

Abstract
Psoriasis is a chronic, autoimmune skin disease affecting approximately 2% of the world's population. Clobetasol propionate which is a superpotent topical corticosteroid is widely used for topical treatment of psoriasis. Conventional dosage forms like creams and ointments are commonly prefered for the therapy. The purpose of this study was to develop a new topical delivery system in order to provide the prolonged release of clobetasol propionate and to reduce systemic absorption and side effects of the drug. Clobetasol propionate loaded-poly(D,L-lactic-co-glycolic acid) (PLGA) microspheres were prepared by oil-in-water emulsion–solvent evaporation technique. Particle size analysis, morphological characterization, DSC and XRD analyses and in vitro drug release studies were performed on the microparticle formulations. Emulgel formulations were prepared as an alternative for topical delivery of clobetasol propionate. In vitro drug release studies were carried out from the emulgel formulations containing pure drug and drug-loaded microspheres. In addition, the same studies were performed to determine the drug release from the commercial cream product of clobetasol propionate. The release of clobetasol propionate from the emulgel formulations was significantly higher than the commercial product. In addition, the encapsulation of clobetasol propionate in the PLGA microspheres significantly delayed the drug release from the emulgel formulation. As a result, the decrease in the side effects of clobetasol propionate by the formulation containing PLGA microspheres is expected.
KEY WORDS: clobetasol propionate; emulgel; emulsion–solvent evaporation method; poly(D,L-lactic-co-glycolic acid) microparticles; psoriasis
INTRODUCTION
Psoriasis is a relatively common skin disease that affects approximately 2% of the world's population. It is a chronic and T-cell-mediated autoimmune disorder with hyperproliferation of the epidermis and inflammatory reactions of the dermis and epidermis (1,2). The treatment of psoriasis varies depending on disease severity and spread. However, topical medications remain the mainstay of psoriasis treatment for most patients. As seen in the literature, topical corticosteroids and particularly super potent ones are the most widely prescribed medications for the topical treatment of psoriasis for decades in the world (3,4). They are available in numerous vehicles including powders, sprays, lotions, solutions, creams, emollient creams, ointments, gels and medicated tapes (3). Although serious cutaneous and systemic side effects of the corticosteroids have limited their use, they are still among the most effective treatments.
Clobetasol propionate is one of the most potent glucocorticoid for topical use and offers effective and rapid healing of psoriatic lesions (5,6). However, incidences of unfavourable side effects are greater than those of related compounds (7). Recently, some studies have been carried out on drug delivery systems as an alternative to the conventional dosage forms. Drug delivery systems such as solid lipid nanoparticles (8), lipid microspheres (9), nanostructured lipid carriers (10) and lecithin/chitosan nanoparticles (11) containing clobetasol propionate were developed and evaluated.
Microparticulate drug delivery systems have been extensively used for oral and parenteral administration and could also be useful to deliver several drugs into the skin. However, there are few researches about dermal application of microparticles. Microparticulate systems provide the possibility to exert an effect over a prolonged period of time and to reduce systemic absorption and toxic side effects (12,13).
Microparticles prepared with synthetic biodegradable and biocompatible polymers such as poly(D,L-lactic-co-glycolic acid) (PLGA) have been increasingly used as drug delivery system, since they do not have most of the problems associated with the natural polymers such as higher cost and questionable purity (14). Several drugs such as paclitaxel (15), retinoic acid (16), flurbiprofen sodium (17) and ganciclovir (18) were successfully encapsulated into PLGA microparticles for several purposes. It has been also shown that PLGA microparticles could be used to deliver acyclovir into the basal epidermis (19). Similar results were obtained in some studies which were carried out with different drugs for topical delivery, such as cidofovir (20) and urea (13).
The aim of this study was to develop a topical drug delivery system in order to provide the prolonged release of clobetasol propionate, minimize the systemic drug absorption and reduce the possible side effects of the drug. For this purpose, PLGA microspheres loaded with clobetasol propionate were prepared and characterized. The effects of different PLGA types, homogenization speeds and times, drug/polymer ratios and polyvinyl alcohol (PVA) concentrations on the characteristics of the microspheres were evaluated. Selected microsphere formulation and pure drug were formulated in an emulgel base. Emulgels contain an oil phase at low percentage in a gel-type vehicle and they have similar characteristics to the lipoprotein structure of the skin. By this way, increase in the permeability of the drugs can be provided. Additionally, emulgels are more easily spreadable bases for the wide surfaces like psoriatic lesions compared to cream or ointment formulations. Release profiles of clobetasol propionate from emulgel formulations were compared with commercial product of the drug.
MATERIALS AND METHODS
Materials
Clobetasol propionate was kindly provided by Sandoz Drug Company (Kocaeli, Turkey). PLGA 50:50 (Mw, 40,000–75,000) and PLGA 75:25 (Mw, 66,000–107,000) were purchased from Sigma (St. Louis, USA); PLGA 50:50 (Mw, 5,000–15,000) and PLGA 85:15 (Mw, 50,000–75,000) were purchased from Aldrich (Steinheim, Germany). Polyvinyl alcohol (PVA) (Mw, 30,000–70,000) was obtained from Sigma–Aldrich (St. Louis, USA). Methanol (high-performance liquid chromatography (HPLC) grade), dichloromethane and ethanol were purchased from Merck (Darmstadt, Germany). All other chemicals were of analytical reagent grade and MilliQ water was used in the experiments. Commercial cream product containing 0.05% clobetasol propionate was also used.
