

Abstract
Homozygous E2F-5 knockout embryos and mice have been generated. Although embryonic development appeared normal, newborn mice developed nonobstructive hydrocephalus, suggesting excessive cerebrospinal fluid (CSF) production. Although the CSF-producing choroid plexus displayed normal cellular organization, it contained abundant electron-lucent epithelial cells, consistent with excessive CSF secretory activity. Moreover, E2F-5 CNS expression in normal animals was largely confined to the choroid plexus. Cell cycle kinetics were not perturbed in homozygous knockout embryo fibroblasts. Thus, E2F-5 is not essential for cell proliferation. Rather, it affects the secretory behavior of a differentiated neural tissue.
Keywords: E2F-5, gene-targeting, hydrocephalus, choroid plexus, transcription, cell cycle
The E2F transcription factor was originally identified as an important regulator of diverse growth-controlling genes, ranging from nucleotide synthesis enzymes to cell cycle regulatory proteins and nuclear proto-oncogenes (Adams and Kaelin 1996; Cobrinik 1996; Slansky and Farnham 1996). E2F functions as a heterodimeric factor, comprising E2F and DP subunits. To date, a family of six distinct E2F and three DP subunits have been identified. The E2Fs can be subgrouped, based on certain structural and functional characteristics. E2F-1, E2F-2, and E2F-3 are structurally similar and form complexes exclusively with the product of the retinoblastoma gene, whereas E2F-4 and E2F-5 interact primarily with the pRb-related “pocket proteins” p107 and p130. In addition, E2F-1, E2F-2, and E2F-3 but not E2F-4 and E2F-5 contain cyclin binding motifs, which facilitate stable binding of cyclin A/CDK2. Recently, a transcriptional repressor with homology to E2F, EMA, which lacks both pocket protein-binding and cyclin-binding sequences, was described (Morkel et al. 1997). The complexity of E2F activity, generated by the formation of different heterodimeric E2F/DP and pocket protein complexes, suggests distinct and diverse functions in cellular growth control.
E2F transcription factors are important for G1- to S-phase entry. In Drosophila, where only one E2F member has been identified, E2F is critical for S-phase progression beyond the seventeenth division in embryogenesis (Duronio et al. 1995). In mammals, overexpression of E2F-1, the best characterized member, is sufficient to induce S-phase entry in quiescent (G0) cells (Johnson et al. 1993; Qin et al. 1994). Furthermore, deregulated expression of E2F-1 to E2F-4 can lead to neoplastic transformation of certain immortalized cell lines (see Adams and Kaelin 1996). E2F-2 and E2F-3 also efficiently induce S-phase entry from quiescence but this is less clear for E2F-4 and E2F-5 (Lukas et al. 1996; DeGregori et al. 1997). In addition to stimulating proliferation, overexpression of E2F-1 in certain conditions can induce apoptosis that is p53-dependent (Qin et al. 1994; Wu and Levine 1994). This proapoptotic function appears specific to E2F-1 (DeGregori et al. 1997). Mice in which the E2F-1 gene has been disrupted display a specific defect in T lymphocyte apoptosis. Moreover, E2F-1−/− mice develop a broad spectrum of unusual epithelial tumors late in adulthood, indicating that E2F-1 has a normal tumor-suppressor function in addition to its proliferative/transforming role (Field et al. 1996; Yamasaki et al. 1996).
Different E2F/pocket protein complexes are identifiable at various phases of the cell cycle suggesting a role for E2F in regulating events beyond G1- to S-phase progression. E2F/p130 complexes, for example, are in greatest abundance in quiescent/G0 cells and E2F/p107 complexes persist beyond S phase (Cobrinik et al. 1993; Vairo et al. 1995; Moberg et al. 1996). Mice lacking both p107 and p130 die shortly after birth and have abnormal long bone development due to unrestricted chondrocyte proliferation, indicating a crucial role for these pocket proteins in bone development and differentiation (Cobrinik et al. 1996). In addition, pRB has been shown to be necessary for the maintenance of the terminally differentiated state in myocytes (Gu et al. 1993) and for normal differentiation of embryonic neuronal and hematopoietic tissue (Jacks et al. 1992; Lee et al. 1992).
The role of E2F-5 in cellular proliferation or differentiation has not been established. First identified by its interaction with p107 and p130 (Hijmans et al. 1995; Sardet et al. 1995) and DP-1 (Buck et al. 1995), E2F-5 shares the greatest homology with E2F-4. It interacts most readily in vivo with the p130 pocket protein. This association raises the possibility that this E2F member (and the more abundant E2F-4) are involved in early events in cell cycle re-entry, or in either commitment to, or maintenance of, cellular differentiation. In keeping with the latter possibility, in situ hybridization studies show increased E2F-5 expression in the superficial, differentiating layers of embryonic epithelial tissues such as skin and intestine (Dagnino et al. 1997a). Furthermore, overexpression of E2F-5 does not induce S-phase entry in quiescent cells, nor the expression of a variety of E2F-regulated cell cycle genes (Lukas et al. 1996; DeGregori et al. 1997).
