

Abstract
IκB kinases (IKKs) IKK1 and IKK2 are two putative IκBα kinases involved in NF-κB activation. To examine the in vivo functions of IKK1, we generated IKK1-deficient mice. The mutant mice are perinatally lethal and exhibit a wide range of developmental defects. Newborn mutant mice have shiny, taut, and sticky skin without whiskers. Histological analysis shows thicker epidermis, which is unable to differentiate. Limbs and tail are wrapped inside the skin and do not extend properly out of the body trunk. Skeleton staining reveals a cleft secondary palate, split sternebra 6, and deformed incisors. NF-κB activation mediated by TNFα and IL-1 is diminished in IKK1-deficient mouse embryonic fibroblast (MEF) cells. The IKK complex in the absence of IKK1 is capable of phosphorylating IκBα and IκBβ in vitro. Our results support a role for IKK1 in NF-κB activation and uncover its involvement in skin and skeleton development. We conclude further that the two related kinases IKK1 and IKK2 have distinct functions and can not be substituted for each other’s functions.
Keywords: IKK1, NF-κB, skin, skeleton, limb, mice
NF-κB transcription factors are dimers composed of various combinations of structurally related proteins p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), c-Rel, and RelB (for review, see Verma et al. 1995; Baeuerle and Baichwal 1997 ). In resting cells, NF-κB complexes are retained in the cytoplasm in association with inhibitory proteins IκBs (IκBα, Iκβ, Iκε). Upon stimulation by TNFα, IL-1α, UV, and γ-irradiation, or bacterial and viral infection, IκBs are phosphorylated at specific sites that lead to their ubiqitination, degradation by the proteosome, and release of NF-κB proteins for translocation to the nucleus where they regulate expression of target genes.
NF-κB proteins play a major role in many physiological and pathological processes. Analyses of mice deficient in different members of the NF-κB and IκB families have revealed essential roles for these transcription factors in lymphocyte development and immune responses (for review, see Attar et al. 1997), fetal liver development (Beg et al. 1995), and osteoclast maturation (Franzoso et al. 1997; Iotsova et al. 1997). Rel/NF-κB genes may also play a role in vertebrate limb development (Bushdid et al. 1998; Kanegae et al. 1998). Additionally, several groups have shown the involvement of NF-κB proteins in anti-apoptotic processes (Beg and Baltimore 1996; Van Antwerp et al. 1996; Wang et al. 1996). Lack of p65 (RelA) results in hepatocyte apoptosis and embryonic lethality at embryonic day 15 (E15), which may reflect its anti-apoptotic function in hepatocytes during development (Beg et al. 1995).
A central step to NF-κB activation is the induced phosphorylation of IκBs (Verma et al. 1995). Recently, the long-sought kinases for signal-induced phosphorylation of IκB have been identified by three independent groups (DiDonato et al. 1997; Mercurio 1997; Regnier et al. 1997; Woronicz et al. 1997; Zandi et al. 1997). Two highly homologous IκB kinases (IKKs), IKK1 (IKKα) and IKK2 (IKKβ), are present in a large 700–900 kD complex and can specifically phosphorylate IκBα and IκBα in response to induction by TNFα and IL-1. The kinetics of induced complex activity matches with the kinetics of induced NF-κB activity. Overexpression of dominant-negative mutants of IKK1 or IKK2 can specifically inhibit the TNFα- and IL-1-induced NF-κB activity. Furthermore, in vitro kinase assays with the purified recombinant proteins, IKK1 and IKK2 site specifically phosphorylate all three known IκBs (IκBα at Ser-32 and Ser-36 , IκBβ at Ser-19 and Ser-23, and IκBε at Ser-18 and Ser-22; Lee et al. 1998; Li et al. 1998; Zandi et al. 1998). Thus, it appears that IKK1 and IKK2 are bona fide IκB kinases. However, the roles played by the individual kinases in NF-κB activation during development remain unknown.
We have generated IKK1-deficient mice by a gene targeting approach. Mice lacking the IKK1 gene are perinatally lethal and reveal a remarkable abnormal appearance including shorter limbs, a fused tail, and a shiny skin. The undifferentiated epidermis in mutant skin lacks degradation of nuclei and keratinized stratified epidermis in their stratum corneum. IKK1-deficient mice have a cleft secondary palate, split sternebra 6, and abnormal incisors. Furthermore, TNFα- and IL-1-induced NF-κB activation is diminished in mouse embryonic fibroblasts (MEFs) isolated from IKK1-deficient mice.