Preparation of PLGA Microspheres
Clobetasol propionate-loaded PLGA microspheres were prepared by oil-in-water (o/w) emulsion–solvent evaporation technique. The method of Ga De Jalón et al. (19) was modified in the study. The codes and composition of the microsphere formulations and the respective homogenization conditions are shown in Table I. Briefly, clobetasol propionate was dissolved in the solution of polymer in dichloromethane, the organic phase obtained was added to aqueous PVA solution and homogenised using UltraTurrax T25 Homogenizer® (Ika, Germany). This mixture was stirred using a propeller type mechanical stirrer (Stir-Pak, Cole-Parmer Ins., USA) at 25°C for 4 h to evaporate the organic solvent. Microspheres were collected by centrifugation at 5,000 rpm for 20 min, washed three times with ultra-purified water and liyophilized for 48 h. The reduced pressure value and the condenser temperature were 0.018 mbar and −90°C, respectively, during the liyophilization process. The effect of different PLGA types (F1–F4-coded formulations), homogenization times (F3-, F5-coded formulations), homogenization speeds (F3-, F6- and F7-, F8-coded formulations), drug/polymer ratios (F3-, F8- and F6-, F7-coded formulations) and PVA concentrations (F8–F12-coded formulations) on the characteristics of microspheres were investigated.
Table I.
The Codes and Compositions of the Microspheres
| Code | Type of PLGA | Drug/polymer ratio | PVA (%) | Homogenization speed (rpm) | Homogenization time (min) |
|---|---|---|---|---|---|
| F1 | 85:15 (Mw, 50,000–75,000) | 1:10 | 0.5 | 8,000 | 1 |
| F2 | 75:25 (Mw, 66,000–107,000) | 1:10 | 0.5 | 8,000 | 1 |
| F3 | 50:50 (Mw, 40,000–75,000) | 1:10 | 0.5 | 8,000 | 1 |
| F4 | 50:50 (Mw, 5,000–15,000) | 1:10 | 0.5 | 8,000 | 1 |
| F5 | 50:50 (Mw, 40,000–75,000) | 1:10 | 0.5 | 8,000 | 2 |
| F6 | 50:50 (Mw, 40,000–75,000) | 1:10 | 0.5 | 9,500 | 1 |
| F7 | 50:50 (Mw, 40,000–75,000) | 1:5 | 0.5 | 9,500 | 1 |
| F8 | 50:50 (Mw, 40,000–75,000) | 1:5 | 0.5 | 8,000 | 1 |
| F9 | 50:50 (Mw, 40,000–75,000) | 1:5 | 1.0 | 8,000 | 1 |
| F10 | 50:50 (Mw, 40,000–75,000) | 1:5 | 2.0 | 8,000 | 1 |
| F11 | 50:50 (Mw, 40,000–75,000) | 1:5 | 0.2 | 8,000 | 1 |
| F12 | 50:50 (Mw, 40,000–75,000) | 1:5 | 0.1 | 8,000 | 1 |
HPLC Analysis
HPLC analyses were carried out according to the method that has been previously applied by Hu et al. (10). Determination of clobetasol propionate was performed using an HPLC system consisting of an Agilent 1,100 model G1311A pump and Agilent 1,100 model G1315B diode array detector (Agilent Tech, Germany). Separation was carried out using a NovaPak® C18 (4 μm, 150 × 3.9 mm) (Waters, Ireland) column operating at 25°C. HPLC analyses were performed by isocratic elution at a flow rate of 1.0 ml/min. The mobile phase composition was methanol:water (74:26 v/v). All solvents were degassed in an ultrasonic bath before use. The samples prepared with methanol were analysed at 240 nm and samples dissolved in phosphate-buffered saline (PBS) pH 7.4:ethanol (70:30) at 242 nm. An aliquot of 20 μl of each sample were injected into the system. The retention time for clobetasol propionate was observed as 3 min. The peak areas were integrated automatically by computer using a software programme (HPCORE ChemStation, Agilent Tech, Germany).
The analytical validation of the HPLC method was performed. The linearity, accuracy, precision and limit of detection–limit of quantification (LOD–LOQ) values of the method were calculated and evaluated.
Drug Loading and Encapsulation Efficiency
Ten miligrams of microspheres were dissolved in 2 ml of dichloromethane and then the volume was completed to 50 ml with methanol to precipitate the polymer. The solution was centrifuged at 15,000 rpm for 15 min. The supernatant was then collected and analysed by HPLC. The drug loading and the encapsulation efficiency values for all microsphere formulations were calculated according to the equations given below.
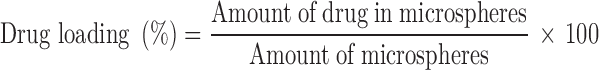 |
1 |
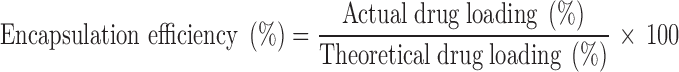 |
2 |
Particle Size Analysis
The mean particle size and size distribution of the microspheres were determined using low-angle laser light diffraction equipment (Sympatec HELOS H0728, Germany). For this purpose, approximately 10 mg of microspheres were suspended in an aqueous solution of 0.1% concentrated Tween® 80 using probe sonicator for 10 min before measurement.
