

Abstract
Background
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited genetic myocardial disease characterized by fibrofatty replacement of the myocardium and a predisposition to cardiac arrhythmias and sudden death. We evaluated the cardiomyopathy gene titin (TTN) as a candidate ARVC gene because of its proximity to an ARVC locus at position 2q32 and the connection of the titin protein to the transitional junction at intercalated disks.
Methods and Results
All 312 titin exons known to be expressed in human cardiac titin and the complete 3’ untranslated region were sequenced in 38 ARVC families. Eight unique TTN variants were detected in 7 families including a prominent Thr2896Ile mutation that showed complete segregation with the ARVC phenotype in one large family. The Thr2896IIe mutation maps within a highly conserved immunoglobulin-like fold (Ig10 domain), located in titin’s spring region. Native gel electrophoresis, NMR, intrinsic fluorescence, and proteolysis assays of wildtype and mutant Ig10 domains revealed that the Thr2896IIe exchange reduces the structural stability and increases the propensity towards degradation of the Ig10 domain. The phenotype of TTN variant carriers was characterized by history of sudden death (5/7 families), progressive myocardial dysfunction causing death or heart transplant (8/14 cases), frequent conduction disease (11/14), and incomplete penetrance (86%).
Conclusions
Our data provide evidence that titin mutations can cause ARVC, a finding that further expands the origin of the disease beyond desmosomal proteins. Structural impairment of the titin spring is a likely cause of ARVC and constitutes a novel mechanism underlying myocardial remodeling and sudden cardiac death.
Keywords: cardiomyopathy, arrhythmia, death, sudden, genetics, arrhythmogenic right ventricular cardiomyopathy
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial disease characterized by fibrofatty replacement of the myocardium and a predisposition to cardiac arrhythmias. The most common presenting symptoms are palpitations, syncope, and sudden death. Structural and functional alterations to the right and left ventricles can also occur, leading to the development of heart failure. ARVC is recognized as a significant cause of sudden death in young persons and athletes and frequently is diagnosed post-mortem. It is a familial disease in up to 50% of cases and the predominant mode of transmission is autosomal dominant, except in the case of Naxos disease where palmoplantar keratosis and unusual wooly hair are also present due to recessive mutations in the JUP and PKP2 genes.1–4 Low penetrance, age dependent expression, and challenges in confirming a clinical diagnosis pre-mortem, along with genetic and allelic heterogeneity have all conspired to complicate the identification of ‘genetic cases’, especially in small pedigrees or where only a single individual is known or suspected to manifest the phenotype. Presently a genetic defect can be confirmed in approximately 40 to 50% of cases.5–7
Currently twelve different ARVC loci are reported in the Online Mendelian Inheritance in Man. Five of these known genes encode proteins integral to cell-cell junctions at the intercalated disk (DSP, PKP2, DSG2, DSC2, and JUP), thereby defining the intercalated disks as a major player in ARVC pathogenesis. The role of three other genes that have emerged as candidates has been less well established to date: the growth factor TGFβ3, the ion channel subunit RYR2, and the transmembrane protein 43 (TMEM43) identified in the Newfoundland founder population.8–10 Of the presently four remaining loci that are genetically linked to ARVC, a locus at chromosomal position 2q31.1-q32.2 (OMIM#602087) is of interest as this 3.4 Mb linkage region defined by the D2S152 and D2S389 microsatellite markers is close to the cardiomyopathy gene titin (TTN) that sits 0.4 Mb centromeric to the ARVC4 locus. Furthermore, there is a functional link between the desmosome and titin, since the titin filament connects to the transitional junction at the intercalated disk 11 and animal models with altered intercalated disks show sarcomere and Z-disk changes. Thus TTN is a candidate gene for ARVC.12
Titin is the largest protein in mammals and is expressed in both cardiac and skeletal muscle. Several isoforms ranging in size from 2970 to 3900 kDa are produced from the single TTN gene, which is located on chromosome region 2q31 and is comprised of 363 exons.13, 14 Titin filaments bridge the sarcomere along its longitudinal axis, overlapping end-to-end at the Z-disk and M-band at the amino and carboxy ends of titin, respectively, and forming a contiguous filament along the myofibril. The role of titin in cellular mechanics and signaling has recently been reviewed.14–16 Specifically, the spring-like properties of the sarcomere that underlie passive and restorative forces occurring after sarcomere lengthening or shortening, respectively, have been attributed to titin and these characteristics promote the restoration of resting sarcomere length.17
The elucidation of the genomic sequence of TTN has allowed for genetic studies in skeletal and cardiac muscle diseases. Several mutations in the terminal exon of TTN result in tibialis muscular dystrophy that does not involve any cardiac signs or symptoms.18 TTN missense mutations have also been reported in hypertrophic cardiomyopathy (Arg740Leu and Ser3799Tyr).19, 20 Additional mutations have been found in dilated cardiomyopathy with nine nonsynonymous and one frameshift mutation reported.21 Although titin’s large size and central position in sarcomeric architecture have made it an attractive candidate for cardiomyopathy studies, the challenges of performing large-scale mutation screening studies of all 363 exons have limited the pace of genetic studies. Our group initiated a DNA resequencing project of TTN in a collection of 38 well-characterized ARVC families.
Methods
Patient population
Probands from 38 ARVC families underwent TTN DNA resequencing provided by the University of Washington, Department of Genome Sciences, under U.S. Federal Government contract number N01-HV-48194 from the National Heart, Lung, and Blood Institute. Families were selected from the Familial Cardiomyopathy Registry, a multi-center, three-decade-long ongoing project studying human hereditary cardiomyopathies; an additional 150 dilated cardiomyopathy probands were also sequenced and will be reported separately. The diagnosis of ARVC was made on the basis of the 2010 update of the 1994 consensus criteria and all available living subjects were evaluated by the investigators.3, 22, 23 Clinical data collected included medical history, family history, physical examination, electrocardiogram, echocardiogram, and whenever available, Holter monitoring, signal average electrocardiogram, and histology from explanted heart or endomyocardial biopsy. Medical records from deceased subjects were reviewed when available. Informed consent was obtained from living subjects and the local institutional review boards approved the protocol.
DNA sequence analysis
Exons and peri-exonic regions of titin isoform N2A (NM_133378) along with additional exons unique to the principal cardiac isoform N2B (NM_003319) were amplified from genomic DNA using polymerase chain reaction (PCR). This covered 312 exons (311 expressed as titin protein) and the complete 3’ untranslated region. The PCR primers were Tm matched and designed from a masked reference sequence. Each primer pair was "tailed" with a universal M13 forward and reverse sequencing primer for subsequent sequencing. Once the regions were amplified, each PCR product was sequenced from the forward and reverse direction to provide double-stranded coverage. Sequencing was carried out with Sanger Big-Dye Terminator sequencing on capillary-based machines (AB 3730, Applied Biosystems, Foster City CA). The sequencing traces were base-called and assembled on the reference sequence, and PolyPhred was applied to identify sequence variants and provide genotypes across the samples.24 Prior genetic studies excluded mutations in the ARVC-linked genes DSC2, DSG2, DSP, and PKP2 (data not shown).