To assess the role of E2F-5 in normal growth and development, we have inactivated the E2F-5 locus in mice by homologous recombination. Mice lacking E2F-5 developed dilated cerebral ventricles with features indicative of cerebrospinal fluid (CSF) overproduction by the choroid plexus. No discrete defects in cellular proliferation were evident. These findings reveal that E2F-5 is not necessary for entry into S phase and indicate a critical, highly tissue-specific, and differentiative function for this member of the E2F family.
Results and Discussion
Generation of mice with null E2F-5 alleles
Homozygous null E2F-5 mice were generated by homologous recombination. Targeted disruption of the E2F-5 locus was performed in 129/Sv CJ7 ES cells by replacement of two exons encoding the carboxy-terminal half of the DNA-binding (H2) and leucine zipper (LZ) domains, respectively, with a neo-resistance gene (Fig. 1A). Four correctly targeted, independent clones were identified by Southern blot analysis (data not shown) and used to generate male chimeric mice. Two of these clones transmitted the E2F-5 null mutation through the germ line (Fig. 1B) and served as templates for the creation of homozygous mutant, ES cell clones. These lines appeared to be complete null alleles by Northern (Fig. 1C) and Western blot analysis (data not shown).
Figure 1.


Targeting strategy, germ-line transmission, and gene expression analysis. (A) Organization of the targeting vector, mouse E2F-5 gene, and the allele resulting from homologous recombination. Coding regions from the DNA-binding (D) and leucine zipper (LZ) domains are boxed. A 1.2-kb XbaI fragment (probe A) was used to screen for recombinant alleles, and the sizes of recombinant, wild-type, or randomly integrated transgene fragments are shown following SphI digestion (broken line). A 0.8-kb EcoRI fragment (probe B) and neo cDNA were used for confirmatory screening. (TK) Thymidine kinase; (neo) neomycin; (Sp) SphI; (P) PstI; (B) BamHI; (N) NotI. (B) Southern blot analysis of SphI-digested tail DNA from a litter of F2 mice confirming germ-line transmission using probe A. (C) Northern blot analysis of total mRNA from ES cell clones and brains of neonatal F2 mice. Genotypes are indicated.
Interbreeding of heterozygous mutant (E2F-5+/−) mice yielded homozygous mutant (E2F-5−/−) pups with a normal Mendelian frequency, indicating that E2F-5 has no role in embryonic viability. The absence of E2F-5 expression in neonatal brain and murine embryonic fibroblasts (MEFs) of these animals was confirmed by Northern (Fig. 1C) and Western blot (MEFs) analysis (data not shown).
E2F-5 knockout mice develop hydrocephalus
E2F-5−/− neonates were normal in size and appearance for 2–3 weeks. However, by 3–4 weeks of age, they began to demonstrate enlarged, domed craniae (Fig. 2A,D,E), reminiscent of animals with congenital hydrocephalus. Enlargement of the calvarium was confirmed by radiography (Fig. 2B,C). The health of most of these mice deteriorated after weaning, when they began to develop ruffled coats, ataxic gait, and dehydration. Most eventually died, although a small proportion survived for several months. Median survival was 6 weeks (Fig. 3A). The craniae of these mice were grossly enlarged, and the cerebral ventricles were consistently enlarged with marked thinning of the cerebral cortices, confirming the diagnosis of hydrocephalus (Fig. 2F,G). Even long-term survivors developed these cranial defects. Hydrocephalus was frequently accompanied by intracerebral hemorrhage. This phenotype, observed in two independent, homozygous knockout strains, was equally penetrant in 129/Sv × C57BL/6 or 129/Sv × BALB/c F2 backgrounds (data not shown). No abnormalities or early mortality were noted in heterozygous littermates (n = 156) followed for a mean of 8 months (range 7–9).
Figure 2.