Morphological Characterization
The shape and surface morphology of the microspheres were examined using a scanning electron microscope (SEM) (Jeol JSM-6,400, Japan). The microspheres were dusted on an adhesive carbon tape and coated with a gold layer at 250 Å. Samples were then imaged at a voltage of 10 kV.
Differential Scanning Calorimetry
Thermal analysis of clobetasol propionate, PLGA 50:50 (Mw,40,000–75,000), drug-loaded and blank microspheres were carried out using a differential scanning calorimeter (DSC) (Shimadzu DSC-60, Japan). The calibration of the heat flow scale was performed. 5 mg of samples were placed in hermetically sealed aluminium pans. The temperature range tested was 5–325°C. Samples were heated at 10°C/min under a nitrogen atmosphere at a flow rate of 40 ml/min.
Powder X-ray Diffractometry
X-ray diffraction patterns of the drug, polymer, drug-loaded and blank microspheres were recorded on a Rigaku D/Max-2,200/PC, Japan using Cu-Kα radiation, 46-kV voltage and 36-mA current for a 2θ diffraction angle with a range of 5–70°.
Preparation of Emulgel Formulations
Emulgel formulations were prepared for topical delivery of clobetasol propionate. Briefly, Carbopol® 934 was dispersed in purified water. Then, propylene glycol and Tween® 60 were added into the solution and this mixture was heated to 70°C. On the other hand, cetyl alcohol, stearyl alcohol, Span® 60, liquid and soft paraffins were melted all together and heated to 70°C. Afterwards, these two phases were mixed and triethanolamine solution in water was slowly poured into this emulsion to achieve gel formation through the neutralisation process. Finally, Cremophor® EL was dissolved in ethanol and added to the formulation when the temperature of the emulgel was approximately 30°C. Clobetasol propionate (0.05%) or clobetasol propionate-loaded microspheres including the same amount of drug were added into this o/w type emulgel formulation and mixed homogenously. The emulgel formulations containing pure clobetasol propionate and microspheres were coded as EC and EM, respectively.
In Vitro Drug Release
In vitro drug release studies of the microspheres, emulgel formulations and commercial cream product were carried out using modified Franz diffusion cell during 24 h. Mixture of PBS pH 7.4:ethanol (70:30) was used as receptor medium and sink condition was determined. The receptor phase was kept at a constant temperature of 37°C and stirred by a magnetic stirrer. Spectra/Por 2® dialysis membrane, MWCO: 12–14 kDa (Spectrum Lab.,USA) was used as the diffusion membrane. At appropriate time intervals, 0.5 ml of samples were collected and replaced by an equal volume of fresh receptor medium. The clobetasol propionate content was analysed by HPLC at a wavelength of 242 nm.
Statistical Analysis
All results were statistically analysed using Student's t test to determine the significance between two groups and using one-way ANOVA test for multiple comparison (SPSS-11.5). The data were considered to be significant at p < 0.05.
RESULTS AND DISCUSSION
Analytical Validation of HPLC Method
Analytical validation of the HPLC method was performed. Excellent linearity was obtained between concentrations of 0.5 and 25 μg/ml in methanol and in PBS pH 7.4:ethanol (70:30) solutions with r2 = 0.999. The accuracy, repeatability and intermediate precision of the method were proved, since the variation coefficient values were found to be less than 2%. While LOQ values in methanol and PBS pH 7.4:ethanol (70:30) solutions were found to be 0.46 and 0.41 μg/ml, LOD values were 0.15 and 0.13 μg/ml, respectively.
Encapsulation Efficiency and Particle Size of the Microspheres
The encapsulation efficiencies for all microsphere formulations prepared by o/w emulsion–solvent evaporation technique were found higher than 90% as seen in Table II. Clobetasol propionate is practically insoluble in water; therefore, this substance was preferred partitioning into the dispersed organic phase of the emulsion. The amount of the drug passing into the aqueous phase was very low; thus, high encapsulation efficiencies were obtained.
Table II.
Encapsulation Efficiencies and Particle Sizes of the Microsphere Formulations (n = 3)
| Formulation code | Drug loading (%) (mean ± SD) | Encapsulation efficiency (%) (mean ± SD) | Particle size (μm) (mean ± SD) |
|---|---|---|---|
| F1 | 8.90 ± 0.07 | 97.98 ± 0.80 | 19.41 ± 1.56 |
| F2 | 8.40 ± 0.04 | 92.41 ± 0.48 | 22.17 ± 1.57 |
| F3 | 8.52 ± 0.02 | 93.73 ± 0.22 | 14.61 ± 1.45 |
| F4 | 8.75 ± 0.11 | 96.26 ± 1.21 | 24.05 ± 2.27 |
| F5 | 8.63 ± 0.05 | 94.94 ± 0.57 | 14.17 ± 1.46 |
| F6 | 8.64 ± 0.08 | 95.09 ± 0.88 | 6.39 ± 1.63 |
| F7 | 16.40 ± 0.08 | 98.34 ± 0.45 | 4.57 ± 2.47 |
| F8 | 16.20 ± 0.10 | 97.06 ± 0.63 | 8.54 ± 1.66 |
| F9 | 16.61 ± 0.03 | 99.66 ± 0.17 | 8.51 ± 2.88 |
| F10 | 16.34 ± 0.18 | 98.02 ± 1.10 | 6.81 ± 2.10 |
| F11 | 15.22 ± 0.01 | 91.32 ± 0.04 | 8.70 ± 2.14 |
| F12 | 15.77 ± 0.02 | 94.62 ± 0.14 | 7.32 ± 2.07 |
SD standard deviation
Particle sizes and size distributions of the microparticles are shown in Table II. The particle size increased with increase in the molecular weight of PLGA for microparticle formulations prepared with different types of PLGA having different molecular weights (p < 0.05). Because the molecular weight of the polymer is directly related to the viscosity of the dispersed phase, high molecular weight resulted in a greater viscosity. As the energy level required to disperse the highly viscous solutions is too high, larger droplets formed during the emulsification process and as a result greater microparticles were obtained. Similar findings were reported previously by Sánchez et al. (21). Unexpectedly, particle diameter of the microparticles prepared with low molecular weight PLGA 50:50 (coded F4) was found to be higher than the other microparticle formulations (coded F1–F3). This was attributed to the tendency of small-size microparticles to aggregation, resulting in the formation of the aggregates. Therefore, the particle size was determined larger than the actual value.