Results
Generation of IKK1-deficient mice
To mutate the IKK1 gene, a neomycin-resistance gene was used to replace a 3-kb genomic DNA fragment containing exon 1 and the upstream promoter region (Fig. 1A). Homologous recombinant ES cell clones were identified by Southern analysis (Fig. 1B) and injected into C57/BL6 blastocysts that, in turn, developed into chimeric mice. Heterozygous mice generated by germ-line transmission of the targeted ES clones into a C57/BL6 genetic background were normal and fertile. Although they could develop to term, the IKK1 homozygous mutant mice were stillborn or died shortly after birth. Examination of E18 and E19 embryos by Caesarean section revealed that all of the mutant embryos were alive in utero but died soon after section. To determine the expression of IKK1 protein in IKK1−/− mice, we performed Western blot analysis on primary MEF cell extracts (Fig. 1D). Anti-IKK1 sera detected IKK1 protein in wild-type MEFs and approximately one-half of that amount in heterozygous MEFs but failed to detect any signal in IKK1−/− MEFs. The relative amount of IKK2, IκBα , and IκBβ proteins was not affected in IKK1−/− MEFs (Fig. 1D). We conclude that homozygous mice are IKK1 null.
Figure 1.

Targeting of the IKK1 gene in mice. (A) Strategy for targeting the IKK1 allele. Simplified restriction maps of the wild-type IKK1 allele, the targeting construct, and the mutated allele are shown. A 3-kb genomic fragment is deleted and replaced by a PGK–neo cassette in an antisense orientation. (B) Southern blot analysis of an ES clone showing the correct insertion of the targeting construct. DNA was digested with BamHI and hybridized to the probe shown in A. The wild-type allele yields an 8.9-kb fragment whereas the mutant allele yields a 4-kb fragment. (C) PCR detection of mouse genotypes. (D) Western blot analysis confirmed the absence of IKK1 in IKK1−/− MEFs, whereas the expression of IKK2, IκBα, and IκBβ was not changed. Forty micrograms of whole-cell protein lysates from IKK2+/+, IKK2+/−, and IKK2−/− MEFs were loaded and immunoblotted with IKK1, IKK2, IκBα, and IκBβ antibodies.
General phenotype of IKK1−/− mice
Newborn IKK1 homozygous mutant mice could easily be identified by the marked malformation of their body morphology (Fig. 2A) and their genotypes were confirmed by PCR analysis (Fig. 1C). The mutant skin was shiny, translucent, sticky, and without whiskers (Fig. 2B,C). All four limbs appeared shorter and, most remarkably, the hind limbs together with the curled tail were embedded in the thick skin. Mutant embryos could be distinguished from their normal littermates as early as E12.5 of gestation because of their short, dumpy limb buds and curled tail (Fig. 2D).
Figure 2.
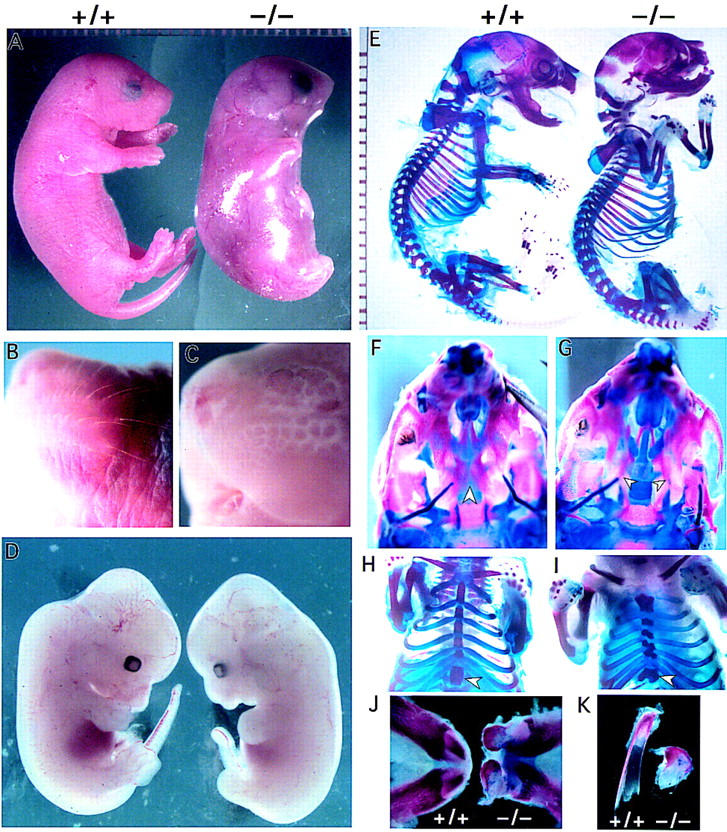
Gross phenotypes of IKK1−/−newborn mice. (A) Wild-type newborn mouse (left) and an IKK1−/− homozygous (right) littermate are shown. The mutant embryos exhibit severe alterations in the overall morphology. Limbs and tail are barely protruding and seem to be shortened. The skin appears translucent and tense. (B,C) A close-up view of a wild-type (B) and mutant (C) head. The mutant embryos lack whiskers, compared with the wild-type embryos. (D) Comparison of E12.5 wild-type (left) and IKK1 (right) mutant embryos. The mutant shows mild phenotypical alterations in morphology of the limb buds, tail and craniofacial region. (E) Bone staining of the embryos shown in A. The mutant skeleton (right) exhibits a few gross alterations. The craniofacial bone structure shows malformation, the tail appears to be shorter, and the bending of the distal limb region is abnormal. However, in contrast to the appearance of a newborn embryo, all long bones of the limbs are formed. (F,G) Ventral view of wild-type (F) and mutant (G) skulls. The skull exhibits a