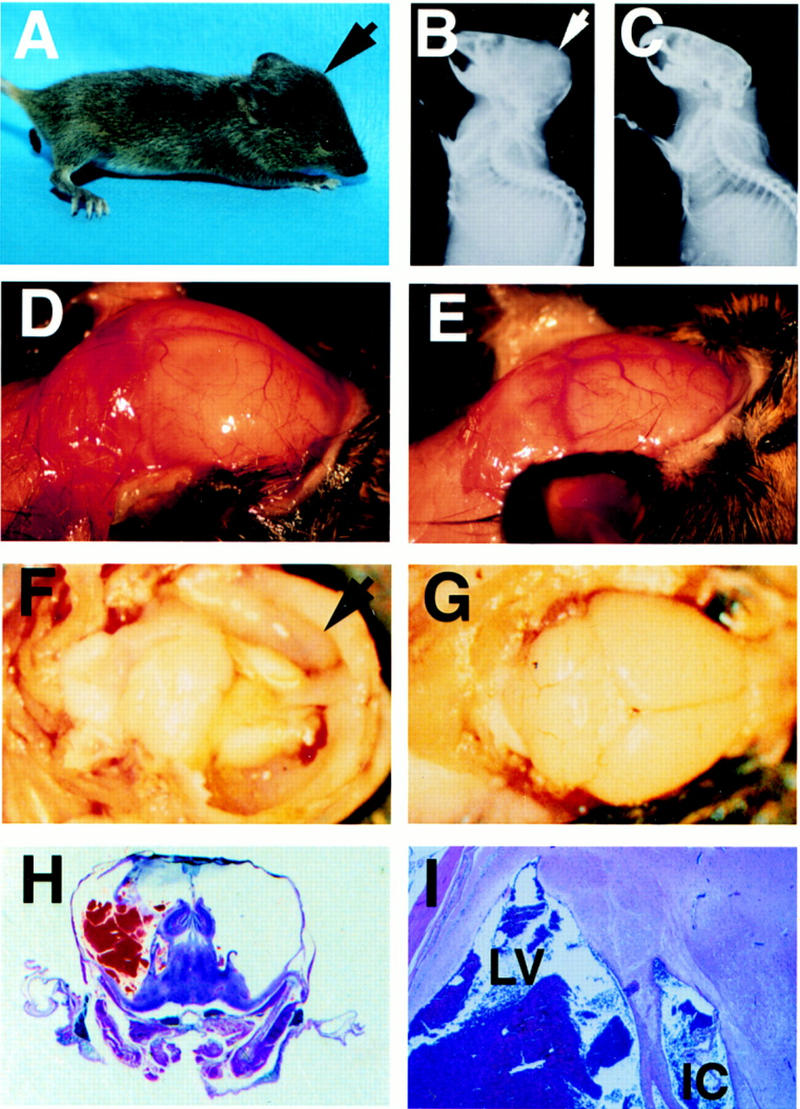
E2F-5−/− mice develop hydrocephalus. (A) Profile of knockout mouse with domed head (arrow). Expanded calvarium (arrow) in homozygous knockout mouse detected by X-radiography (B) compared to a wild-type littermate (C). Expanded calvarium and underlying brain seen in knockout (D) compared to healthy littermate (E). (F) Thin rim of expanded cerebral cortex (anterior cortex arrowed) overlying dilated lateral ventricles; visible underlying structures include (from right to left) hippocampus, thalamus, and cerebellum. (G) Cerebrum of normal, smaller littermate; parietal lobes (right) and cerebellum (left) are visible. (H) H&E-stained coronal section of the cranium revealing massive ventricular dilation, thinning of surrounding neural structures, an intraventricular thrombus, and compression of the hippocampus (superior structures) and underlying thalamus. (I) Dilated lateral ventricle (LV) containing thrombus and intracerebral hemorrhage (IC). (J,L,N) Cerebral cortex of E2F-5−/− pups compared to (K,M,O) the same tissue from a wild-type littermate at D1 (J,K), D4 (L,M), and D7 (N,O). No ventricular dilation was seen at D1, but was apparent by D4. (P,Q), Alcian blue (cartilage) and alizarin red (bone) staining of cleared E16 and E19 E2F-5−/− embryos, respectively. The interparietal bone (i, arrow), supraoccipital bone (s, arrow) and humerus (h, arrow) were normally developed compared to those of wild-type littermates (not shown).
Figure 3.

Lethality from hydrocephalus, MEF proliferation, and cell cycle analysis. (A) Median survival rate for E2F-5−/− mice (strain 33). Animals were sacrificed for autopsy when terminally ill. (B) Cell cycle position of asynchronous wild-type (n = 5) and knockout (n = 4) MEFs determined by BrdU incorporation and FACS analysis. (C) Cumulative growth of primary E2F-5 MEFs. Primary fibroblasts (passage 0) were seeded on 100-mm-diam. plates and sequentially transferred to three plates, each, at 106 cells/plate every 2–3 days. Cell growth was determined after each passage. Cumulative growth was determined and plotted against the number of days of passage. (D) BrdU incorporation of MEFs following synchronization and serum release. Genotypes are indicated.