TTN mutation analysis
Stringent criteria for the classification of a mutation as putatively disease-causing included the following: variants uniquely identified in the patient cohort, predicted alteration of amino acid sequence, evolutionary conservation of the particular residue altered, predicted tolerability of nonsynonyomous changes, segregation among affected family members, and absence in 400 normal control chromosomes. Mutations detected were evaluated against known TTN single nucleotide polymorphisms (SNPs) in available databases and mutations present in multiple other families in the entire cohort were considered common and unlikely to be pathogenic mutations. Nonsynonymous coding mutations were evaluated for putative functional effects using SIFT and PolyPhen analysis and scored as tolerant or intolerant; tolerant mutations were considered unlikely to be pathogenic.25, 26 Testing of mutations in additional ARVC family members and in 400 chromosomes from ethnically similar healthy controls was done using by pyrosequencing (PSQ 96MA, Biotage, Uppsala, Sweden).
Protein analysis
The wildtype and mutated human Ig10 segments of the titin protein were produced as recombinant proteins in E.coli27. WT and mutant proteins were separated on 7% native tris-glycine polyacrylamide gels supplemented with 17% sucrose and were used for NMR studies. Two-dimensional 1H15N HSQC spectra on 0.25 mM samples were acquired at 22 °C on a Bruker DRX600 equipped with cryogenic triple-resonance probes and processed with Topspin. Tryptophane residue fluorescence was studied using 25 µM WT or mutant protein solution (50 mM Tris.HCl, pH 7.5, 150 mM NaCl) to which varying concentrations of guanidinium chloride was added. Fluorescence was measured at 30 °C on a Biotek Synergy 2 plate reader using xenon flash lamp with 284/10 nm excitation and 340/30 nm emission filters. Assays were done in triplicate. Proteolysis assays were carried out with trypsin (25:1 protein:trypsin ratio by weight) at room temperature followed by gel electrophoresis and quantitative gel analysis.
Results
Genetic analysis
Eight unique TTN nonsynonymous variants were identified in seven unrelated ARVC probands (Table 1 and Figure 1) with one proband (TSRVD027) that was a compound heterozygote having two TTN variants. The nonsynonymous variants were all considered ‘intolerant’ by SIFT/Polyphen analysis, were not previously reported in dbSNP, were absent in 400 control chromosomes and an additional 300 chromosomes from patients with dilated cardiomyopathy, and associated with the ARVC phenotype in our analysis of segregation within the families.
Table 1.
RareTTNVariants
| Family Number | Exon | Variant | Amino Acid Change |
Location |
|---|---|---|---|---|
| TSRVD001 | 37 | C29453T | Thr2896Ile | Spring |
| DNRVD006 | 97 | A97341G | Tyr8031Cys | Spring |
| TSRVD030 | 108 | C106734T | His8848Tyr | N2A |
| DNRVD008 | 298 | T215598C | Ile16949Thr | A-Band |
| TSRVD027 | 305 | G221380A | Ala18579Thr | A-Band |
| DNVRD002 | 313 | G226177T | Ala19309Ser | A-Band |
| DNRVD011 | 357 | C272848T | Pro30847Leu | M-Line |
| TSRVD027 | 362 | T281801C | Met33291Thr | M-Line |
Figure 1.
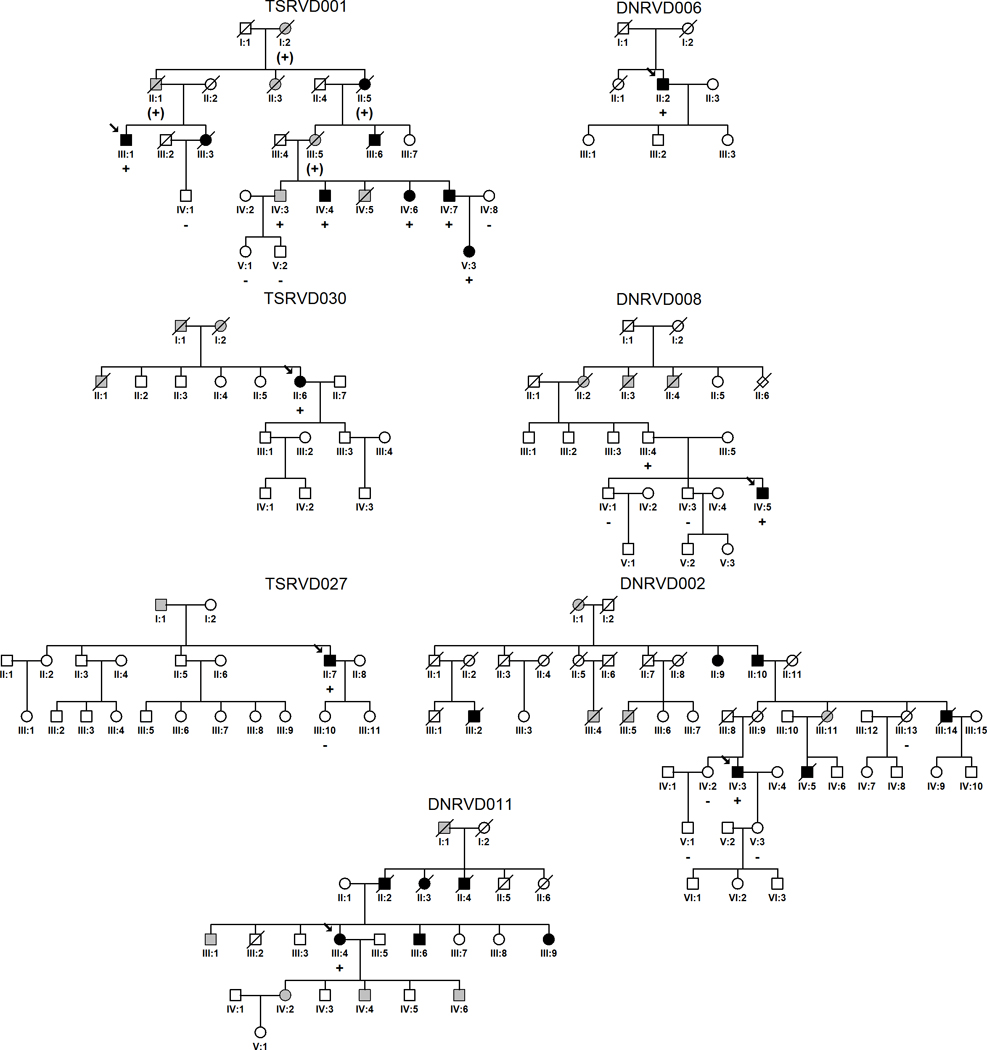
Pedigrees of ARVC families with rare TTN variants. Males and females are indicated by squares and circles respectively. Individuals meeting full ARVC diagnostic criteria are indicated with black shading; grey shading indicates a suggestive cardiac history and/or history of sudden unexplained death (see also Table 2). White squares/circles indicate unaffected individuals based on available family and medical history or, when TTN variant status is indicated, on the basis of full clinical evaluation by the investigators. TTN rare variant status for tested individuals is indicated by ‘+’ (present) and ‘-’ (absent) symbols; parentheses indicates inferred status. Probands are identified with an arrow.
In Family TSRVD001, a C39453T mutation (exon 37, Thr2896Ile, Figure 2 and Figure 3)13, 28, 29 showed complete segregation of the mutation with the ARVC phenotype in six affected individuals (Figure 1).14, 16 The Thr2896Ile mutation, absent in all other tested families and controls, was shared by two fifth degree relatives with ARVC (III-1 and V-5) providing strong genetic evidence that this TTN mutation is linked to the ARVC phenotype. Multiple relatives in this family met diagnostic criteria for ARVC (Table 2) and histological evidence of ARVC was present at autopsy (Figure 3C).
