

Abstract
Tetrandrine (TET) is a poorly water-soluble bisbenzylisoquinoline alkaloid. In this study, TET solid lipid nanoparticles (SLNs) were prepared by a melt–emulsification and ultrasonication technique. Precirol® ATO 5, glyceryl monostearate, and stearic acid were used as the lipid matrix for the SLNs, while Lipoid E80, Pluronic F68, and sodium deoxycholate were used as emulsifying and stabilizing agents. The physicochemical characteristics of the TET–SLNs were investigated when it was found that the mean particle size and zeta potential of the TET–SLNs were 134 ± 1.3 nm and −53.8 ± 1.7 mV, respectively, and the entrapment efficiency (EE) was 89.57% ± 0.39%. Differential scanning calorimetry indicated that TET was in an amorphous state in SLNs. TET–SLNs exhibited a higher release rate at a lower pH and a lower release rate at a higher pH. The release pattern of the TET–SLNs followed the Weibull model. The pharmacokinetics of TET–SLNs after intravenous administration to male rats was studied. TET–SLN resulted in a higher plasma concentration and lower clearance. The biodistribution study indicated that TET–SLN showed a high uptake in reticuloendothelial system organs. In conclusion, TET–SLNs with a small particle size, and high EE, can be produced by the method described in this study. The SLN system is a promising approach for the intravenous delivery of tetrandrine.
Key words: characterization, pharmacokinetics, preparation, solid lipid nanoparticles, tetrandrine
INTRODUCTION
Solid lipid nanoparticles (SLNs) have been widely investigated as an alternative drug delivery system, particularly for poorly water-soluble lipophilic drugs. They consist of a biocompatible lipid core and a surfactant as an outer shell. The nanoparticles are made from biodegradable solid lipids and can be prepared by several methods (1–3). SLNs combine the advantages of polymeric nanoparticles, fat emulsions, and liposomes (4). SLNs can control drug release, change the distribution of the drug in the body, incorporate more drugs, increase the drug solubility and bioavailability, and protect drugs against chemical degradation. Furthermore, SLNs are well tolerated, exhibit low toxicity, as well as good physical stability and biocompatibility (5–8).
Tetrandrine (TET; Fig. 1), the major active constituent of Stephania tetrandra S. Moore, is a bisbenzylisoquinoline alkaloid which is used to treat inflammation, hypertension, and sillicosis. Numerous studies have reported that TET also exhibits antitumor activity and acts as a nonselective calcium channel blocker and calmodulin antagonist (9–13). Although the poor aqueous solubility of TET greatly inhibits its clinical applications, TET formulated in SLN carriers may be a potential system for intravenous administration, with a high targeting efficiency and bioavailability.
Fig. 1.

Chemical structure of tetrandrine
In the present study, TET–SLNs were prepared using a melt–emulsification and ultrasonication method. TET was dissolved in melted solid lipids, and all the emulsifying agents were dispersed in the aqueous phase. Compared with the solvent evaporation method (14), the present method could reduce toxicity because it avoided the use of organic solvents. It was found that TET was very soluble in fatty acids and had a lower solubility in fatty esters. As different solid lipids have different effects on the stability of SLNs, different types and ratios of solid lipids were screened in this study. A combination of Precirol® ATO 5 (PA), glyceryl monostearate (GM), and stearic acid (SA) was used as the lipid matrix, while Lipoid E80, Pluronic F68 and sodium deoxycholate (SDC) were used as emulsifying and stabilizing agents.
The primary aim of the present study is to characterize the factors influencing the preparation of TET–SLNs. In addition, the size, morphology, thermal properties, lyophilization, drug entrapment efficiency (EE), and drug release pattern were studied. A further aim is to investigate the pharmacokinetics of TET–SLNs after intravenous administration to male Wistar rats and evaluate the tissue distribution (to the heart, liver, spleen, lung, kidney, and brain) of TET–SLNs after intravenous administration to mice.
MATERIALS AND METHODS
Materials
TET (Shanchuan Biotechology Co.Ltd., Shanxi, China) has a purity level greater than 98%. PA (Gattefosse, France), SA and GM (Tianjin Bodi Chemicals Co. Ltd., China), Lipoid E80 (Lipoid, Germany), Pluronic F68 (BASF, Germany), and SDC (Sigma-Aldrich, Germany) were obtained from the sources indicated. Distilled water was used in all the experiments. Other chemicals and reagents used were chromatographic or analytical grade.
