

Abstract
Environmentally persistent free radicals (EPFRs) have previously been observed in association with combustion-generated particles and airborne PM2.5 (particulate matter, d < 2.5um). The purpose of this study was to determine if similar radicals were present in soils and sediments at Superfund sites. The site was a former wood treating facility containing pentachlorophenol (PCP) as a major contaminant. Both contaminated and non-contaminated (just outside the contaminated area) soil samples were collected. The samples were subjected to the conventional humic substances (HS) extraction procedure. Electron paramagnetic resonance (EPR) spectroscopy was used to measure the EPFR concentrations and determine their structure for each sample fraction. Analyses revealed a ~30× higher EPFR concentration in the PCP contaminated soils (20.2 × 1017 spins/g) than in the non-contaminated soil (0.7 × 1017 spins/g). Almost 90% of the EPFR signal originated from the Minerals/Clays/Humins fraction. GC-MS analyses revealed ~6500 ppm of PCP in the contaminated soil samples and none detected in the background samples. Inductively coupled plasma-atomic emission spectrophotometry (ICP-AES) analyses revealed ~7× higher concentrations of redox-active transition metals, in the contaminated soils than the non-contaminated soil. Vapor phase and liquid phase dosing of the clays/minerals/humins fraction of the soil with PCP resulted in an EPR signal identical to that observed in the contaminated soil, strongly suggesting the observed EPFR is pentachlorophenoxyl radical. Chemisorption and electron transfer from PCP to transition metals and other electron sinks in the soil are proposed to be responsible for EPFR formation.
Introduction
Soil-pollutant interactions greatly influence the bioavailability of contaminants, which in turn dictates their fate and persistence (1-4). Some soils can retard the contamination of the ecosystem by sequestering and binding contaminants to the clay/mineral and soil organic matter (SOM) components (1, 5, 6). However, this binding can also lead to the formation of additional pollutants, e.g., chlorinated phenols and anisoles, on clay-based systems have been observed to form polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/F) when subjected to thermal treatment (7-12). Chlorinated phenols have also been observed to form chlorinated diphenoquinones through coupling at the para-positions (13, 14). Detection of these pollutants suggests the possible formation of organic radicals as intermediates. Aromatic radical cation formation has been reported for chlorinated phenols on copper(II)-smectite via electron transfer from the aromatic species, reducing Cu(II) to Cu(I) (7, 8). The formation of radicals can affect the toxicity of the matrix in currently unpredictable ways. Thus, it is important to understand both the role of these potential radicals as intermediates in the formation of new pollutants and their toxicity as pollutants in their own right.
We have previously demonstrated that radicals are formed by interactions between substituted aromatic species and transition metals associated with combustion-generated particles under post combustion conditions via the general scheme depicted in Figure 1 (15 - 19). These observed radicals are persistent, i.e., they do not decompose easily and they resist further oxidation and other chemical reactions. Thus, we refer to them as environmentally persistent free radicals (EPFRs). EPFRs were originally thought to only be present in emissions from combustors, where they can be formed by thermally activated reactions in the post-flame, cool zone. However, they have also been observed in airborne fine particulate matter (PM2.5), which is known to be primarily combustion-generated or combustion-derived (17, 20). Their structures and lifetimes were very similar to those generated in our laboratory under combustion conditions. This suggested EPFRs might be more ubiquitous than originally thought.
Figure 1.
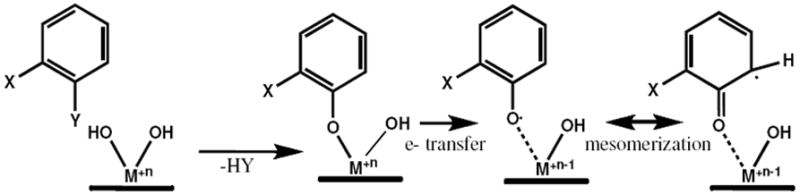
Simplified model of EPFR formation from a substituted aromatic on a metal oxide surface via chemisorption and electron transfer.
The EPFRs of chlorophenols and chlorobenzenes were proposed to form by a mechanism of chemisorption at transition metal oxide sites and electron transfer from the organic adsorbate to the transition metal, resulting in the formation of the organic EPFR and the reduced transition metal. These EPFRs were observed to persist in ambient air for days (15-19). They were also shown to induce pulmonary and cardiovascular dysfunction through induction of oxidative stress (15, 17, 18). In these studies, the EPFRs were formed via thermal reactions at temperatures between 150 and 500 °C, with reaction times of a few seconds. Since soils contain the same transition metals and the reaction times of contaminants within soils are years, rather than seconds, the question was whether EPFRs could also be formed in soils at ambient temperatures.
For this study, soil samples were collected from a Superfund wood treating site, for which pentachlorophenol (PCP) was the major contaminant. PCP is hydrophobic and tends to sorb onto solid matter (14). Because chlorophenols were previously demonstrated to form EPFRs via thermal reactions (19), PCP in a clay soil appeared to be an ideal choice to explore the potential for formation of EPFRs in contaminated soils.
Materials and Methods
Site Description
The contaminated soils were obtained from a 4 acre wood treatment facility used for treating railroad ties and poles from 1946-1991 (21). The facility utilized creosote in the preservation process until the 1970’s, at which point pentachlorophenol (PCP) was added to the process and used until the 1980’s, when PCP was used exclusively until the facility closed. 30,723 gallons of PCP and creosote remained on site in tanks until the Environmental Protection Agency (EPA) removed them in 1994 (21).
Soil Sampling and Preparation
The contaminated soils were randomly collected from nine different locations inside the perimeter of the once-standing complex. At each location, soil samples were collected at three different depths; top (0-10 cm), mid (>10-20 cm), and bottom (>20-30 cm). The background non-contaminated soil samples were collected approximately 500 feet outside of the contaminated area. All samples were placed in sealable plastic bags to prevent outside contamination and assure safe transport back to the lab. Prior to chemical analyses, the soil samples were dried in an oven for 12 hrs at 55°C to remove water. They were then ground to a homogeneous powder and sieved through a USA Standard Testing Sieve No. 120 (125μm opening) to eliminate any coarse-sized mineral and vegetative matter. The soil sample prepared in this way is referred to as the whole soil (WS).
Humic Substances (HS) Extraction Method
The outline of the extraction procedure, the designation of samples, and the testing performed on each sample are summarized in Figure 2. The HS extraction method was in accordance to the procedure recommended by the International Humic Substances Society (IHSS) (22-28). Briefly, 2.0 g of soil sample were extracted with 20 mL of 0.1 M HCl. The pH of the solution was adjusted between 1.0 and 2.0 with 1.0 M HCl. The soil/HCl mixture was shaken for 1 hr and the suspension was allowed to settle. The mixture was then centrifuged at 1478 × g for 10 min, and acid soluble supernatant was separated from the acid insoluble precipitate.
Figure 2.
