

Abstract
Thyroid hormone (T3) has widespread functions in development and homeostasis, although the receptor pathways by which this diversity arises are unclear. Deletion of the T3 receptors TRα1 or TRβ individually reveals only a small proportion of the phenotypes that arise in hypothyroidism, implying that additional pathways must exist. Here, we demonstrate that mice lacking both TRα1 and TRβ (TRα1−/−β−/−) display a novel array of phenotypes not found in single receptor-deficient mice, including an extremely hyperactive pituitary–thyroid axis, poor female fertility and retarded growth and bone maturation. These results establish that major T3 actions are mediated by common pathways in which TRα1 and TRβ cooperate with or substitute for each other. Thus, varying the balance of use of TRα1 and TRβ individually or in combination facilitates control of an extended spectrum of T3 actions. There was no evidence for any previously unidentified T3 receptors in TRα1−/−β−/− mouse tissues. Compared to the debilitating symptoms of severe hypothyroidism, the milder overall phenotype of TRα1−/−β−/− mice, lacking all known T3 receptors, indicates divergent consequences for hormone versus receptor deficiency. These distinctions suggest that T3-independent actions of T3 receptors, demonstrated previously in vitro, may be a significant function in vivo.
Keywords: Thyroid hormone receptor, pituitary, thyroid, growth, bone development
Thyroid hormone (T3) exerts profound control over many functions in development and homeostasis. It is therefore an intriguing challenge to elucidate the T3 receptor (TR) pathways that mediate the diverse responses to this hormone. TRs are ligand-dependent transcription factors (Sap et al. 1986; Weinberger et al. 1986) that belong to the family of nuclear hormone receptors (Mangelsdorf et al. 1995). The presence across vertebrate species of two distinct genes that encode the related receptors TRα1 and TRβ indicates that both TR genes are fundamental components of the T3-signaling pathway. TRα1 and TRβ are expressed in distinct but often overlapping patterns, suggesting that they may mediate both individual and common functions (Lazar 1993; Forrest 1994).
TRα1 and TRβ possess related T3 binding and DNA-binding domains but have divergent amino termini that influence their DNA-binding and transactivation properties. In mammals, the TRα gene encodes a T3 receptor, TRα1, and a splice variant, TRα2, that does not bind T3 and differs from TRα1 in its ability to bind DNA and to heterodimerize with retinoid X receptors (RXRs; Reginato et al. 1996). The role of TRα2 is unclear, although it fails to transactivate and may interfere with transactivation by T3 receptors in transfection assays (Izumo and Mahdavi 1988; Koenig et al. 1989; Katz et al. 1995). The TRβ gene encodes two amino-terminal variants, TRβ1 and TRβ2, both of which bind T3 and transactivate (Hodin et al. 1989; Ng et al. 1995; Sjöberg and Vennström 1995). In vitro, TRα1 and TRβ can transactivate through the same T3 response elements, albeit with subtle differences (Sjöberg and Vennström 1995; Zhu et al. 1997), suggesting that they have the potential to regulate common functions. In vitro, in the absence of T3, TRs may still bind to T3 response elements to repress basal transcription (Damm et al. 1989; Brent et al. 1991; Fondell et al. 1993), suggesting that TRs possess a dual capacity to regulate transcription by both T3-dependent and -independent means.
The derivation of TR-deficient mice has indicated specific in vivo roles for TRα1 and TRβ. TRβ−/− mice, lacking both TRβ1 and TRβ2, overproduce thyroid hormones and thyrotropin (TSH), the pituitary hormone that stimulates thyroid activity (Forrest et al. 1996a). Normally, TSH is suppressed by elevated thyroid hormone levels, such that the TRβ−/− phenotype indicates a key role for TRβ in this feedback control of the pituitary–thyroid axis. Also, TRβ is essential for auditory function (Forrest et al. 1996b; Rüsch et al. 1998). TRβ−/− mice are a model for human resistance to thyroid hormone that is associated with TRβ mutations (Refetoff et al. 1993). TRα1−/− mice (which retain the non-T3 binding TRα2 variant), but not TRβ−/− mice, have a reduced heart rate and low body temperature (Johansson et al. 1998; Wikström et al. 1998). A distinct TRα mutation, deleting both TRα1 and TRα2, results in runted mice with gut malformation, small thyroid glands, and hypothyroidism, which die after weaning (Fraichard et al. 1997). This mutation causes a more severe phenotype than loss of TRα1 (Wikström et al. 1998), but for reasons that remain to be determined.
In view of the many functions of T3, the phenotypes of the single T3 receptor-deficient mice are remarkably limited. For example, congenital hypothyroidism retards growth severely in rodents and humans (Ohlsson et al. 1993; Snyder 1996), whereas there is no overt growth deficiency in TRα1−/− or TRβ−/− mice (Forrest et al. 1996a; Wikström et al. 1998). This raises a fundamental question concerning the receptor pathways that account for the remaining actions of T3. Although cDNA cloning studies have identified only TRα1 and TRβ, other unknown, distantly related receptors could conceivably play a role. Alternatively, TRα1 and TRβ may mediate common functions that compensate, to an extent, for each other’s absence. To discriminate between these possibilities, we derived mice lacking both TRα1 and TRβ, with the objective of removing all T3 receptors from such putative common pathways. The results indicate that TRα1 and TRβ act through common pathways to account for an extended range of T3 functions, including female fertility, control of the pituitary–thyroid axis, and bone development. However, compared to hypothyroid mice, the surprisingly milder phenotype and vitality of TRα1−/−β−/− mice suggests a distinction between thyroid hormone versus TR deficiency. This may point to a physiological significance for T3-independent TR functions that have been described previously in vitro.
Results
Derivation of mice deficient for all known T3 receptors
TRα1−/− and TRβ−/− (Forrest et al. 1996a; Wikström et al. 1998) mouse strains were crossed to yield progeny heterozygous for both mutations, which were then intercrossed to generate litters that included wild-type and doubly homozygous mutant (TRα1−/−β−/−) progeny on the same genetic background. Most TRα1−/−β−/− mice were viable, indicating that the T3 receptors are not required for life.
To ascertain that TRα1−/−β−/− mice lacked TRα1 and TRβ proteins, gel mobility-shift experiments were performed with nuclear extracts from tissues of wild-type, TRα1−/−, TRβ−/−, and TRα1−/−β−/− mice. Depending on the tissue source, two general patterns were detected. In wild-type liver extracts, three TR-containing bands that specifically bound the F2 T3 response element were detected (Fig. 1A). TRα1−/− extracts lacked the lower one and TRβ−/− extracts lacked the upper two of these bands, whereas TRα1−/−β−/− extracts lacked all three bands. These bands represented TR/RXR heterodimers, as antibodies against TR or RXR proteins abrogated or supershifted all three bands. A different pattern was detected in most other tissues, where in addition to TR-specific bands, other non-TR-containing bands were detected, indicating that these tissues expressed other factors that bound the T3 response element. In these tissues (e.g., lung, Fig. 1B), TRα1−/−β−/− extracts again lacked all specific TR-containing bands that were present in corresponding wild-type extracts. Similar results were obtained for kidney, spleen, heart, and brain (not shown). These results were confirmed using an independent (direct repeat 4) T3 response element (not shown). Thus, neither TRα1, TRβ, nor other previously unidentified TR proteins could be detected in a range of tissues from TRα1−/−β−/− mice.
Figure 1.