Figure 2.
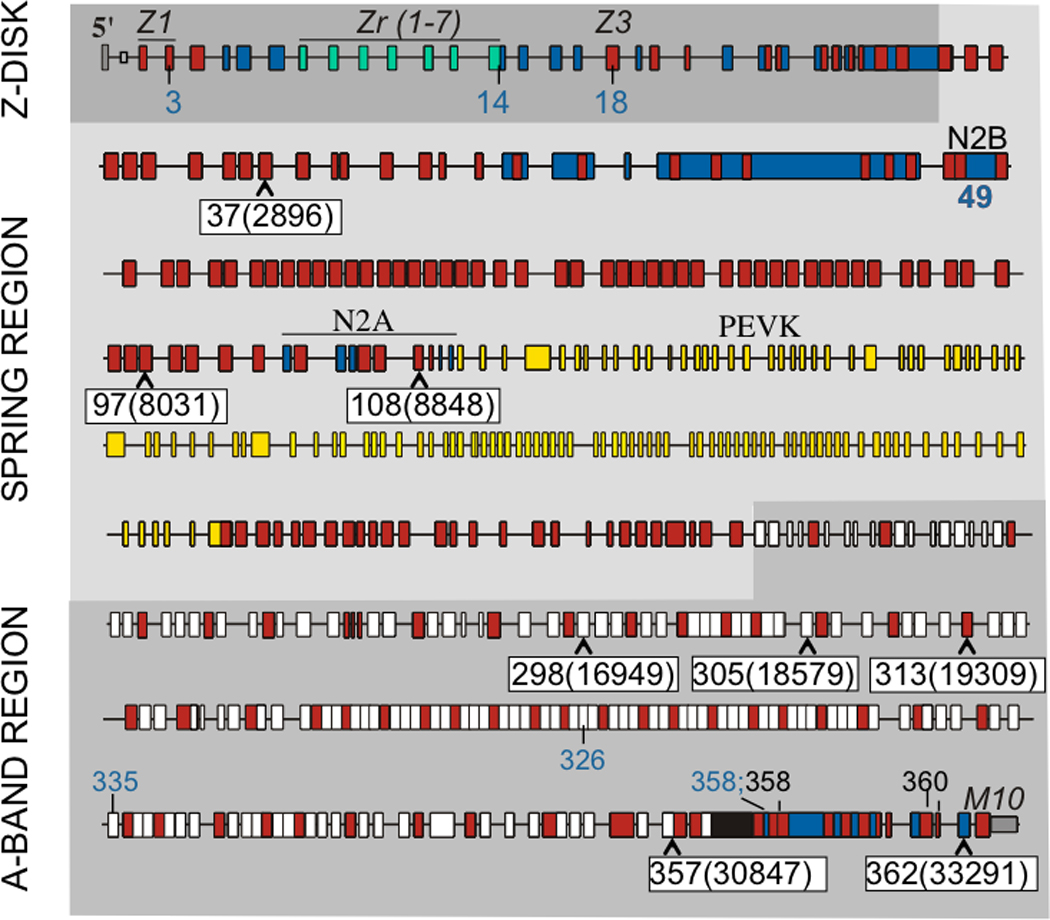
Exon structure of human titin gene with location of rare TTN variants in ARVC families (black font on white background) indicated by exon location and amino acid change (in parentheses). Shown are also previously identified variants associated with other cardiac diseases. Variants in exons 3, 14 (2 different variants), 18, 49 (4 different variants), 326, 335, and 358 have all been associated with dilated cardiomyopathy (blue); additional variants in exons 358 and 360 have been associated with fetal cardiomyopathy (black). For details, original citations, and variants associated with skeletal muscle myopathies, see27. (Red rectangle: immunoglobulin-like domain; white: fibronectin type 3 domain; blue: unique sequence; green: z-repeat domain; yellow: PEVK domain; black: titin kinase domain. (Figure based on Genbank accession AJ277892 and Bang et al13.)
Figure 3.
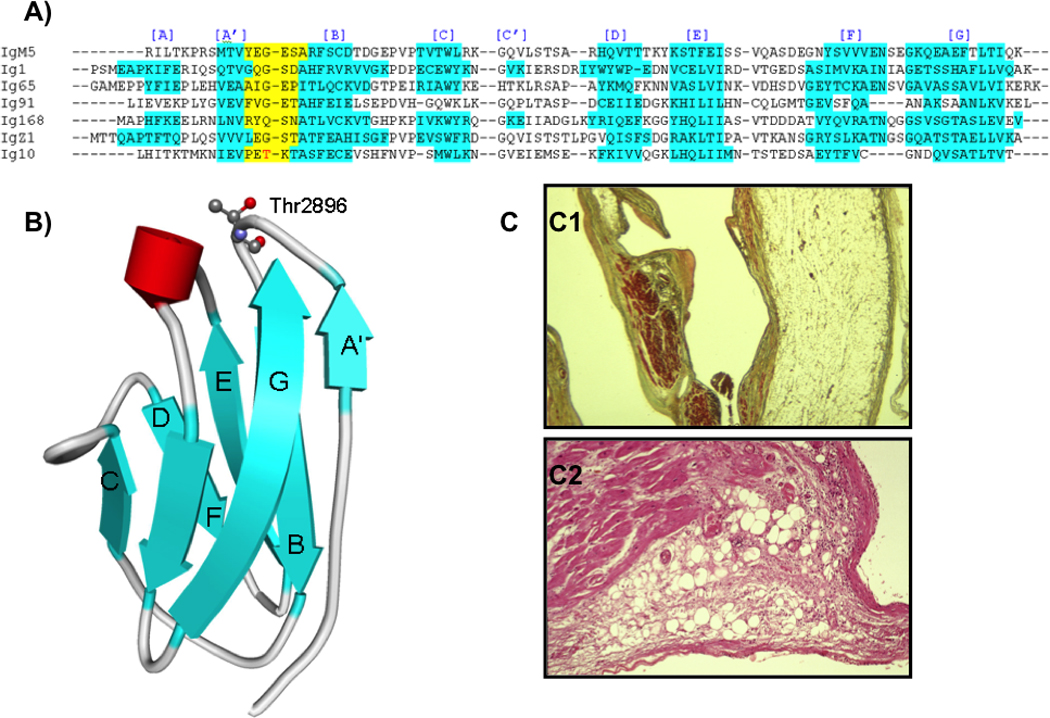
A) Multiple sequence alignments with known Ig structures, indicate that the mutated threonine in Ig10 is located in the short loop connecting the A' and B β-strands. The hydrogen bond network between the A' and G strands is important for Ig mechanical stability, and mutations in the A'B loop have been shown to disrupt this stabilizing network.29, 37 B) Schematic representation of human Ig10 using the homology modeling server ModWeb with Thr2896, shown with a ball-and-stick model. C) Histological section of the right ventricular wall of patient III-3 from family TSRVD001 (Thr2896Ile). C1. The myocardium is substituted by fatty tissue and a layer of subendocardial fibrous tissue (Azan; original magnification x2.5). C2. Fibro-fatty infiltration with presence of inflammatory cells (Hematoxilin-eosin; original magnification x4.5.)
Table 2a, b.