Microscopic analysis of the E2F-5−/− homozygotes revealed severe intracerebral and intraventricular hemorrhage with destruction of much of the cerebral cortices, except for a thin rim composed of neural tissue and meningeal membranes. The lateral ventricles were greatly dilated and filled with bloody CSF (Fig. 2H,I). The ependymal lining of the lateral ventricles was disrupted at multiple sites, and the neuropil was frequently distended and torn. The third ventricle was moderately dilated while the fourth ventricle appeared normal. The hippocampus, thalamus, midbrain, cerebellum, and medulla were generally preserved, although they were compressed. No abnormality was detected in any non-neuronal tissues.
The timing of the onset of hydrocephalus was determined by histologic analysis of both embryos and newborn mice. E15–E19 embryos exhibited normal cortical neuronal development. Neuroepithelial cell proliferation in the ventricular zone was not perturbed, as determined by light microscopy and BrdU analysis (not shown). Although the cerebral cortex and ventricles appeared normal in day 1 (D1) newborn pups (Fig. 2J,K), enlargement of the lateral ventricles was consistently observed by D4 in knockout animals (Fig. 2L,M) with progressive dilation and thinning of the cerebral cortex by D7 (Fig. 2N,O). Necrosis accompanied the cortical atrophy and was likely a result of markedly elevated intracranial pressure. Importantly, marked apoptosis had not developed in the periventricular regions prior to the onset of ventricular dilatation, as determined by TUNEL staining of brain sections from D1 and D2 pups (not shown). Thus, hydrocephalus developed within the first few days of life and could not be attributed to a perturbation of neural/glial cell apoptosis.
Loss of E2F-5 has no effect on cellular proliferation
MEFs were cultivated from knockout and wild-type mice to further investigate a possible proliferative defect secondary to loss of E2F-5. Knockout MEFs all displayed similar cell cycle kinetics compared with wild-type cells in normal (Fig. 3B) or low serum conditions and were capable of contact inhibition of cell growth, as determined by BrdU incorporation and flow cytometric analysis (data not shown). Consistent with this finding, early passage knockout fibroblasts did not differ in their proliferative rate compared to wild-type fibroblasts (Fig. 3C). Moreover, serum-starved E2F-5−/− fibroblasts entered S phase at the same rate as E2F-5+/+ or E25+/− cultures following stimulation with serum (Fig. 3D). Arguably, other E2F members could have compensated for the absence of E2F-5 in these studies. However, E2F-1−/− MEFs do reveal a significantly lengthened G0–S transit interval (Z.Y. Wang, H. Yang, F. Martelli, and D.M. Livingston, in prep.), and E2F-5 did not induce S phase when overproduced in G0 cells (DeGregori et al. 1997). Therefore, it is possible that E2F-5 does not directly participate in the G0 exit process in fibroblastic cells.
Hydrocephalus is nonobstructive
Next, we sought to determine the generic mechanism underlying the hydrocephalus. Underdevelopment of the interparietal and supraoccipital bones in the calvarium of p107−/− × 130−/− mice has been observed (Cobrinik et al. 1996). As these proteins interact with E2F-5, we asked whether there was abnormal cranial development that could perturb or restrict CSF flow. No such abnormality was noted in E16 and E19 skeletal preparations of E2F-5−/− embryos (Fig. 2P,Q). Thus, hydrocephalus is not a product of abnormal cranial or extracranial bone development.
To establish whether an anatomical defect resulted in a CSF drainage block and hydrocephalus therefrom, brain stems caudal to the thalamus from four knockout and four wild-type littermate animals were serially sectioned. The entire CSF drainage pathway was patent in all cases, and no discrete lesion(s) was detected. This was confirmed by intraventricular injection of trypan blue and monitoring for its reappearance beyond the ventricular tract in the atlanto-occipital subarachnoid space (data not shown). Thus, E2F-5−/− mice develop a communicating (nonobstructive) hydrocephalus.
The choroid plexus normally synthesizes E2F-5 and is altered in E2F-5−/− mice
Hydrocephalus can also result from impaired CSF absorption in the subarachnoid space (Davson et al. 1987), but the histology of this region was normal in the knockout animals (data not shown). It can also occur from overproduction of CSF by the choroid plexus, which is a specialized neuroepithelial tissue found in the lateral, third, and fourth ventricles. The choroid plexus is the major site of CSF production and forms an integral part of the blood–brain barrier (Davson et al. 1987). It is first apparent in the mouse at 12.5 days of gestation, when it is already polarized and well differentiated. It is pertinent that E2F-5 is expressed in the embryonic murine choroid plexus from this period (Dagnino et al. 1997b). Consistent with a potential role for E2F-5 in choroid plexus/CSF production, E2F-5 expression was also observed in the choroid plexus of wild-type neonates (1, 3, and 5 days old), as defined by in situ hybridization (Fig. 4A,B). Choroid plexus expression of E2F-4 has been noted previously (Dagnino et al. 1997b). E2F-5 expression was otherwise confined to the embryologically related ependyma and the subependymal areas in the nervous system (Fig. 4C,D). Importantly, no E2F-5 signal was detected in the subarachnoid/meningeal region, where CSF is absorbed. E2F-5 was also expressed in other non-neuronal epithelia, including skin (Fig. 4E,F), bowel, and submandibular glands, which appeared anatomically normal in homozygous knockout animals (not shown). Thus, selective concentration of E2F-5 in the embryonic and neonatal choroid plexus coupled with E2F-5 deficiency-associated hydrocephalus strongly supports a novel role for E2F-5 in the regulation of CSF production.