nearly complete fusion of the palate (F; arrowhead), whereas the mutant bilateral palate shelves (arrowhead) remains unfused, allowing the more dorsal lying vomer and presphenoid to be visible (G). (H,I) A ventral view of the ribcage and the sternum of a wild-type skeleton (H) and an IKK1−/− skeleton (F). The mutant embryo displays a broader sternum and the ribs exhibit a kinky fusion to the sternum and split sternebra 6 (arrowhead). (J) Lower jaw and lower incisors of wild-type (left) and mutant (right) embryos. The morphology of the protruding incisors in mutant jaw appears malformed. (K) A detailed view of the incisors (left, +/+; right , −/−) shows a reduced and distorted mutant incisor.
Gross examination of internal organs revealed alterations of the gastrointestinal tract. IKK1−/− mice had a much smaller stomach and a shorter and narrower intestine (data not shown). All the mutant newborn pups had an expanded bladder that was not observed in wild-type or heterozygous littermates. Delayed umbilical hernia withdrawal was also observed in mutant embryos at E18 (data not shown). No apparent histological abnormalities were noticed in other organs.
Defects in skeleton development
Whole-mount examination of IKK1−/− mice revealed shorter limb buds at mid-gestation and shorter limbs at birth (Fig. 2A). Surprisingly, cartilage and bone staining of newborn mice with alizarin red/alcian blue did not reveal any major changes in the pattern and size of the proximal limb elements (Fig. 2E). However, alterations in the curvature and bending of the most distal limb elements were observed, and the phalanges were retarded and deformed (data not shown).
There were additional defects in skeletal development, including cleft palate (Fig. 2F,G) and unfused sternebra 6 (Fig. 2H,I). In the mutant mice, the vomer and presphenoid were visible because of the cleft secondary palate (Fig. 2F,G). Sternebra 6 was split and showed a reminiscence of its dual origin. Moreover, the sternal bands were shorter and broader and had a kinked shape instead of a straight one as a result of incomplete and asymmetric ossification of the sternum (Fig. 2H,I). However, the sternal bands were well fused and functional. The gross appearance of incisors in newborn wild-type and IKK1 mutant mice is shown in Figure 2 (J,K). Incisors were present but deformed.
Skin defects
Histological examination of newborn (P0) mouse skin showed abnormalities in epidermal morphogenesis. The superficial keratinized squamous layer was missing, and the density of the suprabasal cells was much higher in homozygous mutant embryos (Fig. 3A,B). Dead cells devoid of nuclei, characteristic of the cornified layer (Fig. 3C), were not observed in mutant mouse skin. The granular layer was also not obvious as the granular cells with distinctive keratohyalin granules were not present. Instead, multiple layers of unusual flattened cells were on the surface of mutant skin. In addition, the number of hair follicles was reduced, and they did not invaginate deeply into dermis in the mutant mice (Fig. 3A,B). Although anti-keratin 14 immunostaining showed no difference of the single basal layer in both wild-type and mutant skin (Fig. 3C,D), keratin 10-positive cells increased dramatically in the mutant suprabasal layer (SB) of skin compared with littermates (Fig. 3E,F). The suprabasal cells failed to differentiate into granular cells and cornified cells as determined by the immunohistochemistry with antibodies to filaggrin (Fig. 3G,H) and loricrin (Fig. 3I,J,K). Both filaggrin and loricrin are expressed in differentiated epidermal cells. They are present in the keratohyalin granules of the granular cells and the cornifed layer in normal epidermis. The expression of both differentiation markers in IKK1-deficient epidermis was reduced considerably.