Preparation of TET–SLNs
TET–SLNs were prepared using the melt–emulsification and ultrasonication method. Pluronic F68, SDC, and Lipoid E80 were used as emulsifying agents. A mixture of PA, SA, and GM was heated as the lipid phase in a water bath at 85°C, and TET was dissolved in the melted lipid phase. A mixture of emulsifying agents was simultaneously dissolved in 40 ml distilled water as the aqueous phase, and briefly heated to 85°C. The aqueous phase was then dripped into the lipid phase to prepare the primary emulsion under magnetic stirring at 85°C for 15 min. The primary emulsion was then sonicated using an ultrasonic probe (JY92-IID, Scientz, China) for 5 min with a 3 s pulse-on period and a 3 s pulse-off period. The nanoemulsion was then cooled and diluted to 50 ml with distilled water. The nanoparticles dispersion was subsequently passed through a 0.45-μm membrane filter.
Measurement of Particle Size and Zeta Potential
The mean particle size of the SLNs was measured using a laser particle size analyzer (Coulter LS-230, Beckman Coulter Co., Ltd., USA). Each sample was diluted to a suitable concentration with double-distilled water before measurement.
The zeta potential was measured using a zeta potential and particle size analyzer (DELSA 440 SX, Beckman Coulter Co., Ltd). The SLNs were diluted with double-distilled water to obtain the desired concentration. The entire procedure was performed at room temperature.
Entrapment Efficiency
The entrapped drug concentration was measured with the minicolumn centrifugation method used to calculate the EE (15, 16). The SLNs were separated on Sephadex G-50 mini-columns. The G-50 gel was introduced into 5 cm3 syringes which were used as the mini-columns. Excess water was removed by centrifugation. The amount of TET in the SLNs was determined as follows: each SLN sample was diluted to a suitable concentration with double-distilled water. A total 0.5 ml of the diluted sample was added to the column, which was then centrifuged at 2,000 rpm for 3 min. Following this, 0.5 ml distilled water was added to the minicolumn which was then centrifuged; this process was repeated five times. The centrifuged material was collected and diluted with methanol to 10 ml, followed by sonication for 30 min. The samples were then passed through a 0.22-μm membrane filter, and TET in the supernatant was determined by high-performance liquid chromatography (HPLC) method. The HPLC system consisted of a mobile phase delivery pump (LC-10 AD; Shimadzu, Japan), and a UV–vis detector (SPD-10A; Shimadzu, Japan). HPLC separation was achieved using a Dikma C18 column (4.6 × 250 mm, 5 μm) maintained at 40°C. The mobile phase consisted of 0.1 M potassium dihydrogen phosphate/methanol/acetonitrile/triethanolamine (50/25/25/0.75, v/v/v/v), and the pH was adjusted to 5.0 with phosphoric acid. The mobile phase was delivered at a flow rate of 1 ml/min, and 20 μl samples were injected. The detection wavelength was 280 nm. Another sample was directly diluted with methanol as the total amount of TET was added to the formulations.
The EE of TET–SLNs was calculated using the following equation:
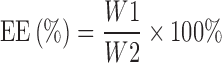 |
where, W1 was the amount of TET in SLNs and W2 was the amount of TET added in the formulations.
Transmission Electron Microscopy
The TET–SLN morphology was examined by transmission electron microscopy (TEM; JEM-1200, JEOL Co., Ltd., Japan). The samples were diluted with purified water to a suitable concentration and stained with 2% (w/v) phosphotungstic acid for 30 s. Then, they were placed on copper grids with films for observation.
Differential Scanning Calorimetry Analysis
Differential scanning calorimetry (DSC) was performed using a differential scanning calorimeter (DSC-60WS; Shimadzu, Japan). The following samples were placed in aluminum pans under a nitrogen purge for analysis: TET, lyophilized TET–SLNs (with or without 10% sucrose), and lyophilized 10% sucrose solution. A scan rate of 10°C/min was used over a temperature range of 25–300°C.