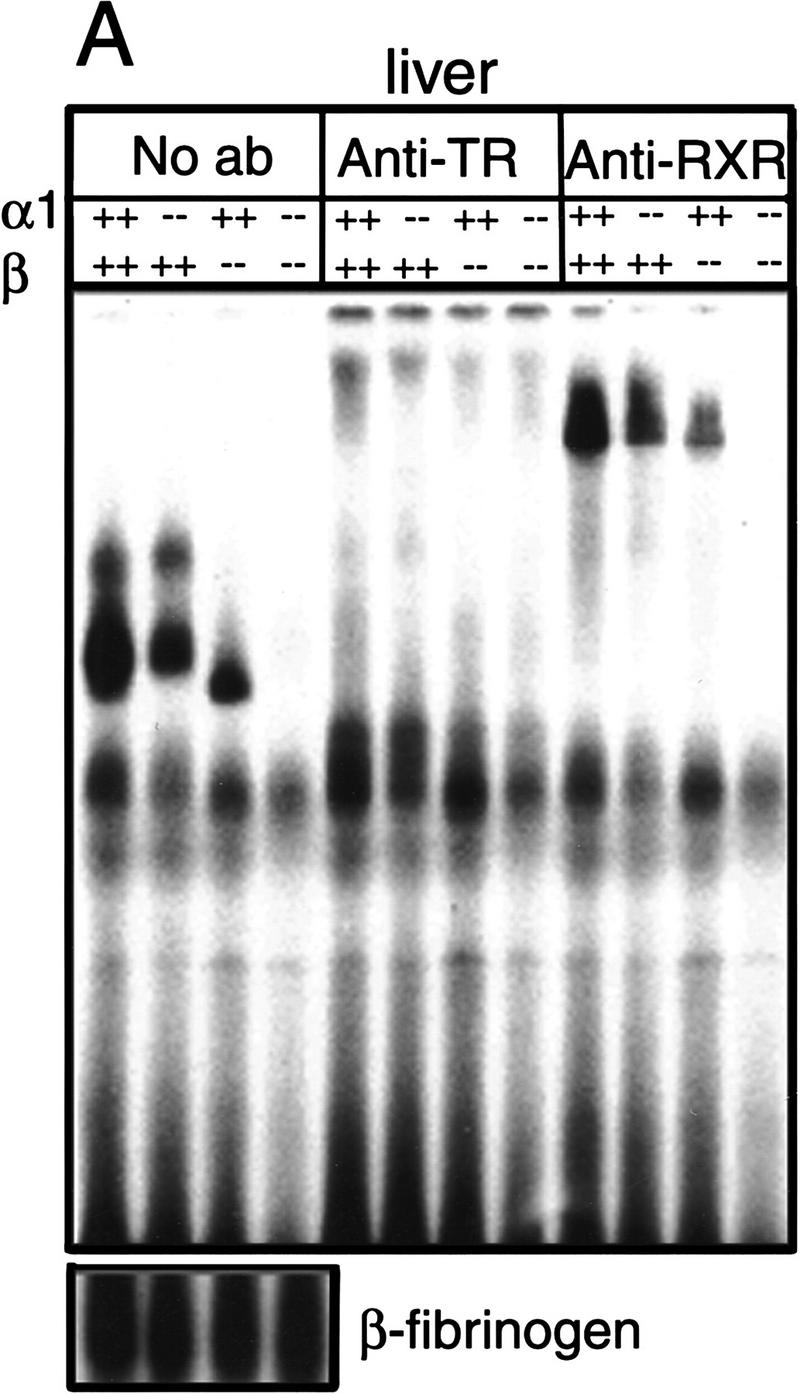
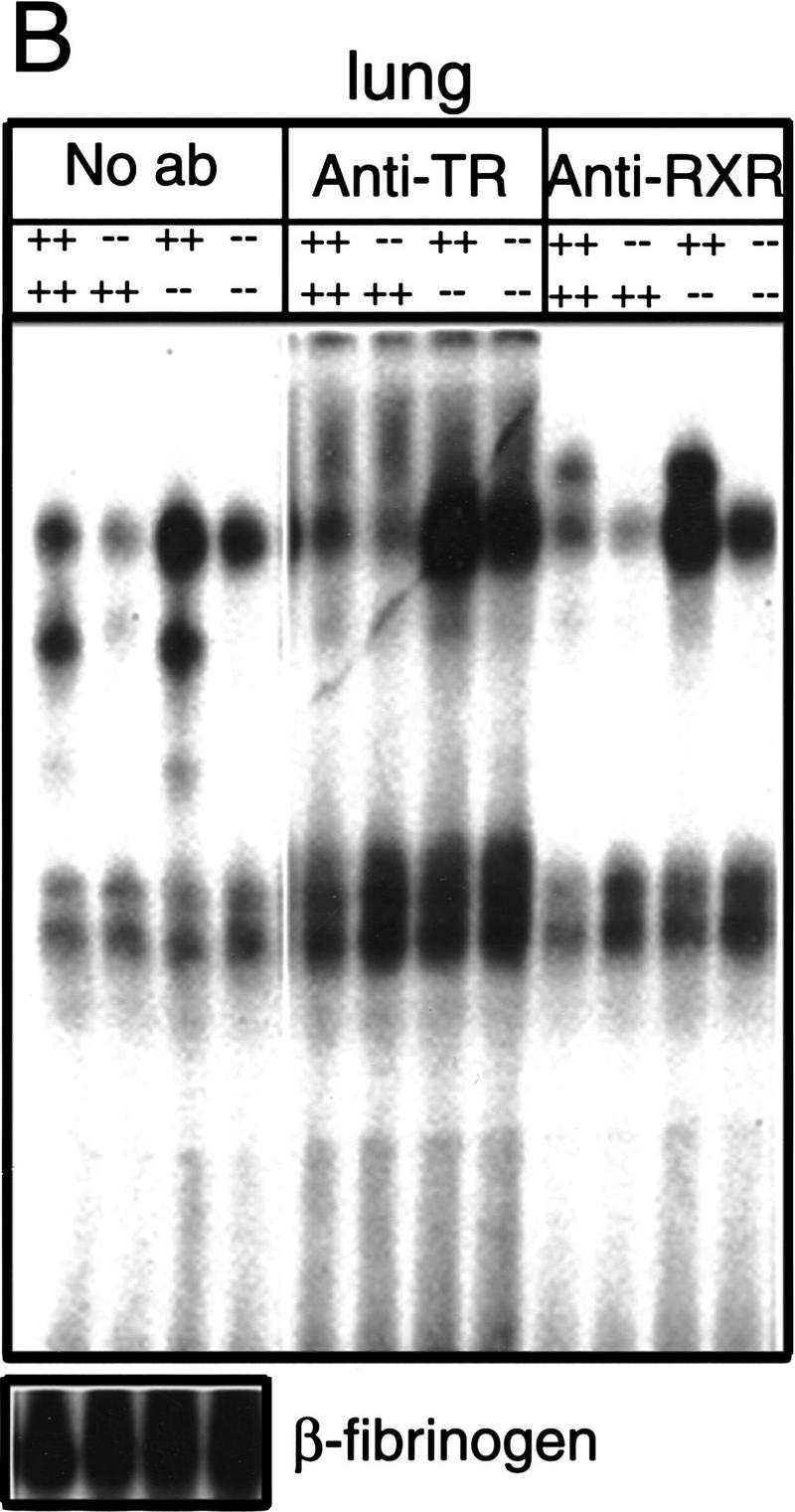
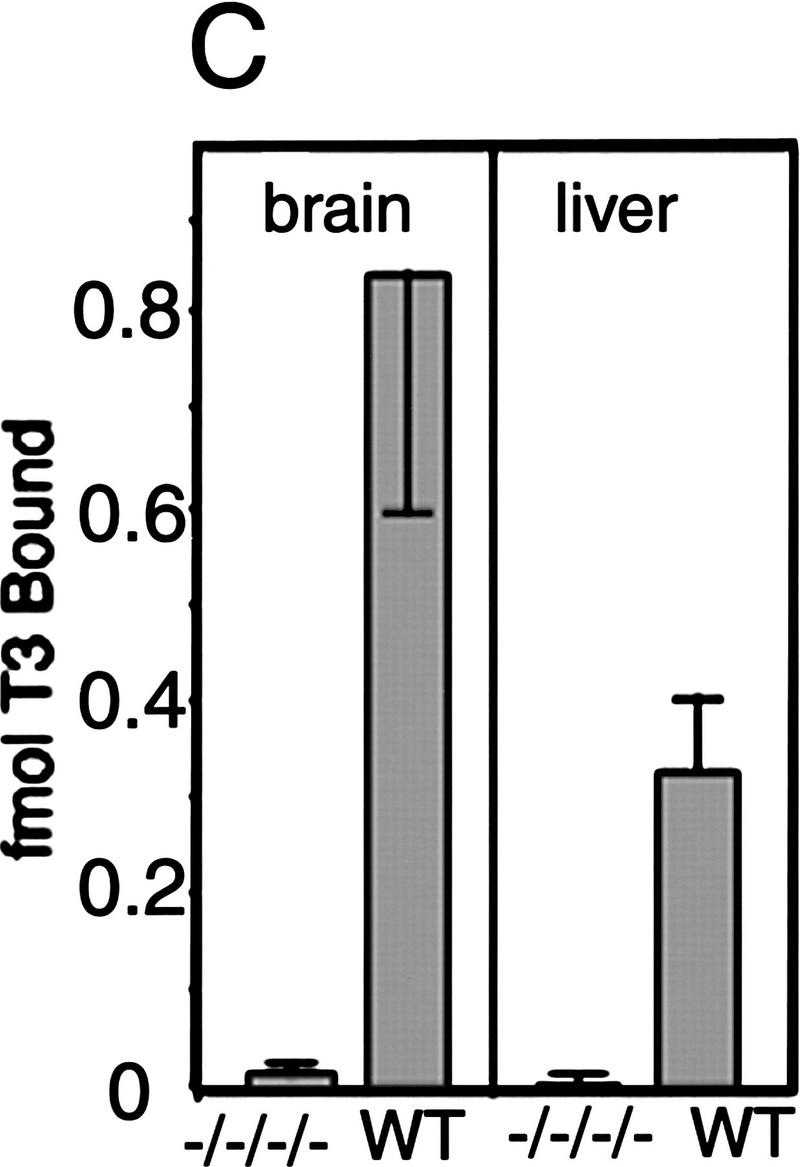
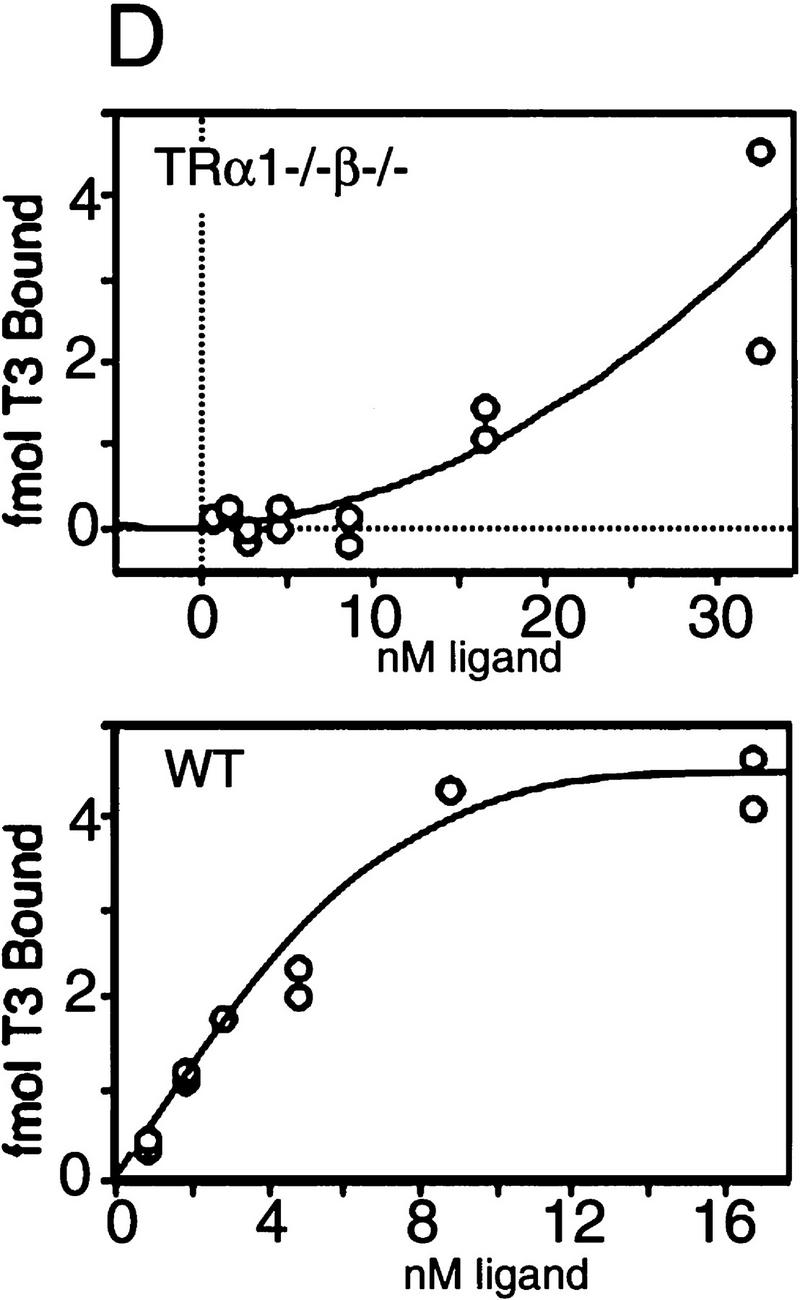
Absence of detectable T3 receptors in nuclear extracts from TRα1−/−β−/− mouse tissues. (A,B) Gel mobility shift experiments showing absence of TR proteins that can bind the F2 T3 response element in nuclear extracts from liver (A) and lung (B) of TRα1−/−β−/− mice. Specific TR-containing bands were identified by addition of antibodies against TR (anti-TR) or RXR (anti-RXR); (No ab) No antibodies added. Parallel analysis of samples with a β-fibrinogen probe demonstrated the integrity of other DNA-binding proteins in TR-deficient samples. (C) Absence of a specific T3 binding capacity in nuclear extracts from brain and liver of TRα1−/−β−/− mice. The 125I-labeled T3 binding capacity of TRα1−/−β−/− and wild-type samples is shown after subtraction of the binding that was not competed by a 1000-fold excess of cold T3 (Torresani and DeGroot 1975). (D) Absence of a saturable, specific T3 binding capacity in nuclear extracts from TRα1−/−β−/− mice. Nuclear extracts were prepared from mice that had been made hypothyroid to exclude the possibility that residual T3 binding sites were present but were masked by prior saturation by the excessive T3 levels in TRα1−/−β−/− mice (see Fig. 2A).