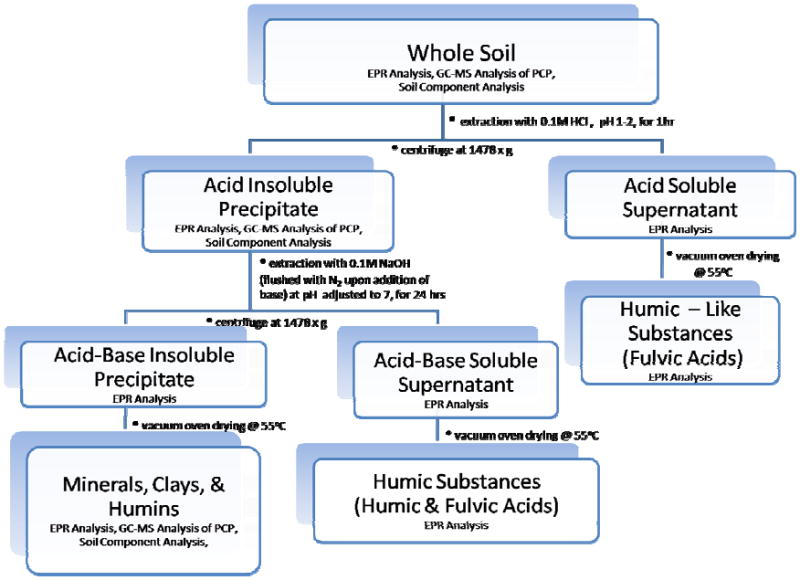
Flow chart for extraction and analysis of contaminated and non-contaminated soils.
The acid insoluble precipitate was neutralized with 1.0 M NaOH to a pH of 7.0, and a 20 mL of 0.1 M NaOH was added under a nitrogen atmosphere (22-28). The mixture was shaken for 24 hrs and allowed to settle overnight. Acid-base soluble (humic and fulvic acid) supernatant and acid-base (clay/minerlal/humin) insoluble precipitate were separated through centrifugation at 1478 × g for 10 min (22-28).
The acid soluble supernatant, the acid insoluble precipitate, the acid-base soluble supernatant, and the acid-base insoluble precipitate were dried and evaporated in a vacuum and oven-dried in circulating air at 55°C prior to EPR analysis (22-28).
Electron Paramagnetic Resonance (EPR) Spectroscopic Analysis
The whole soil samples and the four solid samples obtained from the HS extraction method were placed in a high purity quartz EPR tube and analyzed at room temperature in a Bruker EMX – 10/2.7 EPR Spectrometer with X-band microwave frequency of 9.72 GHz, microwave power of 2.02 mW, spectral window of 1000 Gauss, and modulation amplitude of 4.00 Gauss.
The liquid samples of acid soluble and acid-base soluble supernatants were sampled and analyzed via EPR. To minimize the amount of sample and optimize sensitivity, sample collection was performed using a capillary sealed with critoseal, which was subsequently (the capillary with sample) placed in a high purity quartz EPR tube.
GC-MS Analysis of Pentachlorophenol (PCP)
200 – 250 mg of whole soil, acid insoluble precipitate, or minerals/clays/humins were placed in scintillation vials, and 4-methyl-2-pentanone was added as the extracting solvent. 250 μL aliquots were placed in an amber vial, to which 250 μL of derivatizing agent, N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA), and 500 μL of extracting solvent, tert-butylmethyl ether (TBME), were added, making up a total volume of 1000 μL. The vial was capped using Teflon/Silicone 11 mm crimp caps and inverted to mix. The vial was then placed in a pre-heated heating block for 30 minutes at 76°C (±5°C), and subsequently cooled to room temperature for GC-MS analysis. These sample solutions were verified to contain pentachlorophenol concentration that falls within the range of our calibration curve.
An Agilent 6890 Gas Chromatograph (GC) fitted with a 5973 Mass Selective Detector (MSD) in the manual injection mode was used with the following parameters: column type - J&W DB5 MS 60 m × 0.25 mm i.d. × 0.25 μm, preceded by 5 m of 0.25 mm deactivated retention gap; injection type and temperature - splitless / 250°C; column temperature program - initial 60°C hold for 6 minutes, ramp 10°C/min to 180°C, 15°C/min to 300°C, hold for 2 minutes; total run time was 28.0 minutes; carrier gas - Helium; transfer line temperature - 280°C; injection volume - 1 μL; column flow - 1 μL/min (constant flow); solvent Delay - 14 minutes; MS source temperature - 230°C; MS quadrupole temperature - 150°C; MS mode - SIM; ion dwell time - 100 ms.
Gas Phase Dosing of Soil with PCP
The minerals/clays/humins fraction of the soil samples were exposed to the vapors of the pentachlorophenol adsorbate using a custom-made vacuum exposure system presented in Figure 3, consisting of a vacuum gauge, dosing vial port, equilibration chamber and two reactors. A vacuum valve controlled the adsorbate flow and vacuum of the two outlets from the equilibration chamber. The equilibration chamber was thermocouple controlled to maintain a preset dosing temperature. A detachable two bulb-shaped pyrex reactor with a protruding suprasil quartz EPR tube as a side arm for EPR spectral measurements was attached to the system. The bulb-shaped reactors, containing the soil samples to be dosed, were placed in a vertically oriented small tube furnace. Prior to chemisorption, the samples were evacuated to 10-2 torr in order to remove other interfering organic contaminants. The vapors of pentachlorophenol were introduced at 10 torr at the desired temperatures (25°C - 300°C) for 5 minutes. The samples were then evacuated to 10-2 torr to remove any residual physisorbed dosant and sealed with a vacuum tight PFE stopcock. The samples were cooled to room temperature prior to EPR measurements. Control samples were prepared by exposing clay/mineral/humins to the same conditions, except without dosing of PCP.
Figure 3.
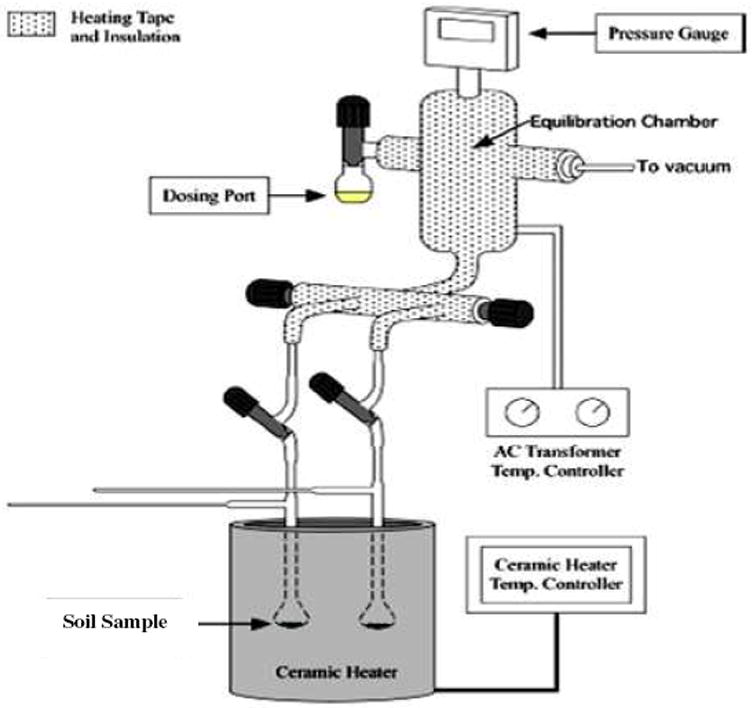
Pictorial diagram of two reactor vacuum dosing set up.
Liquid Phase Dosing of Soil with PCP
1.00 mg/mL of PCP in benzene and 0.008 mg/mL of PCP in water were dosed into the minerals/clays/humins fraction of the contaminated and non-contaminated soil samples. Blanks were produced by dosing with pure benzene and water. Individual 20 mL aliquots of solvent were added to 100 mg of acid-base insoluble precipitates. One set of samples was exposed with the prepared solutions and solvents for 24 hrs and the second set for 26 days. After exposure, the samples were centrifuged, the precipitate separated and dried, and analyzed by EPR as previously discussed.
