

Abstract
Hydroxyzine HCl is used in oral formulations for the treatment of urticaria and atopic dermatitis. Dizziness, blurred vision, and anticholinergic responses, represent the most common side effects. It has been shown that controlled release of the drug from a delivery system to the skin could reduce the side effects while reducing percutaneous absorption. Therefore, the aim of the present study was to produce an effective drug-loaded dosage form that is able to control the release of hydroxyzine hydrochloride into the skin. The Microsponge Delivery System is a unique technology for the controlled release of topical agents, and it consists of porous polymeric microspheres, typically 10–50 μm in diameter, loaded with active agents. Eudragit RS-100 microsponges of the drug were prepared by the oil in an oil emulsion solvent diffusion method using acetone as dispersing solvent and liquid paraffin as the continuous medium. Magnesium stearate was added to the dispersed phase to prevent flocculation of Eudragit RS-100 microsponges. Pore inducers such as sucrose and pregelatinized starch were used to enhance the rate of drug release. Microsponges of nearly 98% encapsulation efficiency and 60–70% porosity were produced. The pharmacodynamic effect of the chosen preparation was tested on the shaved back of histamine-sensitized rabbits. Histopathological studies were driven for the detection of the healing of inflamed tissues.
KEYWORDS: hydroxyzine HCl, microsponges, oil in oil emulsion solvent diffusion, skin delivery
INTRODUCTION
One of the simple reproducible, economic methods of microencapsulation of hydrophilic drugs like hydroxyzine HCl is an emulsion solvent diffusion in oil technique. Solvent diffusion in oil is a preparation technique that is widely preferred for the preparation of controlled release microspheres (1–3). The first stage of this method is to prepare the emulsion by adding the dispersed phase (consisting of the drug, polymer, and appropriate dispersing agent in the organic solvent) to a dispersion medium which is immiscible with the dispersed phase. At the second stage, minimatrix forms are obtained by removing the solvent used at the dispersed phase from the emulsion droplets. The selection of the organic solvents and the dispersion medium is done according to their dielectric constants, which indicates their mutual miscibility (4–6).
Several studies had been previously done leading to the best choice of solvent used in such techniques. It has been reported earlier that methyl acetate with dielectric constant (ε = 6.7), showed nonpolar properties leading to high miscibility with liquid paraffin, which resulted in rapid solvent diffusion during emulsification and consequent agglomeration of microspheres. The use of methanol having high polarity (ε = 32.6), resulted in the absence of diffusion through the microspheres to the external liquid paraffin, and the precipitation of the polymer in the droplets was not observed (4,7). The use of acetone, with medium polarity (ε = 20.7) and medium hydrophilicity resulted in successfully prepared microspheres. This was due to the slow diffusion of the solvent out of the emulsion droplets to the external liquid paraffin medium, further slow precipitation of polymer matrix, and subsequent separation of a microspheres having a spongy structure (4,6).
One of the polymers preferred in the preparation of the microspheres by oil-in-oil (o/o) solvent diffusion is Eudragit RS-100. It is a copolymer of acrylic and methacrylic acid esters containing 5% of functional quaternary ammonium groups. As the polymer contains the ammonium salt groups, its permeability is pH independent (8–10).
Many research workers succeeded to encapsulate water-soluble drugs using Eudragit RS-100 by the application of solvent diffusion in oil technique, such as acetazolamide by Haznedar (11), verapamil hydrochloride by Baykara (12) and terbutaline sulfate by Kim et al. (13).
In the present study, hydroxyzine HCl-loaded microsponges were prepared by o/o emulsion solvent diffusion method. The effects of different variables affecting microencapsulation were studied. The pharmacodynamic effect of the selected hydroxyzine hydrochloride (HYD-HCl) microsponges was evaluated.
MATERIALS AND METHODS
Materials
Hydroxyzine hydrochloride (C.I.D Company, Cairo, Egypt) is soluble 1:1 in water, 1:4.5 in ethanol, slightly soluble in acetone, and practically insoluble in ether. Eudragit RS-100 (Degussa-Rhöm GmbH & Co.). The following materials were used for this study: light liquid paraffin, acetone, n-hexane, magnesium stearate, sucrose, and pregelatinzed starch (PGS), potassium dihydrogen phosphate, disodium hydrogen phosphate, sodium chloride, formaldehyde, ethyl alcohol 95%, methyl alcohol, xylene (ADWIC, El-Nasr Chemical Co., Cairo, Egypt); sorbitan monooleate (span 80), histamine diphosphate, hematoxylin and eosin stain (Sigma-Aldrich, USA); 0.9% sodium chloride (sterile saline solution, Otsuka Co., Egypt); and cellulose nitrate filter with a pore size of 0.45 μm (Sartorius AG, Goettingen, Germany).
Animals
New Zealand white rabbits of male sex with average weight of 3.8 ± 0.5 kg (n = 16) were used in this study.
Method
General Procedure
A specific weight of Eudragit RS-100 was dissolved in acetone. Once a clear solution was obtained, 0.5 g of the drug was added in addition to magnesium stearate (3% w/v of solvent), and the whole mixture was kept in the ultrasonic bath of 70-kHz frequency for 2 min (CREST, Ultrasonic Corporation, Cortland, New York) where homogenous dispersion was obtained (14–16).
The mixture was then poured into 150 ml of liquid paraffin previously cooled to 10 ± 0.5°C while it was stirred by a mechanical stirrer for 45 min. The oil in oil emulsion prepared was gradually heated to 35 ± 2°C and was stirred at this temperature for another 30 min. During this time, the acetone was completely removed by diffusion into liquid paraffin and evaporation through the air/liquid interface. The solidified microsponges were filtered, washed six times with 50 ml of n-hexane, air-dried at room temperature for 12 h, and stored in a desiccator for further investigations (17).
Optimization of Formulation Variables
Microsponges were prepared adopting the above method using the following variables:
Drug/polymer ratio: The effect of four drug/polymer ratios namely 1:1, 1:2, 1:3, and 1:4 was investigated in the preparations F1, F2, F3, and F4, respectively.
The percentage of magnesium stearate: Two additional concentrations of magnesium stearate have been investigated, namely 1.5% w/vol. used in preparations of F5, F7, and 5% w/vol. incorporated in F6 and F8.
Polymer/solvent ratio in the presence and absence of Span 80: The polymer/solvent ratio initially used in the preparation of microsponges was 1:5. However, this ratio has been changed to 1:10, 1:20, and 1:40 in the following formulae F9, F10, and F11. The same formulae were repeated in the presence of 0.1% v/v Span 80 added as emulsifier to the liquid paraffin.