Ligand binding experiments were performed with nuclear extracts from tissues of TRα1−/−β−/− mice to determine the extent to which T3 binding was abrogated in the absence of both TRα1 and TRβ. Nuclear extracts from brain and liver of TRα1−/−β−/− mice failed to bind 125I-labeled T3 above background levels (Fig. 1C), whereas extracts from wild-type brain and liver specifically bound 0.8 and 0.3 fmoles of T3/μg of nuclear extract, respectively. This suggested the absence of T3 binding activity in TRα1−/−β−/− mice. However, it was conceivable that their high serum levels of thyroid hormones (see below) could have saturated any remaining receptors. Therefore, TRα1−/−β−/− and wild-type groups were made hypothyroid by treatment for 6 weeks with methimazole (MMI) and potassium perchlorate (Solé et al. 1996) while on a low iodine diet, resulting in free T3 levels of <1 pM (normal wild-type male range, 6–9 pM; see Fig. 2, below). Nuclear extracts were then tested for T3 binding. Again, in contrast to wild-type extracts, no nuclear T3 binding activity was detectable in liver or brain from TRα1−/−β−/− mice. In a saturation binding experiment with liver nuclear extracts from the hypothyroid groups, there was no evidence of high affinity, low capacity binding activity in TRα1−/−β−/− mice compared to the wild-type control extract (Fig. 1D). Thus, nuclear extracts from TRα1−/−β−/− mouse tissues contained no detectable T3 binding capacity nor TR proteins. There have been reports of transient, minor non-nuclear responses to thyroid hormone (Davis and Davis 1996) that would presumably remain intact in TRα1−/− β−/− mice. In conclusion, although the results cannot entirely exclude the existence of hypothetical unidentified TRs, for example, that may be present in certain cell populations, the data demonstrate that tissues that are major targets for T3 action in TRα1−/−β−/− mice were devoid of nuclear T3 receptors.
Figure 2.
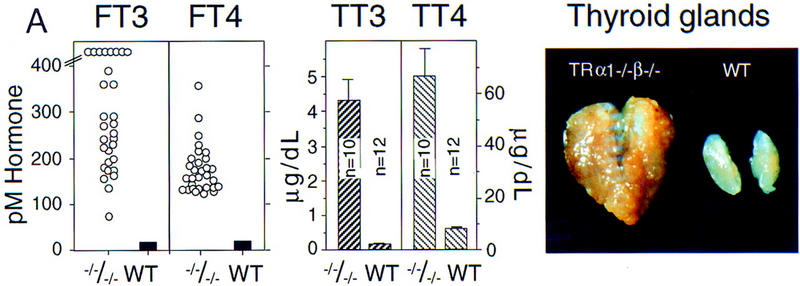
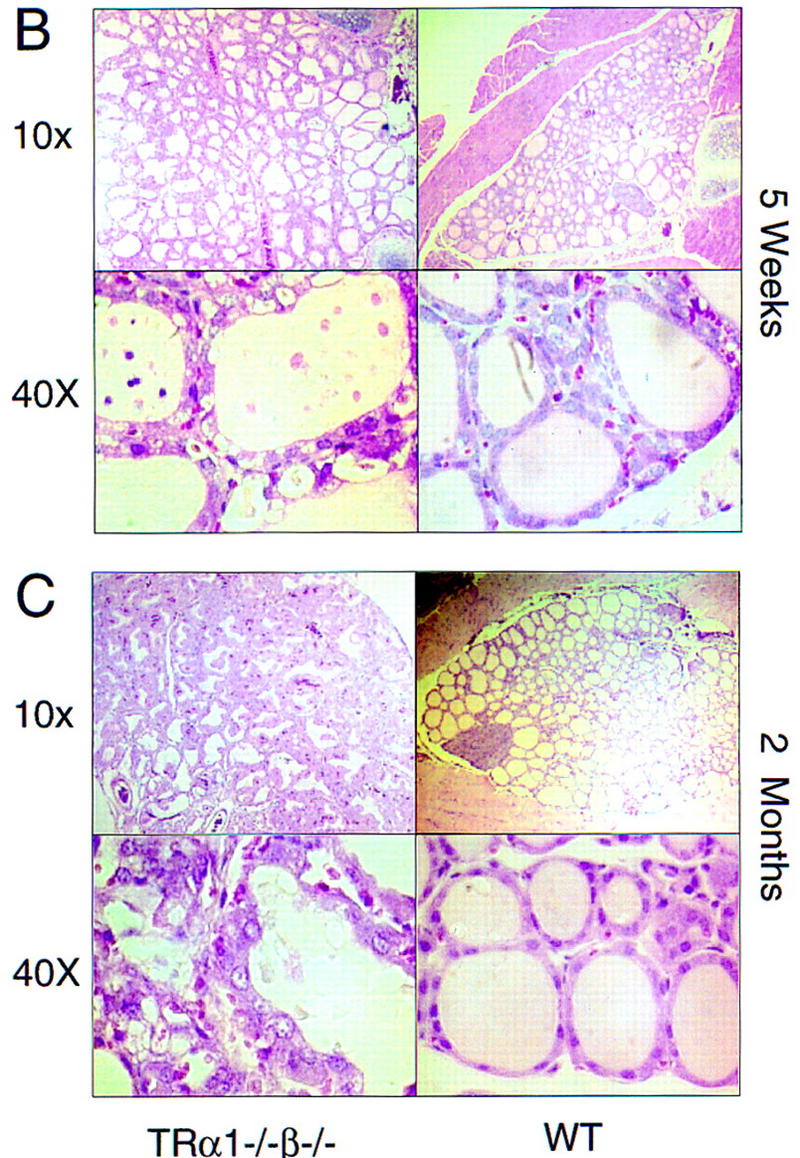
Hyperactive goiter in TRα1−/−β−/− mice. (A) Elevated serum levels of thyroid hormones and goiter in adult TRα1−/−β−/− mice. Free (FT3, FT4) and total (TT3, TT4) levels of thyroid hormones were elevated in TRα1−/−β−/− mice. Values are shown for males; females gave similar results. (Right) The grossly enlarged thyroid gland dissected from an 8-week-old TRα1−/−β−/− mouse. (B,C) Progressive pathological changes in the thyroid glands of TRα1−/−β−/− mice (left) compared to wild-type mice (right) at 5 (B) and 8 (C) weeks. Sections were stained with hematoxylin and eosin. The enlarged follicles often contained degenerating cellular material in the colloid. Hyperproliferation progressed considerably by 8 weeks.
Fertility and survival
TRα1−/−β−/− males were fertile but TRα1−/−β−/− females rarely became pregnant and were deficient in nurturing any pups born. Reduced fertility was not observed in TRα1−/− or TRβ−/− mice (Forrest et al. 1996a; Wikström et al. 1998), indicating that female reproductive processes depend on common pathways regulated by both TRα1 and TRβ. This female-biased deficiency is consistent with observations that hypothyroidism impairs reproductive function to a greater extent in females than in males (Longcope 1996). In view of this deficiency, mice for other analyses were generated by crossing TRα1−/−β−/− males with TRα1−/−β+/− females.
Mean litter sizes derived from matings of TRα1−/−β−/− males × TRα1−/−β+/− females (mean ± s.e.m.: 5.9 pups ± 0.5, n = 52) were not significantly smaller than litters from wild-type male × wild-type female matings (5.9 pups ± 0.6, n = 30). Litters (n = 40) from TRα1−/−β−/− male × TRα1−/− β+/− female matings yielded 98 TRα1−/−β−/− pups and 144 TRα1−/−β−/+ pups (40% and 60%, respectively) with TRα1−/−β−/− pups evenly divided between both sexes. The 10% reduction below the predicted 50% Mendelian frequency was significant (P < 0.01), indicating a reduced rate of survival either during prenatal or early postnatal stages of development. Both male and female TRα1−/−β−/− mice were typically viable for at least 12 months, although they had an increased mortality rate with 30% (13/43) dying before 9 months of age, contrasting with the zero mortality rate of wild-type animals (0/90) over the same period.
These results suggest that in a minority of the TRα1−/− β−/− population, functions required for normal survival are compromised. Other than the defects described below in the pituitary–thyroid axis and in bone, no gross pathological changes were observed in TRα1−/−β−/− mice that would explain this mortality. This suggested the involvement of a subtle defect of partial penetrance. Alternatively, several different defects may be implicated, as the mortality was sporadic throughout the life span of TRα1−/−β−/− mice. Increased mortality was not evident in TRα1−/− or TRβ−/− mice (Forrest et al. 1996a; Wikström et al. 1998), indicating that these survival functions depend on pathways common to both TRα1 and TRβ.