Figure 3.
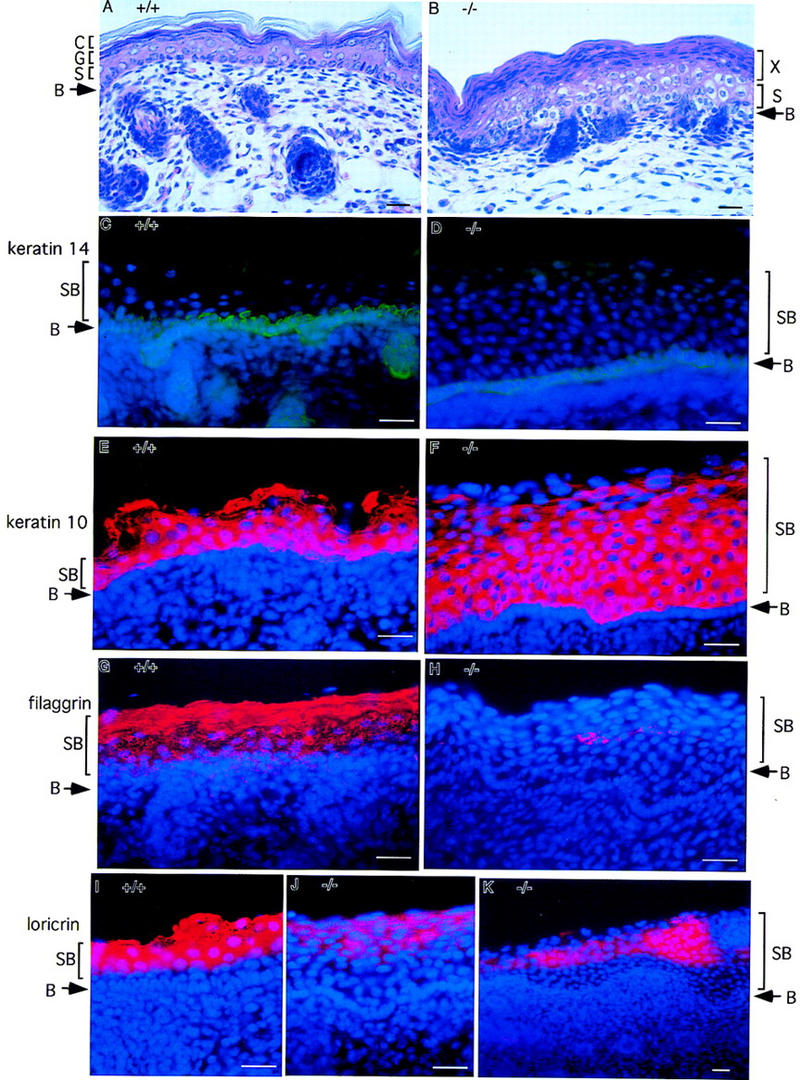
Lack of epidermal differentiation in the skin from IKK1−/− mice. Staining was performed on skin from newborn wild-type (+/+) or mutant mice (−/−). (A,B) Hematoxylin and eosin staining of skin. In the superficial layer of mutant skin, an unusual thick layer of flatten cells (layer X in B) was present instead of cornified layer. (C,D) Keratin 14 immunostaining (green) shows normal staining pattern of positive cells in the stratum basal layer. Suprabasal layer above basal layer was much thicker in mutant skin than in wild-type skin. (E,F) Anti-keratin 10 antibody stains the cells (red) in suprabasal layers in normal epidermis. The number of keratin 10 positive cells (red) in mutant skin increased dramatically. (G,H) Filaggrin is expressed in the granule and cornified layers of normal skin; however, its expression is diminished and limited to small patches of cells (red) in the mutant skin. (I–K) Expression of loricrin (red) is also reduced dramatically in the mutant skin. Nuclei (blue) were labeled by DAPI in C–K. Abbreviations: (B) Basal layer of epidermis; (S) spinous layer; (G) granular layer; (C) cornified layer; (SB) suprabasal layer. Bar size, 24 μm.