Pore inducers: Sucrose and PGS, in two different amounts (0.5 and 1.5 g), were incorporated in the preparation of microsponges as release enhancers. A 4-ml ethanolic solution of 0.5 g of drug was dispersed and levigated with either sucrose or PGS. The wet levigate was left to complete dryness, then incorporated in the internal phase, and the general procedure was performed.
The details of the microsponge preparations and the studied variables were plotted in Table I.
Table I.
The Composition of Different Microsponges Prepared by Oil in Oil Emulsion Solvent Diffusion Method
| Preparations (F) | Eudragit RS-100 (g) | Acetone (ml) | D/P ratioa | P/S ratiob | Span 80 | % w/v Mg stearate | Pore inducers | |
|---|---|---|---|---|---|---|---|---|
| Type | Weight (g) | |||||||
| F1 | 0.5 | 2.5 | 1:1 | 1:5 | − | 3 | − | − |
| F2 | 1 | 5 | 1:2 | 1:5 | − | 3 | − | − |
| F3 | 1.5 | 7.5 | 1:3 | 1:5 | − | 3 | − | − |
| F4 | 2 | 10 | 1:4 | 1:5 | − | 3 | − | − |
| F5 | 0.5 | 2.5 | 1:1 | 1:5 | − | 1.5 | − | − |
| F6 | 0.5 | 2.5 | 1:1 | 1:5 | − | 5 | − | − |
| F7 | 1 | 5 | 1:2 | 1:5 | − | 1.5 | − | − |
| F8 | 1 | 5 | 1:2 | 1:5 | − | 5 | − | − |
| F9 | 0.5 | 5 | 1:1 | 1:10 | − | 3 | − | − |
| F10 | 0.5 | 10 | 1:1 | 1:20 | − | 3 | − | − |
| F11 | 0.5 | 20 | 1:1 | 1:40 | − | 3 | − | − |
| F12 | 0.5 | 5 | 1:1 | 1:10 | + | 3 | − | − |
| F13 | 0.5 | 10 | 1:1 | 1:20 | + | 3 | − | − |
| F14 | 0.5 | 20 | 1:1 | 1:40 | + | 3 | − | − |
| F15 | 0.5 | 2.5 | 1:1 | 1:5 | + | 3 | − | − |
| F16 | 2 | 10 | 1:4 | 1:5 | − | 3 | Sucrose | 0.5 |
| F17 | 2 | 10 | 1:4 | 1:5 | − | 3 | Sucrose | 1.5 |
| F18 | 2 | 10 | 1:4 | 1:5 | − | 3 | PGS | 0.5 |
| F19 | 2 | 10 | 1:4 | 1:5 | − | 3 | PGS | 1.5 |
All microsponges were prepared by using 0.5 g of drug and 150 ml of liquid paraffin. Magnesium stearate was added as a percentage weight relative to the acetone volume
aDrug/polymer ratio
bPolymer/solvent ratio
Characterization and Evaluation of Microsponges
Differential Scanning Calorimetric Analysis
Thermal analysis using a differential scanning calorimetric (DSC) method was carried out on HYD-HCl, Eudragit RS-100, and microsponges were prepared using a 1:1 drug/polymer ratio. Samples were submitted to DSC analysis using a differential scanning calorimeter (Schimadzu, model TA-50 WSI, Kyoto, Japan) calibrated with indium. The analysis was carried out on 4-mg samples sealed in standard aluminum pans. Thermograms were obtained at a scanning rate of 10°C/min using a dry nitrogen flow of 25 ml/min. Each sample was scanned between 0°C and 400°C.
Photomicroscopic Analysis
A sample of microsponge powder was examined for morphological characteristics under a binocular image analyzer microscope (Leica DMLB, Berlin, Germany) and photographed at a magnification of ×50.
Scanning Electron Microscopy
The detailed surface topography of the selected microsponges was observed using a scanning electron microscope (Model JEM-100S, JEOL, Tokyo, Japan). The microsponge sample was attached to the specimen holder using a double-coated adhesive tape and was gold-coated (∼20-nm thickness) under vacuum using a sputter coater (Model JFC-1100, JEOL, Japan) for 5–10 min at 40 mA and then investigated at 30 kV (18).
Particle Size Analysis
The particle size of the selected microsponges was determined by light scattering based on laser diffraction using the laser diffraction particle size analyzer, Malvern Master Sizer (Malvern Instruments Ltd., Worcestershire, UK) (19), and the distribution modal size was estimated. Measurements were performed using a 45-mm focus objective, a beam length of 2.4 mm, and obscuration levels from 5% to 10%.
Determination of Encapsulation Efficiency
An accurately weighed amount of loaded microsponges (20 mg) was dissolved in 10 ml of methylene chloride, 30 ml of distilled water was added, and the mixture was stirred for 24 h till complete solvent evaporation. The solution was filtered in a volumetric flask; the volume was completed with water (20,21). The concentration of hydroxyzine HCl in water was determined spectrophotometrically (Shimadzu, model UV-1601 PC, Kyoto, Japan) at λmax = 231 nm after appropriate dilution. Unloaded microsponges produced no significant absorbance values at the same wavelength. Each tested formulation was analyzed in triplicate. The encapsulation efficiency was calculated through the following relationship:
Encapsulation Efficiency
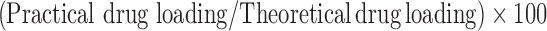 |
1 |
Viscosity Measurement
Viscosities of internal oily phase were determined with rotation viscometer (Brookfield model DV-I, MA, USA) by using small sample adapter, S18 spindle for dispersed phases at the rate of 50 rpm and 25 ± 0.5°C.
Flowability Testing
`The flowability of the prepared powder microsponges was tested by measuring their angle of repose and the calculation of Carr’s compressibility index and Hausner’s ratio.
Pore Volume and Pore Size Determination of MDS
A pore analysis of the microparticles was carried out using mercury intrusion porosimetry (Pascal 140, Italy). During the test, a sample of the microsponges was placed in a vacuum chamber and was submerged under a pool of mercury contained within a volume-calibrated cell. As pressure was gradually increased in the cell, mercury was forced into progressively smaller pores of the microsponges. Thus, the apparent volume of mercury within the calibrated cell was reduced as it penetrated into the microsponges.