Results and Discussion
Detection of EPFRs in Contaminated Soil
The EPR spectrum of the contaminated, whole soil exhibited a large singlet superimposed on a weaker six-line signal, with hyperfine splitting of 89:91:94:96:97 as depicted in Figure 4. The most striking feature of this figure is the strong central signal exhibited by the contaminated soil. The non-contaminated whole soil exhibited a much weaker doublet signal at the center of the spectrum. The narrow singlet and doublet signals observed for the contaminated and non-contaminated soils have g factors 2.00309 and 2.00340 with ΔHp-p of 6.4 G and 8.0 G, respectively; these values are typical of organic radicals.
Figure 4.
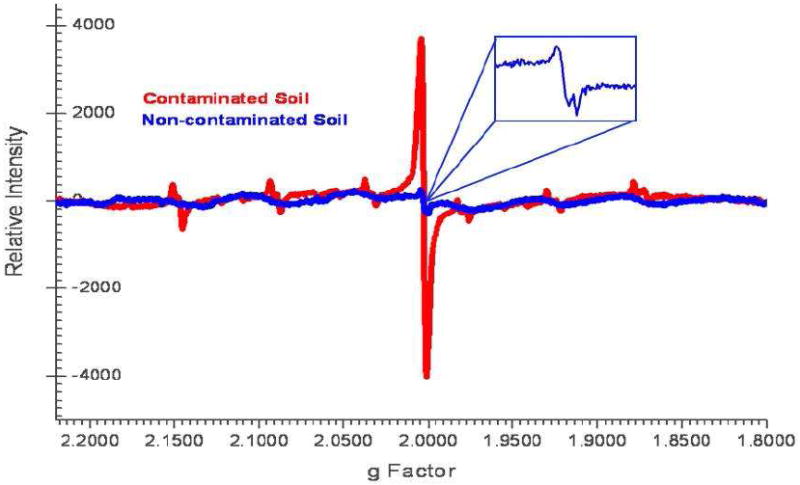
EPR spectra of the whole soils of contaminated (red) and non-contaminated (blue) soils.
The six-line background signal is characteristic of manganese(II) ion ([Ar]3d5 configuration, nuclear spin, I=5/2) (5, 29) and is similar to that of octahedral Mn(H2O)62+ (4, 30). This suggests the Mn(II) ion is bound predominantly to the soil surface acid sites (the carboxylate and/or phenolate groups), forming an outer sphere complex via van der Waals interactions with fulvic acids (5).
Figure 5 depicts EPR spectra of various fractions of the contaminated soils. In the contaminated soils, approximately 90% of the measurable organic radicals were found in the minerals/clays/humins (acid-base insoluble precipitate) fraction, ~5% in the humic/fulvic acid (acid-base soluble supernatant) fraction, and barely detectable levels in the acid soluble fraction (humic-like substances) (cf. Figures 4 and 5). The large singlet observed for the contaminated soils exhibited a g-factor of 2.0030-2.0039 and ΔHp-p ~ 6 G. This is typical of an organic radical that is either carbon-centered with a nearby heteroatom, such as oxygen or halide, which increase the spin-orbit coupling constant or a purely oxygen-centered radical (31-42). Even after being subjected to a strong acid and strong base extraction, the radical signal in the contaminated soil was almost unchanged, indicating the radicals’ stability and persistency.
Figure 5.
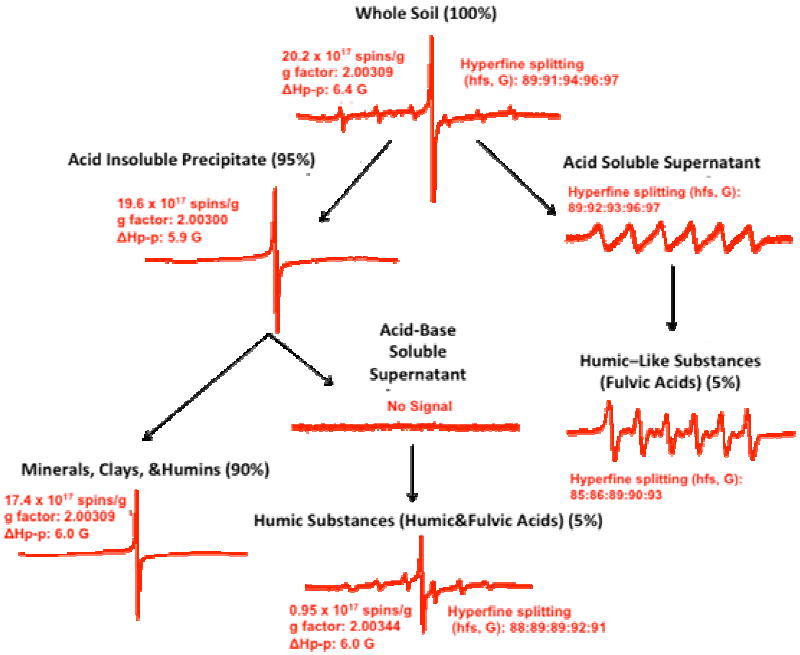
EPR spectra of various soil fractions of contaminated soils. (Individual spectra are not on the same scales, percentages in parenthesis are the approximate percent mass recoveries of extraction.) Acid-base soluble and acid soluble supernatants are solution spectra and have poorer resolution than the powder spectra of the Humic Substances (HA&FA) and Humic Like Substances (FA) respectively. The remaining figures on the left hand side of the diagram are all powder spectra.
As depicted in Figure 6, the concentration of organic radicals was ~30× higher for the contaminated soils, 20.2(+/-0.20) × 1017spins/g of soil, than non-contaminated soils, 0.7(+/-0.08) × 1017spins/g. Based on our previous laboratory studies of EPFR formation from 2-monochlorophenol and subsequent experiments performed for this study (vide infra), the radicals above the background levels of the non-contaminated soils were attributed to pentachlorophenoxyl.
Figure 6.
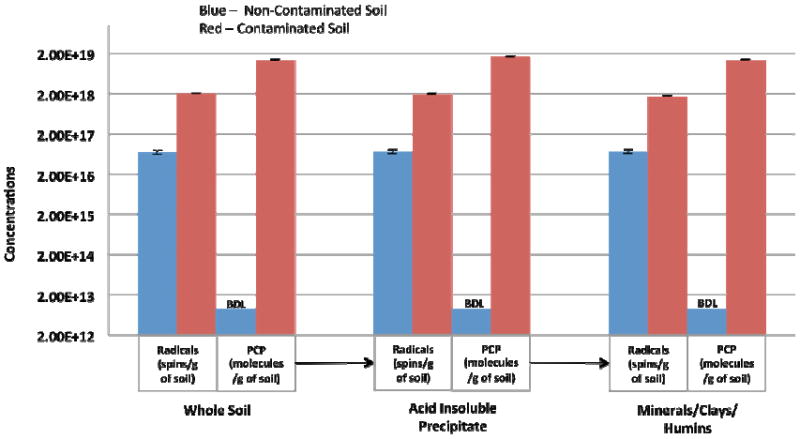
Comparisons of the radical concentrations (spins/g of soil) and PCP concentrations (molecules/g of soil) for the three insoluble isolates of contaminated and non-contaminated soils. The corresponding PCP concentrations in ppm (mg/kg of soil) for the contaminated soils’ fractions are as follows: Whole Soil = 6054(±280) ppm; Acid Insoluble Precipitate = 7485(±248) ppm; Minerals/Clays/Humins = 6212(±150) ppm. The PCP analyses of for the three fractions of non-contaminated soil are below detection limit (BDL) of the method (8.9×1012 spins/g).