Figure 4.
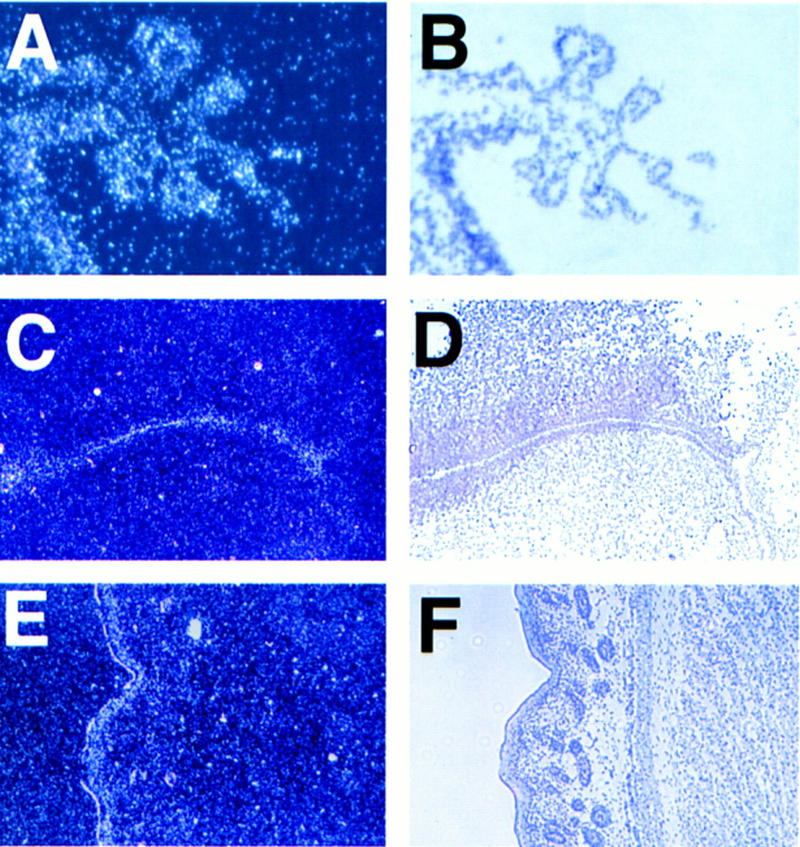
E2F-5 is expressed in the choroid plexus epithelium and periventricular region in newborn mice. Dark-field (A,C,E) and corresponding light-field (B,D,F) micrographs hybridized to an E2F-5 antisense probe. The relevant tissues were D1 choroid plexus (A,B) derived from fourth ventricle (sagittal section); D1 cortex (C,D) showing positive grains in the ependymal cells lining the lateral ventricles (coronal section); D3 skin (E,F) with positive grains in the suprabasal epidermis (sagittal section). A control, sense probe did not hybridize to these tissues (data not shown).
Although the choroid plexus from the lateral and fourth ventricles appeared normal in E2F-5−/− animals (Fig 5A,B), electron microscopy revealed a dramatic reduction in the electron density of numerous choroid plexus cells (Fig. 5C) consistent with their participation in excessive secretory activity (Sato 1977; Schultz et al. 1977). Electron-lucent “light” cells were rarely seen in littermate controls (Fig. 5D). Electron lucency is characteristic of cells that are water-laden (Ghadially 1988). The normal complement of mitrochondria, Golgi, and RER in light cells (Fig. 5E) suggests that they are engaged in active secretion. The fenestrated basement membrane that comprises the blood–brain barrier was normal and intercellular apical tight junctions were intact, indicating that increased CSF flow was unlikely to result from “leaky” epithelium (not shown). Taken together with the E2F-5 expression profile in brain, the absence of ventricular obstruction and the finding of abundant light choroid plexus cells in E2F-5−/− but not wild-type animals implicate dysfunction of the choroid plexus epithelium in the pathogenesis of the hydrocephalus.