In Vitro Release
The release rate of TET from SLNs was determined in 0.1 N HCl and phosphate buffer (pH 6.8 and 7.4). For this, 1 ml TET–SLN dispersion was transferred to a dialysis bag with a molecular weight cutoff between 8,000 and 14,000 kDa. The dialysis bags were soaked in double-distilled water for 24 h before use. Each bag was introduced into an Erlenmeyer flask filled with 50 ml dialysis medium and shaken at 100 rpm at 37°C. At fixed time intervals, 2 ml of sample was taken and replaced with 2 ml fresh medium. The filtrate was analyzed using the HPLC method described above.
Lyophilization
The TET–SLNs were lyophilized using a programmable freeze dryer (FDU-1100; Eyela, Japan). Before freezing, cryoprotectants were added to the SLN dispersion. Maltose, trehalose, and sucrose were screened at the levels of 5% and 10% (w/v) for their cryoprotectant ability, while lactose was screened at the levels of 2.5% and 5% (w/v). The freeze drying was conducted as follows: primary drying of the dispersion was at −35°C for 1 h. The shelf temperature was then raised to −25°C for 12 h. Secondary drying was performed at 15°C for 4 h. The lyophilized products were reconstituted by dilution with distilled water and shaking.
Intravenous Administration
All animals used in this study were obtained from the Experiment Animal Center (Shenyang Pharmaceutical University, Shenyang, China). The experimental procedures complied with the University Animal Ethics Committee Guidelines.
The HPLC method described above was used in the pharmacokinetic study. Male Wistar rats (200–220 g) were used for in vivo studies. Six rats were given tetrandrine solution (16 mg/kg, dispersed in HCl, pH adjusted to 5 with NaOH) via the caudal vein. Another six rats were given TET–SLNs (16 mg/kg) via the caudal vein. The rats were acclimatized in the animal facility for 7 days. Prior to the experiment, the rats were fasted for 12 h with free access to water. Blood samples of about 0.3 ml were collected in heparinized test tubes via the post-orbital venous plexus at 0 (pre-dose), 2, 5, 10, 15, 30, 45, 60, 120, 180, 240, 360, 480, 720 min after administration. Plasma was separated by centrifugation at 12,000 rpm for 10 min and stored at −20°C until analysis.
Tissue Distribution
To study the tissue distribution, 72 Kunming mice (20 ± 2 g) were divided into two groups of 36 mice. They were given TET–SLNs and TET solution at a dose of 24 mg/kg via the tail vein. Six rats per formulation were sacrificed at 2, 5, and 15 min and at 1, 4, and 12 h post-dosing and samples were collected from the heart, liver, spleen, lungs, kidneys, and brain. All of the tissue specimens were rinsed with sterile physiological saline and immediately stored at −20°C for later analysis.
Statistical Analysis
All pharmacokinetic parameters were calculated by DAS software (Ver. 2.0, Mathematical Pharmacology Professional Committee of China). All statistical analyses were performed using statistical package for social sciences (SPSS, version 16.0). The data were presented as mean ± SD, and the Student’s t test was used to analyze differences between the two groups.
RESULTS AND DISCUSSION
Preparation of TET–SLNs
Selection of Lipid Phase
The solubility of TET in the lipid matrix was an important parameter affecting drug incorporation and the stability of SLNs. Different lipid materials were screened, including myristic acid, palmitic acid, SA, behenic acid, GM, Compritol® 888 ATO, PA, medium-chain triglycerides, soybean oil, and aethylis oleas (17–25). The lipids were heated in a water bath at 85°C. A certain amount of TET was then dissolved in the melted lipid phase under constant stirring for 30 min. The maximum amount of TET dissolved in the lipids was recorded as the solubility of TET in lipids. TET exhibited better solubility in fatty acids and a poorer solubility in fatty esters. It did not dissolve well in the liquid lipid. Because TET was an alkaloid, fatty acids may chemically react with TET, producing a higher solubility in fatty acids. For the first group of lipids tested, myristic acid, palmitic acid, SA, behenic acid, GM, Compritol® 888 ATO, and PA were screened as the potential components of the lipid phase.
