some cell types, ATM appears to maintain the fidelity of DNA recombination via a different pathway. Given the link demonstrated here between Atm and V(D)J-driven lymphoma suppression, further molecular studies will be required to determine the extent to which this function represents a new activity for Atm.
Materials and methods
Mice
Homozygous Atm−/− mice are infertile (Barlow et al. 1996; Elson et al. 1996; Xu et al. 1996). Thus, Atm+/− mice [129/SvEv-C57BL/6J; kindly provided by A. Wynshaw-Boris (Barlow et al. 1996)] were bred with Rag-1−/− mice (C57BL/6J-sv/129; Jackson Labs) to generate Atm+/−Rag-1+/− (F1) mice. F1 mice were backcrossed to Rag-1−/− mice to generate both Atm+/−Rag-1−/− and Atm+/−Rag-1+/− mice. These mice were crossed further to generate Atm−/−Rag-1−/− and Atm−/−Rag-1+/− mice. TgN(TcrLCMV) mice (C57BL/6J-DBA2; Jackson Labs) were bred with Atm+/−Rag-1−/− mice to generate Atm+/−TCR+Rag-1+/− mice, which were further bred with Atm+/−Rag-1−/− mice to generate Atm−/−TCR+Rag-1−/− mice. PCR primers were designed for genotyping Atm−/− mice. Primer pairs Atm-F2 (5′-AAACCGACTTCTGTCAGATGTTGC-3′) and Atm-B2 (5′-TTTGCAGGAGTTGCTGAGCG-3′) were used to identify the wild-type Atm allele. Atm-F (5′-GACTTCTGTCAGATGTTGCTGCC-3′) and Atm-Neo (5′-GGGTGGGATTAGATAAATGCCTG-3′) were used to identify the knockout allele in 35 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min. The Atm-F2 and Atm-B2 pair generates a 152-bp PCR product, and the Atm-F and Atm-Neo pair generates a 441-bp PCR product. PCR was used for genotyping Rag-1 and TgN(TcrLCMV) mice as described previously (Liao et al. 1998). Mice were maintained under sterile conditions with autoclaved microisolator cages and autoclaved water, food, and bedding.
Immunophenotyping
Thymocytes were isolated as described previously (Liao et al. 1998) and counted using a hemocytometer. Peripheral blood was collected from tail vein into heparin (Elkins-Sinn, Inc.) containing tubes. Red blood cells were lysed to obtain peripheral blood lymphocytes (PBLs) using the Unopette solution (Becton-Dickinson). CD4, CD8, CD3, α/β or γ/δ TCR markers were stained as described previously using antibodies from PharMingen (Liao et al. 1998). H-2Db in PBLs was detected using FITC-conjugated anti-mouse H-2Db and FACS analysis. PBLs were treated with anti-mouse CD16/CD32 (PharMingen) before anti-H-2Db staining to block nonspecific binding to Fc receptors.
Acknowledgments
We thank Tony Wynshaw-Boris for Atm+/− mice, Lishan Su for helpful discussions, and Dale Ramsden and William Kaufmann for critical reading of the manuscript. The UNC Lineberger Comprehensive Cancer Center Flow Cytometry Center was utilized for FACS studies in this report. This work was supported by a grant from the National Cancer Institute to T.V.D. (CA65773).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
A similar inhibition of tumorigenesis has been observed in Atm-deficient/RAG-deficient mice in studies by Y. Xu (pers. comm.; for review, see Xu, Y. 1999. ATM in lymphoid development and tumorigenesis. Adv. Immunol. 72: 179–189.)
Footnotes
E-MAIL tvdlab@med.unc.edu; FAX (919) 962-4296.
References
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: A paradigm of Ataxia Telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barlow C, Brown K, Deng C, Tagle D, Wynshaw-Boris A. Atm selectively regulates distince p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat Genet. 1997;17:453–456. doi: 10.1038/ng1297-453. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Danska JS, Guidos CJ. Essential and perilous: V(D)J recombination and DNA damage checkpoints in lymphocyte precursors. Semin Immunol. 1997;9:199–206. doi: 10.1006/smim.1997.0072. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: Preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty C, Morton C, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: A possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- Jeggo PA, Carr AM, Lehmann AR. Splitting the ATM: Distinct repair and checkpoint defects in ataxia-telangiectasia. Trends Genet. 1998;14:312–316. doi: 10.1016/s0168-9525(98)01511-x. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh W V, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in Ataxia-Telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kirsch IR. V(D)J recombination and ataxia-telangiectasia: A review. Int J Radiat Biol. 1994;66:S97–S108. [PubMed] [Google Scholar]
- Kirsch IR, Lista F. Lymphocyte-specific genomic instability and risk of lymphoid malignancy. Semin Immunol. 1997;9:207–215. doi: 10.1006/smim.1997.0071. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Tycko B, Soreng AL, Sklar J. Transrearrangements between antigen receptor genes in normal human lymphoid tissues and in ataxia telangiectasia J. Immunol. 1991;147:3201–3209. [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- Lewis SM. The mechanism of V(D)J joining: Lessons from molecular, immunological, and comparative analyses. Adv Immunol. 1994;56:27–150. doi: 10.1016/s0065-2776(08)60450-2. [DOI] [PubMed] [Google Scholar]
- Liao M-J, Zhang X, Hill R, Gao J, Qumsiyeh M, Nichols W, Van Dyke T. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol. 1998;18:3495–3501. doi: 10.1128/mcb.18.6.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkowitz S, Stern MH, Kirsch IR. Hybrid T cell receptor genes formed by interlocus recombination in normal and ataxia-telangiectasis lymphocytes J. Exp Med. 1990;172:409–418. doi: 10.1084/jem.172.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- Meyn MS. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res. 1995;55:5991–6001. [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou V. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Nacht M, Jacks T. V(D)J recombination is not required for the development of lymphoma in p53-deficient mice. Cell Growth Differ. 1998;9:131–138. [PubMed] [Google Scholar]
- Nowell PC. Genetic alterations in leukemias and lymphomas: Impressive progress and continuing complexity. Cancer Genet Cytogenet. 1997;94:13–19. doi: 10.1016/s0165-4608(96)00227-0. [DOI] [PubMed] [Google Scholar]
- Pandita TK, Hittelman WN. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat Res. 1992;130:94–103. [PubMed] [Google Scholar]
- Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- Plug A, Peters A, Xu Y, Keegan K, Hoekstra M, Baltimore D, de Boer P, Ashley T. ATM and RPA in meiotic chromosome synapsis and recombination. Nat Genet. 1997;17:457–461. doi: 10.1038/ng1297-457. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Rotman G. Ataxia-telangiectasia and the ATM gene: linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints J. Clin Immunol. 1996;16:254–260. doi: 10.1007/BF01541389. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam K, Oltz E, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall A, Alt F. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Koyasu S, Nakayama K, Murphy K, Loh D, Reinherz E, Alt F. Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science. 1993;259:822–825. doi: 10.1126/science.8430336. [DOI] [PubMed] [Google Scholar]
- Taylor AMR, Metcalfe JA, Thick J, Mak Y-F. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87:423–438. [PubMed] [Google Scholar]
- Westphal C, Rowan S, Schmaltz C, Elson A, Fisher D, Leder P. atm and p53 cooperate in apoptosis and suppression of tumorigenesis, but not in resistance to acute radiation toxicity. Nat Genet. 1997;16:397–401. doi: 10.1038/ng0897-397. [DOI] [PubMed] [Google Scholar]
- Xu Y, Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes & Dev. 1996;10:2401–2410. doi: 10.1101/gad.10.19.2401. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]