No significant difference was observed between the particle sizes of F3 and F5-coded microspheres prepared by homogenization at 8,000 rpm for 1 and 2 min, respectively (p > 0.05). It was concluded that increasing the homogenization time from 1 to 2 min had no significant effect on the particle size of the microspheres.
The effect of the drug/polymer ratio on the particle size of the microparticles was also investigated. The particle size of F3-coded microspheres prepared at 1:10 drug/polymer ratio was found as 14.61 μm, whereas the microspheres prepared at 1:5 ratio was 8.54 μm in F8-coded formulation. It was evident that the size of the microspheres increased significantly with increasing PLGA concentration, because of the higher viscosity of the internal organic phase. These results are in accordance with the findings of other researchers (22–24).
The particle size of the microspheres prepared with 1:10 drug/polymer ratio at 9,500 rpm (F6) were significantly lower than the microspheres prepared at 8,000 rpm (F3) (p < 0.05). Similar results were obtained for the microsphere formulations prepared with 1:5 drug/polymer ratio at two different homogenization speeds. The results of the particle size analysis showed that mean diameter of the particles significantly decreased when the homogenization rate was increased from 8,000 rpm to 9,500 rpm. This situation was explained by the fact that energy level enabled the maximum dispersion of the organic phase and smaller droplets were obtained at high homogenization speeds. Similar results were previously reported (21,25).
PVA concentration in the external water phase is well known to influence the particle size of microspheres (26). Since the presence of PVA in the external phase stabilises emulsion droplets against coalescence, increasing the PVA concentration usually leads to a decrease in the size of microspheres (27,28). However, we did not find any linear relationship between the PVA concentration and particle size.
Morphology of the Microspheres
Scanning electron micrographs showed that all the microparticles were spherical shape and no drug crystals were observed on the surface of the microspheres as seen in Fig. 1. It was evident that clobetasol propionate was completely encapsulated in the polymer matrix and unencapsulated drug was not attached to the surface of the microparticles.
Fig. 1.

Scanning electron micrographs of F3 (1), F8 (2), F9 (3), F10 (4), F11 (5) and F12 (6) coded microspheres (the scale size is 10 μm for general appearance and 1 μm for surface appearance)
F3-coded microspheres prepared with 1:10 drug/polymer ratio had very smooth and non-porous surfaces, whereas the surface of the F8-coded microspheres prepared at 1:5 drug/polymer ratio was highly porous. These results suggested that porosity of the microspheres decreased when the polymer concentration in the organic phase was increased. Schlicher et al. (29) and Arıca et al. (30) have also reported similar findings for desferrioxamine and bromocryptine mesylate-loaded microspheres, respectively. It is considered that the highly porous surface of F8-coded microspheres might have occured because of the high solvent evaporation rate which is a result of the decreasing viscosity of the organic phase. It has been previously indicated that the surface of the microparticle loaded with fentanyl is generally smooth when the polymer precipitates slowly, due to slow removal of the organic solvent (31).
The surface morphology of the microspheres prepared with different PVA ratios were also evaluated. It was determined that F8- and F11-coded microspheres had highly porous surfaces; however F9-, F10- and F12-coded microparticles showed dimples but non-porous surface property.
Differential Scanning Calorimetry
DSC analysis was carried out to evaluate the thermal behaviour of the drug, polymer and microspheres. DSC thermograms obtained for pure drug, polymer, drug-loaded and blank microspheres were given in Fig. 2. The endothermic peak of PLGA at 52.77°C was attributed to the glass transition temperature (Tg) of the polymer. On the other hand, the pure clobetasol propionate showed a sharp endothermic peak of melting at about 204.12°C, but no related peak was observed for drug-loaded microsphere formulations. These results suggest that clobetasol propionate was totally entrapped in the polymer and the drug was in an amorphous state in the microspheres. These results are similar to that mentioned previously by some researchers (24,32).
Fig. 2.