In Vitro Release Studies
The in vitro release of hydroxyzine HCl from the microsponges was performed by the membrane diffusion technique using cellulose nitrate membrane (22). An accurately measured amount of microsponges, equivalent to a 10-mg drug, was suspended in 1 ml of phosphate buffer (pH 5.5) and transferred to a glass cylinder having a length of 7 cm and 2.5 cm in diameter. This cylinder was previously fitted with the buffer-soaked cellulose membrane and was suspended in the dissolution flask of the USP dissolution tester (Pharma Test, Hainburg, Germany), containing 200 ml phosphate buffer (pH 5.5). The apparatus was run at 50 rpm at a temperature of 32 ± 0.5°C. An aliquot of 3-ml sample was collected over a period of 9 h and was assayed spectrophotometrically for the drug at λmax = 231 nm. Each time sample was withdrawn, a fresh buffer was added to keep the dissolution medium volume constant. Experiments were done in triplicate. A kinetic treatment of the data was performed to determine the mechanism of the release of the drug.
In Vivo Study
The most common comparative tool to detect pharmacodynamic activity of antihistamines is the percutaneous injection of histamine in human or animal skin. This test is based on the ability of these drugs to block the histamine-induced wheal and flare response, by competing with histamine on its binding site (23). Any experiments involving the use of animals were conducted in accordance with the principles of Laboratory Animal Care and were approved by the Institutional Ethics Committee, Faculty of Pharmacy, Cairo University. The study was directed as follows:
- Preliminary Study:
- Two days before the study, a 12 × 12-cm area on the back of each rabbit was shaved using an electric razor. One day before the study, a depilatory cream was applied for 15 min to the shaved area on the back and to both ears and then thoroughly washed off to ensure complete removal of the hair in the respective areas.
-
Induction of histamine-induced wheal and flare response:A sterile aqueous solution, 0.1 ml, of histamine diphosphate containing 10, 50, 100, 200, and 300 mg/ml was subcutaneously injected into the shaved back of a rabbit for five consecutive days. The initial wheal area was traced after 10 min of histamine injection (24). Wheal diameter was traced, and its area was calculated according to the following law:
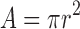
2 The wheal area of each dose was an average of the triplicate.
Animal Group Design
In this experimental design, eight rabbits were used in parallel group design. They were divided into two groups (n = 4/group) as follows:
Group I: animals topically treated with plain F2 microsponges
Group II: animals topically treated with medicated preparation F2
Evaluation of Antihistaminic Activity of Hydroxyzine Hydrochloride
The topical antihistaminic effect of the prepared formulae was assessed by the determination of the onset and extent of wheal suppression (25).
For dosing, an amount of powder microcapsules containing 10 mg of drug was applied by rubbing to the defined area on the rabbit’s back. The rabbit was placed in a wooden cage with approved collar allowing the head out during the 5-h study to prevent it from licking its back.
The wheal area was traced after 0.5, 1, 2, 3, and 4 h.
- The percentage suppression of the histamine-induced wheals was used as an indication of the peripheral H1-antihistaminic activity, and it was calculated using the following equation:
where E is the efficacy of the medication, W0 is the baseline wheal area, and Wt is the wheal area after time (t) of medication application.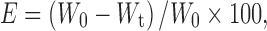
3
A one-way analysis of variance (ANOVA) was applied to detect the significance in the difference of the percentage wheal area suppression after 4 h at (p ≤ 0.05).
Histopathological Study
Autopsy samples were taken from the skin in different groups of rabbits and fixed in 10% formal saline (10% formaldehyde in normal saline) for 24 h. Washing was done using tap water then serial dilutions of alcohol (methyl and absolute ethyl alcohol) were used for dehydration. Specimens were cleared in xylene embedded in paraffin at 56°C in a hot air oven for 24 h. Paraffin beeswax tissue blocks were prepared for sectioning at 4-μm thickness by sledge microtome (Hacker-Bright, Windsor, USA). The obtained tissue sections were collected on glass slides, deparaffinized, and stained by hematoxylin and eosin stain for histopathological examination through the electric light microscope (National model 138, China) (26).
RESULTS AND DISCUSSION
By using the oil in oil emulsion solvent diffusion method, free flowing microsponges were prepared. The microsponges as illustrated in Fig. 1, had regular, spherical shape with roughness on the surface and several pores. The DSC thermogram of HYD- HCl obtained exhibited a wide range endotherm from 190°C to 225°C. No shift in the Hydroxyzine HCl peak in the loaded microsponges, accordingly no interaction was ascertained between the drug and the polymer. Different variables had been studied for their effect on the microsponges prepared.
Fig. 1.

Photomicrograph of the microsponges prepared by o/o solvent diffusion method
Effect of Increasing Polymer Amount
Results obtained in Table II showed that the encapsulation efficiencies of the drug were greater than 98% in all experiments (F1–F4). There was no significant difference between the encapsulation efficiencies at P ≤ 0.05 as shown in Table III. Accordingly, the variation of the drug/polymer ratio in the formulations investigated was not an influencing variable on the efficiency of drug loading. That might be due to the presence of the nonpolar liquid paraffin in the external phase, inhibiting the water-soluble drug to escape from the prepared microsponges.
Table II.
Characteristics of Microsponges Obtained with Different Drug/Polymer Ratios
| Parameter evaluated | Drug/polymer ratio | |||
|---|---|---|---|---|
| 1:1 (F1) | 1:2 (F2) | 1:3 (F3) | 1:4 (F4) | |
| EE% (% ±SD)a | 99 ± 0.075 | 98.08 ± 0.7 | 98.64 ± 1.4 | 98.12 ± 1.5 |
| Mean particle size (μm ± SD) | 57.1 ± 1.26 | 42.6 ± 1.7 | 35.2 ± 2.3 | 32 ± 2.1 |
| Angle of repose | 27 | 25 | 20 | 19.3 |
| Carr’s index | 20 | 18.03 | 17.355 | 16.66 |
| Hausner ratio | 1.25 | 1.22 | 1.21 | 1.2 |
aEncapsulation efficiency
Table III.
ANOVA of the Percentage of Drug Encapsulated in F1–F4
| Sum of squares | df | Mean square | F | Significance | |
|---|---|---|---|---|---|
| Between groups | 2.48 | 3 | 0.82 | 6.25 | p < 0.05 |
| Within groups | 0.53 | 4 | 0.13 | ||
| Total | 3.01 | 7 |
The results from the laser diffractometry study illustrated in Fig. 2 showed that the particle size decreased with increasing polymer amount. A one-way ANOVA was done at level p < 0.05 and a significant decrease in the particle size was associated with the increase in polymer (Tables IV and V).
Fig. 2.