Figure 5.

Light microscopic and ultrastructural analysis of E2F-5 mutant choroid plexus. Choroid plexus within the dilated lateral ventricle of a homozygous knockout animal showing normal morphology by light microscopy (A) compared to choroid plexus (arrow) from a wild-type littermate (B). (C)E2F-5−/− choroid plexus showing numerous electron lucent (light) cells, one of which is arrowed. The microvillus border is irregular and disorganized (original magnification 2940×). (D) In contrast, a wild-type littermate shows uniform cellular electron density, apart from an occasional light cell (2940×). (E) Magnified view showing light cells in E2F-5−/− choroid plexus. Ultrastructural organelles appear intact (4750×). EM sections were from 4-day-old neonates.
Interestingly, E2F-5 and the highly homologous E2F-4 display largely disparate embryonic expression patterns. E2F-4 is primarily concentrated in early proliferative epithelium, whereas E2F-5 is located in terminally differentiating or differentiated cells (Dagnino et al. 1997a), implying that they effect different functions despite their high degree of structural similarity. Even when their expression profiles overlap, as they do in the embryonic choroid plexus (Dagnino et al. 1997b), our results suggest that they have distinct in vivo functions.
On the other hand, that E2F-5 knockout mice were healthy, except for the hydrocephalus, suggests that either E2F-5 has no role in tissues other than choroid plexus or that the absence of E2F-5 is compensated by the presence of low levels of E2F-4 and/or other E2F species. A limited repertoire of E2F and DP proteins is available in the choroid plexus to substitute for the absence of E2F-5: E2F-1 and E2F-3 transcripts are absent, whereas E2F-2 is barely above background in in situ hybridization assays. Intriguingly, DP-1 transcripts are not detectable in normal embryonic choroid plexus, despite abundant expression elsewhere implicating DP-2 or DP-3 in heterodimer formation (Dagnino et al. 1997b).
In summary, it appears that E2F-5 is essential for postnatal survival by virtue of its role in normal choroid plexus function. The E2F-5 knockout phenotype most likely represents the failure of an E2F-5-specific developmental or postproliferative, organ physiology control function. The critical target gene(s) and mechanism of CSF overproduction remain to be identified. Our findings do not provide evidence of a proliferation control defect associated with E2F-5 deficiency. Conceivably, it was obscured by functional redundancy between E2F-5 and other E2F family members. This notwithstanding, the phenotype of E2F-5−/− mice contrasts with the proliferative and apoptotic defects observed in tumor-prone E2F-1−/− mice (Field et al. 1996; Yamasaki et al. 1996) and supports the view that different E2Fs regulate distinct genes. More specifically, the data presented here reveal that the complexity of the E2F family, in part, reflects the existence of individuated biological roles for otherwise closely related family members.
Materials and methods
Gene targeting
To generate an E2F-5 targeting vector, a 15-kb clone was isolated from a Lambda Fix II library derived from 129Sv genomic DNA (Stratagene) using a human E2F-5 cDNA probe (nucleotide sequences 1–246). Two exons, comprising the 3′ portions of the H2 and LZ domains, were identified by Southern blotting and sequence analysis. A 3.6-kb PstI fragment, containing upstream intronic sequences and the PstI site found in the H2 exon, and a 4.4-kb BamHI fragment, containing intronic sequences downstream of the LZ exon, were inserted into the targeting vector pPNT, which contained a thymidine kinase and a neo gene (Tybulewicz et al. 1991). If properly recombined with the host chromosome, the targeting vector should replace the H2 and LZ exons with a neo cassette, inserted in the opposite orientation. A null allele was predicted following successful homologous recombination, with an in frame termination codon after residue 71.
Homologous recombination and generation of germ-line chimeras
The NotI-linearized recombinant was electroporated into 129/Sv CJ7 ES cells (Swiatek and Gridley 1993), which were seeded on feeder layers of γ-irradiated (3000 cGy) MEFs and selected in medium containing 300 μg/ml G418 and 2 μm gancyclovir. Four correctly targeted clones were obtained from 65 selected ES clones, as determined by Southern blot analysis. They were each injected into C57BL/6 and Balb/c blastocysts to generate chimeras. Chimeric males from two independent clones (28 and 33) passed their mutation to offspring. F2 progeny of heterozygous intercrosses were screened by Southern blot analysis or PCR to determine their genotype, using genomic tail DNA. Primers were P1, 5′-GATCGGCCATTGAACAAGATGG; P2, 5′-CCTGATGCTCTTCGTCCAGATC; P3, 5′-TGCAGATACCTTGGCTGTGAGGCAAAAGCG; and P4, 5′-CTTTAGTATTACAGCCAGCACCTACACCCC. Primers P1 and P2 amplify a neo cDNA sequence (∼0.5 kb), indicating the presence of a mutant allele, and P3 and P4 amplify sequences present only in a wild-type allele (∼1.0 kb). Homozygous deletion of E2F-5 in ES cell clones was achieved by selection in a high concentration of G418 (1.5–2.0 mg/ml) (Mortensen et al. 1992) and detected by PCR.