Dysfunction of the pituitary–thyroid axis
Thyroid hormone production is strictly regulated by a feedback mechanism whereby pituitary thyroid-stimulating hormone (TSH) is suppressed by elevated thyroid hormone levels. To assess the combined function of TRα1 and TRβ in this regulation of the pituitary–thyroid axis, serum levels of thyroid hormones were determined in TRα1−/−β−/− mice (Fig. 2A). Free T3 (FT3) levels were 20- to 60-fold higher in TRα1−/−β−/− mice compared to wild-type mice. Free T4 (FT4) levels were similarly elevated, whereas total T3 and T4 levels were ∼30- and 11-fold elevated, respectively, over wild-type values. Similar results were recorded in both male and female mice.
The excessive thyroid hormone levels in TRα1−/−β−/− mice were accompanied by pronounced enlargement of the thyroid gland (Fig. 2A), which weighed 15-fold more than in wild-type mice (mean wet weights ± s.e.m.: 97.0 ± 18.1 mg and 6.4 ± 0.3 mg, respectively; n = 6/group). Histological analysis of adults at 2 months of age revealed extensive hyperproliferation of the follicular epithelial cells (Fig. 2C). The colloid in the residual follicles was vacuolated and in a degenerative state. At later ages studied (8 and 12 months) the gland structure had further deteriorated with disorganized and often vacuolated epithelial cell layers. Abnormalities were present in TRα1−/−β−/− mice from birth when the thyroid gland, although not increased in size, already contained enlarged follicles. By 5 weeks of age the gland was enlarged and contained numerous large follicles with degenerating cellular material in the colloid (Fig. 2B). Thus, hyperproliferation of the thyroid gland from early stages led rapidly to goiter in the young adult. With age, the hyperplasia worsened and was accompanied by pronounced vascularization.
Despite the elevated T4 and T3 levels, serum TSH was not suppressed but was, in contrast, highly elevated. Figure 3A shows ∼60-fold elevated levels in representative male TRα1−/−β−/− mice at 5 months of age (mean ± s.e.m.: 6959 ± 882 vs. 114 ± 3.6 ng/ml in TRα1−/−β−/− and wild-type mice, respectively; P < 0.001). Up to 160-fold elevated levels were recorded in younger mice between 4 and 8 weeks of age (e.g., 20,259 ± 1450 vs. 124 ± 2.7 ng/ml in TRα1−/−β−/− and wild-type mice, respectively, at 4 weeks of age; P < 0.001; Fig. 3D). Thus, values declined slightly with age but still remained extremely high (>30-fold elevated) even in TRα1−/−β−/− mice at ∼1 year of age. Northern blot analysis revealed that levels of mRNA for the TSHα and TSHβ subunits were increased 3.3- and 26.0-fold, respectively, in pituitaries of TRα1−/−β−/− adult male mice compared to wild-type mice. In contrast, TRα1−/− and TRβ−/− mice exhibited comparatively marginal (≤3-fold) alterations of TSHα and TSHβ mRNA levels (Forrest et al. 1996a; Wikström et al. 1998). Thus, in TRα1−/−β−/− mice, the defect in TSHβ mRNA and serum TSH levels is exacerbated, suggesting a highly interactive role for TRα1 and TRβ in the negative control of TSH production. The greater elevation of TSHβ mRNA is consistent with TSHβ being more sensitive than TSHα to T3 regulation (Shupnik et al. 1985).
Figure 3.
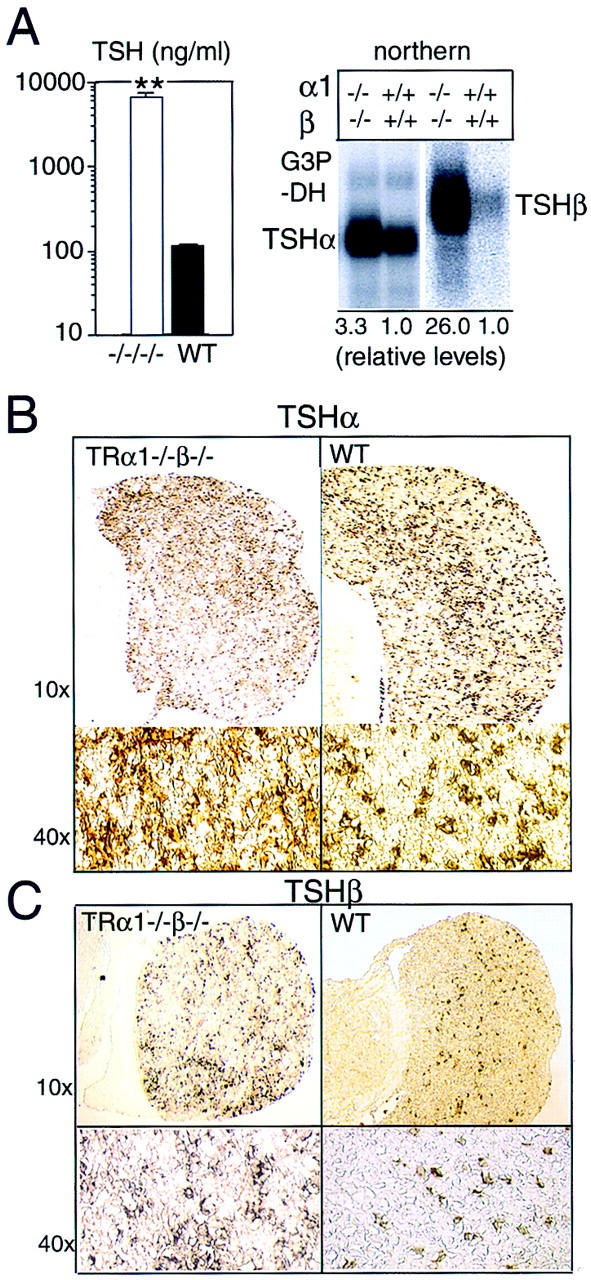

Overproduction of TSH in TRα1−/−β−/− mice. (A) Serum TSH levels were 60-fold elevated (left) in 5-month-old adult male mice, and the pituitary glands contained increased levels of TSHα and TSHβ subunit mRNAs (right). As a control for the amount and integrity of RNA loaded, the same Northern blot was hybridized with a probe for G3PDH, as shown in the TSHα lanes. (B,C) Immunostaining showed increased numbers of TSHα-positive cells (1.9-fold; B) and TSHβ-positive cells (2.8-fold; C) in the anterior lobe of the pituitary gland. Cell counts were determined for n = 2 wild-type and n = 3 TRα1−/−β−/− female mice at 5 weeks of age. (D) Resistance of TSH levels to change under MMI-induced hypothyroid conditions. Serum TSH levels were sequentially determined in groups of n = 4 wild-type or TRα1−/−β−/− male mice (4 weeks old) provided with normal diet (ND), low iodine diet with MMI/perchlorate in drinking water for 4 weeks (MMI), and finally under MMI with the addition of 5.0 μg/ml L-T3 in the drinking water for 8 days (MMI+T3). MMI resulted in hypothyroidism (T4 < 0.4 μg/dl) in both groups. MMI+T3 resulted in serum total T3 levels of 9440 ± 2110 and 14,937 ± 1,347 ng/dl, respectively in wild-type and TRα1−/−β−/− mice (116- and 5.5-fold elevated above the respective levels under normal diet). One wild-type mouse died under MMI+T3 conditions. TSH detection limit, 25 ng/ml.
Pituitary gland morphology
The pituitary gland in TRα1−/−β−/− mice, although overproducing TSH, was not grossly malformed or enlarged over a range of postnatal stages examined, even in mice at 1 year of age. Histological analysis revealed a somewhat disorganized cellular structure with some irregularly shaped and vacuolated cells. Immunostaining showed a 1.9-fold increase in numbers of cells positive for the TSHα subunit in TRα1−/−β−/− versus wild-type mice (27.8 ± 1.8 vs. 14.8 ± 0.8, respectively; mean percent ± s.e.m. of positively staining cells/total cells; P < 0.001) (Fig. 3B). Numbers of TSHβ subunit-positive cells were 2.8-fold increased and showed a normal distribution in TRα1−/−β−/− mice compared to wildtype mice (22.7 ± 1.9 vs. 8.2 ± 0.7, respectively; mean percent ± s.e.m. of positively staining cells/total cells; P < 0.001) (Fig. 3C). Thus, the overproduction of TSH in TRα1−/−β−/− mice correlated with a moderate increase in numbers of TSH-containing cells but was not due to a pituitary tumour or malformation. These results suggested that the thyrotropes, although not grossly abnormal, were in a constitutively hyperactive state.
Response to hypothyroidism
To investigate if the excessive thyroid hormone levels, acting through hypothetical residual T3 receptors, contributed to the phenotype of TRα1−/−β−/− mice, groups of wild-type and TRα1−/−β−/− male mice (n = 4/group) were made hypothyroid using low iodine diets and MMI/perchlorate treatment for 4 weeks. This reduced levels of total T4 in both wild-type and TRα1−/−β−/− mice from 9.5 and 48.2 μg/dl, respectively, to <0.8 μg/dl; total T3 levels were reduced from 100 (wild-type) and 3600 (TRα1−/−β−/−) to <16 ng/dl. Figure 3D shows that this resulted in 161-fold increased TSH levels in wild-type mice from 124 ± 5 to 20,064 ± 1665 ng/ml; P < 0.01. In contrast, in TRα1−/−β−/− mice, TSH which was already at 20,259 ± 1450 ng/ml, changed insignificantly to 24,432 ± 1752 ng/ml (P > 0.05). This indicated that in the absence of T3 receptors, TSH production was already maximal in TRα1−/−β−/− mice, thus precluding its further elevation.
TRα1−/−β−/− mice survived MMI-induced hypothyroid conditions indicating that thyroid hormones are not essential for their viability. Furthermore, a significant 7.8% loss of weight was observed in adult male wild-type mice during 3 weeks of MMI treatment with mean weights ± s.d. decreasing from 35.9 ± 2.5 to 33.1 ± 1.9 grams after 3 weeks (P < 0.001). In contrast, significant weight loss was not observed in the already smaller TRα1−/−β−/− mice (starting and ending weights during 3 weeks of MMI treatment: 22.9 ± 1.4 to 22.0 ± 1.3 grams, respectively; P > 0.05). These results suggest that TRα1−/− β−/− mice are largely impervious to the actions of thyroid hormone, regardless of hormone levels, suggesting that TRα1 and TRβ together provide the principal means for nuclear T3 responses. In conclusion, neither the existing great excess of thyroid hormones nor MMI-induced hypothyroidism produced any significant responses, indicating an absence of detectable T3 receptor functions, whether T3-dependent or T3-independent, in TRα1−/− β−/− mice.