Clinical phenotype of study subjects
| Family | TSRVD001 | TSRVD001 | TSRVD001 | TSRVD001 | TSRVD001 | TSRVD001 | TSRVD001 |
|---|---|---|---|---|---|---|---|
| Individual | III-1 | III-3 | IV-3 | IV-4 | IV-6 | IV-7 | V-3 |
| Gender | Male | Female | Male | Male | Female | Male | Female |
| Patient Mutation | Thr2896Ile | NA | Thr2896Ile | Thr2896Ile | Thr2896Ile | Thr2896Ile | Thr2896Ile |
| ARVD CRITERIA | 3M 2m | 3M 3m | No | 3M 1m | 3M 1m | 3M 2m | 1M 4m |
| Age at onset | 49 | 51 | -- | 41 | 30 | 29 | 19 |
| Age at diagnosis | 50 | 58 | 52 | 41 | 39 | 37 | 19 |
| Exertional Dyspnea | Yes | No | Yes | No | Yes | No | No |
| Palpitations | No | Yes | No | No | Yes | No | Yes |
| Syncope | Yes | No | No | No | Yes | Yes | No |
| ECG | AF, LAFB, PM | AF, 3AVB, EW, PM |
AF, LBBB, 2AVB, PM |
PVCs, EW | AF, 1–3AVB, PM |
SND, 1AVB, EW, PM |
SND, AF, LBBB, PM |
| RV Dilation | Severe | Severe | No | Mild | Moderate | Moderate | Moderate |
| RV Bulging | Yes | Yes | No | Yes | Yes | Yes | No |
| RV hypokinesia | Regional | Regional | No | Regional | Diffuse | Diffuse | No |
| RV Function | RVFS 12% | RVFS 15% | Normal | RVFS 33% | RVFS 31% | RVFS 19% | RVFS 41% |
| LV Dilation (LVEDD) | No | No | No | Yes | Yes | No | No |
| LV EF% | 43 | 69 | Normal | 55 | 45 | 57 | 66 |
| Late Potentials on SAECG |
Yes | NA | No | Yes | NA | Yes | No |
| LBBB type VT (NSVT) |
Yes | Yes | No | No | Yes | Yes | No |
| LBBB type VT (Sustained VT) |
No | No | No | No | Yes | No | No |
| Ventricular extrasystoles >1000/24 hours |
Yes | No | No | No | No | No | Yes |
| Age and cause of death or transplant |
HF (66) | HF (62) | Stable (72) | OHT 64) | OHT (57) | OHT (58) | Stable (36) |
| Fibrous or fibrofatty replacement |
Yes | Yes | NA | NA | Yes | Yes | NA |
| Family | DNRVD002 | DNRVD008 | DNRVD008 | DNRVD006 | TSRVD030 | DNRVD011 | TSRVD027 |
|---|---|---|---|---|---|---|---|
| Individual | IV-3 | III-4 | IV-5 | II-2 | II-6 | III-4 | II-7 |
| Gender | Male | Male | Male | Male | Female | Female | Male |
| Patient Mutation | Ala19309Ser | Ile16949Thr | Ile16949Thr | Tyr8031Cys | Gly1137Arg | Pro30847Leu | Ala18579Thr Met33291Thr |
| ARVD CRITERIA | 2M 3m | No | 1M 2m | 2M, 1m | 2M | 2M,2m | 2M,1 m |
| Age at onset | 19 | -- | 35 | 44 | 59 | 15 | 35 |
| Age at diagnosis | 44 | 66 | 36 | 46 | 59 | 33 | 40 |
| Exertional Dyspnea | Yes | No | Yes | Yes | No | Yes | Yes |
| Palpitations | Yes | No | YES | Yes | Yes | Yes | Yes |
| Syncope | Yes | No | No | Yes | No | Yes | Yes |
| ECG | AF, AFL, SND, 1AVB, PM |
NSR | SB | AFL, 1AVB, RBBB |
PAF, iRBBB,SB, PM |
NSR | SR,RBBB, 3AVB,PM |
| RV Dilation | Severe | No | Mild | Severe | Mild | Moderate-Severe | Severe |
| RV Bulging | Yes | No | No | No | Yes | No | Yes |
| RV hypokinesia | Diffuse | No | Regional | Diffuse | Diffuse | Diffuse | Diffuse |
| RV Function | RVEF 21% | Normal | RVEF 43% | RVFS6% | RVFS33% | RVFS17% | Mild reduction |
| LV Dilation (LVEDD) | No | No | No | No | No | No | No |
| LV EF% | 35 | Normal | 40 | 66 | 57 | 63 | 52 |
| Late Potentials on SAECG |
NA | No | Yes | No | No | NA | Yes |
| LBBB type VT (NSVT) |
Yes | No | No | Yes | No | Yes | No |
| LBBB type VT (Sustained VT) |
Yes | No | No | Yes | No | Yes | No |
| Ventricular extrasystoles >1000/24 hours |
NA | No | No | Yes | No | Yes | No |
| Clinical status (Age) | AICD OHT (50) |
Stable (67) | AICD Stable (39) |
AICD OHT (65) |
Stable (82) | AICD OHT (47) |
HF (55) AICD |
| Fibrous or fibrofatty replacement |
Yes | NA | NA | Yes | Yes | Yes | NA |
Family and Individual numbers from Figure 1; Abbreviations in table: AF, atrial fibrillation; LAFB, left anterior fascicular block; PM, pacemaker; 1AVB, 2AVB and 3AVB, 1st, 2nd and 3rd degree atrio-ventricular block; EW, epsilon waves; PVCs, premature ventricular contractions; 1AVB, 1st degree atrio-ventricular block; SSS, sick sinus syndrome; SB, sinus bradycardia; NSR, normal sinus rhythm; AFL, atrial flutter; SND, sinus node dysfunction; RBBB, right bundle branch block; iRBBB, incomplete RBBB; RV, right ventricular; RVFS, right ventricular fractional shortening; RVEF, right ventricular ejection fraction; LV, left ventricular; LVEF, left ventricular ejection fraction; SAECG, signal averaged ECG; LBBB, left bundle branch block; VT, ventricular tachycardia; HF, heart failure; AICD, automated implantable cardioverter defibrillator; OHT, orthotopic heart transplant; NA, not applicable/no data; M, major criteria; m, minor criteria; PAF, paroxysmal atrial fibrillation; HFd, death for heart failure.
Phenotype analysis
Among the seven families with TTN variants, a total of fourteen individuals (9 males and 5 females) were found to be carriers with twelve meeting 2010 criteria for ARVC.23 Details of the phenotype features are reported in Table 2 and the pedigrees of the seven kindreds are shown in Figure 1. Two of the fourteen TTN variant carriers did not meet ARVC criteria: DNRVC008-III:4 and TSRVD001-IV:3, who had atrial fibrillation and conduction disease necessitating a pacemaker, indicating incomplete penetrance.