Reduced NF-κB activity in IKK1−/− MEFs
Because IKK1 functions as an IκB kinase, loss of IKK1 may impair signal-induced phosphorylation and degradation of IκBs and, therefore, block NF-κB nuclear translocation and subsequent DNA binding. To assess whether NF-κB activation in IKK1−/− MEFs was impaired, we performed a gel mobility shift analysis on nuclear extracts with a HIV-κB probe. A significant reduction of the NF-κB binding activity in IKK1 mutant MEFs was observed upon induction with TNFα or IL-1 (Fig. 4A). Next, we examined the significance of the reduced NF-κB DNA-binding activity by measuring the expression of NF-κB-targeted genes such as those encoding IκBα, macrophage colony-stimulating factor (M-CSF), and IL-6. Consistent with the decreased NF-κB-binding activity, the IκBα mRNA level in IKK1−/− MEF was reduced (Fig. 4B). We measured the basal and induced expression of M-CSF and IL-6 in IKK1+/+ and IKK1−/− MEFs. The induced expression of M-CSF mRNA was reduced considerably in IKK1−/− MEFs, whereas its basal expression level was not affected (Fig. 4C). Similarly, the TNFα-induced IL-6 expression was also reduced (Fig. 4C). Together, these results support a role for IKK1 as an IκB kinase, although a considerable amount of NF-κB binding activity remains in the absence of IKK1.
Figure 4.

NF-κB activity in IKK1−/− MEFs. (A) NF-κB binding activity is reduced in IKK1−/− MEF. Gel mobility shift analysis was conducted on 5 μg of nuclear extract from IKK1+/+ and IKK1−/− MEFs with or without 10 ng/ml TNFα induction for the indicated time. (B) TNFα-induced IκBα RNA synthesis is reduced in MEFs lacking IKK1. Northern blot analysis was performed on 10 μg of total RNA from each sample, using probe from a full-length IκBα cDNA. (C) RNase protection shows that TNFα-induced mRNA expression of M-CSF and IL-6 are diminished in IKK1−/− MEF. (D) Phosphorylation of IκBα, IκBβ, and p65 by NEMO immunocomplexes from IKK1−/− MEFs is not affected. Cells from three 15-cm plates were lysed and immunoprecipitated with anti-NEMO serum. Equal amounts of NEMO immunoprecipitates were incubated with the substrates indicated for the in vitro kinase assay. NEMO immunoprecipitation samples were loaded on a 10% SDS–polyacrylamide gel for anti-IKK1 and anti-IKK2 immunoblotting (D, bottom).
In addition to IKK1 and IKK2, two other proteins, NEMO (IKKγIKKAP1) and IKAP, have been identified in the large IKK complex (Cohen et al. 1998; Rothwarf et al. 1998; Yamaoka et al. 1998; Mercurio et al. 1999). NEMO is not a kinase but is necessary for NF-κB activity induced by TNFα and other external stimuli (Yamaoka et al. 1998). To examine the activity of IKK complex in the absence of the IKK1, we performed an in vitro kinase assay with immunocomplexes precipitated with NEMO antibody (Fig. 4D). Interestingly, both IκBα and IκBβ, as well as p65 (RelA) were phosphorylated efficiently by NEMO immunocomplexes from both IKK1+/+ and IKK1−/− MEFs (Fig. 4D). Thus, it appears that IKK1 may not be involved directly in IκBα or IκBβ phosphorylation but has another and yet unknown function for full NF-κB activation.
Discussion
NF-κB/Rel signaling pathways have been studied extensively in the past decade. Knockout mice for individual members of the NF-κB family have been generated and showed no major developmental defects except liver degeneration in p65-deficient mice (Beg et al. 1995; Attar et al. 1997). To our surprise, the phenotypes of mice deficient for the putative IκB kinase IKK1 revealed multiple defects involving skin and skeleton development. Severe defects in epidermal differentiation were observed, which may be the cause of perinatal death. Detailed examination of the epidermis revealed an arrest in stratum corneum differentiation and a total lack of squames. Furthermore, IKK1-deficient mice do not have whiskers (Fig. 2B,C), indicating a role of IKK1 in skin morphogenesis.
The most striking phenotype of IKK1 mutant mice was a bottle-shaped body morphology with a failure of the upper limb (humerus) and middle limb elements (radius, and ulna) to protrude out of the body trunk in a normal way. Upon close examination, the pattern of the limbs and associated muscles appeared normal, however, the skin was not shaped and attached to the limbs, but instead covered the body like a bag. Interestingly, the altered morphology became apparent as early as E12.5, when the skin was not yet differentiated. The skin defects in IKK1 mutant mice are similar to a human skin disorder termed lamellar ichthyosis (LI) that is frequently evident at birth as a collodion baby (Roop 1995). Absence of functional stratum corneum in IKK1-deficient mice is probably the major cause of the perinatal lethality as the stratum corneum serves as a barrier to protect the internal body from the external environment. Neonatal lethality has been previously associated with skin defects (Matsuki et al. 1998; Xie et al. 1998).