Several formulations with different types and ratios of the lipids were prepared to develop an optimal TET–SLN and the results were listed in Table I. In the single-factor experiments, fatty acids were found to be unable to produce stable formulations: formulations using fatty acids alone flocculated within 1 h. When the SLNs were prepared with Compritol® 888 ATO using the same level of emulsifying agents, a homogeneous dispersion was not achieved. On the other hand, although TET exhibited a low solubility in GM and PA, stable nanoparticle formulations of TET could be prepared with these lipids. Mixed lipids containing a fatty acid with either GM or PA were further studied to increase the drug loading. GM/SA mixtures at ratios of 1/1, 2/1, 3/1, 4/1, and 5/1 were evaluated. When the GM/SA ratio was equal to or higher than 4/1, stable SLNs were obtained. Mixtures including GM and other fatty acids at a ratio of 4/1 were also examined. Among the screened fatty acids, myristic acid and palmitic acid formed a homogeneous system and encapsulated more TET than SA. However, the dispersions began to sediment after 2 days. When behenic acid was used, a homogeneous preparation was not achieved. The particle sizes of the dispersions ranged from 40 nm to 20 μm. Finally, SA was selected as the lipid core based on these results.
Table I.
Results of Different Types and Ratios of the Lipids
| Lipid raw materials | Appearance at room temperature | Different ratios of fatty acids | Appearance at room temperature |
|---|---|---|---|
| Compritol® 888 ATO | Precipitation | GM/SA (1/1) | Phase separation |
| GM | Stable | GM/SA (2/1) | Phase separation |
| PA | Stable | GM/SA (3/1) | Phase separation |
| SA | Flocculation within 1 h | GM/SA (4/1) | Stable |
| Myristic acid | Flocculation within 1 h | GM/SA (5/1) | Stable |
| Palmitic acid | Flocculation within 1 h | PA/SA (4/1) | Stable |
| Behenic acid | Precipitation | GM/myristic acid (4/1) | Precipitation after 2 days |
| GM/palmitic acid (4/1) | Precipitation after 2 days | ||
| GM/behenic acid (4/1) | Phase separation |
The stability levels of SLNs prepared with PA and GM were studied. Mixtures of GM/SA (1/4) and PA/SA (1/4) were investigated at 25°C for 1 month. Flocculation was observed in SLNs prepared with GM. The drug-free SLNs prepared with PA were stable, whereas the TET–SLNs exhibited sedimentation. Therefore, the mixture of GM and PA was selected for the formulation. Formulations with different PA/GM ratios were tested; and of these, the 3/1 ratio exhibited excellent stability. Thus, the final lipid composition had a PA/GM/SA ratio of 3/1/1.
Selection of Emulsifying Agents
Lipoid E80, Pluronic F68, and SDC were screened as emulsifying agents. A mixture of different surfactants was able to improve the physical characteristics of SLNs (26, 27). Combinations of Lipoid E80 and Pluronic F68 were investigated first.
Pluronic F68 was able to reduce the mean diameter of the preparation compared with Lipoid E80. Long-term stability testing (storage at 4°C and 25°C) demonstrated that the preparation with Pluronic F68 had the propensity to gelatinize, especially at 4°C. The lipid concentration should be reduced to improve the stability of SLNs and prevent gelatinization. SDC was added because the ionic surfactant had a synergistic effect on electrostatic repulsion and stabilization of the formulation (28, 29).
Drug Concentration
When the TET concentration was greater than 8 mg/ml, SLNs sedimented within 10 h. Investigation showed that 6 mg/ml was the optimal concentration of TET in this study. The optimal composition of the TET–SLN formulation was as follows: PA/GM/SA/Lipoid E80/Pluronic F68/SDC/TET/water at a ratio of 2.4/0.8/0.8/3.0/3.0/0.4/0.6/100 m/m.
Measurement of Size and Zeta Potential
The mean diameter of drug-free SLNs was 129 ± 0.6 nm (n = 3), whereas the mean diameter of TET–SLNs was 134 ± 1.3 nm (n = 3). The mean zeta potential of TET–SLNs and drug-free SLNs were −58.7 ± 1.3 and −53.8 ± 1.7 mV (n = 3), respectively.