Mean age of onset of the TTN carrier cohort was 38 years, ranging from 19 to 59 years, thus indicating variability of expression. Most patients presented with symptoms related to ventricular arrhythmias (palpitations 9, syncope 7) and heart failure (dyspnea on exertion 8). Five patients (36% of carriers) had evidence of left-ventricular involvement with decreased left-ventricular ejection fraction or enlargement on echocardiogram, and 9 (64%) showed progressive myocardial dysfunction (Table 2), leading to heart transplant in 6 (43%), or causing death for progressive intractable heart failure at a mean age of 55 years (range 50 to 66, Table 2). Indications for heart transplant were intractable predominantly right heart failure (5 cases) and intractable ventricular arrhythmias (1 case). Two patients were resuscitated from cardiac arrest (DNRVD002-IV:3, and DN006-II:2). There was a history of sudden cardiac death in 5 out of 7 families. The electrocardiogram showed epsilon waves in 3 cases, all belonging to the large kindred TSRVD001 (III-3, IV:4, IV:7, Figure 4). Remarkably, TTN variant carriers had frequent conduction disease (11/14 patients in 6 families) in the course of their follow-up, leading to a permanent pacemaker in 8/14 patients (3/7 probands) (Table 2). On the contrary, in the group of ARVC patients non-carriers of TTN variants, only 2 developed bradyarrhythmias requiring a pacemaker (2/31 probands, 2/50 affected family members, p<0.001, Fisher’s exact test). Atrial arrhythmias (atrial fibrillation and atrial flutter) were also common (8/14 in 6 families) and were associated with right atrial dilation in three cases where measurement data were available (TSRVD001-V:3, DNRVD002-IV:3, DNRVD006-II:2). Five patients received implanted defibrillators due to histories of ventricular arrhythmias. A detailed description of the phenotype of family TSRVD001 is reported in the online supplement.
Figure 4.
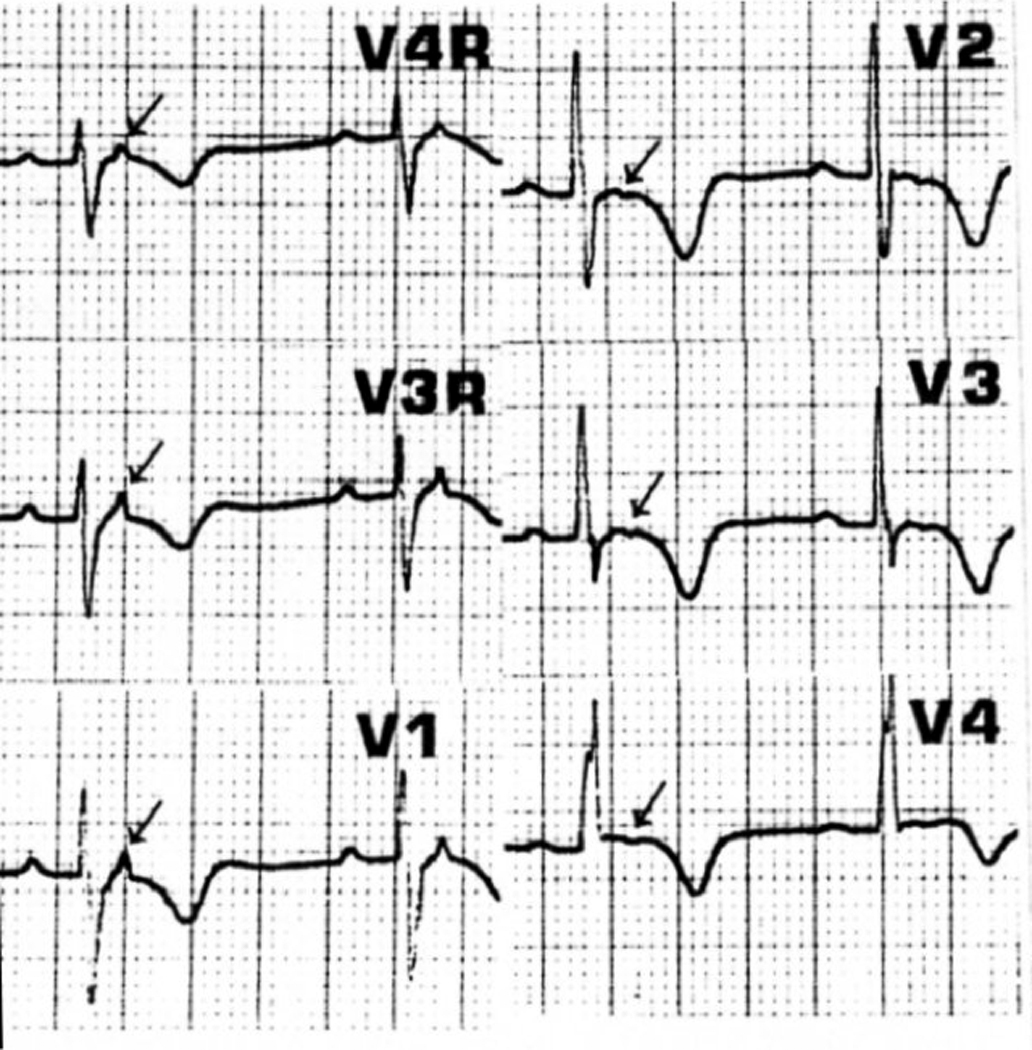
Electrocardiogram from individual IV-7 from family TSRVD001 (Thr2896Ile) with arrows indicating epsilon waves.
Protein Analysis
The Thr2896IIe residue exchange substitutes a hydrophobic isoleucine for the hydrophilic threonine in the Ig10 immunoglobulin domain; sequence alignment with known Ig domain structures shows that the mutated residue is located near the A’ β-strand of Ig10 (Figure 3A and B). Ig10 is found in the proximal tandem Ig repeat region near the Z-disk30, 31 (Figure 5) a region in titin that is not known to contain binding sites for sarcomeric or regulatory proteins, but instead is critical for the generation of passive cardiomyocyte tension13, 32. Evidence suggests that passive tension results from unbending of sequences that link the Ig domains, whereas the Ig domains themselves remain folded33, 34. Steered molecular dynamics simulations have shown that the hydrogen bond network between parallel A’ and G β-strands is crucial for determining the mechanical stability of Ig domains35 and atomic force microscopy experiments have shown that point mutations in A’ strand residues can reduce the force needed to unfold Ig domains.29, 36, 37 Therefore, we hypothesized that the mutation near the A’ β-strand in Ig10 reduces the mechanical stability of Ig10, and increases the probability of domain unfolding. To examine this experimentally, we produced the wild-type (WT) and the Thr2896Ile mutant version of Ig10 in E.coli.27 Expressed proteins were separated on 7% native tris-glycine polyacrylamide gels. The reduced electrophoretic mobility of mutant Ig10 that was found indicates a larger hydrodynamic radius in the mutant, suggestive of a less compact folded state (Figure 6A). To study this further, we determined next the intrinsic fluorescence of the domain’s single tryptophan residue (W), located in the hydrophilic core fold of Ig10, as a function of the concentration of the chemical denaturant guanidine chloride (GuCl). In the presence of 3M GuCl the fluorescence levels of the two protein types were both low, indicating that W was exposed to a hydrophilic surrounding in a non-folded state (Figure. 6B). In the absence of denaturant, the fluorescence of the mutant Ig10 was lower than for WT Ig10, suggesting that the central W was less buried inside the Ig10 hydrophobic core (Figure. 6B). We also applied HSQC (Heteronuclear single quantum correlation) NMR to determine if indeed mutant Ig10 has a less well-folded state. The 13C15N labelled mutant Ig10 exhibited two sets of NMR signals: one set with chemical shifts similar to those present in the WT domain and a second set with less chemical shift dispersion (Figure 6C; spectra of WT (red) and mutant (blue) Ig10). Therefore, the mutant Ig10 domain coexists as a mixture of well-structured and less structured sequences in solution. Taken together, these data demonstrate that the mutant domain is less stable than the WT domain, and is more likely to adopt an unfolded state. Finally we also compared the susceptibility for proteolytic degradation of the WT and mutant titin peptides. Incubation of Ig7 to Ig13 spanning fragments with a low level of the protease trypsin was found to cause more prominent proteolysis in the sample containing the centrally located mutant Ig10 (Fig. 6D and E).