What is the molecular explanation for defects observed in IKK1-deficient mice? The simplest interpretation is that defects in the NF-κB activation pathway in IKK1 mutant mice are responsible for those phenotypes, or at least some of those phenotypes. We demonstrated that induced NF-κB activation was impaired in the primary IKK1−/− MEFs, although we failed to detect any defect in phosphorylation of the IκBα or IκBβ by IKK immunocomplexes (Fig. 4D). The involvement of NF-κB pathway in skin morphogenesis has been reported previously (Seitz et al. 1998). Transgenic mice producing a transdominant IκBα under the control of the keratin 14 promoter resulted in hyperplastic epidermis (Seitz et al. 1998). Alternatively, the observed phenotypes in IKK1−/− mice may be due to novel functions of IKK1 unrelated to its IκB kinase activity. We have also generated IKK2-deficient mice that have reduced NF-κB activity and kinase activity of the IKK complex. These mutant mice have a phenotype similar to p65-deficient mice, confirming its function as an IκB kinase (Li et al. 1999). Distinct phenotypes observed in IKK1 and IKK2 mutant mice suggest that they have different functions during embryonic development. IKK1 and IKK2 may be part of a large complex but respond differentially to specific inducers or have different substrates. Alternatively, IKK1 has additional functions independent of the NF-κB pathway. It will be of particular interest to identify specific genes involved in skin and skeletal phenotypes and determine whether those genes are regulated by NF-κB. Availability of mice deficient in specific NF-κB or IκB genes will allow for the further study of the functions of the IKKs with a genetic approach.
Materials and methods
Generation of IKK1−/− mice
Genomic fragments of the mouse IKK1 were screened from a mouse 129/SvJ genomic DNA library in P1 vector (Genomic Systems) by PCR with mouse IKK1-specific primers. The positive clone was subcloned and used to generate a IKK1-targeting construct. The IKK1-targeting vector was constructed by insertion of a 1.8-kb BglII fragment as 5′ homologous arm and a 5.8-kb EcoRV fragment as 3′ homologous arm into the pPNT vector. A 3-kb IKK1 genomic fragment containing the promoter region (−500 to −1), coding region (cDNA 1–105), and intron sequence was deleted and replaced by a PGK-neo expression cassette in an antisense orientation. Therefore, it was predicted that no functional IKK1 protein would be expressed from this IKK1-targeted allele.
Twenty-five micrograms of NotI-linearized targeting construct was electroporated into J1 ES cells, and the targeted clones were selected with G418 (0.2 mg/ml, active form) and FIAU (200 μm) in DMEM medium. The resistant colonies were screened for homologous recombination by Southern blot analysis with a 240-bp 5′-external PstI fragment as a probe. A 4-kb BamHI fragment from the targeted allele was distinguished from a 8.9-kb wild-type BamHI fragment.
Two independent targeted ES cell clones (5B5 and 2D1) with only a single insertion were identified and injected into C57BL/6 blastocysts. Heterozygous mutant mice were generated from one line (5B5). Crossing of the chimeras with C57BL/6J mice generated animals with a mixed genetic background.
F1 heterozygous mice were intercrossed to generate F2 offspring that were genotyped by PCR analysis to determine IKK1-knockout status. For PCR genotyping, the following primers were used: forward primer a (5′-AGGAGTTGAAGGATCTCTTGTG-3′) annealing to deleted part of genomic sequences; forward primer b (5′-GGGAACTTCCTGACTAGGGG-3′) located within the PGK promoter in the neomycin-resistance cassette; reverse primer c (5′-TCAGAACCAAAAAAGGCTATAC-3′) annealing to the genomic sequences in 3′ arm homologous region. A 400-bp PCR fragment was generated from the wild-type allele with primers a and c, whereas an ∼300-bp fragment was generated from the targeted allele with primers b and c.
Histology analysis, skeleton staining, and in situ immunostaining
Newborn mice or embryos at E12.5, E13.5, E18, and E19 were harvested and fixed in 4% paraformaldehyde (PFA) at 4°C for 24 hr. Genotyping was performed on genomic DNAs from yac sacs or tails. Embryos were grossly examined and photographed before and after fixation. Fixed embryos were then dehydrated, paraffin-embedded, and serially sectioned at 7 μm. The selected sections were stained with hematoxylin and eosin for routine histologic examination. Cartilage and bone staining was performed as described (Martin et al. 1995).