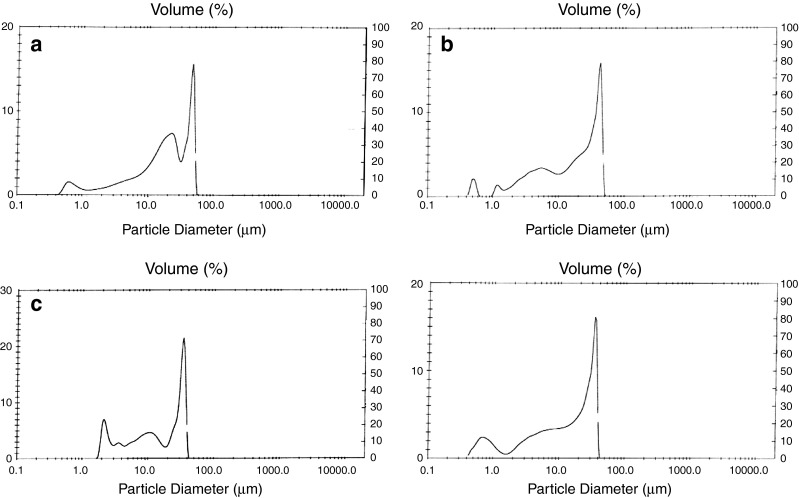
Particle size distribution of F1–F4
Table IV.
ANOVA of the particle size of microsponges between F1–F4
| Sum of Squares | df | Mean Square | F | Significance | |
|---|---|---|---|---|---|
| Between groups | 692.5 | 3 | 230.8 | 102.59 | p < 0.0001 |
| Within groups | 9 | 4 | 2.25 | ||
| Total | 701.5 | 7 |
Table V.
LSD of the Particle Size of Microsponges Between F1–F4
| (I) VAR00001 | (J) VAR00001 | Mean difference (I − J) | Standard error | Significance | 95% confidence interval | |
|---|---|---|---|---|---|---|
| Lower bound | Upper bound | |||||
| 1.00 | 2.00 | 14.0000a | 1.335 | .000 | 10.2945 | 17.7055 |
| 3.00 | 19.2500a | 1.335 | .000 | 15.5445 | 22.9555 | |
| 4.00 | 24.5000a | 1.335 | .000 | 20.7945 | 28.2055 | |
| 2.00 | 1.00 | 14.0000a | 1.335 | .000 | −17.7055 | −10.2945 |
| 3.00 | 5.2500a | 1.335 | .017 | 1.5445 | 8.9555 | |
| 4.00 | 10.5000a | 1.335 | .001 | 6.1945 | 14.2055 | |
| 3.00 | 1.00 | −19.2500a | 1.335 | .000 | −22.9555 | −15.5445 |
| 2.00 | −5.2500a | 1.335 | .017 | −8.9555 | −1.5445 | |
| 4.00 | 5.2500a | 1.335 | .017 | 1.5445 | 8.9555 | |
| 4.00 | 1.00 | −24.5000a | 1.335 | .000 | −28.2055 | −20.7945 |
| 2.00 | 10.5000a | 1.335 | .001 | −14.2055 | −6.7945 | |
| 3.00 | −5.2500a | 1.335 | .017 | −8.9555 | −1.5445 | |
aSignificant at the 0.05 level
Such results were contradictory to many previous studies which conclude that increasing in particle size is concomitant to the increase in polymer amount. This might be due to the larger volume of solvent associated with an increasing amount of polymer (the ratio of polymer to solvent is 1:5) resulting in diluting the internal phase and decreasing its viscosity. Some researchers pointed out that the particle size of microparticles was observed to be proportional with the dispersed phase viscosities, in such a way that decreasing the dispersed phase viscosity resulted in decreasing the particle size of microparticles (27–29).
The viscosities of dispersed phases of each of the four microsponge preparations (F1–F4) and that of liquid paraffin were measured, and the values were represented in Table VI. It was observed that when the viscosities of the dispersed phases decreased from 25.6 cP in F1 to 18 cP in F4, the particle sizes of the microsponges were found to decrease.
Table VI.
Viscosity of Dispersed Phase and Dispersion Medium of Microsponge Preparations
| Dispersed and continuous phase | F1 | F2 | F3 | F4 | Liquid paraffin |
|---|---|---|---|---|---|
| Viscosity (cP) | 25.6 | 22 | 20.5 | 18 | 30.5 |
The larger the difference in viscosities between the dispersed phase and the dispersion medium (30.5 cP), the lower the mean particle sizes resulted and vice versa. This was attributed to the fact that when the dispersed phase with higher viscosity was poured into the dispersion medium of high viscosity itself, the emulsion was hardly broken into small droplets, and the bigger ones were formed.
Flowability test results (Table II) revealed that the values of angles of repose for the formulae (F1, F2) were nearly 25 while those of F3 and F4 were 20 and 19.3, respectively. According to their Carr’s index, all formulae showed good flow properties. The values of the Hausner ratio for all formulae were nearly around 1.2 indicating the presence of interparticle friction. Such friction was attributed to the presence of liquid paraffin as continuous phase of emulsion, and despite of the use of several amounts of n-hexane as washing solvent, a portion of mineral oil still remained causing adhesion of the microsponges.
The emulsion solvent diffusion method could fuel interest in the preparation of microsponges, thus providing a high percentage of porosity. By studying the effect of the drug/polymer ratio on the porosity of microsponges, the results presented in Table VII were observed.
Table VII.
The Effect of Drug/Polymer Ratio on Porosity of Microsponge Formulations
| Preparation | Drug/polymer | Total intrusion (ml/g)a | Total pore area (m2/g)b | Average pore diameter (μm) | Bulk density (g/ml)a | Apparent density (g/ml)a | Porosity%b |
|---|---|---|---|---|---|---|---|
| F4 | 1:4 | 1.27 ± 0.14 | 24.01 ± 0.43 | 0.21 ± 0.02 | 0.48 ± 0.08 | 1.23 ± 0.14 | 60.91 ± 2.11 |
| F3 | 1:3 | 1.42 ± 0.16 | 25.27 ± 0.39 | 0.24 ± 0.03 | 0.44 ± 0.06 | 1.17 ± 0.11 | 62.56 ± 1.94 |
| F3 | 1:2 | 1.60 ± 0.10 | 26.94 ± 0.42 | 0.26 ± 0.05 | 0.41 ± 0.09 | 1.17 ± 0.10 | 65.24 ± 2.01 |
| F1 | 1:1 | 2.10 ± 0.13 | 125.33 ± 0.51 | 0.07 ± 0.01 | 0.34 ± 0.04 | 1.21 ± 0.13 | 71.83 ± 2.05 |
aNo significant differences between all formulations (p > 0.05)
bNo significant differences between F2, F3 and F4 (p > 0.05), and significant differences between F1 and other formulations (F2–F4) (p < 0.05)
According to the comparison of F1–F4 formulations, total intrusion volume, average pore diameter, total pore area, and porosity values showed a small increase due to the decrease in the drug/polymer ratio, but no significant difference was seen between these results (p > 0.05). On the other hand, F1 exhibited a fivefold increase in the total pore area and a fourfold decrease in the average pore diameter when compared with F2–F4 (p < 0.05). The percentage of porosity of F2–F4 ranged between 60.9% and 65.2% (p > 0.05), of F1 was 71.8%, and the difference between F1 and the other formulations was statistically significant (p < 0.05). The previous data indicated that as the drug/polymer ratio decreased, the percentage porosity of microsponges decreased. The in vitro release profile of the drug from microsponges possessing different drug/polymer ratios were illustrated in Fig. 3, and the kinetic treatment of release data were presented in Table VIII. It was revealed that the drug diffused through the microsponges’ pores and found its way out to the dissolution medium following fickian mechanism of diffusion (Table VIII).