Northern blot analysis
Total RNAs were prepared from ES cells or 3-day-old mouse brain using RNAzol B (Tel-Test Inc.) for Northern blot analysis (15 μg per lane). Transcripts were detected using murine E2F-5 and rat GAPDH cDNA probes.
MEFs
MEFs were prepared from 14-d.p.c embryos derived from a mating between E2F-5 heterozygote knockout mice essentially as described (Robertson 1987). The heads of the embryos were removed and processed for genotyping by PCR, and the embryos were washed in PBS, minced, and incubated with 0.25% trypsin/EDTA (GIBCO BRL) for 20–30 min at 37°C. Cells were resuspended in DME with 10% FBS (GIBCO BRL), plated on 100-mm-diam. petri dishes, and maintained at 37°C in 10% CO2. These were considered passage 0 (P0) cells. MEFs were growth arrested by incubation for 48 hr in DME containing 0.1% FBS, stimulated by addition of FBS to a final concentration of 10%, and harvested at appropriate times thereafter. For cell cycle analysis, 10 μm BrdU (Boehringer Mannheim) was added 1 hr prior to harvesting. Cells were then stained with anti-BrdU FITC-conjugated antibody and subjected to FACS analysis, as recommended by the manufacturer (Becton-Dickinson). P3 and P4 MEFs were used for all experiments.
Histology, in situ hybridization, electron microscopy, and radiology
Embryos or tissues were fixed in either 10% neutral-buffered formalin or Bouin’s solution prior to preparation of 6-μm paraffin sections, stained with hematoxylin and eosin (HE). E16 and E19 embryos were stained with cartilage-specific alcian blue and bone-specific alizarin red. Soft tissue was cleared with alkali, essentially as described (Cobrinik et al. 1996). In situ hybridization using previously established conditions was performed with 8-μm frozen sections from C.B-17 mice aged 1, 3, and 5 days, as described (Dagnino et al. 1997b). Non-cross-reactive mE2F-5 nucleotides 399–1069 were used to prepare the E2F-5 radiolabeled riboprobe. For electron microscopy, choroid plexus was microdissected from lateral and fourth ventricles of neonatal mice. Tissues were fixed in 2.5% glutaraldehyde in cacodylate buffer containing 1% CaCl2 at 4°C, postfixed with 1% osmium tetroxide, 0.1 m potassium ferricyanide, and processed as described (Gilligan et al. 1996). Ultrathin sections (∼50 nm) were prepared with an MT5000 Sorvall ultramicrotome, stained with lead citrate and uranyl citrate, and examined by transmission electron microscopy (100CX; JEOL U.S.A. Inc, Peabody, MA) at 60 kV using a 20-μm objective aperture. Whole body radiographs were performed using the LoRad MIII Mammography Unit at 24 kG, 20 MaS. All comparisions between wild-type and mutants were performed on age-matched littermates.
Acknowledgments
CJ7 ES cells and advice were generously provided by S.H. Orkin, C.P. Browne, and Y. Fujiwara. We are grateful to A. Sharpe and L. Du for the blastocyst injections, E. Meluleni for histologic work, L. Callahan and C. Bianchi at the Redstone animal facilities, and B.N. Weissman and D. Major for mouse radiography. We thank all of our divisional colleagues for numerous, helpful discussions. This work was supported by funds provided to D.M.L. by the National Cancer Institute and the Dana-Farber/Sandoz Drug Discovery Program. G.J.L. was a recipient of a Neil-Hamilton Fairley Postdoctoral Fellowship from The National Health and Medical Research Council of Australia. S.G. is supported by a European Molecular Biology Organization Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL david_livingston@dfci.harvard.edu; FAX (617) 632-4381.