Entrapment Efficiency Determination
The entrapment efficiency of SLNs was determined using the mini-column centrifugation method. The TET concentration in the system ranging from 2 to 8 mg/ml exhibited EE values ranging from 95.17% to 71.46%. A high concentration of TET was associated with a low entrapment efficacy. The effect of lipid on the entrapment efficiency was also studied by maintaining the amount of the surfactants constant while varying the amount of lipids. The EE value increased as the amount of lipids increased, and this was due to the good solubility of TET in SA. However, a high lipid concentration would increase the particle size of SLNs and lead to gelatination. For the optimal formulation, the concentration of TET was set at 6 mg/ml, with an EE% of 89.57% ± 0.39%.
TEM Investigation
The TEM image of the TET–SLNs was shown in Fig. 2. The shape of SLNs could be seen to be almost spherical. The particle size ranged from about 40–100 nm. The size of the SLNs determined by the Laser Particle Size Analyzer was not consistent with the TEM results. The diameter determined by TEM was about 50 nm; while using the Laser Particle Size Analyzer, the size was 134 ± 1.3 nm. It was reasonable to believe that these two methods were based on different mechanisms which might lead to this disparity (6, 30).
Fig. 2.

TEM photography of TET–SLN (×40,000 magnification)
Effect of Cryoprotectants During Lyophilization
Although lyophilization provided SLNs with good chemical and physical stability by preventing Ostwald ripening and hydrolysis reactions (6), lyophilization may lead to particle aggregation and drug leakage. Cryoprotectants have been used to prevent SLN aggregation during the lyophilization process (6, 31). Table II showed that an increase in particle size and a decrease in EE were significant without a cryoprotectant. The cryoprotectant effect of maltose, trehalose, sucrose, and lactose was studied with regard to preventing SLN aggregation. Given that 10% sucrose was the most effective cryoprotectant among the compounds tested, sucrose was chosen as the cryoprotectant in the subsequent experiments.
Table II.
Characteristics of Lyophilized TET–SLNs
| Sample | PS (nm) | ZP (mV) | EE% |
|---|---|---|---|
| Without cryoprotectants | 283 ± 2.4 | −45.3 ± 2.2 | 82.71 |
| Contain 10% sucrose | 142 ± 1.2 | −40.3 ± 1.4 | 88.66 |
The thermograms of TET, lyophilized TET–SLNs (with or without 10% sucrose), and lyophilized sucrose solution were shown in Fig. 3. For the lyophilized TET–SLNs without cryoprotectant, the melting point was 51°C, which was the endothermic peak of the mixed lipid. The melting peak of the freeze-dried sucrose solution and the TET–SLN without sucrose were at 191°C and 228°C, respectively, which represented diffraction peaks of the lyophilized sucrose. The endothermic peak of TET could not be obtained from the heating curve of the lyophilized TET–SLNs without a cryoprotectant. TET was shown to be in an amorphous state in SLNs and not in a crystalline state (24, 32–34).
Fig. 3.

DSC thermograms: a TET, b lyophilized TET–SLN without sucrose, c lyophilized TET–SLN with 10% sucrose, and d lyophilized 10% sucrose solution
In Vitro Release Study
The release of TET from the TET–SLNs (6 mg/ml) was shown in Fig. 4, and it could be seen that TET had a very poor aqueous solubility. The TET release rate from the SLNs was affected by the pH value of the dissolution medium and it increased as the pH decreased. As shown in Fig. 4, approximately 60%, 10%, and 5% of TET was released from TET–SLNs within 8 h in 0.1 N HCl, phosphate buffer at pH 6.8 and 7.4, respectively. No TET was released in the double-distilled water.
Fig. 4.

In vitro release of TET from TET–SLNs in different medium (n = 3)
An effective anti-cancer drug delivery system should be able to kill cancer cells without harming normal cells. Given that cancer cells had a more acidic microenvironment (35), a delivery system with good release profile similar to the physiological pH would be invaluable for anticancer therapy. As shown in Fig. 4, TET was released at a higher rate at a lower pH, whereas a lower release rate was observed at a higher pH. Given that the pKa of TET is 7.8, it was more soluble at a lower pH. Thus, TET entrapped in SLN had a greater ability to enter an acidic medium, resulting in a higher release rate of TET in cancer cells.