Fig. 3.

The in vitro release profile of HYD-HCl from microsponges F1–F4
Table VIII.
Kinetic Treatment of the Release Data of the Drug from F1 to F4
| Preparation number | Correlation coefficient (r 2) | ||||
|---|---|---|---|---|---|
| Zero | First | Second | Diffusion | Order of release | |
| F1 | 0.3512 | 0.2898 | 0.2621 | 0.4777 | Diffusion |
| F2 | 0.5504 | 0.4683 | 0.3615 | 0.7253 | Diffusion |
| F3 | 0.8362 | 0.7244 | 0.5627 | 0.9443 | Diffusion |
| F4 | 0.8500 | 0.6774 | 0.4472 | 0.9546 | Diffusion |
It is reasonable to conclude that the release profiles of hydroxyzine HCl from the microsponges in all cases showed two distinct phases. An initial burst release phase occurs within the first hour, followed by a gradual release phase. The release profiles could be explained by two diffusion processes, one occurring after the other. The diffusion of the aqueous dissolution medium into the matrix results in the dissolution of the drug, which is then followed by drug diffusion through the pores into the dissolution medium.
According to the rate constant of the release (Table IX), almost similar constant rates of release were observed in F1 and F2, while nearly similar lower rate constants appeared to be associated with F3 and F4. In the case of low polymer-containing preparations, a thin matrix was encapsulating the drug. Accordingly, a shorter diffusion path length resulted, and the drug was easily released in the dissolution medium.
Table IX.
Kinetic Parameters of the Drug Release Data from Microsponge Preparations
| Preparation number | Rate constant of release (mg/min1/2; slope) | Burst effect (%) (intercept) | Half-life (min) | Half-life (h) |
|---|---|---|---|---|
| F1 | 7.115 | 28.60 | 9.05 | 0.15 |
| F2 | 6.88 | 20.1 | 48.47 | 0.80 |
| F3 | 3.77 | 14.10 | 90.67 | 1.5 |
| F4 | 3.9 | 6.08 | 157.78 | 2.63 |
Effect of Changing the Percentage of Magnesium Stearate
Magnesium stearate was added to the formulations as droplet stabilizer to overcome the problem of coalescence during solvent evaporation as it reduces the phase tension between Eudragit microcapsules and liquid paraffin and prevents flocculation of microsponges (30–32).
Two additional concentrations 1.5% and 5% w/v Mg stearate were used in preparing the microsponges possessing (F5, F6) 1:1 and (F7, F8) 1:2 drug/polymer ratio.
The characteristics of the prepared microsponges represented in Table X show that low concentration of Mg stearate (1.5%) resulted in aggregation of microsponges. It has been reported that metal stearate, such as magnesium and aluminum, reduces the interfacial tension and prevents electrification and flocculation during the preparation of microspheres in general. However, increasing its percentage to 5% neutralized the electric charge of Eudragit RS-100 due to accumulation of magnesium stearate on the surface, leading to aggregates as those previously reported by Kim et al. (13).
Table X.
Effect of Different Concentrations of Mg Stearate on the Microsponge Preparations
| Parameter evaluated | Magnesium stearate concentration | |||
|---|---|---|---|---|
| 1.5% (F5) | 5% (F6) | 1.5% (F7) | 5% (F8) | |
| Particle shape | Aggregates | |||
Magnesium stearate was previously reported to produce smooth nonporous surfaces when dissolved in the dispersed phase solvent. However, the porous microsponges which resulted in the study were due to its insolubility and just its dispersion in the solvent of the dispersed phase (33).
Effect of the Polymer/Solvent Ratio and Span 80 on the Particle Size of Selected Formula
F1 was detected as the formula with the highest mean particle size. In an attempt to decrease its mean particle size, the viscosity of the dispersed phase was decreased by increasing the amount of solvent. The decrease in polymer/solvent ratio in preparations F9–F11, in the absence of Span 80 or in its presence like in preparations F12–F15, resulted in a drastic decrease in the particle size to reach the nano size, which is not accepted as topically applied powder (Table XI).
Table XI.
Effect of Increasing Polymer/Solvent Ratio in the Absence or Presence of Span 80
| Parameter evaluated | Absence (−) or presence (+) of Span 80 | ||||||
|---|---|---|---|---|---|---|---|
| F9 (−) | F10 (−) | F11 (−) | F12 (−) | F13 (−) | F14 (−) | F15 (−) | |
| Mean particle size | D < 1 μ | D < 1 μ | D < 1 μ | Nanoparticle | |||
For more confirmatory results, Span 80 was added during the preparation of F1 (1:5) polymer to solvent ratio, and similar results were obtained. Such nano size microspheres obtained after the addition of Span 80 to liquid paraffin is due to the higher stabilizing effect that Span 80 imparts to the small emulsion droplets which prevents them from collision with larger ones.
Study of the Effect of Pore Inducers on the Release Profile of Selected Formula
The formula containing the 1:4 drug/polymer ratio was chosen to enhance the release of the drug by the addition of a pore inducer. Pore inducers are hydrophilic powders that can be added during the formulation of microspheres to obtain a desirable release of active ingredients; they are liable to drain water from the dissolution medium either by osmotic effect like sucrose or by their adsorbing and disintegrant effect like pregelatinized starch (34).
It was obvious from results obtained in Table XII and Fig. 4, that increasing the amount of either sucrose or PGS from 0.5 g in F16 and F18 to 1.5 g in F17 and F19, increased the encapsulation efficiency and the particle size of the microsponges prepared. This could be attributed to the large amount of pore inducers which provided a large surface area for the drug to be evenly adsorbed onto its particles, resulting in the preparation of microsponges adequately loaded with hydroxyzine HCl. PGS microsponges possessed a larger particle size than those prepared by sucrose, which was due the initial large particle size of PGS (Fig. 5).