DSC thermograms of clobetasol propionate (a), PLGA 50:50 (40,000–75,000) (b), blank microspheres (c), F3 (d) and F8 (e) coded microspheres
Powder X-ray Diffractometry
As shown in Fig. 3, the X-ray diffraction peaks of clobetasol propionate were consistent with the characteristic pattern of crystalline substances. However, these peaks disappared in the diffractogram of drug-loaded microparticles. On the other hand, the diffraction patterns of the blank microspheres were similar to that of the amorphous polymers. These findings were in agreement with the results of DSC analysis and indicated that clobetasol propionate was dissolved in the polymer matrix and the drug was not in the crystalline form in the microparticle formulations (33,34).
Fig. 3.

X-ray diffractograms of clobetasol propionate (a), PLGA 50:50 (40,000–75,000) (b), blank microspheres (c), F3 (d) and F8 (e) coded microspheres
In Vitro Drug Release from Microspheres
There was no initial burst effect observed for any of the microsphere formulations. In vitro release profiles of clobetasol propionate from microspheres prepared with different types of PLGA were indicated in Fig. 4. The drug release from F3-coded microspheres prepared with PLGA 50:50 (Mw, 40,000–75,000) was higher than that of the microparticles containing other types of PLGA. The released amount of the drug reached 52.09% at the end of 24 h. It was explained by the fact that the polymer used to prepare F3-coded microspheres had lower molecular weight and higher glycolic acid content than the other PLGA types. It is well known that the molecular weight of the polymer significantly affects the release rate of the drug. The extent of the drug release also depends on the glycolic acid content of the polymer. Since glycolic acid units are more hydrophilic than lactic acid units, the increase in the glycolic acid ratio leads to an increase in the water uptake into the polymer and an acceleration in the hydrolytic cleavage. Barakat and Radwan (35) also indicated that carbamazepine release was faster from the microspheres prepared with a polymer that had a higher glycolic acid content and lower molecular weight. On the other hand, the drug release from F2-coded microspheres prepared with PLGA 75:25 was slower than F1-coded microspheres containing PLGA 85:15 (p < 0.05), although the glycolic acid content of F2-coded microspheres was higher. This finding was related to the higher molecular weight of PLGA 75:25 (Mw, 66,000–107,000) compared to PLGA 85:15 (Mw, 50,000–75,000). Some researchers also declared that drug release decreased when the molecular weight of the polymer increased (36,37). However, the release rate of clobetasol propionate from the F4-coded microspheres prepared with the PLGA 50:50 (Mw, 5,000–15,000) was significantly and unexpectedly lower than from the other formulations. It was considered that the particle size increased due to the formation of the aggregates between the small size microspheres, and this increase led to a decrease in the release rate.
Fig. 4.

Effect of the polymer type on the in vitro drug release profiles from PLGA microspheres (n = 3)
Using Fig. 5, the effect of homogenization time on the drug release was evaluated for F3- and F5-coded microspheres. There is no significant difference were observed (p > 0.05) between them as in the particle size results.
Fig. 5.

Effect of the homogenization time on the in vitro drug release profiles from PLGA microspheres (n = 3)
The release of clobetasol propionate from PLGA microspheres prepared at two different homogenization speeds are shown in Fig. 6. The drug release from the F6-coded microspheres prepared at 9,500 rpm was higher than the F3-coded formulation. This finding was attributed to the smaller particle size of the F6-coded formulation. While decrease in the size of microspheres, an increase in drug release was obtained. This kind of relationship between the particle size and drug release was also reported in other articles previously (38,39). Klose et al. (40) also explained that the relative lidocaine release rate decreased with increasing the dimension of the PLGA microparticles. Unexpectedly, the amount of the drug released from the F7-coded microspheres prepared at 9,500 rpm was lower than the F8-coded microparticles. It was concluded that, the release of the drug decreased because of the possible aggregation between the small size microspheres of the F7-coded formulation (4.57 μm). Furthermore, F8-coded microspheres were determined to have a highly porous surface characteristic as seen in Fig. 1. The porosity of the microspheres play an important role in the drug release characteristics and release rates generally increase as a result of higher porosity of the microparticles (41).
Fig. 6.

Effects of the homogenization speed and drug/polymer ratio on the in vitro drug release profiles from PLGA microspheres (n = 3)
The effect of the drug/polymer ratio on the release profiles of clobetasol propionate from PLGA microspheres was also evaluated (Fig. 6). The drug release from the F8-coded formulation prepared at 1:5 drug/polymer ratio was significantly faster than the F3-coded formulation prepared at 1:10 drug/polymer ratio (p < 0.05). This results can be explained by the particle size and surface morphology characteristics of the formulations which were prepared at different drug/polymer ratios. As it can be seen in Fig. 1, the F3-coded microspheres have a smooth and non-porous surface while the F8-coded microspheres possess an extremely porous surface. Moreover, the particle size of the F8-coded microspheres (8.54 μm) was smaller than the F3-coded formulation (14.61 μm) (p < 0.05). In this respect, the smaller size and porous surface of the F8-coded microspheres led to increase in the release rate.
In vitro drug release profiles from microspheres containing different concentrations of PVA are shown in Fig. 7. The highest drug releases were obtained for the PVA concentrations of 0.5% (F8) and 0.2% (F11). The amount of clobetasol propionate released at the end of the 24 h was found to be 64.43% and 59.80% for F8- and F11-coded formulations, respectively. On the other hand, no significant difference was determined between the release profiles of F9-, F10- and F12-coded microspheres (p > 0.05). These results can be explained by the fact that, F8- and F11-coded microspheres had highly porous surfaces whereas the surface of the other microparticles were dimples but non-porous as seen in Fig. 1.