Decreased body growth and deficiency of growth-promoting hormones
Both male and female TRα1−/−β−/− mice were smaller than wild-type mice at all ages examined. Newborn TRα1−/−β−/− pups weighed 11% less than wild-type pups (mean ± s.d.: TRα1−/−β−/−, 1.35 ± 0.05 gram, n = 10; wild-type, 1.52 ± 0.12 gram, n = 9; P < 0.001). TRα1−/−β−/− pups also displayed a retarded growth spurt during the first 3 postnatal weeks (Fig. 4A). By weaning, these mice were distinctly smaller (P < 0.001) than TRα1−/−β−/+ littermates, indicating that the growth deficiency was intrinsic to the TRα1−/−β−/− genotype and was not due to maternal factors. Size and weight gain remained chronically retarded beyond weaning with TRα1−/−β−/− adults being ∼30% lighter than wild-type mice (e.g., mean weight ± s.d. for TRα1−/−β−/− and wild-type groups at 15–18 weeks of age: 20.2 ± 3.7 vs. 27.9 ± 1.7 grams; n = 40 and 12, respectively; P < 0.0001).
Figure 4.
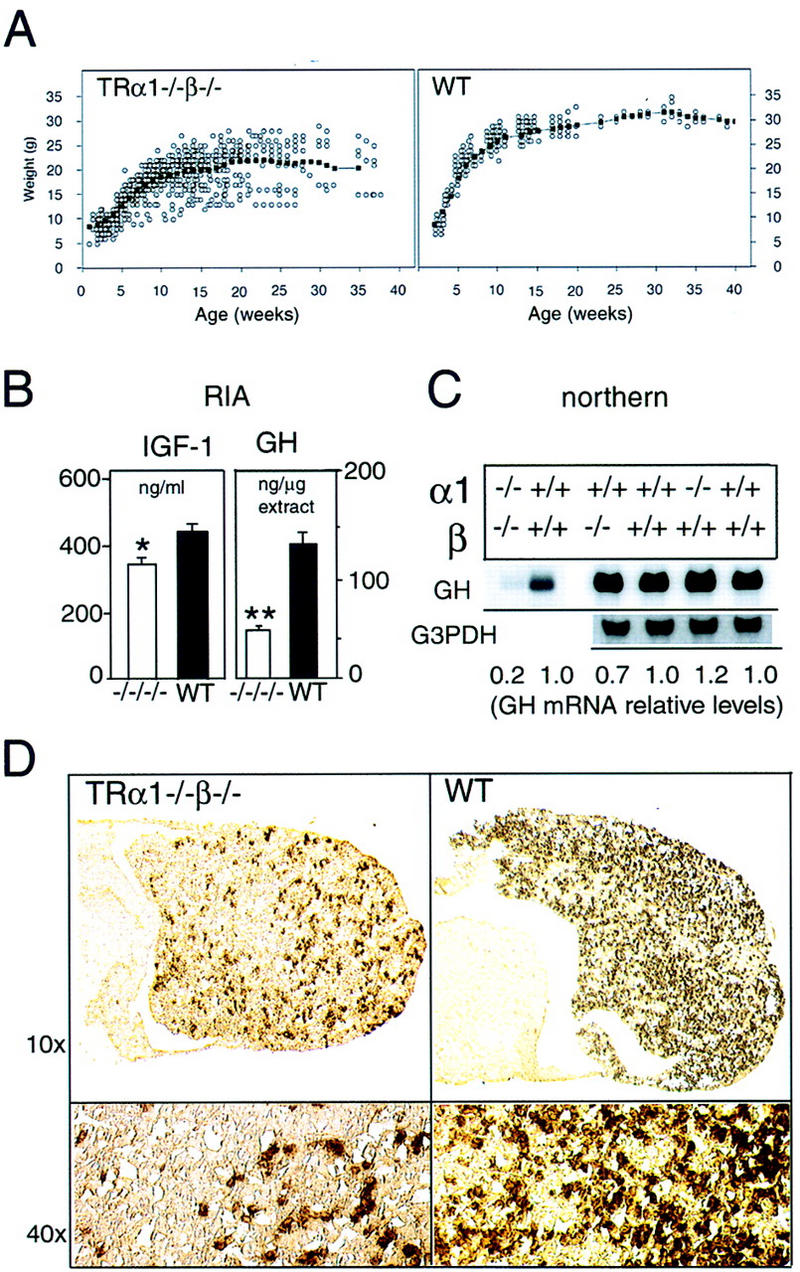

Retarded growth and reduced expression of IGF-I and GH in TRα1−/−β−/− mice. (A) Weight gain of TRα1−/−β−/− and wild-type mice measured over the first 40 postnatal weeks. The average weight gain curve, determined as a moving three-point average, is shown by the linked, black points. (B) Reduced serum levels of IGF-I and of pituitary GH content in TRα1−/−β−/− mice, determined by radioimmunoassay. (C) Northern blot analysis of GH mRNA in TRα1−/−β−/−, TRα1−/−, and TRβ−/− mice. Adult male samples are shown alongside wild-type samples of the corresponding genetic background. Quantitation was determined by PhosphorImager with values normalized to G3PDH signals; for TRα1−/−β−/− mice, the same Northern blot was used as in Fig. 3A; thus, normalization was to the G3PDH bands shown in Fig. 3A. (D) Immunostaining of pituitary gland sections showed 4.5-fold reduced numbers of GH-positive cells in the anterior lobe, determined on the same pituitaries as for TSH immunostaining in Fig. 3. (E) Groups of adult male mice were pretreated on low iodine diet for 3 weeks and were then supplemented with MMI/perchlorate (LID/MMI) in the drinking water for 3 weeks, starting at 0 weeks. Over 3 weeks, weight decreased significantly in wild-type but not TRα1−/−β−/− mice. (*) P < 0.01 vs. weight at time 0 weeks. MMI treatment resulted in free T3 levels ≤ 2.6 pM and free T4 ≤ 3.5 pM in both groups (normal wild-type levels are 6–9 pM for FT3 and 12–16 pM for FT4).
It is known that thyroid hormone can regulate growth indirectly by stimulation of growth hormone (GH) and that the growth-promoting actions of GH occur in part by stimulation of insulin-like growth factor-I (IGF-I) synthesis (Ohlsson et al. 1998). Therefore, levels of GH and IGF-1 were determined. Figure 4B shows that serum levels of IGF-I were reduced significantly by 21% (P = 0.013) in TRα1−/−β−/− mice. In the pituitary, the content of GH protein was 2.7-fold lower (P < 0.01) and levels of GH mRNA were 5-fold lower in TRα1−/−β−/− than in wild-type mice (Fig. 4B,C). In contrast, GH mRNA levels were not altered significantly in TRα1−/− or TRβ−/− mice (Fig. 4C). Immunostaining of anterior pituitary sections revealed a 4.5-fold decrease in numbers of GHpositive cells compared to wild-type mice (9.0 ± 0.4 and 41.0 ± 2.5, respectively; mean percent ± s.e.m. of positively staining cells per total cells; P < 0.001) (Fig. 4D). Similar reductions in GH levels and GH-positive cell numbers occurred in both male and female TRα1−/−β−/− mice. These results implicate GH deficiency as a proximal cause of retarded growth in TRα1−/−β−/− mice.
Retardation of bone development and mineralization
The growth-retardation reflected a pronounced decrease in bone lengths. For newborn TRα1−/−β−/− mice this is illustrated by the significantly decreased lengths of femur, tibia, and the sixth lumbar vertebra (L6) as shown in Table 1. Decreased bone lengths persisted into adulthood, as shown by representative measurements of femur, tibia, and L6. Adult TRα1−/−β−/− mice at ∼20 weeks of age showed a disproportional growth retardation of long bones with the most marked retardation in the femur. Histological examination revealed disorganized growth plates and a delayed ossification of the epiphysis (Fig. 5). In the epiphysis of 5-week-old TRα1−/−β−/− mice, the cartilage area was increased, whereas the bone area was decreased compared to wild-type mice (cartilage areas for TRα1−/−β−/− vs. wild-type, 61.0 ± 9.8% vs. 12.0 ± 5.0%, respectively; bone areas, 19.0 ± 4.4 vs. 38.0 ± 0.5%, respectively; n = 3 per group). Adult TRα1−/− β−/− mice also had several large areas of cartilage in the epiphysis, whereas no cartilage was found in the epiphysis of adult wild-type mice (Fig. 5B). These defects resembled those caused by hypothyroidism, although in contrast to hypothyroidism, in which the width of the proximal growth plate in the tibia is decreased (Lewinson et al. 1989), this dimension was increased in TRα1−/−β−/− mice. The reason for the disorganization in the growth plates in TRα1−/−β−/− mice is presently being investigated in further detail. In 5-week-old wild-type and TRα1−/−β−/− mice, the width of the tibial growth plate was 258 ± 8 and 348 ± 20 μm, respectively, and in adult mice, it was 111 ± 7 and 141 ± 5 μm, respectively.
Table 1.
Dimensions of bones in TRα1−/−β−/− mice
|
|
|
mm
|
Wild type
|
TRα1−/−β−/−
|
Difference (%)a
|
|---|---|---|---|---|---|
| Newborns | femur | length | 4.10 ± 0.04 | 3.66 ± 0.04 | −11.0** |
| tibia | length | 4.09 ± 0.04 | 3.58 ± 0.04 | −12.0** | |
| vertebral height | (L6) | 0.741 ± 0.007 | 0.699 ± 0.019 | −5.6* | |
| Adult mice | femur | length | 16.6 ± 0.1 | 14.0 ± 0.3 | −16** |
| diaphyseal diam. | 2.02 ± 0.05 | 1.82 ± 0.02 | −9.9** | ||
| tibia | length | 18.40 ± 0.02 | 17.70 ± 0.10 | −3.7** | |
| diaphyseal diam. | 2.15 ± 0.04 | 1.98 ± 0.09 | −7.8 | ||
| vertebral height | (L6) | 3.16 ± 0.06 | 2.86 ± 0.04 | −9.5** | |
| crown–rump | length | 86.3 ± 0.5 | 80.0 ± 1.2 | −7.2** |
(*) P < 0.05; (**) P < 0.01 vs. wild type; no asterisk indicates no significant difference (P > 0.05); determined by Student’s t-test.
Figure 5.