Table XII.
Evaluation of Microsponges Containing Different Types of Pore Inducers
| Parameters evaluated | Type of pore inducer | |||
|---|---|---|---|---|
| Sucrose (F16) | Sucrose (F17) | PGS (F18) | PGS (F19) | |
| Encapsulation efficiency (% ±SD) | 69 ± 3.4 | 85 ± 2.2 | 75 ± 2.25 | 90 ± 1.05 |
| Mean particle size (μm ± SD) | 33.2 ± 3.3 | 35.2 ± 3.3 | 45.2 ± 3.3 | 51.2 ± 0.3 |
Fig. 4.

Encapsulation efficiency of microsponges prepared using different amounts of sucrose and PGS
Fig. 5.

Particle size of microsponges prepared using 1.5-g pore inducers
F17 and F19 were chosen for the in vitro release study. The release profile of hydroxyzine HCl in phosphate buffer at pH 5.5 was illustrated in Fig. 6. In order to determine the order of release, the data were analyzed according to zero order, first order, and diffusion controlled mechanism according to simplified the Higuchi model as shown in Tables XIII and XIV.
Fig. 6.

In vitro release profile of HYD-HCl from microsponges containing pore inducers
Table XIII.
Kinetic Treatment of the Release Data of the Drug from F17 and F19
| Preparation number | Correlation coefficient (r 2) | ||||
|---|---|---|---|---|---|
| Zero | First | Second | Diffusion | Order of release | |
| F17(sucrose) | 0.6405 | 0.5772 | 0.5088 | 0.7893 | Diffusion |
| F19 (PGS) | 0.7961 | 0.7270 | 0.6455 | 0.9106 | Diffusion |
Table XIV.
Kinetic Parameters of the Dissolution Data of the Drug from F17 and F19
| Preparation number | Rate constant of release (mg/min1/2) | Burst effect (%) | Half-life (min) | Half-life (h) |
|---|---|---|---|---|
| F17 (sucrose) | 4.25 | 30.611 | 20.8 | 0.34 |
| F19 (PGS) | 5.72 | 32.24 | 9.64 | 0.166 |
As illustrated in Fig. 6, F19 (containing PGS) showed 100% of the drug released after 180 min while F17 (containing sucrose) reached a steady-state concentration of 78% after 90 min. The drug was released from all formulae according to fickian diffusion mechanism (Table XIII).
According to Table XIV, the rate of drug released from PGS containing microsponges was faster than containing sucrose. That might be due to the efficiency of the disintegrant effect of PGS in the faster rupture of the microsponge matrix. Initial burst effect was associated with both release patterns. The present pore inducers in large amounts (1.5 g) lead to the absorption of larger volumes of dissolution medium which passes through the pores of microsponges and consequently bursts, releasing the drug carried onto their surface. PGS provided shorter half-life of drug release than sucrose due to its highly disintegrant property in the presence of water containing media.
From the release profile of F2 previously discussed, microsponges showed nearly 100% of the drug released after 180 min; this was quite similar to the release of the drug from the preparation containing the PGS pore inducer. Other properties have been compared to detect the best formulae among F2 and F19. Data obtained in Tables XV and XVI, revealed that F2 had a higher drug entrapment and smaller particle size than F19.
Table XV.
Comparison Between the Characteristics of F2 and F19
| Parameter evaluated | Preparation number | |
|---|---|---|
| F2 | F19 | |
| *EE% (% ±SD) | 98.08 ± 0.7 | 90 ± 1.05 |
| Mean particle size (μm) (±SD) | 42.6 ± 1.7 | 57.2 ± 0.3 |
| Angle of repose | 24.61 | 24 |
| Carr’s index | 18.03 | 21.87 |
| Hausner ratio | 1.22 | 1.28 |
Table XVI.
ANOVA Test for the Percentage HYD-HCl Encapsulated in F2 and F19
| Sum of squares | df | Mean square | F | Significance | |
|---|---|---|---|---|---|
| Between groups | 79.21 | 1 | 79.21 | 74.72 | p < 0.01 |
| Within groups | 2.12 | 2 | 1.06 | ||
| Total | 81.33 | 3 |
The scanning electron micrograph of F2 illustrated in Fig. 7 showed microspheres with roughly cracked surfaces containing pores. The solvent diffusion of acetone to the external liquid paraffin resulted in cracking and channeling in the surface of microspheres, owning such surface topography; the microspheres prepared were referred to as microsponges.
Based on such results F2 was chosen for further study.
Fig. 7.

Scaning electron micrograph of microsponges F2 prepared by o/o emulsion solvent diffusion method
In Vivo Study
The preliminary study for the determination of the dose of histamine that produced optimum wheal area has led to the choice of 300 mg/ml. That optimum dose resulted in a wheal of swollen tissue, having a radius of 8.5 mm and an area of 226.865 mm2 that persisted for 6 h as illustrated in Fig. 8.
Fig. 8.

Relationship between wheal area and histamine dose 10 min after its intradermal injection
Comparative Pharmacodynamic Activities of Hydroxyzine Hydrochloride Microsponge Preparations
After topical application of the chosen microsponge preparations (plain or medicated) on the inflamed back of rabbits, the mean wheal areas obtained at different time intervals were calculated in each group of rabbit. The correlation between mean areas and time were represented in Fig. 9. The peripheral H1-antihistaminic activity of the drug over time interval was illustrated in Fig. 10 as mean percentage suppression of histamine-induced wheal area.
Fig. 9.

Wheal area of the tested preparations at different time intervals
Fig. 10.

Percentage wheal area suppression for the tested preparations after 4 h
It was obvious that the wheal disappeared completely in the group treated with medicated microsponges F2; whereas, the group receiving plain microsponges showed 21.2% reduction in the inflamed area after the same time interval.
In comparison with the initial wheal areas, all the medicated preparations showed obvious wheal area suppression after 4 h. The wheal suppression was about 80% after the first 2 h in the case of the rabbits treated with F2, while its maximum suppression (99.46%) was reached after 4 h. No detectable healing of the backs of rabbits in groups receiving the nonmedicated preparations, which indicated the absence of therapeutic effect of plain microsponges. Further histopathological study was conducted to demonstrate the therapeutic effect of medicated F2.
Histopathological Study of Rabbit Skin
Histological Structure of the Intact Rabbit Skin Layers
Normal histological structures were seen in the epidermal cell layers (germinativum, spinosum or prickle cell layer, granulosum, lucidum, and corneum) and the underlying dermal connective tissue layer (Fig. 11a, b).