The concentrations of PCP within the various soil extraction fractions are also depicted in Figure 6. These results indicate the PCP remains associated with the minerals/clays/humins fraction and persists after acid/base exposure. Our previous work has demonstrated that EPFRs can be formed and stabilized by interactions with Cu(II) and Fe(III), thus the metal content of the soil was analyzed by ICP-AES. This ICP-AES analyses yielded average concentrations of 16,266(±55) mg/kg and 60(±1) mg/kg and 25,200(±1590) mg/kg and 9(±0.5) mg/kg of iron and copper for the contaminated and non-contaminated soils, respectively (results not shown).
Based on the PCP and EPFR analyses, the EPFR concentration was high when the PCP concentration was also high. Assuming the singlet EPR signal detected_was due solely to the EPFR of PCP, 15 %, 13 %, and 14 % of the PCP exists as its EPFR for the whole soil, acid insoluble precipitate, and minerals/clays/humins (acid-base insoluble precipitate) fractions, respectively.
PCP Incubation studies
To further investigate whether the observed EPR signal was due to pentachlorophenoxyl radical, ~100 ppm of PCP vapor was dosed onto samples of the minerals/clays/humins fraction of both the contaminated and non-contaminated soils for a reaction time of 5 min at temperatures ranging from 25 to 300 °C. This resulted in an increase in the signal of the organic radical ranging from ~30% at 25 °C to 300% at 100 °C for the contaminated soil (results not shown). The resulting additional signal was still detectable with unaltered g-value and width after one month storage in air, demonstrating the stability and persistence of this radical. As depicted in Figure 7, the EPR signal for PCP vapor dosed non-contaminated soil exhibited striking similarity to the EPR signals observed on original soil contaminated with PCP. This suggests that the observed EPR signal is that of pentachlorophenoxyl radical.
Figure 7.
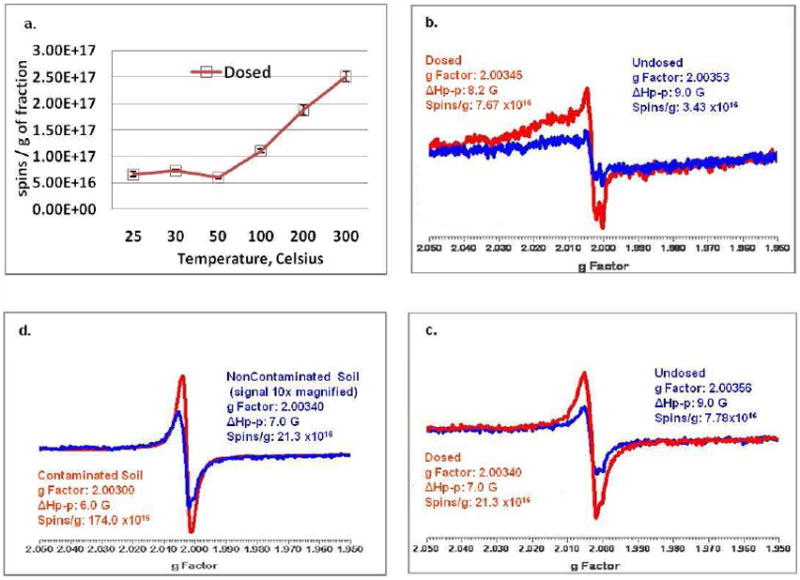
Temperature dependence of radical formation from dosing the minerals/clays/humins fraction of noncontaminated soil with gas phase PCP. a. Temperature dependence of pentachlorophenoxyl radical formation. EPR dosing spectra at: b. 30C and c. 200C. d. Comparison of EPR spectra of noncontaminated soil dosed with PCP at 200C with original soil contaminated with PCP.
In addition to the vapor phase dosing, 8 ppm of PCP in water and 1000 ppm of PCP in benzene solvents were added to the minerals/clays/humins fraction of contaminated and non-contaminated soils and allowed to age for 26 days at ambient temperature of 22 °C. (see Figure 8). An increase in radical concentration of 1.9 × 1017+/-0.008 spins/g was observed for the contaminated soils dosed with PCP in water and 0.8 × 1017+/-0.009 spins/g of radicals for the non-contaminated soils dosed with PCP in benzene. Although the radical concentration increase was not as dramatic as for the high temperature, gas-phase dosing, these data clearly demonstrate pentachlorophenoxyl radical was generated under conditions emulating ambient conditions for exposures of only 26 days, rather than to the ~ 26 years for the actual site.
Figure 8.
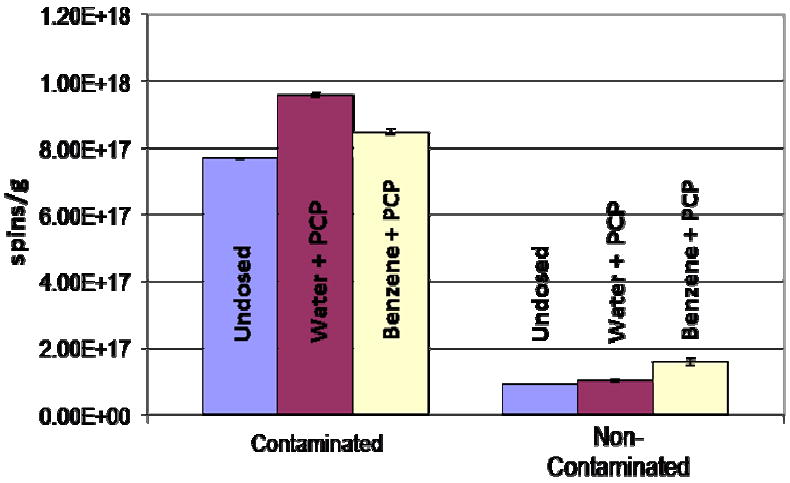
Concentration of organic radicals after dosing minerals/clays/humins fractions of soils with 8 ppm & 1000 ppm of PCP in water and benzene, respectively.
EPFR Formation
Sorption of pentachlorophenol in the organic and inorganic fractions of soils and sediments is well documented (3, 6, 43-48). The inorganic fraction of soil, such as smectite, is a 2:1 layered aluminosilicate clay mineral possessing structural negative charges that are compensated by exchangeable cations in the interlayer regions due to the isomorphic substitution in the tetrahedral Si and/or octahedral Al layers. Transition metals can be present in cationic positions via the cation exchange capacity of the soil and have been reported to be capable of formation of radical cations upon adsorption of chlorophenols (7-12, 49). The biological component of soil, e.g., white rot fungi, may also produce radicals as it degrades various soil contaminants (50-52). Peroxidase and laccase enzymes are implicated in phenol degradation, yielding phenoxyl radicals in the presence of either hydrogen peroxide–for peroxidase, or oxygen–for laccase, respectively.