The obtained release data were then fitted into first-order, Higuchi, Hixcon–Crowell, Nibergull, and Weibull equations. The regression results indicated that, among these equations, the Weibull model best fitted the release data (R = 0.9949).
Pharmacokinetic Study
The mean plasma concentration–time profiles for the TET–SLNs and solution were shown in Fig. 5. The pharmacokinetic parameters of TET were calculated using a noncompartmental analysis and the parameters were summarized in Table III. For TET–SLN, the AUC(0–∞) was 6,581.50 ± 1,539.41 μgh/L, which was higher than that of the solution, and the Cmax was also increased (p < 0.05). Compared with the TET solution, the t1/2 and MRT were prolonged while the CL was reduced (p < 0.05). The results of the pharmacokinetics study suggested that the pharmacokinetic behavior of SLNs was significantly different from that of the solution.
Fig. 5.

Plasma concentration–time curves after intravenous administration TET solution and SLNs with dose of 16 mg/kg in rats (n = 6)
Table III.
Pharmacokinetic Parameters of Tetrandrine after Intravenous Administration of SLNs and Solution with Dose of 16 mg/kg in Rats (n = 6)
| Formulation | AUC(0-∞) (μgh/L) | C max (μg/L) | t 1/2 (h) | MRT(0-∞) (h) | CL (L/h/kg) |
|---|---|---|---|---|---|
| TET–SLNs | 6,581.50 ± 1,539.41 | 1,103.43 ± 94.31 | 13.86 ± 3.27 | 19.37 ± 4.60 | 2.52 ± 0.50 |
| TET solution | 3,081.94 ± 728.21 | 630.18 ± 183.45 | 7.22 ± 1.13 | 10.06 ± 1.57 | 5.41 ± 1.22 |
Tissue Distributions
The tissue concentrations of TET after intravenous administration of TET–SLNs and TET solution were shown in Fig. 6. The total concentration of TET for SLNs was in the order of lung > kidney > liver > spleen > heart > brain. The uptake by the reticuloendothelial system (RES) organs, especially the spleen, was observed to be higher for TET–SLN compared with TET solution (p < 0.05). Similar behaviors have been widely reported in the literatures for nanostructure lipid carriers (36, 37). It has been reported that after intravenous administration, lipid emulsions are rapidly taken up by the RES in the liver and spleen. The mechanism has been demonstrated to be either by phagocytosis or endocytosis (38). TET–SLN produced a lower drug concentration in the heart and kidney than the TET solution. For example, the concentrations of TET in the heart and kidney were 24.08 ± 9.84 and 26.81 ± 9.33 μg/g for TET–SLNs, while the corresponding values were 87.84 ± 20.36 and 83.30 ± 16.92 μg/g for the TET solution at 2 min.
Fig. 6.

Concentrations of TET in mice tissues after intravenous administration of TET–SLNs and solution (n = 6)
CONCLUSIONS
In this study, TET–SLNs were successfully prepared using an ultrasonic and emulsification technique. The physicochemical characteristics of the preparation were investigated. The type and ratio of the solid lipids were important factors for the formulation. TET–SLNs with a small particle size and high EE were obtained. The results of the in vitro experiments indicated a higher release rate of TET in an acidic medium. The in vivo study demonstrated that the TET–SLNs changed the pharmacokinetic parameters and biodistribution. The described system may offer an alternative approach to intravenous delivery of tetrandrine.
ACKNOWLEDGMENT
Dr. David B. Jack is gratefully thanked for correcting the manuscript. The authors acknowledged HX Pang for helping with the biodistribution experiment. These studies were supported by the National Basic Research Program of China (973 Program), no. 2009CB903302.