Fig. 7.

Effect of the PVA concentration on the in vitro drug release profiles from PLGA microspheres (n = 3)
Two possible mechanisms could explain the drug release from PLGA microspheres. These are dissolution/diffusion from the spherical matrices and the matrix erosion resulting from degradation/dissolution of PLGA polymers. Since the bulk degradation of PLGA is trivial during the initial period and the drug release time in this study, 24 h is considerably shorter than the degradation lifetime of PLGA. The polymer backbone may maintain its integrity without significant degradation/dissolution (42). Therefore, clobetasol propionate release from the PLGA microspheres may be explained by the diffusion mechanism.
Studies on the Emulgel Formulations
Creams and ointments are most widely used conventional semi-solid dosage forms for the topical treatment of psoriasis depending on the size and location of the psoriatic lesions. In the study, formulation of an alternative dosage form for topical treatment of psoriasis was investigated. For this purpose, emulgel formulations were evaluated because they are more easily spreadable vehicles for the wide surfaces like psoriatic lesions compared to cream or ointment formulations. The permeability of the drugs can be also increased by using emulgels.
The pH value of the emulgel formulation prepared was determined and it was found to be 6.53 ± 0.01 (n = 3). It was concluded that the pH value of this formulation is reasonable for dermal application.
The release profiles of the emulgel formulations containing pure clobetasol propionate (EC), the F8-coded microspheres (EM) and also the commercial clobetasol propionate cream were compared in Fig. 8. While the amount of clobetasol propionate released from commercial product after 24 h was found to be 27.91 μg/cm2 (7.12%), the released amount from emulgel formulations at the end of 24 h were 44.97 μg/cm2 (12.21%) and 34.37 μg/cm2 (9.86%) for EC and EM, respectively. In other words, the released amount of drug decreased in the order of EC > EM > commercial cream. For the EM formulation, firstly the drug releases from the microparticles to the emulgel base, and then diffuses from the semi solid vehicle. However, only the diffusion of clobetasol propionate from emulgel base to the receptor medium occurs for the EC formulation. The different thermodynamic activities between formulations could also have accounted for these results.
Fig. 8.

The release profiles of clobetasol propionate from the EC- and EM-coded emulgel formulations and commercial cream product (n = 3)
The results of the in vitro release studies revealed that the release of clobetasol propionate was significantly increased with both of the emulgel formulations developed, compared to the commercial product. It was also observed that, the encapsulation of clobetasol propionate in the PLGA microspheres significantly delayed the drug release from the emulgel formulation compared to the emulgel containing pure drug. It is expected that, the topical and systemic side effects of the drug may be reduced and the efficiency of treatment may be increased as a result of the prolonged release of clobetasol propionate from the emulgel containing PLGA microparticles. In vitro antipsoriatic efficiency of the microparticulate based emulgel formulation will be evaluated with further cell culture studies that will be performed on the human epidermal keratinocyte cell line.
CONCLUSIONS
Clobetasol propionate-loaded PLGA microspheres were successfully prepared using oil-in-water emulsion–solvent evaporation technique. The F8-coded formulation prepared with PLGA 50:50 (Mw, 40,000–75,000) at 1:5 drug/polymer ratio and homogenised at 8,000 rpm for 1 min was selected as the best formulation. F8-coded microparticles were formulated in an emulgel base and a topical delivery system was developed for psoriasis treatment. In order to evaluate the in vitro antipsoriatic efficiency of the emulgel formulation containing PLGA microspheres, further cell culture studies will be carried out on the human epidermal keratinocyte cell line.
Acknowledgments
DECLARATION OF INTEREST
This study was supported by the Management of Scientific Research Projects of Ankara University (Project Number: 07B3336002). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Footnotes
ANSWERS TO REVIEWER 3
1. The use of English in this manuscript was thoroughly checked by a native speaker.
2. The explanation (“The different thermodynamic activities between formulations could also have accounted for these results”) was added into the “RESULTS AND DISCUSSION” section on the advice of the reviewer.