Fig. 11.

a Normal histological structure of the epidermis (E). b normal histological structure of the dermis (D)
Histological Structure of the Inflamed Rabbit Skin Layers
When histamine was injected, a spontaneous flare and swollen spherical area was obtained and was clearly detected by the naked eye. The histological structure of the skin showed hyperkeratosis which indicated thickening of the stratum corneum and acanthosis (inflammation and increased number of the prickle cell layer) of the epidermis. Such epidermal inflammation was associated with inflammatory cell infiltration, edema, and dilatation of the blood vessels occurring in the dermal layer as illustrated in Fig. 12a, b, c.
Fig. 12.

a Thickening of the epidermis by acanthosis (a) and hyperkeratosis (E). b, c Inflammatory cell infiltration with edema (m) and dilated blood vessels (D) in the dermal layer
Histological Structure of the Treated Rabbit Skin
The histopathological patterns of the inflamed and treated skin varied greatly according to the type of the applied formula. As shown in Fig. 13 a, a focal area of mild acanthosis was observed in the prickle cell layer of the epidermis of the skin of rabbits treated with F2. Figure 13b showed invaginated acanthosis of the prickle area of the skin; it was associated with blood cell infiltration in the dermal layer, in the case of skin treated with plain F2.
Fig. 13.

a Skin treated with F2, showing mild acanthosis with normal dermal histology. b Skin treated with plain microsponges, showing invaginated epidermal acanthosis and blood cell infiltration in the dermis
It was obvious that an antihistaminic effect was exerted from the medicated formulae. HYD-HCl despite of its ionic properties, succeeded to pass through the stratum corneum to reach the lower epidermal layers, inhibiting the action of histamine. Such passage could be realized through channels in protein molecules embedded in the lipid bilayer of the cell membrane (35).
A previous study on the topical antiviral effect of cidofovir-loaded microparticles (36) suggested the passage of the drug at higher concentrations to the first skin layers to an occlusive effect exerted by microparticles. That effect produced a film on the skin surface which reduced the transepidermal water loss and favored drug penetration into the skin.
ccording to the results previously reported, we could assume the relevance and the reliability of the application of microsponges containing hydroxyzine hydrochloride for topical drug delivery.
CONCLUSION
From the previous results, it was possible to conclude the following:
Microencapsulation by oil in oil solvent diffusion represented a successful method of encapsulation of water-soluble drugs as it led to more than 95% of the drug entrapped.
Solvent diffusion of acetone from the internal phase to the external liquid paraffin resulted in channels and pores formation on the surface of microspheres; such structure was referred to as microsponges.
Decreasing the viscosity of internal oily phase in comparison to that of liquid paraffin decreased the mean particle size of the prepared microsponges.
The addition of magnesium stearate in a specified concentration resulted in successful microencapsulation and free-flowable powder. Such concentration was dependent on the solvent volume and amount of polymer; in a manner that 3% w/v Mg stearate added to formulae prepared using 1:5 polymer/solvent ratio succeeded to prepare free-flowable powder microsponges.
The addition of Span 80 to liquid paraffin caused a drastic decrease in particle size to a nano size.
Inappropriate washing of liquid paraffin increased cohesion between microsponges.
The addition of PGS as pore inducer enhanced the release of the drug from microsponges due to its water absorption and disintegrant properties.
The F2 preparation chosen to evaluate the pharmacodynamic activity of hydroxyzine HCl showed effective therapeutic response.
Footnotes
The protocol of the present work was approved by the Experiments and Advanced Pharmaceutical Research Unit (EAPRU), Faculty of Pharmacy, Cairo University, Cairo, Egypt.
Any experiments involving the use of animals were conducted in accordance with the principles of Laboratory Animal Care and were approved by the institutional ethics committee.
REFERENCES
- 1.Villamizar L, Barrera G, Marina CA, Martínez F. Eudragit S100 microparticles containing Spodoptera frugiperda nucleopolyehedrovirus: physicochemical characterization, photostability and in vitro virus release. J Microencapsulation. 2010;27(4):314–24. doi: 10.3109/02652040903191826. [DOI] [PubMed] [Google Scholar]
- 2.Mana Z, Pellequer Y, Lamprecht A. Oil-in-oil microencapsulation technique with an external perfluorohexane phase. Int J Pharm. 2007;338:231–237. doi: 10.1016/j.ijpharm.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Viswanathan NB, Thomas PA, Pandit JK, Kulkarni MG, Mashelkar RA. Preparation of non-porous microspheres with high entrapment efficiency of proteins by a (water-in-oil)-in-oil emulsion technique. J Contr Rel. 1999;58:9–20. doi: 10.1016/S0168-3659(98)00140-0. [DOI] [PubMed] [Google Scholar]
- 4.Bogataj M, Mrhar A, Kristl A, Kozjek F. Eudragit E microspheres containing bacampicillin: preparation by solvent removal methods. J Microencapsulation. 1991;8:401–406. doi: 10.3109/02652049109069567. [DOI] [PubMed] [Google Scholar]
- 5.Benoit JP, Marchais H, Rolland H, Velde VV. Biodegredable microspheres: advances in production technology. In: Benita S, editor. Microencapsulation methods and industrial applications. New York: Marcel Dekker; 1996. pp. 35–72. [Google Scholar]
- 6.Thies C. Formation of degradable drug-loaded micro-particles by in-liquid drying processes. In: Donbrow M, editor. Microcapsules and nanoparticles in medicine and pharmacy. London: CRC; 1992. pp. 47–71. [Google Scholar]
- 7.Bogataj M, Mrhar A. Determination of microsphere solidification time in the solvent evaporation process. J Microencapsulation. 2005;22(1):81–90. doi: 10.1080/02652040400026301. [DOI] [PubMed] [Google Scholar]
- 8.Kibbe AH. Handbook of pharmaceutical excipients. 3. Washington, DC: American Pharmaceutical Association and Pharmaceutical; 2000. pp. 401–406. [Google Scholar]
- 9.Eudragit Data Sheets, Industrial Products Division, Röhm Pharma GmbH, Weiterstadt, Germany.
- 10.US Pharmacopeia 24, US Pharmacopeial Convention, Rockville, MD, 2000, pp. 2477–2478 and 1945–1946.