References
- 1.Jores K, Mehnert W, Drechsler M, Bunjes H, Johann C, Mäder K. Investigations on the structure of solid lipid nanoparticles (SLN) and oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95(2):217–227. doi: 10.1016/j.jconrel.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Müller RH, Keck CM. Challenges and solutions for the delivery of biotech drugs—a review of drug nanocrystal technology and lipid nanoparticles. J Biochem. 2004;113(1–3):151–170. doi: 10.1016/j.jbiotec.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Müller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. doi: 10.1016/S0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 4.Schwarz C, Mehnert WJ, Lucks S, Müller RH. Solid lipid nanoparticles(SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30:83–96. doi: 10.1016/0168-3659(94)90047-7. [DOI] [Google Scholar]
- 5.Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN(TM)) dispersions. Int J Pharm. 1998;168(2):221–229. doi: 10.1016/S0378-5173(98)00092-1. [DOI] [Google Scholar]
- 6.Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Delivery Rev. 2001;47(2–3):165–196. doi: 10.1016/S0169-409X(01)00105-3. [DOI] [PubMed] [Google Scholar]
- 7.Olbrich C, Müller RH. Enzymatic degradation of SLN—effect of surfactant and surfactant mixtures. Int J Pharm. 1999;180(1):31–39. doi: 10.1016/S0378-5173(98)00404-9. [DOI] [PubMed] [Google Scholar]
- 8.Marengo E, Cavalli R, Caputo O, Rodriguez L, Gasco MR. Scale-up of the preparation process of solid lipid nanospheres. Part I. Int J Pharm. 205;3(1–2). [DOI] [PubMed]
- 9.Fu LW, Zhang YM, Liang YJ, Yang XP, Pan QC. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. Eur J Cancer. 2002;38(3):418–426. doi: 10.1016/S0959-8049(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 10.Kwan C, Leung Y, Kwan TK, Daniel EE. Tetrandrine inhibits Ca2+ release-activated Ca2+ channels in vascular endothelial cells. Life Sci. 2001;68(7):841–847. doi: 10.1016/S0024-3205(00)00988-7. [DOI] [PubMed] [Google Scholar]
- 11.Liu B, Wang T, Qian X, Liu G, Yu L, Ding Y. Anticancer effect of tetrandrine on primary cancer cells isolated from ascites and pleural fluids. Cancer Letters. 2008;268(1):166–175. doi: 10.1016/j.canlet.2008.03.059. [DOI] [PubMed] [Google Scholar]
- 12.Wang G, Lemos J. Tetrandrine: a new ligand to block voltage-dependent Ca2+ and Ca2+-activated K+ channels. Life Sci. 1994;56(5):295–306. doi: 10.1016/0024-3205(94)00952-X. [DOI] [PubMed] [Google Scholar]
- 13.Wu J-M, Chen Y, Chen J-C, Lin T-Y, Tseng S-H. Tetrandrine induces apoptosis and growth suppression of colon cancer cells in mice. Cancer Letters. 2010;287(2):187–195. doi: 10.1016/j.canlet.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Dong L, Jia A, Chang X, Xue H. Preparation and characterization of solid lipid nanoparticles loaded traditional chinese medicine. Int J Biol Macromol. 2006;38(3–5):296–299. doi: 10.1016/j.ijbiomac.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Fry DW, White JC, Goldman ID. Rapid separation of low molecular weight solutes from liposomes without dilution. Anal Biochem. 1978;90(2):809–815. doi: 10.1016/0003-2697(78)90172-0. [DOI] [PubMed] [Google Scholar]
- 16.Han F, Li S, Yin R, Shi X, Jia Q. Investigation of nanostructured lipid carriers for transdermal delivery of flurbiprofen. Drug Dev Ind Pharm. 2008;34:453–458. doi: 10.1080/03639040701833708. [DOI] [PubMed] [Google Scholar]
- 17.Cavalli R, Caputo O, Carlotti ME, Trotta M, Scarnecchia C, Gasco MR. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int J Pharm. 1997;148(1):47–54. doi: 10.1016/S0378-5173(96)04822-3. [DOI] [Google Scholar]