Immunohistochemical staining for keratin 14 (Noco Castra), keratin 10 (BAbCO Berkeley Antibody Company), filaggrin (BAbCO Berkeley Antibody Company), and loricrin (BAbCO Berkeley Antibody Company) was performed on 4% PFA-fixed cryosections as recommended by the manufacturer. Briefly, fresh frozen slides were fixed in 4% PFA for 10 min, washed with PBS, and blocked for 15 min. Then, slides were incubated with primary antibodies for 1 hr at room temperature, washed three times with PBS buffer, and probed with FITC-conjugated goat anti-mouse (for keratin 14) or Cy3-conjugated goat anti-rabbit secondary antibody (for keratin 10, filaggrin, and loricrin).
RNA isolation, Northern blot analysis, and RNase protection
Cultured MEFs from IKK2+/+, IKK2+/−, and IKK2−/− embryos were stimulated with or without 10 ng/ml of human TNFα for 30 and 60 min. Cells were lysed in 2 ml of RNazol B buffer (Tel-Test, Inc., Friendswood, TX). Total RNA was prepared from MEFs following the manufacturer’s instruction (Tel-Test, Inc.). Ten micrograms of total RNA was fractionated on formaldehyde–agarose gels, blotted onto GeneScreen Plus membrane (Biotechnology Systems), and hybridized with a 32P-labeled probe from full-length IκBα cDNA. Quick Hyb (Stratagene, San Diego, CA) was used for Northern analysis. Prehybridization, hybridization, and washes were performed as recommended by the manufacturer (Stratagene).
RNase protection was performed by use of mCK-4 RiboQuant Multi-Probe Template Set (PharMingen, San Diego, CA), containing multiple probes including IL-3, IL-11, IL-7, GM-CSF, M-CSF, G-CSF, LIF, IL-6, SCF, L32, and GAPDH. Ten micrograms of total RNAs from IKK1+/+ and IKK1−/− MEFs treated with or without 10 ng/ml human TNFα for 1 hr were used. The RNase protection assay was conducted following the manufacturer’s protocol.
Western blot analysis, gel mobility shift assays, immunoprecipitation, and kinase assay
Forty micrograms of each extract was separated on a 10% SDS–polyacrylamide gel and transferred onto a nitrocellulose membrane, blocked in 1× PBST (PBS plus 0.2% Tween) and 5% milk powder for 15–30 min, washed twice in PBST, and probed further with primary antibodies to IKK1, or IKK2, or IκBα , or IκBβ (Santa Cruz Biotechnology) for 1–2 hr at room temperature. After several washes in PBST, the filters were incubated with a horseradish peroxidase-conjugated secondary antibody for 30–60 min in PBST, washed several times in PBST, and eventually developed with ECL kit (Amersham).
To assay effects of IKK1 inactivation on the NF-κB activation pathway, we subjected MEFs from all three genotypes to a variety of stimuli. MEFs at ∼90% confluency on a 10-cm plate were either untreated or treated with 10 ng/ml human TNFα (Calbiochem) or 2 ng/ml human IL-1α at the indicated times. After treatment, cells were washed with cold PBS, the cytoplasmic and nuclear extracts were prepared, and binding assays were performed as described (Miyamoto et al. 1994; Van Antwerp et al. 1996).
Whole-cell extracts from >95% confluent MEFs in 10-cm plates were used for immunoprecipitation with anti-NEMO. The NEMO antibody (anti-C-IKKAP1) raised against the synthetic peptide from the carboxyl terminus of human NEMO was a gift from Signal Pharmaceutical Inc. (San Diego, CA; Mercurio et al. 1999). Immunocomplexes precipitated from cell lysates with anti-NEMO antibody and then were eluted off the antibody by the synthetic peptide. NEMO immunocomplexes were subjected to an in vitro kinase assay as described (Mercurio et al. 1997). Equal amounts of NEMO immunocomplex from different MEFs were used for kinase assays with 3 μg of GST–IκBα(1–54) (amino acids 1–54), 1.5 μg of GST–IκBα(1–54)S/T (amino acids 1-54, Ser-32 → Thr-32 and Ser-36 → Thr-36), 2.5 μg of GST–IκBβ(1–44) (amino acids 1–44), 2.5 μg of GST–IκBβ(1–44)S/A (amino acids 1–44, Ser-19 → Ala-19 and Ser-23 → Ala-23), 1 μg GST–IκBα(1–317) (Santa Cruz Biotechnology), or 1 μg of p65/RelA as a substrate. Equal amounts of eluates were analyzed, and the amount of IKK1 and IKK2 proteins was determined by Western blot analysis with anti-IKK1 and anti-IKK2 antibodies.