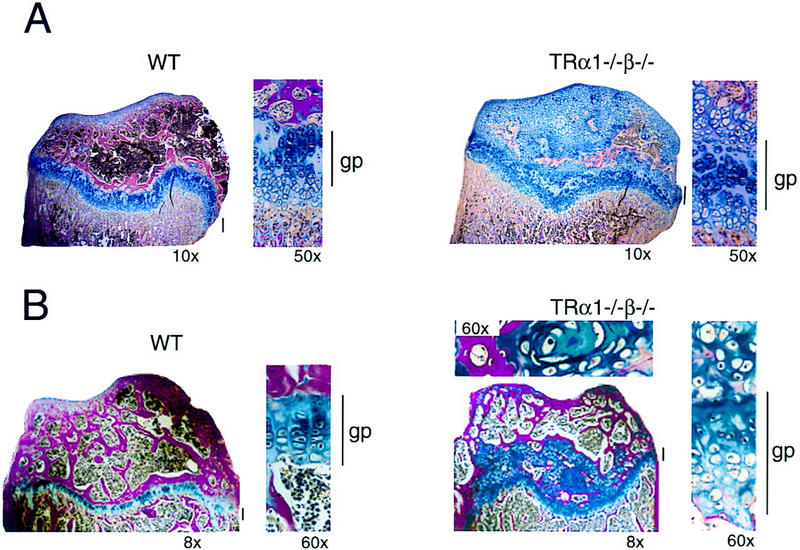
Retarded bone growth and maturation in TRα1−/−β−/− mice. (A) Histological section showing disorganized growth plates of the proximal tibia in pubertal (5-week-old) TRα1−/−β−/− and wild-type mice. (B) Histological section of the proximal tibia in adult (4-month-old) TRα1−/−β−/− and wild-type mice. For the TRα1−/−β−/− sample, the top shows a higher magnification of a cartilage area from the epiphysis. Note the extensive areas of remaining cartilage (blue) in the epiphysis above the growth plate in both pubertal and adult TRα1−/−β−/−mice. (gp) Growth plate (also indicated by vertical bars). Sections were stained with alcian blue/Van Gieson (cartilage area, blue; bone area, red). Magnifications are indicated.
Dual x-ray absorptiometry (DXA) measurements of adult TRα1−/−β−/− mice revealed significant decreases in bone area and in bone mineral content (BMC), as illustrated by measurements on femur, tibia, and vertebra L6 shown in Table 2. Bone mineral density was not significantly different, suggesting that the decreased bone mineral content was caused by a decrease in dimensions and not by a decrease in bone mineral density as measured by DXA. Mid-diaphyseal pQCT scans of femora showed that the cortical material density was decreased significantly by 4.2% in adult TRα1−/−β−/− compared to wild-type mice (Table 3). The mid-diaphyseal cortical area of femora was decreased significantly and this decrease was due to a decrease of the periosteal circumference. This resulted in a 27.1% decrease in the cross-sectional moment of inertia. Metaphyseal pQCT scans demonstrated that the trabecular bone mineral density was not changed in adult TRα1−/−β−/− mice (TRα1−/−β−/−, 237 ± 6.0 mg/cm3; wild-type, 230 ± 6.3 mg/cm3, n = 6). In conclusion, the bone abnormalities in TRα1−/−β−/− mice included decreased longitudinal bone growth associated with disorganized growth plates and impaired ossification and mineralization. Most of these defects resemble those occurring in congenital hypothyroidism, suggesting a high degree of concordance between the combined roles of TRα1 and TRβ and that of T3 in bone development.
Table 2.
DXA measurements of BMC in adult mice
|
|
|
Wild type (n = 6)
|
TRα1−/−β−/− (n = 6)
|
Difference (%)a
|
|---|---|---|---|---|
| Total body | bone mineral density (mg/cm2) | 68.2 ± 5.2 | 67.0 ± 4.8 | −1.7 |
| bone area (cm2) | 11.8 ± 1.8 | 10.6 ± 1.0 | −9.7 | |
| BMC (mg) | 761 ± 69 | 689 ± 29 | −9.5 | |
| Femur | Bone mineral density (mg/cm2) | 62.2 ± 1.6 | 63.2 ± 0.8 | 1.6 |
| Bone area (cm2) | 0.517 ± 0.010 | 0.418 ± 0.008 | −19.0** | |
| BMC (mg) | 32.2 ± 1.2 | 26.5 ± 0.8 | −18.0** | |
| Tibia | Bone mineral density (mg/cm2) | 57.3 ± 1.4 | 54.2 ± 0.6 | −5.5 |
| Bone area (cm2) | 0.435 ± 0.010 | 0.397 ± 0.006 | −8.8** | |
| BMC (mg) | 25.0 ± 1.1 | 21.5 ± 0.5 | −14.0* | |
| Vertebrae L6 | Bone mineral density (mg/cm2) | 61.8 ± 0.7 | 64.7 ± 0.9 | 4.6 |
| Bone area (cm2) | 0.132 ± 0.004 | 0.100 ± 0.004 | −24.0** | |
| BMC (mg) | 8.15 ± 0.22 | 6.47 ± 0.28 | −21.0** |
(*) P < 0.05; (**) P < 0.01 vs. wild type; no asterisk indicates no significant difference (P > 0.05); determined by Student’s t-test.
Table 3.
Mid-diaphyseal pQCT scans of femora in adult mice
|
|
Wild type (n = 6)
|
TRα1−/−β−/− (n = 6)
|
Difference (%)a
|
|---|---|---|---|
| Cortical density (mg/mm3) | 1.301 ± 0.013 | 1.246 ± 0.017 | −4.2* |
| Cortical area (mm2) | 1.225 ± 0.039 | 1.031 ± 0.039 | −15.8** |
| BMC (mg/mm) | 1.596 ± 0.063 | 1.286 ± 0.057 | −19.4** |
| Cortical thickness (mm) | 0.348 ± 0.010 | 0.319 ± 0.010 | −8.3 |
| Periosteal circumference (mm) | 5.448 ± 0.084 | 5.063 ± 0.137 | −7.1* |
| Endosteal circumference (mm) | 3.360 ± 0.179 | 3.267 ± 0.119 | −2.8 |
| Cross-sectional moment of inertia (mm4) | 0.798 ± 0.029 | 0.582 ± 0.046 | −27.1** |
| Moment of resistance (mm3) | 0.665 ± 0.022 | 0.547 ± 0.025 | −17.8** |
(*) P < 0.05; (**) P < 0.01; vs. wild type; no asterisk indicates no significant difference (P > 0.05); determined by Student’s t-test.
Discussion
Diversity in T3 receptor pathways
The identification of TRα1 and TRβ, but not other T3 receptor types, across vertebrate species suggests that both receptors are necessary and sufficient for mediating the many actions of T3. Studies of TRα1−/− and TRβ−/− mice, however, revealed only limited roles that could be ascribed individually to TRα1 or TRβ: TRα1 is important for setting basal body temperature and heart rate (Johansson et al. 1998; Wikström et al. 1998), whereas TRβ has a role in pituitary function and hearing (Forrest et al. 1996a; Rüsch et al. 1998). Even in sum, these phenotypes account for a only small proportion of T3 functions. The present study demonstrates that mice lacking both, rather than only one, of the T3 receptors, exhibit a novel array of phenotypes, including poor female fertility, hyperactivity of the pituitary–thyroid axis, and retardation of growth and bone development. These findings establish the existence of physiological pathways in which TRα1 and TRβ cooperate with, or substitute for, each other and indicate that such pathways facilitate an extended spectrum of T3 actions beyond those governed by TRα1 or TRβ alone.
The absence of a detectable T3 binding capacity or of TR proteins that could bind T3 response elements in nuclear extracts from TRα1−/−β−/− mice supports the view that TRα1 and TRβ represent the full complement of nuclear T3 receptors. Functionally, if unidentified T3 receptors remained, TRα1−/−β−/− mice would be expected to exhibit thyrotoxic symptoms, such as hyperactivity, tachycardia, and hypermetabolism due to their great excess of thyroid hormones. On the contrary, TRα1−/−β−/− mice have normal activity levels, their heart rate is not excessive (Johansson et al. 1999), and liver and kidney T3 responses are defective rather than excessive (L. Amma, A. Campos Barros, Z. Wang, B. Vennström, and D. Forrest, unpubl.). Furthermore, TSH levels and weight did not change significantly following MMI-induced changes in thyroid hormone levels. In sum, this evidence argues against the existence of other hypothetical receptors and suggests that TRα1−/−β−/− mice are devoid of nuclear T3 receptors.
The existence of different pathways that depend on either one or both receptors may provide a means of specifying distinct cellular responses to T3. Two underlying mechanisms may be considered. First, the structural differences in the DNA-binding domains or amino termini of TRα1 and TRβ may confer distinct properties in the recognition or regulation of subsets of target genes, consistent with in vitro findings that the transactivation properties of TRα1 and TRβ are closely, although not entirely, related (Sjöberg and Vennström 1995; Zhu et al. 1997). The differences, however, would be sufficiently permissive to allow regulation of overlapping gene networks that underlie those functions common to both TRα1 and TRβ. Alternatively, if TRα1 and TRβ are fully interchangeable, a given T3 response may depend merely on attainment of a ‘critical mass’ of T3 receptors, whether of the TRα1 or TRβ type. In this case, two TR genes, rather than one, may facilitate fine tuning in the control of expression of receptor levels. Further studies are warranted to explore these models. Similar findings of individual and common roles have been made for the retinoid receptor family (Kastner et al. 1995), suggesting a possible common operational code for determining diversity in the functions of multimember families of nuclear receptors.
Common functions for TRα1 and TRβ
This study clearly indicates common roles for TRα1 and TRβ in the control of the pituitary–thyroid axis and growth. The dysfunction of the pituitary–thyroid axis is markedly worse in TRα1−/−β−/− than in TRβ−/− or TRα1−/− mice: TRα1−/−β−/− mice greatly overproduce thyroid hormones, whereas TSH is not suppressed but is itself up to 160-fold increased, indicating a profound pituitary resistance to T3. In contrast, TRβ−/− mice have comparatively minor pituitary resistance with only 2.5-fold increased thyroid hormone and TSH levels (Forrest et al. 1996a; Campos-Barros et al. 1998), and there are only mild abnormalities in TRα1−/− mice, restricted to males, which have <30% decreases in free T4 and TSH (Wikström et al. 1998). The masking of this major role for both receptors in the single receptor deletions may be explained if there is a high degree of functional overlap between TRα1 and TRβ in the feedback suppression of the pituitary–thyroid axis. Given the necessity of maintaining appropriate thyroid hormone levels in development and homeostasis, it is possible that a dynamic interplay between two distinct TR genes enhances the sensitivity of the pituitary–thyroid axis in response to fluctuating physiological conditions.