The soil organic matter (SOM) fraction is a highly complex heterogeneous mixture composed of weakly associated molecular assemblies (53), with molecular moieties displaying both hydrophobic and hydrophilic characteristics akin to those of proteins (54). Hence, they also play a key role in the fate and transport of organic pollutants within soils (55-60). Additionally, SOM is known to be both an electron sink and source in soil processes (61-71) that can form polyphenol radicals (7, 8, 72, 73). Thus, the inorganic, organic, and biological components of the soil, as well as their combined interactions, must be considered in developing a mechanism of pentachlorophenoxyl radical formation. The combined effect of the mineral components, i.e. clays and metals, binding onto SOM significantly influence the retention of organic pollutants, such as chlorinated phenols, especially in soils with low organic matter content or if the sorbate is highly polar (2, 74).
PCP Component
Pentachlorophenol has a pKa of 5.3 and is an ionizable hydrophobic organic compound (74-76). Thus, pentachlorophenol may be present in both molecular (neutral) and ionized forms, which results in its sorption capacity being highly dependent on pH (43, 75-79). While the non-contaminated soil had a pH of ~5, the contaminated soil for this experiment exhibited a pH of ~8 indicating that the majority of the PCP contaminant in the contaminated soil existed in ionized form, with a lesser fraction in the neutral form. Data on the sorption of ionized and neutral PCP in a surface soil suggested the neutral form was partitioned through hydrophobic interactions, while the ionized form was sorbed through more specific exothermic adsorption reactions, such as hydrogen bonding and charge transfer (76). This resulted in the neutral PCP sorbing to the organic humic substances and the ionized pentachlorophenolate interacting with the clay/mineral surfaces fractions (2). However, due to the high organic carbon content of our contaminated soil, it is likely that the majority of the soil’s active mineral surface will be coated with the soil organic carbon, which then serves as the site for the sorption of PCP.
Mineral Component
Due to the high concentration of iron in the soil under study here, our previously proposed mechanism for formation of the EPFRs of 2-monochlorophenoll on Cu(II) is adapted to the formation of the EPFRs of PCP on Fe(III) (cf. Figure 9)(18, 19). PCP first physisorbs to an Fe(III) oxide surface, then chemisorbs, by loss of water, to form phenolate, with simultaneous or rapid sequential electron transfer from the phenolate to the Fe(III), thus producing Fe(II) and the pentachlorophenoxyl EPFR. Oxygen-centered and carbon-centered, keto- mesomers both contribute to the true structure of the radical. If chemisorption occurs through loss of HCl, then a tetrachlorosemiquinone radical is formed. If both H2O and HCl are eliminated, then a tetrachlorosemiquinone radical or radical-anion is formed. Of these pathways, only the first and second were observed under post-combustion, cool-zone conditions, where the reaction times were 30 s and the reaction temperatures ranged between 150 and 600 °C (16-19). However, we cannot rule out the possibility of additional formation of tetrachlorosemiquinone radical in the soil at ambient temperatures and reaction times of years.
Figure 9.
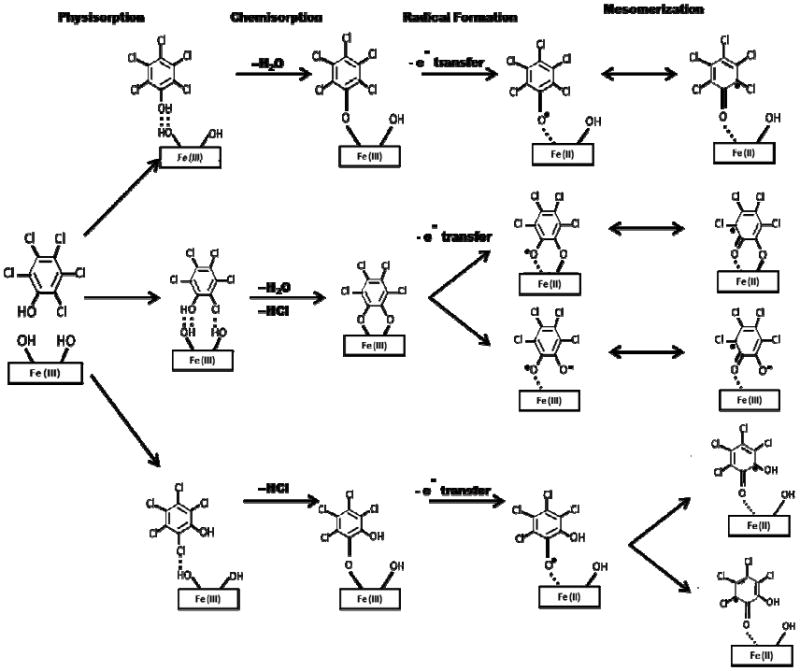
Possible pathways of formation of EPFRs from PCP on an Fe(IIl) oxide surface. The upper pathway depicts formation of pentachlorophenoxyl radicals as both the oxygen-centered and carbon-centered mesomer. The middle pathway depicts the formation of tetrachlorosemiquinone radical-anion.
Organic Matter Component
Due to the high organic matter content of these soils, this component cannot be ignored. Judging by the deep brown-to- black color (due to organic matter) of the contaminated soil, it can be assumed that there is a high concentration of aromatic moieties (high aromaticity) within the SOM for this site (89-90). Organic matter is known to be redox active, including acting as a bidirectional electron shuttle (63, 65, 71, 80-84). Quinone moieties within the organic matter have been suggested to be active components in the redox processes; however, other moieties have been discussed (63). For the purpose of this paper we will focus on the ability of organic matter to act as an electron sink or withdrawer. In particular, the recent work by Aeschbacher et al. (71) unambiguously showed that organic matter, ranging from terrestrial humic acids to aquatic fulvic acids, have electron accepting capacities spanning from 490 to 1960 μmole- g-1. The electron accepting capacity of the studied organic matters were found to closely correlate (R2=0.82) with both the C/H ratio and aromaticity (via 13C NMR) of organic matter. A similar correlation between electron accepting ability and aromaticity has been reported by Scott et al (63). It can be therefore postulated that since metal-free SOM can act as an electron acceptor, in a fashion analogous to the abovementioned metal-centered mechanism, a metal center is not necessarily required.
In addition to acting alone, SOM can serve as an electron conduit between the pollutant and the metal center. In other words, the metal center does not need to be in direct contact with a pollutant in order to withdraw an electron from it. Hence, even when the mineral/clay surface is completely covered by organic matter, as can be assumed given the high organic matter content of the soil in question, the metal center at the mineral/clay surface can act as the final electron sink. The metal center can also donate the acquired electron to the SOM, partially catalyzing the formation of radicals from the PCP. Both mechanisms support the idea of SOM stabilizing the formed radical by such local effects as π-stacking and hydrophobic associations (85). In fact, it has been found that the humic fraction has the highest concentration of radicals in non-polluted soils (86, 27).
Biological Component
It is well known that white rot fungi reduces a range of pollutants, including PCP (51, 52, 50). Both, the peroxidases and laccases are implicated in phenol degradation. Both classes of enzymes yield phenoxyl radicals in the presence of hydrogen peroxide and oxygen for peroxidase and laccase, respectively, mediated by single electron oxidation. With the use of other enzymes, white rot fungi produce extracellular hydrogen peroxide from a range of available molecules, such as glucose. This means fungi can produce phenoxyl radicals either via laccase or peroxidase enzymes. Organic matter can again act as an electron conduit.
Environmental Implications
Recognition that EPFRs can be formed in PCP contaminated soils indicates EPFRs are not confined to combustion-generated PM and are more environmentally prevalent than originally suspected. At uncontrolled sites, human exposure can occur by using water containing contaminated sediments or inhalation of airborne dust from wind erosion. EPFRs similar to pentachlorophenoxyl have been shown to generate ROS, oxidative stress, and cardiopulmonary dysfunction in rat pups exposed by inhalation (15, 87, 88). The existence of potentially toxic EPFRs questions the long held belief that sorption of an organic pollutant to a soil matrix is a method of mitigating its environmental impact.