REFERENCES
- 1.Bhosle MJ, Kulkarni A, Feldman SR, Balkrishnan R. Quality of life in patients with psoriasis. Health and Quality of Life Outcomes. 2006;4:35. doi: 10.1186/1477-7525-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fluhr JW, Cavallotti C, Berardesca E. Emollients, moisturizers and keratolytic agents in psoriasis. Clin Dermatol. 2008;26:380–386. doi: 10.1016/j.clindermatol.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Lebwohl M, Ting PT, Koo JYM. Psoriasis treatment: traditional therapy. Ann Rheum Dis. 2005;64(Suppl II):ii83–ii86. doi: 10.1136/ard.2004.030791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van de Kerkhof PCM. The topical treatment of psoriasis. Clin Exp Dermatol. 2005;30:205–208. doi: 10.1111/j.1365-2230.2005.01719.x. [DOI] [PubMed] [Google Scholar]
- 5.Austad J, Bjerke JR, Gjertsen BT, Helland S, Livden JK, Morken T, et al. Clobetasol propionate followed by calcipotriol is superior to calcipotriol alone in topical treatment of psoriasis. JEADV. 1998;11:19–24. [PubMed] [Google Scholar]
- 6.Tsai J-C, Cheng C-L, Tsai Y-F, Sheu H-M, Chou C-H. Evaluation of in vivo bioequivalence methodology for topical clobetasol 17-propionate based on pharmacodynamic modeling using chinese skin. J Pharm Sci. 2004;93(1):207–217. doi: 10.1002/jps.10536. [DOI] [PubMed] [Google Scholar]
- 7.Fang J-Y, Fang C-L, Sung KC, Chen H-Y. Effect of low frequency ultrasound on the in vitro percutaneous absorption of clobetasol 17-propionate. Int J Pharm. 1999;191:33–42. doi: 10.1016/S0378-5173(99)00230-6. [DOI] [PubMed] [Google Scholar]
- 8.Hu FQ, Yuan H, Zhang HH, Fang M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int J Pharm. 2002;239:121–128. doi: 10.1016/S0378-5173(02)00081-9. [DOI] [PubMed] [Google Scholar]
- 9.Campisi G, Giandalia G, De Caro V, Di Liberto C, Arıcò P, Giannola LI. A new delivery system of clobetasol-17-propionate (lipid-loaded microspheres 0.025%) compared with a conventional formulation (lipophilic ointment in a hydrophilic phase 0.025%) in topical treatment of atrophic/erosive oral lichen planus. A phase IV, randomized, observer-blinded, parallel group clinical trial. Br J Dermatol. 2004;150:984–990. doi: 10.1111/j.1365-2133.2004.05943.x. [DOI] [PubMed] [Google Scholar]
- 10.Hu F-Q, Jiang S-P, Du Y-Z, Yuan H, Ye Y-Q, Zeng S. Preparation and characteristics of monostearin nanostructured lipid carriers. Int J Pharm. 2006;314:83–89. doi: 10.1016/j.ijpharm.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 11.Şenyiğit T, Sonvico F, Barbieri S, Özer Ö, Santi P, Colombo P. Lecithin/chitosan nanoparticles of clobetasol-17-propionate capable of accumulation in pig skin. J Cont Rel. 2010;142:368–373. doi: 10.1016/j.jconrel.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Ga De Jalón E, Blanco-Prieto MJ, Ygartua P, Santoyo S. PLGA microparticles: possible vehicles for topical drug delivery. Int J Pharm. 2001;226:181–184. doi: 10.1016/S0378-5173(01)00811-0. [DOI] [PubMed] [Google Scholar]
- 13.Haddadi A, Farboud ES, Erfan M, Aboofazeli R. Preparation and characterization of biodegradable urea-loaded microparticles as an approach for transdermal delivery. J Microencapsulation. 2006;23(6):698–712. doi: 10.1080/02652040600789328. [DOI] [PubMed] [Google Scholar]
- 14.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. doi: 10.1016/S0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Ng CW, Win KY, Shoemakers P, Lee TKY, Feng SS, et al. Release of paclitaxel from polylactide-co-glycolide (PLGA) microparticles and discs under irradiation. J Microencapsulation. 2003;20(3):317–327. doi: 10.1080/0265204021000058401. [DOI] [PubMed] [Google Scholar]
- 16.Çırpanlı Y, Ünlü N, Çalış S, Hıncal AA. Formulation and in vitro characterization of retinoic acid loaded poly (lactic-co-glycolic acid) microspheres. J Microencapsulation. 2005;22(8):877–889. doi: 10.1080/02652040500273878. [DOI] [PubMed] [Google Scholar]
- 17.Samatlı Y, Yüksel N, Tarımcı N. Preparation and characterization of poly(D, L-lactic-co-glycolic acid) microspheres containing flurbiprofen sodium. Drug Delivery. 2006;13:105–111. doi: 10.1080/10717540500313331. [DOI] [PubMed] [Google Scholar]
- 18.Janoria KG, Mitra AK. Effect of lactide/glycolide ratio on the in vitro release of ganciclovir and its lipophilic prodrug (GCV-monobutyrate) from PLGA microspheres. Int J Pharm. 2007;338:133–141. doi: 10.1016/j.ijpharm.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 19.Ga De Jalón E, Blanco-Prieto MJ, Ygartua P, Santoyo S. Topical application of acyclovir loaded microparticles: quantification of the drug in porcine skin layers. J Cont Rel. 2001;75:191–197. doi: 10.1016/S0168-3659(01)00395-9. [DOI] [PubMed] [Google Scholar]
- 20.Santoyo S, Ga De Jalón E, Ygartua P, Renedo MJ, Blanco-Prieto MJ. Optimization of topical cidofovir penetration using microparticles. Int J Pharm. 2002;242:107–113. doi: 10.1016/S0378-5173(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 21.Sánchez A, Vila-Jato JL, Alonso MJ. Development of biodegradable microspheres and nanospheres for the controlled release of cyclosporin A. Int J Pharm. 1993;99:263–273. doi: 10.1016/0378-5173(93)90369-Q. [DOI] [Google Scholar]