TSH suppression is a complex process involving interactions between thyroid hormone and thyrotropin-releasing hormone, such that both direct and indirect defects may cause the overproduction of TSH in TRα1−/−β−/− mice. However, the 26- and 3-fold increases in TSHβ and TSHα mRNA levels, respectively, in TRα1−/−β−/− mice show that a major defect resides in the transcriptional regulation of the TSH subunit genes. Thus, in accord with their coexpression in the anterior pituitary (Bradley et al. 1992), both TRα1 and TRβ may have a role in the transcriptional repression of the TSH subunits (Shupnik et al. 1985; Wondisford et al. 1996). Interestingly, the dramatic elevation of TSH in TRα1−/− β−/− mice parallels that caused by hypothyroidism and indicates that any putative T3-independent activities of TRβ and TRα1 are not necessarily required to achieve high level TSH expression.
The growth retardation of TRα1−/−β−/− mice from early postnatal stages resembles that arising in congenital hypothyroidism (Snyder 1996). This retardation reflects defects in long bone development that include impaired maturation of the epiphyses and retarded elongation. The absence of overt growth deficiency in TRα1−/− or TRβ−/− mice suggests that there is substantial overlap in the underlying TRα1 and TRβ pathways in the control of growth. As in hypothyroidism, the GH deficiency is likely to be a major cause of the growth retardation. The marked decrease in GH mRNA levels in the pituitary of TRα1−/−β−/− but not TRα1−/− or TRβ−/− mice suggests that, consistent with the presence of T3 response elements in the GH gene, both TRα1 and TRβ may regulate GH gene transcription.
In addition, there may be intrinsic functions for TRs in bone, as TR expression has been detected in growth plate chondrocytes (Williams et al. 1998). Also, the formation of hypertrophic cells in the growth plate of thyroidectomized and hypophysectomized rats is stimulated by administration of thyroid hormones but not GH (Ray et al. 1954; Lewinson et al. 1989), and the maturation of growth plate chondrocytes in vitro can be regulated by thyroid hormones (Carrascosa et al. 1992; Ohlsson et al. 1992). Thus, our results support a model whereby TRα1 and TRβ interact at different levels in bone development, including in the direct responses to T3 in bone and cartilage tissue as well as an indirect role through the common regulation of GH expression.
The TRα mutation reported by Fraichard et al. (1997), which deletes both TRα1 and TRα2, results in retarded bone development resembling that of TRα1−/−β−/− mice. Unlike TRα1−/−β−/− mice that overproduce thyroid hormones, however, the mice described by Fraichard et al. (1997) are hypothyroid. It is possible that the bone abnormalities in these mice reflect this hypothyroidism, whereas the defects in TRα1−/−β−/− mice, despite their high T4 and T3 levels, are probably due to a ‘functional hypothyroidism’ caused by the absence of all nuclear T3 receptors.
Comparison of thyroid hormone vs. T3 receptor deficiency
T3 has profound roles throughout vertebrate development. Human congenital hypothyroidism is associated with retardation of mental and physical development, whereas adult hypothyroidism has debilitating consequences on metabolism, behavior, and cardiac function. Furthermore, combined maternal and fetal hypothyroidism, as occurs in areas of endemic iodine deficiency, worsens the developmental deficiencies and illustrates the importance of T3 during development in utero (Morreale de Escobar et al. 1989; Xue-Yi et al. 1994). It is remarkable therefore that TRα1−/−β−/− mice that lack all known T3 receptors throughout life from conception thrive relatively well.
As may be predicted in the absence of T3 receptors, the phenotype of TRα1−/−β−/− mice includes a number of the typical features of congenital hypothyroidism. These include poor female fertility, retardation of growth and bone development, TSH elevation, and deficiency of growth-promoting hormones. In contrast to hypothyroidism, however, fatigue and lethargy are not evident, as TRα1−/−β−/− mice display normal locomotor activity levels that vary normally over complete circadian cycles (Johansson et al. 1999). Comparison with genetically hypothyroid mice further suggests that the growth retardation of TRα1−/−β−/− mice is less severe than that caused by hypothyroidism. Hypothyroidism in hyt, cog, and αGSU mutant mice results in body weights ∼50% below normal (Beamer et al. 1981, 1987; Kendall et al. 1995), contrasting with the ∼30% reduction in TRα1−/−β−/− mice. Recent studies also indicate that thyroid hormone is required to sustain autonomous life after weaning, as profound hypothyroidism in Pax8-deficient mice, which lack thyroid follicular cells, retards growth and causes abrupt postweaning lethality (Mansouri et al. 1998). In contrast, apart from the minority that die sporadically, TRα1−/−β−/− mice have normal longevity, suggesting an absence of serious problems with most vital functions.
Thus, the comparatively mild overall phenotype of TRα1−/−β−/− mice recapitulates the hypothyroid phenotype only partially, suggesting that receptor and hormone deficiencies have different consequences. One explanation may invoke the ability of TRs, in the absence of T3, to bind T3 response elements and repress transcription (Damm et al. 1989; Sap et al. 1989). In hypothyroidism, T3 would not be available to alleviate the transcriptional repression exerted by TRs. Thus, chronic, deleterious repression of gene networks may lead to the more severe phenotype occurring in hypothyroidism than in TRα1−/−β−/− mice. There has been considerable interest in the mechanism of T3-independent repression involving TR/corepressor interactions, as described in vitro (Chen and Evans 1995; Hörlein et al. 1995). The present study provides evidence that there may be physiological consequences for such T3-independent TR functions.
Materials and methods
Mouse strains
All animal experiments were performed under approved protocols at each institution. Mice were genotyped by Southern blot or PCR analysis using primers specific for the TRα1 and TRβ mutations, as described (Forrest et al. 1996b; Wikström et al. 1998). TRβ−/− mice (on a hybrid background of 129/Sv × C57BL/6J strains) and TRα1−/− mice (on a hybrid background of 129OlaHsd × BALB/c strains) were intercrossed to generate both wild-type and TRα1−/−β−/− mice on the same mixed background for analysis. Mice were housed under l2-hr light/12-hr dark cycles in a controlled environment with 40%–50% relative humidity at 22°C. Because of their small size, TRα1−/−β−/− pups were generally not weaned until they were ∼4 weeks of age. After weaning, TRα1−/−β−/− pups were usually housed with their littermates. When TRα1−/−β−/− mice were housed alone at 18°C, no increased mortality was evident, suggesting that these mice were not susceptible to hypothermia under these conditions. Mice were made hypothyroid by providing a low iodine diet (R584, AnalyCen Nordic AB, Lidköping, Sweden for mice in Stockholm; custom defined diet, ICN Biochemicals, Cleveland, OH for mice in New York; iodine content 0.05 mg/kg) and by inclusion of 0.05% methimazole and 1% potassium perchlorate in the drinking water (Solé et al. 1996). For the weight determination experiment (Fig. 4E), mice were provided with low iodine diet for 21 days prior to addition of MMI and perchlorate to the drinking water. Similar weight data were obtained in an additional experiment on groups of n = 4 male mice.
Nuclear T3 binding determinations
Nuclear extracts from liver and brain tissue were prepared as described (Vivanco Ruiz et al. 1991). Extracts from untreated mice or mice that had been made hypothyroid (see above) were prepared from frozen tissue as described (Schreiber et al. 1989). Binding assays with 125I-labeled T3 were performed as described (Torresani and DeGroot 1975).
Gel-mobility shift assays
Nuclear protein extracts were prepared as described (Schreiber et al. 1989). DNA probe sequences were as described: F2 and DR4 (Ng et al. 1995); β-fibrinogen (Aguanno et al. 1996). DNA probes were labeled, and gel-mobility shift assays were performed as described with slight modification (Ng et al. 1995). Ten micrograms of nuclear protein was incubated with 2.5 μg of poly[d(I-C)] for 10 min at room temperature. Probe was added and the mixture incubated on ice for 15 min before electrophoresis at room temperature. Supershift assays were performed with a monoclonal antibody against a conserved domain of mouse RXR (4RX-1D12) that detects all three mouse RXR gene products (Rochette-Egly et al. 1994) and polyclonal antisera against full-length chick TRβ (B01; M. Sjöberg and B. Vennström, pers. comm.) or TRα (Demczuk et al. 1993). Each anti-TR antisera detected both TRα and TRβ proteins. The specificity of the antibodies was further established, as none cross-reacted with endogenous proteins in tissue nuclear extracts that bound the β-fibrinogen probe.
Northern blot analyses
Polyadenylated mRNA was prepared from pituitary glands and Northern blots were hybridized with cDNA probes for mouse TSHα, TSHβ (a gift from J. Gurr, Temple University School of Medicine, Philadelphia, PA), GH (a gift from D. Linzer, Northwestern University, Evanston, IL), and G3PDH, as an independent control for equivalent loading and integrity of mRNA. Levels of expression of TSH and GH mRNAs were normalized to that of G3PDH mRNA. Quantification was performed using a Molecular Dynamics PhosphorImager.
Radioimmunoassays
Serum was obtained from mice after sacrifice or by anesthetizing mice with CO2 and bleeding from the retro-orbital sinus. Free T4 and T3 were measured using the Amerlex-MAB kit, as described (Wikström et al. 1998). Total T3, T4, and TSH were determined as described (Campos-Barros 1998). TSH was measured using a mouse TSH/LH reference preparation (AFP51718mp), a mouse TSH antiserum (AFP98991), and rat TSH antigen for radioiodination (NIDDK-rTSH-I-9). IGF-I levels in serum were determined after acid ethanol extraction according to the manufacturer’s protocol (Nicols Institute Diagnostics, San Juan Capistrano, CA). GH radioimmunoassays used a purified mouse GH antigen for iodination (AFP-10783B), rat GH antiserum (NIDDK–anti-rGH-5) and a mouse GH reference preparation (AFP-10783B). GH tracer was iodinated with 125I using lactoperoxidase/glucose oxidase and purified by Sephadex G-50 chromatography. GH tracer was stored at −80°C and was further purified by Sephadex G-100 chromatography immediately before use. Pituitary glands were homogenized in distilled water and centrifuged at 14,000 rpm for 7 min (Hervás et al. 1975). The supernatant was stored in aliquots at −80°C. For radioimmunoassay, soluble extract from single pituitaries was diluted ∼1:50,000 with 1% BSA in PBS. Pituitary extracts from GH-deficient dwarf mice (Jackson Laboratory no. JR0643) were used as an internal standard for the low range of detection of GH in this assay.