Acknowledgments
Support for this research was provided by the NIEHS Superfund Research Program through grant # 1P42ES013648-01A2 and the LSU Patrick F. Taylor Chair is gratefully acknowledged. We also thank Mr. Scott Miller of EPA for providing access to the site. For the technical support and helpful comments on soil chemistry, we thank Dr. Charisma Lattao.
Literature Cited
- 1.Senesi N. Binding mechanisms of pesticides in soil humic substances. Sci Total Environ. 1992;123:63–76. doi: 10.1016/0048-9697(92)90133-d. [DOI] [PubMed] [Google Scholar]
- 2.Stipicevic S, Fingler S, Drevenkar V. Effect of organic and mineral soil fractions on sorption behavior of chlorophenol and triazinemicropollutants. Arh Hig Rada Toksikol. 2009;60:43–52. doi: 10.2478/10004-1254-60-2009-1898. [DOI] [PubMed] [Google Scholar]
- 3.Pandey AK, Pandey SD, Misra V, Viswanathan PN. Role of free radicals in the binding of organochlorine pesticides and heavy metals with humic acid. Sci Total Environ. 1999;231:125–133. [Google Scholar]
- 4.Flogeac K, Guillon E, Aplincourt M. Adsorption of several metal ions onto a model soil sample: Equilibrium and EPR studies. J Colloid Interface Sci. 2005;286:596–601. doi: 10.1016/j.jcis.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 5.Senesi N. Application of electron spin resonance (ESR) spectroscopy in soil chemistry. Adv Soil Sci. 1990;14:117–130. [Google Scholar]
- 6.Spark KM, Swift RS. Effect of soil composition and dissolved organic matter on pesticide sorption. Sci Total Environ. 2002;298:147–161. doi: 10.1016/s0048-9697(02)00213-9. [DOI] [PubMed] [Google Scholar]
- 7.Boyd SA, Mortland MM. Dioxin radical formation and polymerization on Cu(II)-smectite. Nature. 1985;316:532–535. [Google Scholar]
- 8.Boyd SA, Mortland MM. Radical formation and polymerization of chlorophenols and chloroanisole on Copper(II)-Smectite. Eviron Sci Technol. 1986;20:1056–1058. doi: 10.1021/es00152a017. [DOI] [PubMed] [Google Scholar]
- 9.Rana K, Boyd SA, Teppen BJ, Li H, Liu C, Johnston CT. Probing the microscopic hydrophobicity of smectite surfaces. A vibrational spectroscopic study of dibenzo-p-dioxin sorption to smectite. Phys Chem Chem Phys. 2009;11:2976–2985. doi: 10.1039/b822635k. [DOI] [PubMed] [Google Scholar]
- 10.Liu C, Li H, Teppen BJ, Johnston CT, Boyd SA. Mechanisms associated with the high adsorption of dibenzo-p-dioxin from water by smectite clays. Environ Sci Technol. 2009;43:2777–2783. doi: 10.1021/es802381z. [DOI] [PubMed] [Google Scholar]
- 11.Boyd SA, Shaobai S, Lee J-F, Mortland MM. Pentachlorophenol sorption by organo-clays. Clays Clay Min. 1988;36(2):125–130. [Google Scholar]
- 12.Govindraj N, Mortland MM, Boyd SA. Single electron transfer mechanism of oxidative of oxidative dechlorination of 4-chloroanisole on copper(II)-smectite. Environ Sci Technol. 1987;21:1119–1123. [Google Scholar]
- 13.Otto F, Leupold G, Parlar H, Rosemann R, Bahadir M, Hopf H. Chlorinated diphenoquinones: A new class of dioxin isomeric compounds discovered in fly ashes, slags, and pyrolysis oil samples by using HPLC/ELCD and HRGC/MS. Anal Chem. 1998;70(14):2831–2838. [Google Scholar]
- 14.Dudal Y, Jacobson AR, Samson R, Deschenes L. Modelling the dynamics of pentachlorophenol bioavailability in column experiments. Water Res. 2004;38:3147–3154. doi: 10.1016/j.watres.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 15.Cormier SA, Lomnicki S, Backes W, Dellinger D. Origin and health impacts of emissions of toxic by-products and fine particles from combustion and thermal treatment of hazardous wastes and materials. Environ Health Perspect. 2006;114(6):810–817. doi: 10.1289/ehp.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dellinger B, Lomnicki S, Khatchatryan L, Maskos Z, Hall RW, Adounkpe J, McFerrin C, Truong H. Formation and stabilization of persistent free radicals. Proc Combust Inst. 2007;31:521–528. doi: 10.1016/j.proci.2006.07.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dellinger B, Pryor W, Cueto R, Squadrito GL, Hedge V, Deutsch WA. Role of free radicals in the toxicity of airborne fine particulate matter. Chem Res Toxicol. 2001;14:1371–1377. doi: 10.1021/tx010050x. [DOI] [PubMed] [Google Scholar]
- 18.Dellinger B, Lomnicki S, Pryor W, Cueto R, Squadrito GL, Deutsch WA. The role of combustion-generated radicals in the toxicity of PM2.5. Proc Combust Inst. 2000;28:2675–2681. [Google Scholar]
- 19.Lomnicki S, Truong H, Vejerano E, Dellinger B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environ Sci Technol. 2008;42:4982–4988. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- 20.Truong H, Lomnicki S, Dellinger B. Potential for misidentification of environmentally persistent free radicals as molecular pollutants in particulate matter. Environ Sci Technol. 2010;44(6):1933–1939. doi: 10.1021/es902648t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Superfund, United States Environmental Protection Agencty (EPA) Official Website; http:///www.epa.gov/superfund/
- 22.International Humic Substances Society (IHSS) Official Website; http://ihss.gatech.edu/ihss2/
- 23.Rosa AH, Simoes ML, de Oliveira LC, Rocha JC, Martin-Neto L, Milori DMBP. Multimethod study of the degree of humification of humic substances extracted from different tropical soil profiles in Brazil’s Amazonian region. Geoderma. 2005;127:1–10. [Google Scholar]
- 24.Rosa AH, Vicente A, Rocha JC, Trevisan HC. A new application of humic substances: activation supports for invertase immobilization. Fres J Anal Chem. 2000;368:730–733. doi: 10.1007/s002160000535. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-Perez M, Martin-Neto L, Colnago LA, Milori DMBP, de Camargo OA, Berton R, Bettiol W. Characterization of humic acids extracted from sewage sludge-amended oxisols by electron paramagnetic resonance. Soil Tillage Res. 2006;91:95–100. [Google Scholar]
- 26.Novotny EH, Martin-Neto L. Effects of humidity and metal ions on the free radicals analysis of peat humus. Geoderma. 2002;106:305–317. [Google Scholar]
- 27.Saab SC, Martin-Neto L. Studies of semiquinone free radicals by ESR in the whole soil, HA, FA, and humin substances. J Braz Chem Soc. 2004;15(1):34–37. [Google Scholar]
- 28.Saab SC, Martin-Neto L. Characterization by electron paramagnetic resonance of organic matter in whole soil (Gleysoil) and organic-mineral fractions. J Braz Chem Soc. 2008;19(3):413–417. [Google Scholar]