- 22.Ghaderi R, Sturesson C, Carlfors J. Effect of preparative parameters on the characteristics of poly(D, L-lactide-co-glycolide) microspheres made by the double emulsion method. Int J Pharm. 1996;141:205–216. doi: 10.1016/0378-5173(96)04639-X. [DOI] [Google Scholar]
- 23.Sehra S, Dhake AS. Formulation and evaluation of sustained release microspheres of poly-lactide-co-glycolide containing tamoxifen citrate. J Microencapsulation. 2005;22(5):521–528. doi: 10.1080/02652040500162170. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Gao S. Temozolomide/PLGA microparticles and antitumor activity against glioma C6 cancer cells in vitro. Int J Pharm. 2007;329:122–128. doi: 10.1016/j.ijpharm.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 25.Gabor F, Ertl B, Wirth M, Mallinger R. Ketoprofen- poly(D, L-lactic-co-glycolic acid) microspheres: influence of manufacturing parameters and type of polymer on the release characteristics. J Microencapsulation. 1999;16(1):1–12. doi: 10.1080/026520499289266. [DOI] [PubMed] [Google Scholar]
- 26.Mao S, Xu J, Cai C, Germershaus O, Schaper A, Kissel T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int J Pharm. 2007;334:137–148. doi: 10.1016/j.ijpharm.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 27.Freitas S, Merkle HP, Gander B. Microencapsulation by solvent extraction/evaporation: reviewing the state of the art of microsphere preparation process technology. J Cont Rel. 2005;102:313–332. doi: 10.1016/j.jconrel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y-Y, Chung T-S, Ng NP. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–241. doi: 10.1016/S0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 29.Schlicher EJAM, Postma NS, Zuidema J, Talsma H, Hennink WE. Preparation and characterization of poly(D, L-lactic-co-glycolic acid) microspheres containing desferrioxamine. Int J Pharm. 1997;153:235–245. doi: 10.1016/S0378-5173(97)00116-6. [DOI] [Google Scholar]
- 30.Arıca B, Kaş HS, Orman MN, Hıncal AA. Biodegredable bromocryptine mesylate microspheres prepared by a solvent evaporation technique. I: evaluation of formulation variables on microspheres characteristics for brain delivery. J Microencapsulation. 2002;19(4):473–484. doi: 10.1080/02652040210144216. [DOI] [PubMed] [Google Scholar]
- 31.Choi HS, Seo S-A, Khang G, Rhee JM, Lee HB. Preparation and characterization of fentanyl-loaded PLGA microspheres: in vitro release profiles. Int J Pharm. 2002;234:195–203. doi: 10.1016/S0378-5173(01)00968-1. [DOI] [PubMed] [Google Scholar]
- 32.Jagadeesh HG, Devi VK. Tamoxifen loaded poly (ε-caprolactone) based injectable microspheres for breast cancer. Int J Pharm Pharmaceut Sci. 2010;2(4):189–195. [Google Scholar]
- 33.Fernandez-Carballido A, Herrero-Vanrell R, Molina-Martinez IT, Pastoriza P. Biodegradable ibuprofen-loaded PLGA microspheres for intraarticular administration: effect of labrafil addition on release in vitro. Int J Pharm. 2004;279:33–41. doi: 10.1016/j.ijpharm.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Mahajan HS, Gattani SG. Gellan gum based microparticles of metoclopromide hydrochloride for intranasal delivery: development and evaluation. Chem Pharm Bull. 2009;57(4):388–392. doi: 10.1248/cpb.57.388. [DOI] [PubMed] [Google Scholar]
- 35.Barakat NS, Radwan MA. In vitro performance of carbamazepine loaded to various molecular weights of poly(D, L-lactide-co-glycolide) Drug Delivery. 2006;13:9–18. doi: 10.1080/10717540500308992. [DOI] [PubMed] [Google Scholar]
- 36.Heya T, Okada H, Ogawa Y, Toguchi H. Factors influencing the profiles of TRH release from copoly(d, l-lactic/glycolic acid) microspheres. Int J Pharm. 1991;72:199–205. doi: 10.1016/0378-5173(91)90108-Z. [DOI] [Google Scholar]
- 37.Zolnik BS, Leary PE, Burgess DJ. Elevated temperature accelerated release testing of PLGA microspheres. J Cont Rel. 2006;112:293–300. doi: 10.1016/j.jconrel.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Bezemer JM, Radersma R, Grijpma DW, Dijkstra PJ, Van Blitterswijk CA, Feijen J. Microspheres for protein delivery prepared from amphiphilic multiblock copolymers 2. modulation of release rate. J Cont Rel. 2000;67:249–260. doi: 10.1016/S0168-3659(00)00212-1. [DOI] [PubMed] [Google Scholar]
- 39.Fu X, Ping Q, Gao Y. Effects of formulation factors on encapsulation efficiency and release behaviour in vitro of huperzine A-PLGA microspheres. J Microencapsulation. 2005;22(7):705–714. doi: 10.1080/02652040500162196. [DOI] [PubMed] [Google Scholar]
- 40.Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int J Pharm. 2006;314:198–206. doi: 10.1016/j.ijpharm.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 41.Mao S, Shi Y, Li L, Xu J, Schaper A, Kissel T. Effects of process and formulation parameters on characteristics and internal morphology of poly(D, L-lactide-co-glycolide) microspheres formed by the solvent evaporation method. Eur J Pharm Biopharm. 2008;68:214–223. doi: 10.1016/j.ejpb.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Yen S-Y, Sung KC, Wang J-J, Hu OY-P. Controlled release of nalbuphine propionate from biodegradable microspheres: in vitro and in vivo studies. Int J Pharm. 2001;220:91–99. doi: 10.1016/S0378-5173(01)00649-4. [DOI] [PubMed] [Google Scholar]