Figure 5.
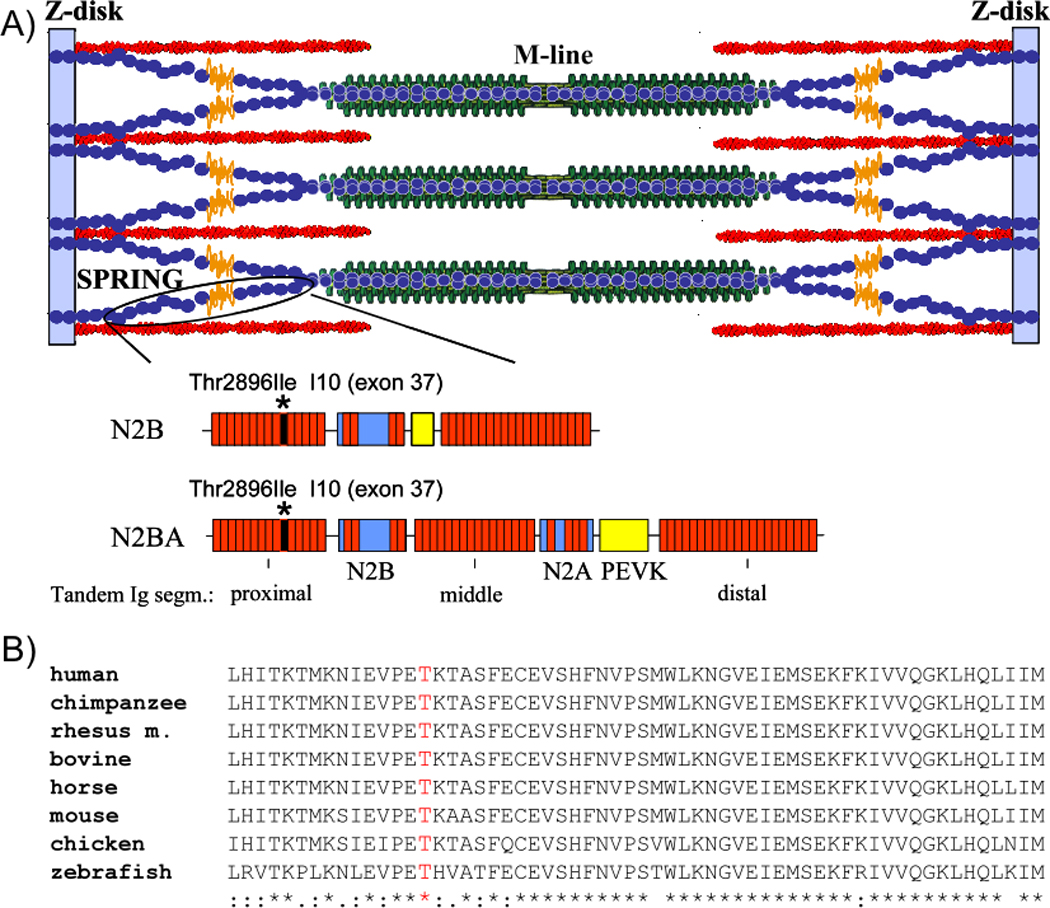
A) Schematic of titin in the sarcomere; sequences of spring regions of the two main cardiac isoforms (N2B and N2BA titin) are indicated schematically at bottom. In both isoforms the spring consists of tandemly arranged immunoglobulin-like domains (red), the N2B element and the PEVK. The N2BA isoform also contains the N2A element. The main ARVC mutation identified in this work (Thr2896Ile) is located in Ig10 (encoded by exon 37). B) Ig10, including T2896, is highly conserved in a wide range of species.
Figure 6.
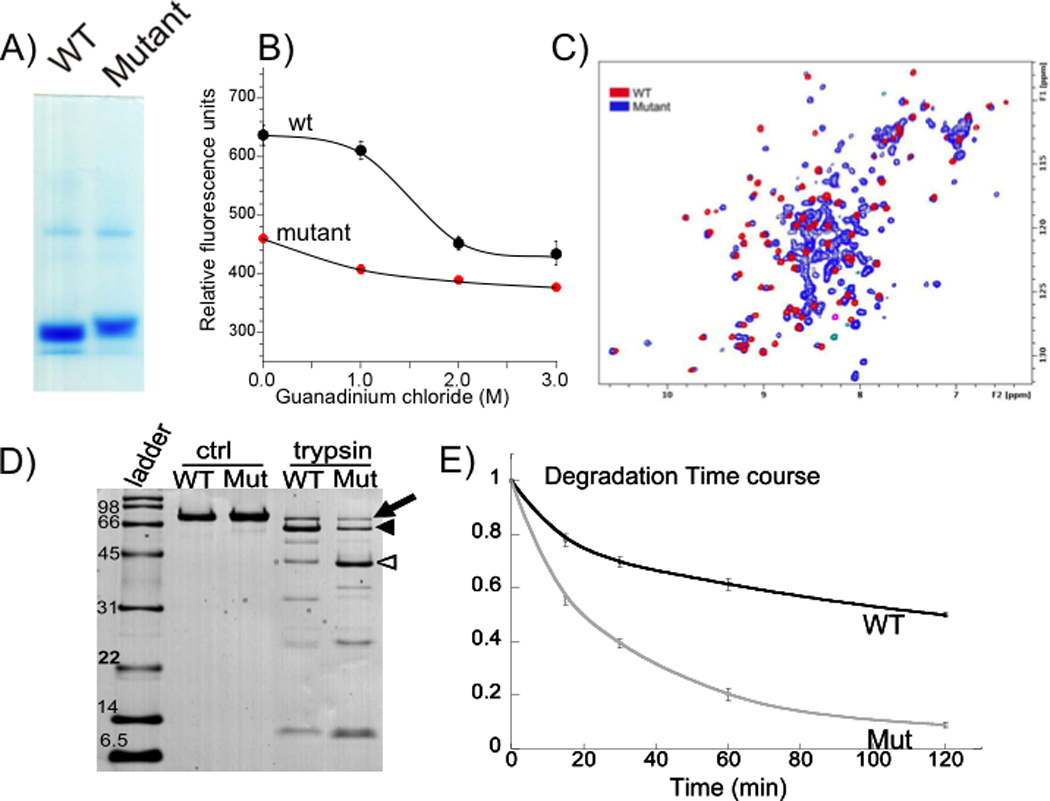
A) Reduced electrophoretic mobility of mutant Ig10 Thr2896Ile domain in native gel indicates a larger hydrodynamic radius, which is typical for unfolded proteins. B) Tryptophan fluorescence at various GuCl concentrations. Fluorescence in the absence of GuCl is much lower in mutant Ig10 than WT Ig10, but this difference is reduced as denaturant concentration increases. This suggests that under native conditions tryptophan is more exposed to hydrophilic conditions in mutant than WT Ig10. C) 15N HSQC NMR spectra of WT (red) and mutant Ig10 (blue). In the WT protein spectrum, we observe all expected peaks (one peak per backbone NH, Trp indole NH and two peaks per Gln/Asn NH2 moieties). The chemical shift dispersion is in accord with that expected for an IG domain fold protein. In contrast, the mutant exhibits at least two sets of NMR signals: one set with chemical shifts (and structure) similar to that of the WT domain and one or more additional sets with less chemical shift dispersion, which indicates the presence of additional unstructured molecules in the mutant domain sample. D) SDS-PAGE of recombinant WT and mutant Ig 7–13 fragments (I10 with Thr2896Ile mutation), in the absence of trypsin (ctrl) and after one hour of trypsin incubation (~25:1 Ig:trypsin by weight). The full-length peptides (arrow) have a MW of ~75 kDa. Trypsin treatment causes severe proteolysis and the appearance of a ~ 60 kDa fragment (filled arrowhead) which likely reflects the cleavage of one of the terminal Ig domains. In addition there is a prominent ~35–40 kDa degradation product (open arrowhead) that must result from a cleavage site somewhere in the middle of the Ig 7–13 protein; this fragment is much more prominent in the mutant sample. A protein MW ladder is shown for reference. E) Ratio of large Ig fragments (full-length protein + 60 kDa fragment) to all Ig fragments, by OD, as a function of trypsin incubation time. Degradation is more severe in the mutant protein. Error bars: ± SE; n = 4 for all time points except t = 120 min (n =3).