- 11.Haznedar S, Dortunç B. Preparation and in vitro evaluation of Eudragit microspheres containing acetazolamide. Int J Pharm. 2004;269:131–140. doi: 10.1016/j.ijpharm.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Kiliçarslan M, Baykara T. The effect of the drug/polymer ratio on the properties of the verapamil HCl loaded microspheres. Int J Pharm. 2003;252:99–109. doi: 10.1016/S0378-5173(02)00630-0. [DOI] [PubMed] [Google Scholar]
- 13.Kim CK, Kim MJ, Oh KH. Preparation and evaluation of sustained release microspheres of terbutaline sulfate. Int J Pharm. 1994;106:213–219. doi: 10.1016/0378-5173(94)90004-3. [DOI] [Google Scholar]
- 14.Goto S, Kawata M, Nakamura M, Maekawa K, Aoyama T. Eudragit E, L and S (acrylic resins) microcapsules as pH sensitive release preparations of ketoprofen. J Microencapsulation. 1986;3(b):305–316. doi: 10.3109/02652048609021800. [DOI] [PubMed] [Google Scholar]
- 15.Kawata M, Nakamura M, Goto S, Aoyama T. Preparation and dissolution pattern of Eudragit RS microcapsules containing ketoprofen. Chem Pharm Bull. 1986;34:2618–2623. doi: 10.1248/cpb.34.2618. [DOI] [PubMed] [Google Scholar]
- 16.Yüksel N, Baykara T. Preparation of polymeric microspheres by the solvent evaporation method using sucrose stearate as droplet stabilizer. J. Microencapsulation. 1997;14:725–733. doi: 10.3109/02652049709006822. [DOI] [PubMed] [Google Scholar]
- 17.Bhardwaj SB, Shukla AJ, Collins CC. Effect of varying drug loading on particle size distribution and drug release kinetics of verapamil hydrochloride microspheres prepared with cellulose esters. J Microencapsulation. 1995;12:71–81. doi: 10.3109/02652049509051128. [DOI] [PubMed] [Google Scholar]
- 18.El Gibaly, Abdel-Ghaffar SK. Effect of hexacosanol on the characteristics of novel sustained release allopurinol solid lipospheres (SLS): factorial design application and product evaluation. Int J Pharm. 2005;294:33–51. doi: 10.1016/j.ijpharm.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 19.Toongsuwan S, Li L, Erickson BK, Chang H. Formulation and characterization of bupivacaine lipospheres. Int J Pharm. 2004;280:57–65. doi: 10.1016/j.ijpharm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Iannuccelli V, Sala N, Tursilli R. Influence of liposphere preparation on butyl-methoxydibenzoyl methane photostability. Eur J Pharm Biopharm. 2006;63:140–145. doi: 10.1016/j.ejpb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Tursilli R, Casolari A, Iannuccelli V, Scalia S. Enhancement of melatonin photostability by encapsulation in lipospheres. J Pharm Biomed Anal. 2006;40:910–914. doi: 10.1016/j.jpba.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 22.Nasr M, Mansour S, Mortada N, El Shamy AA. Lipospheres as carriers for topical delivery of aceclofenac: preparation, characterization and in vivo evaluation. AAPS PharmSciTech. 2008;9(1):154–62. doi: 10.1208/s12249-007-9028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pauli G, Frossard N. Étude des antihistaminiques par la technique du « wheal and flare »:quelle signification faut-il lui accorder ? Revue française d’allergologie et d’immunologie Clinique. 2004;44:194–198. [Google Scholar]
- 24.El zainy A, Simons KJ. Hydroxyzine from topical phospholipid liposomal formulations: evaluation of peripheral antihistaminic activity and systemic absorption in a rabbit model. AAPS PharmSci. 2003;5(4), Article 28. [DOI] [PMC free article] [PubMed]
- 25.Simons FER, Silver NA, Gu X, Simons KJ. Skin concentrations of H1-receptor antagonists. J Allergy Clin Immunol. 2001;107:526–530. doi: 10.1067/mai.2001.113080. [DOI] [PubMed] [Google Scholar]
- 26.Banchroft JD, Stevens A, Turner DR. Theory and practice of histological techniques. 4. New York: Churchil Livingstone; 1996. pp. 445–455. [Google Scholar]
- 27.Pongpaibul Y, Price JC, Whitworth CW. Preparation and evaluation of controlled release indomethacin microspheres. Drug Dev Ind Pharm. 1984;10:1597–1616. doi: 10.3109/03639048409039070. [DOI] [Google Scholar]
- 28.Malamataris S, Avgerinos A. Controlled release indomethacin microspheres prepared by using an emulsion solvent-diffusion technique. Int J Pharm. 1990;62:105–111. doi: 10.1016/0378-5173(90)90223-Q. [DOI] [Google Scholar]
- 29.Chiao CSL, Price JC. Formulation, preparation and dissolution characteristics of propranolol hydrochloride microspheres. J Microencapsulation. 1994;11:153–159. doi: 10.3109/02652049409040446. [DOI] [PubMed] [Google Scholar]
- 30.Goto S, Kawata M, Nakamura M, Aoyama T. Effect of magnesium stearate in the preparation of Eudragit RS microcapsules containing drugs. Yakugaku Zasshi. 1985;105:1087–1095. doi: 10.1248/yakushi1947.105.11_1087. [DOI] [PubMed] [Google Scholar]
- 31.Goto S, Kawata M, Nakamura M, Maekawa K, Aoyama T. Eudragit RS and RL (acrylic resins) microcapsules as pH insensitive and sustained release preparations of ketoprofen. J Microencapsulation. 1986;3(4):293–304. doi: 10.3109/02652048609021799. [DOI] [PubMed] [Google Scholar]
- 32.Arshady R. Preparation of biodegradable microspheres and microcapsules 2. Polylactides and related polyesters. J Contr Rel. 1991;17:1–22. doi: 10.1016/0168-3659(91)90126-X. [DOI] [Google Scholar]
- 33.Horoz BB, Kiliçarslan M, Yüksel N, Baykara T. Influence of aluminum tristearate and sucrose stearate as the dispersing agents on physical properties and release characteristics of Eudragit RS microspheres. AAPS PharmSciTech. 2006;7(1):E111–E117. doi: 10.1208/pt070116. [DOI] [PubMed] [Google Scholar]
- 34.Song M, Li N, Sun S, Tiedt LR. Effect of viscosity and concentration of wall former, emulsifier and pore-inducer on the properties of amoxicillin microcapsules prepared by emulsion solvent evaporation. П Farmaco. 2005;60:261–267. doi: 10.1016/j.farmac.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175(23):720–31. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 36.Santoyo S, Ga de Jalón E, Ygartua P, Renedo MJ, Blanco-Príeto MJ. Optimization of topical cidofovir penetration using microparticles. Int J Pharm. 2002;242(1–2):107–13. doi: 10.1016/S0378-5173(02)00178-3. [DOI] [PubMed] [Google Scholar]