Acknowledgments
We thank Yelena Marchuk and Bertha Dominguez for their excellent technical assistance, Frank Mercurio for IKK1 cDNA, GST–IκBα, GST–IκBβ and NEMO antibody, Steven Crone for helpful advice in ES technology, Daniel Van Antwerp and Wen Xie for discussion, Conchi Rodriguez Esteban for helping with skeleton staining, and Beth Coyne for her help with this manuscript. Q.L. is supported by a training grant from the National Institutes of Health (NIH); D.B. is supported by a fellowship from Deutsche Forschungsgemeinschaft, K.-F.L is a Pew Scholar and is supported by the NIH and the March of Dimes Foundation; J.C.I.B. is supported by NIH and Mathers Foundation grants; and I.M.V. is an American Cancer Society Professor of Molecular Biology and is supported by the Valley Foundation and grants from the NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL verma@salk.edu; FAX (619) 558-7454.
References
- Attar RM, Caamano J, Carrasco D, Iotsova V, Ishikawa H, Ryseck RP, Weih F, Bravo R. Genetic approaches to study Rel/NF-κB/I κB function in mice. Semin Cancer Biol. 1997;8:93–101. doi: 10.1006/scbi.1997.0060. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Baichwal VR. NF-κ B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol. 1997;65:111–137. [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-κ in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κ B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Bushdid PB, Brantley DM, Yull FE, Blaeuer GL, Hoffman LH, Niswander L, Kerr LD. Inhibition of NF-κB activity results in disruption of the apical ectodermal ridge and aberrant limb morphogenesis. Nature. 1998;392:615–618. doi: 10.1038/33435. [DOI] [PubMed] [Google Scholar]
- Cohen L, Henzel WJ, Baeuerle PA. IKAP is a scaffold protein of the IκB kinase complex. Nature. 1998;395:292–296. doi: 10.1038/26254. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-κB in osteoclast and B-cell development. Genes & Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- Kanegae Y, Tavares AT, Izpisua Belmonte JC, Verma IM. Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Nature. 1998;392:611–614. doi: 10.1038/33429. [DOI] [PubMed] [Google Scholar]
- Lee FS, Peters RT, Dang LC, Maniatis T. MEKK1 activates both IκB kinase α and IκB kinase β. Proc Natl Acad Sci. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Peet GW, Pullen SS, Schembri-King J, Warren TC, Marcu KB, Kehry MR, Barton R, Jakes S. Recombinant IκB kinases α and β are direct kinases of IκBα. J Biol Chem. 1998;273:30736–30741. doi: 10.1074/jbc.273.46.30736. [DOI] [PubMed] [Google Scholar]
- Li Q, Van Antwerp D, Mercurio F, Lee K-F, Verma IM. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes & Dev. 1995;9:1237–1249. doi: 10.1101/gad.9.10.1237. [DOI] [PubMed] [Google Scholar]
- Matsuki M, Yamashita F, Ishida-Yamamoto A, Yamada K, Kinoshita C, Fushiki S, Ueda E, Morishima Y, Tabata K, Yasuno H, Hashida M, Iizuka H, Ikawa M, Okabe M, Kondoh G, Kinoshita T, Takeda J, Yamanishi K. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase) Proc Natl Acad Sci. 1998;95:1044–1049. doi: 10.1073/pnas.95.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: Cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, Pascual G, Motiwala A, Zhu H, Mann M, Manning AM. IκB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Tumor necrosis factor α-induced phosphorylation of I κB α is a signal for its degradation but not dissociation from NF-κB. Proc Natl Acad Sci. 1994;91:12740–12744. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Roop D. Defects in the barrier. Science. 1995;267:474–475. doi: 10.1126/science.7529942. [DOI] [PubMed] [Google Scholar]
- Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc Natl Acad Sci. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/I κB family: Intimate tales of association and dissociation. Genes & Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- Xie W, Wu X, Chow LT, Chin E, Paterson AJ, Kudlow JE. Targeted expression of activated erbB-2 to the epidermis of transgenic mice elicits striking developmental abnormalities in the epidermis and hair follicles. Cell Growth Differ. 1998;9:313–325. [PubMed] [Google Scholar]
- Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Zandi E, Chen Y, Karin M. Direct phosphorylation of IκB by IKKα and IKKβ: Discrimination between free and NF-κB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]