References
- Adams PD, Kaelin WG., Jr The cellular effects of E2F overexpression. Curr Top Microbiol Immunol. 1996;208:79–93. doi: 10.1007/978-3-642-79910-5_4. [DOI] [PubMed] [Google Scholar]
- Buck V, Allen KE, Sørensen T, Bybee A, Hijmans EM, Voorhoeve PM, La Thangue N. Molecular and functional characterisation of E2F-5, a new member of the E2F family. Oncogene. 1995;11:31–38. [PubMed] [Google Scholar]
- Cobrinik D. Regulatory interactions among E2Fs and cell cycle control proteins. Curr Top Microbiol Immunol. 1996;208:31–61. doi: 10.1007/978-3-642-79910-5_2. [DOI] [PubMed] [Google Scholar]
- Cobrinik D, Whyte P, Peeper D, Jacks T, Weinberg RA. Cell cycle-specific association of E2F with the p130 E1A-binding protein. Genes & Dev. 1993;7:2392–2404. doi: 10.1101/gad.7.12a.2392. [DOI] [PubMed] [Google Scholar]
- Cobrinik D, Lee M-H, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes & Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- Dagnino L, Fry C, Bartley S, Farnham P, Gallie B, Phillips RA. Expression patterns of the E2F family of transcription factors during murine epithelial development. Cell Growth Differ. 1997a;8:553–563. [PubMed] [Google Scholar]
- Dagnino L, Fry C, Bartley S, Farnham P, Gallie B, Phillips RA. Expression of the E2F family of transcription factors during mouse nervous system development. Mech Dev. 1997b;66:13–25. doi: 10.1016/s0925-4773(97)00083-x. [DOI] [PubMed] [Google Scholar]
- Davson H, Welch K, Segal M. Physiology and pathophysiology of the cerebrospinal fluid. Edinburgh, UK: Churchill Livingstone; 1987. [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio R, O’Farrell P, Xie J, Brook A, Dyson N. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes & Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai F-Y, Kuo F, Zubiaga AM, Kaelin WG, Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Ghadially F. Ultrastructural pathology of the cell and matrix. II. London, UK: Butterworths; 1988. pp. 954–958. [Google Scholar]
- Gilligan H, Bredy B, Brady H, Hébert M-J, Slayter H, Xu Y, Rauch J, Shia M, Koh J, Levine J. Antineutrophil cytoplasmic autoantibodies interact with primary granule constituents on the surface of apoptotic neutrophils in the absence of neutrophil priming. J Exp Med. 1996;184:2231–2241. doi: 10.1084/jem.184.6.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Schneider J, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Hijmans EM, Voorhoeve PM, Beijersbergen RL, van’t Veer LJ, Bernards R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol Cell Biol. 1995;15:3082–3089. doi: 10.1128/mcb.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fateli A, Schmitt E, Bronson R, Goodell M, Weinberg RA. Effects of an Rb-mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Johnson D, Schwarz J, Cress W, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S-phase. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Lee E-H, Chang C-Y, Hu N, Wang Y-C, Lai C-C, Herrup K, Lee W-H, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lukas J, Petersen B, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morkel M, Wenkel J, Kouzarides T, Bannister A, Hagemeier C. An E2F-like repressor of transcription. Nature. 1997;390:567–568. doi: 10.1038/37507. [DOI] [PubMed] [Google Scholar]
- Mortensen R, Conner D, Chao S, Geisterfer-Lowrance A, Seidman J. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Livingston DM, Kaelin WG, Adams P. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Nat Acad Sci. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson E. Embryo-derived stem cell lines. In: Robertson E, editor. Teratocarcinomas and embryonic stem cells: A practical approach. Oxford, UK: IRL Press; 1987. pp. 71–112. [Google Scholar]
- Sardet C, Vidal M, Cobrinik D, Geng Y, Onufryk C, Chen A, Weinberg RA. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc Natl Acad Sci. 1995;92:2403–2407. doi: 10.1073/pnas.92.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K. Pharmacology and function of the myoepithelial cell in the eccrine sweat gland. Experientia. 1977;33:631–633. doi: 10.1007/BF01946542. [DOI] [PubMed] [Google Scholar]
- Schultz W, Brownfield M, Kozlowski G. The hypothalamo-choroidal tract II. Ultrastructural response of the choroid plexus to vasopressin. Cell Tissue Res. 1977;178:129–141. doi: 10.1007/BF00232830. [DOI] [PubMed] [Google Scholar]
- Slansky J, Farnham P. Introduction to the E2F family protein structure and gene regulation. Curr Top Microbiol Immunol. 1996;208:1–30. doi: 10.1007/978-3-642-79910-5_1. [DOI] [PubMed] [Google Scholar]
- Swiatek P, Gridley T. Perinatal lethality and defects in hindbrain development in mice homozygous for a targeted mutation of the zinc finger gene Krox20. Genes & Dev. 1993;7:2071–2084. doi: 10.1101/gad.7.11.2071. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V, Crawford C, Jackson P, Bronson R, Mulligan R. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl protooncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Vairo G, Livingston DM, Ginsberg D. Functional interaction between E2F-4 and p130: Evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes & Dev. 1995;9:869–881. doi: 10.1101/gad.9.7.869. [DOI] [PubMed] [Google Scholar]
- Wu X, Levine A. P53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]