Discussion
Titin is the largest protein known and its 363 exons, located at chromosome position 2q31, span ~0.3 Mb.13, 14, 16 Given the huge size of the titin gene, a large number of disease causing mutations are expected, but compared to other and much smaller sarcomeric proteins (for example >100 mutations in the 15 times smaller myosin heavy chain38), relatively few titin mutations have been reported until now. Gerull et al. described TTN mutations segregating with DCM, a 2 base insertion and a 1-bp deletion both causing a truncated A-band titin, and a Trp930Arg mutation in the Z-disk.39, 40 Matsumoto et al. reported DCM TTN mutations with Arg743Val and Val54Met Z-disk region mutations at the site where titin interacts with several Z-disk proteins, Glu4053ter and Ser4465Asn mutations, which were hypothesized to alter the N2B cardiac-specific spring element, and an Arg25618Gly mutation mapping to the inextensible A-band.19, 41 The investigators suggested a decreased binding (loss-of-function) of mutant titin to titin-binding proteins. On the other hand, HCM-causing mutations Arg25618Gly and Ser3799Tyr appear to increase binding to alpha-actinin and FHL2, respectively (gain-of-function).20, 41 Several mutations in and near the terminal exon of TTN cause tibial muscular dystrophy that does not involve any apparent cardiac signs or symptoms. The mechanisms by which constitutively expressed titin mutations induce muscle type-specific myopathies are currently unknown and warrant future study.
In the present work we sequenced titin in a cohort of ARVC families and discovered novel TTN variants in 18% of the families. The most prominent variant was Thr2896Ile which showed strong segregation evidence for being pathogenic as it was present in 9 confirmed/obligate ARVC subjects including 2 fifth-degree relatives, absent in 300 cardiomyopathy and 400 control chromosomes, and scored ‘intolerant’ by SIFT and PolyPhen predictive algorithms. The phenotype of ARVC TTN variant carriers was characterized by progressive right ventricular dilatation and dysfunction, leading to death or cardiac transplantation. Right ventricular pathology, when available, showed fibrous or fibrofatty replacement. Common among TTN-ARVC families were reports of syncope and sudden unexplained death, which were often the presenting symptom in affected individuals. One subject who had normal biventricular function and did not meet full ARVC diagnostic criteria developed atrial fibrillation and early conduction system disease (TSRVD001-IV:3), findings more typical of advanced ARVC.42 This could suggest that conduction system disease and atrial arrhythmia could be early features of the TTN phenotype. Atrial fibrillation and conduction disease, which have been described previously in ARVC, 43 were more common in our population among TTN variant carriers than non-carriers. We speculate that these phenotypic findings in ARVC might therefore implicate underlying TTN defects. The unusually high proportion of these findings in our study could suggest that there may be phenotypic overlap between ARVC and other conduction system disorders in carriers of TTN mutations. The most remarkable finding was the high prevalence of conduction disease in these patients, ranging from sinus node dysfunction to atrio-ventricular and intraventricular blocks, requiring a permanent pace-maker in over 50% of affected carriers. These findings along with the presence of atrial arrhythmia suggest pathologic changes beyond the right ventricular wall, possibly extending to the atrial myocardium. No evidence of involvement of other organs or systems was found in our cohort, as reported for non-desmosomal genes.44, 45 Segregation data on the remaining seven TTN variants was extremely limited in other families in our study and although it is likely that some of these variants may represent pathogenic changes, to conclusively establish this requires future studies.
The Thr2896Ile variant appears to have a functional consequence and based on our in vitro data we predict that at low sarcomere stretch where passive force is low, the mutant Ig10 is partially folded, and as sarcomere stretch increases and force rises, the likelihood of unfolding will exceed that of the wild-type domain. The effect of unfolding of a single domain on passive force will be small considering that there are a large number of Ig domains (38 in N2B titin and ~50 in N2BA titin) that make up the extensible spring region, in addition to the N2B and PEVK spring elements.32 Using a serially-linked wormlike chain model for simulating force – extension curves of titin we calculated that at a sarcomere length of 2.2 µm the reduction is <3 % of the total passive force (for details, see34). This is a relatively small reduction and it seems unlikely that force reduction due to unfolding of a single domain initiates the disease pathology. Instead the trypsin degradation experiments (Fig. 6 D and E) suggest that the mutant domain is more vulnerable to proteolysis and degradation, and this increased vulnerability might be an essential step in the disease pathology. Unfolding of mutant Ig10 and ensuing titin proteolysis will be most prominent in those regions of the heart where titin’s spring elements are most extended. The thinner-walled stress-susceptible right ventricle might be such a region, especially at the transitional junction of the intercalated disk, where titin’s strain might far exceed that encountered elsewhere in the heart.11 Thus we propose that titin mutations found in ARVC lower Ig domain stability, and that this leads to titin degradation that initiates the pathological process that eventually leads to ARVC. Future work is needed to test this hypothesized disease mechanism.
Conclusions
We discovered novel variants in the giant sarcomeric protein titin that are associated with ARVC. The phenotype of TTN variant carriers includes severe biventricular dysfunction, conduction disease, and sudden death. The Thr2896Ile ARVC TTN mutation has increased susceptibility to proteolysis and we propose that this is a primary step in the disease pathology. Considering titin’s critical role in multiple processes, including increased calcium sensitivity with stretch, cell signaling, and protein turnover17, 46, 47, there are multiple pathways by which titin proteolysis might trigger the ARVC disease pathology, warranting future follow-up research. The involvement of titin and sarcomeric dysfunction in ARVC further expands the causes of ARVC beyond desmosomal proteins. Interestingly, pathological changes with fibrofatty replacement of the myocardium were previously reported in non-ARVC patients with desmin, myosin heavy chain and PRKAG2 mutations. 44, 45, 48 That extra-desmosomal mutations can induce fibrofatty replacement suggests a more expanded view of the triggers of this myocardial reparative process. Importantly, the discovery of titin mutations in ARVC might allow the design of novel therapeutic strategies, such as the design of drugs that reduce proteolytic degradation or enhance Ig domain stability.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a serious inherited myocardial disease characterized by fibrofatty replacement of the myocardium and a predisposition to cardiac arrhythmias and sudden death. Although genetic mutations affecting a number of protein components of intercalated discs, providing structural and electrical connections between contracting myocytes, have been implicated in ARVC in the majority of cases, the underlying genetic defect is unknown. In this study we evaluated the giant muscle protein titin as a candidate ARVC gene because of the known functional link between titin and elements of the intercalated disc and the prior finding of an ARVC genetic locus mapping to the titin region on chromosome 2. Screening of all 312 cardiac titin gene exons detected several variants including one (Thr2896Ile) which showed strong genetic segregation evidence for being pathogenic. In vitro studies of the Thr2896Ile mutation support that structural impairment of the titin spring is a likely cause of ARVC and that this constitutes a novel mechanism underlying myocardial remodeling and sudden cardiac death.