- 18.Hu L, Xing Q, Meng J, Shang C. Preparation and enhanced oral bioavailability of cryptotanshinone-loaded solid lipid nanoparticles. AAPS PharmSciTech. 2010;11(2):582–587. doi: 10.1208/s12249-010-9410-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battaglia L, Gallarate M, Cavalli R, Trotta M. Solid lipid nanoparticles produced through a coacervation method. J Microencapsul. 2010;27(1):78–85. doi: 10.3109/02652040903031279. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Mira E, Egea MA, Garcia ML, Souto EB. Design and ocular tolerance of flurbiprofen loaded ultrasound-engineered NLC. Colloids Surf B. 2010;81(2):412–421. doi: 10.1016/j.colsurfb.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Chang X, Du D, Li J, Xu H, Yang X. Microemulsion-based hydrogel formulation of ibuprofen for topical delivery. Int J Pharm. 2006;315(1–2):52–58. doi: 10.1016/j.ijpharm.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Patel M, Patel R, Parikh J, Solanki A, Patel B. Effect of formulation components on the in vitro permeation of microemulsion drug delivery system of fluconazole. AAPS PharmSciTech. 2009;10(3):917–923. doi: 10.1208/s12249-009-9286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casadei MA, Cerreto F, Cesa S, Giannuzzo M, Feeney M, Marianecci C, et al. Solid lipid nanoparticles incorporated in dextran hydrogels: a new drug delivery system for oral formulations. Int J Pharm. 2006;325(1–2):140–146. doi: 10.1016/j.ijpharm.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Wang Y, Liu Z, Lin X, He H, Tang X. Formulation and characterization of bufadienolides-loaded nanostructured lipid carriers. Drug Dev Ind Pharm. 2010;36(5):508–517. doi: 10.3109/03639040903264397. [DOI] [PubMed] [Google Scholar]
- 25.Bhalekar M, Pokharkar V, Madgulkar A, Patil N, Patil N. Preparation and evaluation of miconazole nitrate-loaded solid lipid nanoparticles for topical delivery. AAPS PharmSciTech. 2009;10(1):289–296. doi: 10.1208/s12249-009-9199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui Z, Mumper RJ. Intranasal administration of plasmid DNA-coated nanoparticles results in enhanced immune responses. J Pharm Pharmacol. 2002;54:1195–1203. doi: 10.1211/002235702320402035. [DOI] [PubMed] [Google Scholar]
- 27.Souto EB, Müller RH. The use of SLN and NLC as topical particulate carriers for imidazole antifungal agents. Pharmazie. 2006;61:431–437. [PubMed] [Google Scholar]
- 28.Reich G. In vitro stability of poly (d, l-lactide) and poly (d, l-lactide)/poloxamer nanoparticles in gastrointestinal fluids. Drug Dev Ind Pharm. 1997;23:1191–1198. doi: 10.3109/03639049709146156. [DOI] [Google Scholar]
- 29.Souto EB, Müller RH. Investigation of the factors influencing the incorporation of clotrimazole in SLN and NLC prepared by hot high-pressure homogenization. J Microencapsul. 2006;23:377–388. doi: 10.1080/02652040500435295. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Gong T, Wang C, Zhong Z, Zhang Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: preparation and characterization. Int J Pharm. 2007;340(1–2):153–162. doi: 10.1016/j.ijpharm.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Shahgaldian P, Gualbert J, Aïssa K, Coleman AW. A study of the freeze-drying conditions of calixarene based solid lipid nanoparticles. Eur J Pharm Biopharm. 2003;55(2):181–184. doi: 10.1016/S0939-6411(02)00196-0. [DOI] [PubMed] [Google Scholar]
- 32.Yang SC, Zhu JB. Preparation and characterization of camptothecin solid lipid nanoparticles. Drug Dev Ind Pharm. 2002;28:265–274. doi: 10.1081/DDC-120002842. [DOI] [PubMed] [Google Scholar]
- 33.Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95(3):627–638. doi: 10.1016/j.jconrel.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Zhang W-L, Gu X, Bai H, Yang R-H, Dong C-D, Liu J-P. Nanostructured lipid carriers constituted from high-density lipoprotein components for delivery of a lipophilic cardiovascular drug. Int J Pharm. 2010;391(1–2):313–321. doi: 10.1016/j.ijpharm.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Research. 1989;49(23):6449. [PubMed] [Google Scholar]
- 36.Li F, Weng Y, Wang L, He H, Yang J, Tang X. The efficacy and safety of bufadienolides-loaded nanostructured lipid carriers. Int J Pharm. 2010;393(1–2):204–212. doi: 10.1016/j.ijpharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release. 2005;107(2):215–228. doi: 10.1016/j.jconrel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Mizushima Y, Hamano T, Yokoyama K. Tissue distribution and anti-inflammatory activity of corticosteroids incorporated in lipid emulsion. Ann Rheum Dis. 1982;41(3):263–267. doi: 10.1136/ard.41.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]