- 29.Riffaldi R, Schnitzer M. Electron spin resonance spectrometry of humic substances. Soil Sci Soc Am Proc. 1972;36:301. [Google Scholar]
- 30.Senesi N. Molecular and quantitive aspects of the chemistry of fulvic acid and its interactions with metal ions and organic chemicals. Part I. The electron spin resonance approach. Anal Chim Acta. 1990;232:51–75. [Google Scholar]
- 31.Senesi N, Loffredo E. The chemistry of soil organic matter. In: Sparks DL, editor. Soil Physical Chemistry. CRC Press; Boca Raton: 1999. pp. 242–345. [Google Scholar]
- 32.Jezierski A, Skrzypek G, Jezierski P, Paul D, Jedrysek MO. Electron paramagnetic resonance (EPR) and stable isotope records of paleoenvironmental conditions during peat formation. Spectrochim Acta Part A. 2008;69(5):1311–1316. doi: 10.1016/j.saa.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 33.Polewski K, Slawinska D, Slawinski J, Pawlak A. The effect of UV and visible light radiation on natural humic acid: EPR spectral and kinetic studies. Geoderma. 2005;126:291–299. [Google Scholar]
- 34.Jezierski A, Czechowski F, Jerzykiewicz M, Chen Y, Drozd J. Electron paramagnetic resonance (EPR) studies on stable and transient radicals in humic acids from compost, soil, peat and brown coal. Spectrochim Acta Part A. 2000;56(2):379–385. doi: 10.1016/s1386-1425(99)00249-8. [DOI] [PubMed] [Google Scholar]
- 35.Christofiridis KC, Un S, Deligiannakis Y. High field 285 GHz electron paramagnetic resonance study of indigenous radicals of humic acids. J Phy Chem A. 2007;111(46):11860–11866. doi: 10.1021/jp0717692. [DOI] [PubMed] [Google Scholar]
- 36.Jezierski A, Czechowski F, Jerzykiewicz M, Golonka I, Drozd J, Bylinska E, Chen Y, Seaward MRD. Quantitative EPR study on free radicals in the natural polyphenols interacting with metal ions and other environmental pollutants. Spectrochim Acta Part A. 2002;58(6):1293–1300. doi: 10.1016/s1386-1425(01)00718-1. [DOI] [PubMed] [Google Scholar]
- 37.Delhaes P, Marchand A. Analyse de la formeet de la position de signaixrpe observes sur des carbonesgraphitiquespulverulents. Carbon. 1968;6(2):257–266. [Google Scholar]
- 38.Szent-Gyorgi A, Isenberg I, Baird SL., Jr On the electron donating properties of carcinogens. Proc Natl Acad Sci USA. 1960;46:1444–1449. doi: 10.1073/pnas.46.11.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barclay LRC, Vinqvist MR. Membrane peroxidation: Inhibiting effects of water- soluble antioxidants on phospholipids of different charge types. Free Radic Biol Med. 1994;16:779–788. doi: 10.1016/0891-5849(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 40.Hales B. Immobilized radicals. I. Principal electron spin resonance parameters of the benzosemiquinone radical. J Am Chem Soc. 1975;97(21):5993–5997. [Google Scholar]
- 41.Graf F, Loth K, Gunthard H. Chlorine hyperfine splittings and spin density distributions of phenoxy radicals. An ESR and quantum chemical study. Helv Chim Acta. 1977;60(3):710–721. [Google Scholar]
- 42.Soma Y, Soma M, Harada I. Reactions of aromatic molecules in the interlayer of transition-metal ion-exchanged montmorillonite studies by resonance Raman spectroscopy. 2. Monosubstituted benzenes and 4,4’-disubstituted biphenyls. J Phys Chem. 1985;89:738–742. [Google Scholar]
- 43.Schellenberg K, Leuenberger C, Schwarzenbach RP. Sorption of chlorinated phenols by natural sediments and aquifer materials. Environ Sci Technol. 1984;18:652–657. [Google Scholar]
- 44.Frobe Z, Fingler S, Drevenkar V, Juracic M. Sorption behavior of some chlorophenols in natural sorbents. 1. Validity of the partition model for sorption of phenolates. Sci Total Environ. 1994;155:199–213. [Google Scholar]
- 45.Fingler S, Drevenkar V, Frobe Z. Sorption of chlorophenolates in soils and aquifer and marine sediments. Arch Environ Contam Toxicol. 2005;48:32–39. doi: 10.1007/s00244-003-0185-3. [DOI] [PubMed] [Google Scholar]
- 46.Huang CP, Ehrlich RS. Adsorption of chlorophenols onto soil and kaolinite: effects of the properties of adsorbent and adsorbate. J Chin Inst Environ Eng. 1993;3(3):143–152. [Google Scholar]
- 47.Calvet R. Adsorption of organic chemicals in soils. Environ Health Perpect. 1989;83:145–177. doi: 10.1289/ehp.8983145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedlander HZ, Saldick J, Frink CR. Electron spin resonance spectra in various clay minerals. Nature. 1963;199:61–62. [Google Scholar]
- 49.Gu C, Li H, Teppen BJ, Boyd SA. Octachlorodibenzodioxin formation on Fe(III)-montmorillonite clay. Eviron Sci Technol. 2008;42:4758–4763. doi: 10.1021/es7029834. [DOI] [PubMed] [Google Scholar]
- 50.Field JA, Sierra-Alvarez R. Microbial degradation of chlorinated phenols. Rev Environ Sci Biotechnol. 2008;7:211–241. [Google Scholar]
- 51.Barr DP, Aust SD. Mechanisms white rot fungi use to degrade pollutants. Environ Sci Technol. 1994;28:78A–87A. doi: 10.1021/es00051a724. [DOI] [PubMed] [Google Scholar]
- 52.Rabinovich ML, Bolobova AV, Vasil’chenko LG. Fungal decomposition of natural aromatic structures and xenobiotics: a review. Appl Biochem Microbiol. 2004;40:1–17. [PubMed] [Google Scholar]
- 53.Sutton R, Sposito G. Molecular structure in soil humic substances: the new view. Environ Sci Technol. 2005;39:9009–9015. doi: 10.1021/es050778q. [DOI] [PubMed] [Google Scholar]
- 54.Stevenson FJ. Humus Chemistry: Genesis, Composition, Reactions. 2. John Wiley & Sons; New York: 1994. [Google Scholar]
- 55.Chiou CT, Peters LJ, Freed VH. A physical concept of soil-water equilibriums for nonionic organic compounds. Science. 1979;206:831–832. doi: 10.1126/science.206.4420.831. [DOI] [PubMed] [Google Scholar]
- 56.Chiou CT, Porter PE, Schmedding DW. Partition equilibriums of nonionic organic compounds between soil organic matter and water. Environ Sci Technol. 1983;17:227–231. doi: 10.1021/es00110a009. [DOI] [PubMed] [Google Scholar]
- 57.Chiou CT, McGroddy SE, Kile DE. Partition characteristics of polycyclic aromatic hydrocarbons on soils and sediments. Environ Sci Technol. 1998;32:264–269. [Google Scholar]
- 58.Karickhoff SW, Brown DS, Scott TA. Sorption of hydrophobic pollutants on natural sediments. Wat Res. 1979;13:241–248. [Google Scholar]
- 59.Weber WJ, Jr, Huang W. A distributed reactivity model for sorption by soils and sediments. 4. Intraparticle heterogeneity and phase-distribution relationships under nonequilibrium conditions. Environ Sci Technol. 1996;30:881–888. [Google Scholar]