Supplementary Material
Acknowledgments
Funding Sources: NIH N01-HV-48194, RO1 HL69071, RO1 HL062881, MO1 #RR00051-1575, 1K23Hl67915-01A1; American Heart Association 0150453N and 0250271N, Muscular Dystrophy Association PN0007-056, Wilhelm-Müller Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none
References
- 1.Awad MM, Dalal D, Tichnell C, James C, Tucker A, Abraham T, Spevak PJ, Calkins H, Judge DP. Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum Mutat. 2006;27:1157. doi: 10.1002/humu.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 3.McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lunqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br. heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Maertini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia. A study involving nine families. J Am Coll Cardiol. 1988;12:1222–1228. doi: 10.1016/0735-1097(88)92603-4. [DOI] [PubMed] [Google Scholar]
- 5.den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP. Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. doi: 10.1161/CIRCGENETICS.109.858217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Simon F, Bessirard V, Roux-Buisson N, Hebert JL, Azarine A, Casset-Senon D, Rouzet F, Lecarpentier Y, Fontaine G, Coirault C, Frank R, Hainque B, Charron P. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 7.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 11.Bennett PM, Maggs AM, Baines AJ, Pinder JC. The transitional junction: a new functional subcellular domain at the intercalated disc. Mol Biol Cell. 2006;17:2091–2100. doi: 10.1091/mbc.E05-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 13.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 14.LeWinter MM, Wu Y, Labeit S, Granzier H. Cardiac titin: structure, functions and role in disease. Clin Chim Acta. 2007;375:1–9. doi: 10.1016/j.cca.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. J Physiol Sci. 2008;58:151–159. doi: 10.2170/physiolsci.RV005408. [DOI] [PubMed] [Google Scholar]
- 16.LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 18.Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500. doi: 10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, Hiroe M, Marumo F, Imaizumi T, Yasunami M, Kimura A. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 20.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun. 1999;262:411–417. doi: 10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 21.Greaser ML. Stressing the giant: a new approach to understanding dilated cardiomyopathy. J Mol Cell Cardiol. 2009;47:347–349. doi: 10.1016/j.yjmcc.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, Sachdev B, Rowland E, Elliott PM, McKenna WJ. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. doi: 10.1016/s0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 23.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–814. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet. 2006;38:375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- 25.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogomolovas J, Simon B, Sattler M, Stier G. Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein Expr Purif. 2009;64:16–23. doi: 10.1016/j.pep.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Fraternali F, Amodeo P, Musco G, Nilges M, Pastore A. Exploring protein interiors: the role of a buried histidine in the KH module fold. Proteins. 1999;34:484–496. doi: 10.1002/(sici)1097-0134(19990301)34:4<484::aid-prot8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE, Fernandez JM. Point mutations alter the mechanical stability of immunoglobulin modules. Nat Struct Biol. 2000;7:1117–1120. doi: 10.1038/81964. [DOI] [PubMed] [Google Scholar]
- 30.Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res. 2000;86:1114–1121. doi: 10.1161/01.res.86.11.1114. [DOI] [PubMed] [Google Scholar]
- 31.LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121:2137–2145. doi: 10.1161/CIRCULATIONAHA.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trombitas K, Wu Y, Labeit D, Labeit S, Granzier H. Cardiac titin isoforms are coexpressed in the half-sarcomere and extend independently. Am J Physiol Heart Circ Physiol. 2001;281:H1793–H1799. doi: 10.1152/ajpheart.2001.281.4.H1793. [DOI] [PubMed] [Google Scholar]
- 33.Trombitas K, Wu Y, McNabb M, Greaser M, Kellermayer MS, Labeit S, Granzier H. Molecular basis of passive stress relaxation in human soleus fibers: assessment of the role of immunoglobulin-like domain unfolding. Biophys J. 2003;85:3142–3153. doi: 10.1016/S0006-3495(03)74732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe K, Nair P, Labeit D, Kellermayer MS, Greaser M, Labeit S, Granzier H. Molecular mechanics of cardiac titin's PEVK and N2B spring elements. J Biol Chem. 2002;277:11549–11558. doi: 10.1074/jbc.M200356200. [DOI] [PubMed] [Google Scholar]
- 35.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fowler SB, Best RB, Toca Herrera JL, Rutherford TJ, Steward A, Paci E, Karplus M, Clarke J. Mechanical unfolding of a titin Ig domain: structure of unfolding intermediate revealed by combining AFM, molecular dynamics simulations, NMR and protein engineering. J Mol Biol. 2002;322:841–849. doi: 10.1016/s0022-2836(02)00805-7. [DOI] [PubMed] [Google Scholar]
- 37.Fraternali F, Pastore A. Modularity and homology: modelling of the type II module family from titin. J Mol Biol. 1999;290:581–593. doi: 10.1006/jmbi.1999.2876. [DOI] [PubMed] [Google Scholar]
- 38.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 39.Gerull B, Atherton J, Geupel A, Sasse-Klaassen S, Heuser A, Frenneaux M, McNabb M, Granzier H, Labeit S, Thierfelder L. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med. 2006;84:478–483. doi: 10.1007/s00109-006-0060-6. [DOI] [PubMed] [Google Scholar]
- 40.Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S, Frenneaux M, Thierfelder L. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–204. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto Y, Hayashi T, Inagaki N, Takahashi M, Hiroi S, Nakamura T, Arimura T, Nakamura K, Ashizawa N, Yasunami M, Ohe T, Yano K, Kimura A. Functional analysis of titin/connectin N2-B mutations found in cardiomyopathy. J Muscle Res Cell Motil. 2005;26:367–374. doi: 10.1007/s10974-005-9018-5. [DOI] [PubMed] [Google Scholar]
- 42.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 43.Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G, Di Lenarda A, Morgera T, Mestroni L, Sinagra G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry. Eur Heart J. 2011;32:1105–1113. doi: 10.1093/eurheartj/ehr040. [DOI] [PubMed] [Google Scholar]
- 44.van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, Boven L, van den Heuvel F, Veenstra-Knol HE, Saffitz JE, Hofstra RM, van den Berg MP. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6:1574–1583. doi: 10.1016/j.hrthm.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 45.Roberts JD, Veinot JP, Rutberg J, Gollob MH. Inherited cardiomyopathies mimicking arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2010;19:316–320. doi: 10.1016/j.carpath.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Linke WA, Kruger M. The giant protein titin as an integrator of myocyte signaling pathways. Physiology (Bethesda) 2010;25:186–198. doi: 10.1152/physiol.00005.2010. [DOI] [PubMed] [Google Scholar]
- 47.Witt CC, Ono Y, Puschmann E, McNabb M, Wu Y, Gotthardt M, Witt SH, Haak M, Labeit D, Gregorio CC, Sorimachi H, Granzier H, Labeit S. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J Mol Biol. 2004;336:145–154. doi: 10.1016/j.jmb.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 48.Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2009;7:1058–1064. doi: 10.1016/j.hrthm.2010.04.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.