- 60.Pignatello JJ, Xing B. Mechanisms of slow sorption of organic chemicals to natural particles. Environ Sci Technol. 1996;30:1–11. [Google Scholar]
- 61.Osterberg R, Shirshova L. Oscillating, nonequilibrium redox properties of humic acids. Geochim Cosmochim Acta. 1997;61:4599–4604. [Google Scholar]
- 62.Chen R, Pignatello JJ. Role of quinone intermediates as electron shuttles in fenton and photoassisted fenton oxidation of aromatic compounds. Environ Sci Technol. 1997;31:2399–2406. [Google Scholar]
- 63.Scott DT, McKnight DM, Blunt-Harris EL, Kolesar SE, Lovely DD. Quinone moieties acts as electron acceptors in the reduction of humic substances by humics-reducing microorganisms. Environ Sci Technol. 1998;32:2984–2989. [Google Scholar]
- 64.Struyk Z, Sposito G. Redox properties of standard humic acids. Geoderma. 2001;102:329–346. [Google Scholar]
- 65.Ratasuk N, Nanny MA. Characterization and quantification of reversible redox sites in humic substances. Environ Sci Technol. 2007;41:7844–7850. doi: 10.1021/es071389u. [DOI] [PubMed] [Google Scholar]
- 66.Kang SH, Choi W. Oxidative degradation of organic compounds using zero-valent iron in the presence of natural organic matter serving as an electron shuttle. Environ Sci Technol. 2009;43:878–883. doi: 10.1021/es801705f. [DOI] [PubMed] [Google Scholar]
- 67.Gentleman DJ. Biology meets geology through chemistry. Environ Sci Technol. 2010;44:1–2. doi: 10.1021/es9036176. [DOI] [PubMed] [Google Scholar]
- 68.Borch T, Campbell K, Kretzschmar R. How electron flow controls contaminant dynamics. Environ Sci Technol. 2010;44:3–4. doi: 10.1021/es903264z. [DOI] [PubMed] [Google Scholar]
- 69.Borch T, Kretzschmar R, Kappler A, Van Cappellen P, Ginder-Vogel M, Voegelin A, Campbell K. Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol. 2010;44:15–23. doi: 10.1021/es9026248. [DOI] [PubMed] [Google Scholar]
- 70.Heimann A, Jakobsen R, Blodau C. Energetic constraints on H2-dependent terminal electron accepting processes in anoxic environments: a review of observations and model approaches. Environ Sci Technol. 2010;44:24–33. doi: 10.1021/es9018207. [DOI] [PubMed] [Google Scholar]
- 71.Aeschbacher M, Sanders M, Schwarzenbach RP. Novel electrochemical approach to assess the redox properties of humic substances. Environ Sci Technol. 2010;44:87–93. doi: 10.1021/es902627p. [DOI] [PubMed] [Google Scholar]
- 72.Sawhney BL, Kozloski RK, Isaacson PJ, Gent MPN. Polymerization of 2,6-dimethylphenol on smectite surfaces. Clays Clay Min. 1984;32:108–114. [Google Scholar]
- 73.Sawhney BL. Vapor-phase sorption and polymerization of phenols by smectite in air and nitrogen. Clays Clay Min. 1985;33:123–127. [Google Scholar]
- 74.He Y, Xu J, Wang H, Zhang Q, Muhammad A. Potential contributions of clay minerals and organic matter to pentachlorophenol retention in soils. Chemosphere. 2006;65:497–505. doi: 10.1016/j.chemosphere.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 75.World Health Organization (WHO) Pentachlorophenol Environmental Health Criteria No 71. Geneva: WHO; 1987. [Google Scholar]
- 76.Galil NI, Novak JT. Pentachlorophenol-induced release of soil organics and colloids. Wat Res. 1995;29(6):1533–1540. [Google Scholar]
- 77.DiVincenzo JP, Sparks DL. Sorption of the neutral and charged forms of pentachlorophenol on soil: evidence for different mechanisms. Arch Environ Contam Toxicol. 2001;40:445–450. doi: 10.1007/s002440010196. [DOI] [PubMed] [Google Scholar]
- 78.Lagas P. Sorption of chlorophenols in the soil. Chemosphere. 1988;17:205–216. [Google Scholar]
- 79.Westal JC, Leuenberger C, Schwarzenbach RP. Influence of pH and ionic strength on aqueous-nonaqueous distribution of chlorinated phenols. Environ Sci Tech. 1985;19:193–198. [Google Scholar]
- 80.Zhu BZ, Shan GQ. Potential mechanism for pentachlorophenol-induced carcinogenicity: A novel mechanism for metal-independent production of hydroxyl radicals. Chem Res Toxicol. 2009;22(6):969–977. doi: 10.1021/tx900030v. [DOI] [PubMed] [Google Scholar]
- 81.Dunnivant FM, Schwarzenbach RP, Macalady DL. Reduction of substituted nitrobenzenes in aqueous solutions containing natural organic matter. Environ Sci Technol. 1992;26:2133–2141. [Google Scholar]
- 82.Lovley DR, Coates JD, BluntHarris EL, Phillips EJP, Woodward JC. Humic substances as electron acceptors for microbial respiration. Nature. 1996;382(6590):445–448. [Google Scholar]
- 83.Collins R, Picardal F. Enhanced anaerobic transformation of carbon tetrachloride by soil organic matter. Environ Toxicol Chem. 1999;18:2703–2710. doi: 10.1002/etc.5620181005. [DOI] [PubMed] [Google Scholar]
- 84.Lovley DR, Frage JL, Coates JD, Blunt-Harris EL. Humics as an electron donor for anaerobic respiration. Environ Microbiol. 1999;1:89–98. doi: 10.1046/j.1462-2920.1999.00009.x. [DOI] [PubMed] [Google Scholar]
- 85.Coates JD, Cole KA, Chakraborty R, O’Connor SM, Achenbach LA. Diversity and ubiquity of bacteria capable of utilizing humic substances as electron donors for anaerobic respiration. Appl Environ Microbiol. 2002;68:2445–2452. doi: 10.1128/AEM.68.5.2445-2452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giannakopoulos E, Drosos M, Deligiannakis Y. A humic-acid-like polycondensate produced with no use of catalyst. J Coll & Int Sci. 2009;336:59–66. doi: 10.1016/j.jcis.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 87.Choudhry GG. Humic substances. Part II: photophysical, photochemical and free radical characteristics. Tox & Environ Chem. 1981;4:261–295. [Google Scholar]
- 88.Walsh M, Cormier S, Varner K, Dellnger B. By products of the thermal treatment of hazardous wastes formation and health effects. EM Magazine. 2010 April;:26–30. [PMC free article] [PubMed] [Google Scholar]
- 89.Spielvogel S, Knicker H, Kogel-Knabner I. Soil organic matter composition and soil lightness. J Plant Nut & Soil Sci. 2004;167(5):545–555. [Google Scholar]
- 90.Mendez E, Castillanos D, Alba GI, Hernandez G, Solis S, Levresse G, Vega M, Rodriguez F, Urbina E, Cuevas MC, Garcia MG, Bustos E. Effect in the physical and chemical properties of gleysoil after an electro-kinetic treatment in presence of surfactant Triton X – 114 to remove hydrocarbon. Int J Electrochem Sci. 2011;6:1250–1268. [Google Scholar]