Histology and immunostaining
Pituitary and thyroid glands were embedded in paraffin and 5-μm-thick sections prepared for staining with hematoxylin and eosin or immunostaining, as described (Forrest et al. 1996a). Mice were anesthetized with ketamine and perfused transcardially with 4% paraformaldehyde in PBS before preparation of pituitary sections for immunostaining using antibodies against TSHα (AFP-66P9986), TSHβ (AFP-1274789), and GH (AFP-411S) at 1:8000 dilution, followed by visualization with Vectastain ABC Elite reagents. For any given hormone, quantification was performed by analysis of four to six independent sections at different levels throughout the pituitary gland of each mouse. Positively staining cells were counted and numbers were related to the total cell numbers, determined using phase contrast microscopy or hematoxylin and eosin staining, in the anterior pituitary lobe of the same mouse. Several immunostaining experiments were performed on different groups of male and female mice, which yielded similar results.
Skeletal preparation and bone histology
Newborn mice and embryos were immersed at 65°C for 1 min, fixed in ethanol for 3 days, kept in acetone for 3 days, then rinsed with water. Preparations were stained with alcian blue and counterstained for bone with alizarin red, as described (Zhang et al. 1995). Right tibias were embedded in paraffin, sectioned, and stained with alcian blue/Van Gieson stain. Widths of the growth plates were measured using an image-processing system (Easy Image, Bergströms Instruments, Stockholm, Sweden) coupled to a microscope. The average of 30 growth plate measurements (three sections, 10 measurements per section) was calculated for each tibia. The same image system was used for determining the cartilage and bone areas in the epiphysis.
DXA
Bone mineral content (BMC) and areal bone mineral density (BMC/cm2) were measured using the Norland pDEXA Sabre (Fort Atkinson, WI) with Sabre Research 3.6 software. Ex vivo measurements of the left femur and tibia and vertebrae L6 were performed on excised bones placed on a 1-cm-thick Plexiglas table. All excised bones compared were measured simultaneously in the same scan. High-resolution scans were performed (line spacing; tibia and femur 0.02 cm; vertebrae 0.01 cm). In vivo measurements of animals were performed to measure total body BMC. Medium resolution scans were performed (line spacing 0.05 cm). A maximum of three mice could be analyzed simultaneously in the same scan. Therefore, a wild-type mouse was included as an internal standard in each scan to avoid interscan variations.
Peripheral quantitative computerized tomography
Tomographic measurements were performed by using the Stratec peripheral quantitative computerized tomography (pQCT XCT) Research M from Norland Corporation, specifically modified for use on small bone specimens (software v. 5.4B; resolution 70 μm). Mid-diaphyseal pQCT scans of the left femora were performed to determine cortical bone mineral density, cortical cross-sectional area, cortical thickness, periosteal circumference, endosteal circumference, moment of resistance, and the cross-sectional moment of inertia. The mid-diaphyseal region of femora in mice contains only cortical bone. Metaphyseal pQCT scans of the left femora were performed to measure trabecular bone density. The metaphyseal scan was positioned 4% of the total length of the femur proximal to the distal growth plate (an area consisting of a central portion of trabecular bone). The trabecular bone region was defined by setting an inner threshold to 400 mg/mm3. The interassay coefficients of variation for the pQCT measurements were <2%.
Acknowledgments
We thank P. Chambon for anti-RXR antibodies, J. Gurr for TSHα and TSHβ plasmids, D. Linzer for the GH plasmid, and A.F. Parlow, the National Institutes of Health (NIH) Pituitary Hormone Program, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Child Health Development (NICHD), and U.S. Department of Agriculture (USDA) for immunological reagents for hormone detection. We thank I. Lisoukov and M.-L. Spångberg for technical assistance. We are grateful to L. Amma, M. Andersson, and G. Morreale for advice, to K. Nordström for pituitary RNA analysis, and to A. Gow and T. Perlmann for comments on the manuscript. This work was supported in part by a March of Dimes Basil O’Connor Scholarship and NIH grant DC-03441 (D.F.), the Swedish Medical Research Council and the Lundberg Foundation (C.O.), the Swedish Cancer Foundation, Hedlund’s Stiftelse and the Karolinska Institute (B.V.), and the Human Frontiers Science Program. A.C.B. received a fellowship (PF 00679951) from the Spanish Ministry of Education and Culture.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note
During revision of this manuscript, Gauthier et al. (1999) reported mice carrying the TRα mutation described by Fraichard et al. (1997) combined with a TRβ mutation: “Different functions for thyroid hormone receptors TRα and TRβ in the control of thyroid hormone production and postnatal development.” EMBO. J. 18: 623–631. These mice have >100-fold elevated TSH but like the TRα mutant mice of Fraichard et al. (1997), they die after weaning for reasons that remain to be determined.
Footnotes
E-MAIL dforrest@msvax.mssm.edu, bjorn.vennstrom@cmb.ki.se; FAX (212) 849-2508.
References
- Aguanno A, Afar R, Albert V. Tissue-specific expression of the non-neuronal promoter of the aromatic l-amino acid decarboxylase gene is regulated by hepatocyte nuclear factor 1. J Biol Chem. 1996;271:4528–4538. doi: 10.1074/jbc.271.8.4528. [DOI] [PubMed] [Google Scholar]
- Beamer W, Eicher E, Maltais L, Southard J. Inherited primary hypothyroidism in mice. Science. 1981;212:61–63. doi: 10.1126/science.7209519. [DOI] [PubMed] [Google Scholar]
- Beamer W, Maltais L, DeBaets M, Eicher E. Inherited congenital goiter in mice. Endocrinology. 1987;120:838–840. doi: 10.1210/endo-120-2-838. [DOI] [PubMed] [Google Scholar]
- Bradley DJ, Towle HC, Young WS., III Spatial and temporal expression of α- and β-thyroid hormone receptor mRNAs, including the β2-subtype, in the developing mammalian nervous system. J Neurosci. 1992;12:2288–2302. doi: 10.1523/JNEUROSCI.12-06-02288.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent G, Moore D, Larsen P. Thyroid hormone regulation of gene expression. Annu Rev Physiol. 1991;53:17–35. doi: 10.1146/annurev.ph.53.030191.000313. [DOI] [PubMed] [Google Scholar]
- Campos-Barros A, Erway LC, Krezel W, Curran T, Kastner P, Chambon P, Forrest D. Absence of thyroid hormone receptor β-retinoid X receptor interactions in auditory function and in the pituitary-thyroid axis. Neuro Report. 1998;9:2933–2937. doi: 10.1097/00001756-199809140-00003. [DOI] [PubMed] [Google Scholar]
- Carrascosa A, Fernandez M, Audi L, Ballabriga A. Effects of triiodothyronine (T3) and identification of specific nuclear T3-binding sites in cultured human fetal epiphyseal chondrocytes. J Clin Endocrinol Metab. 1992;75:140–144. doi: 10.1210/jcem.75.1.1619002. [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–458. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Damm K, Thompson CC, Evans RM. Protein encoded by v-erbA functions as a thyroid-hormone receptor antagonist. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- Davis P, Davis F. Nongenomic actions of thyroid hormone. Thyroid. 1996;6:497–504. doi: 10.1089/thy.1996.6.497. [DOI] [PubMed] [Google Scholar]
- Demczuk S, Harbers M, Vennström B. Identification and analysis of all components of a gel retardation assay by combination with immunoblotting. Proc Natl Acad Sci. 1993;90:2574–2578. doi: 10.1073/pnas.90.7.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondell JD, Roy AL, Roeder RG. Unliganded thyroid hormone receptor inhibits formation of a functional preinitiation complex: implications for active repression. Genes & Dev. 1993;7:1400–1410. doi: 10.1101/gad.7.7b.1400. [DOI] [PubMed] [Google Scholar]
- Forrest D. The erbA/thyroid hormone receptor genes in development of the central nervous system. Semin Cancer Biol. 1994;5:167–176. [PubMed] [Google Scholar]
- Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: Evidence for tissue-specific modulation of receptor function. EMBO J. 1996a;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor β is essential for development of auditory function. Nat Genet. 1996b;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Plateroti M, Roux J, Trouillas J, Dehay C, Legrand C, Gauthier K, Kedinger M, Malaval L, Rousset B, Samarut J. The T3Rα gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J. 1997;16:4412–4420. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervás F, Morreale de Escobar G, Escobar del Ray F. Rapid effects of single small doses of L-thyroxine and triiodothyronine on growth hormone, as studied in the rat by radioimmunoassay. Endocrinology. 1975;97:91–101. doi: 10.1210/endo-97-1-91. [DOI] [PubMed] [Google Scholar]
- Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ, Larsen PR, Moore DD, Chin WW. Identification of a thyroid hormone receptor that is pituitary-specific. Science. 1989;244:76–78. doi: 10.1126/science.2539642. [DOI] [PubMed] [Google Scholar]
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Izumo S, Mahdavi V. Thyroid hormone receptor a isoforms generated by alternative splicing differentially activate myosin HC gene transcription. Nature. 1988;334:539–542. doi: 10.1038/334539a0. [DOI] [PubMed] [Google Scholar]
- Johansson C, Vennström B, Thorén P. Evidence that decreased heart rate in thyroid hormone receptor α1-deficient mice is an intrinsic defect. Am J Physiol. 1998;275:R640–646. doi: 10.1152/ajpregu.1998.275.2.R640. [DOI] [PubMed] [Google Scholar]
- Johansson, C., S. Göthe, D. Forrest, B. Vennström, and P. Thorén. 1999. Cardiovascular phenotype and temperature control in mice lacking thyroid hormone receptor β or both α1 and β. Am. J. Physiol. (in press). [DOI] [PubMed]
- Kastner P, Mark M, Chambon P. Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life. Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- Katz D, Reginato MJ, Lazar MA. Functional regulation of thyroid hormone receptor variant TRα2 by phosphorylation. Mol Cell Biol. 1995;15:2341–2348. doi: 10.1128/mcb.15.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SK, Samuelson LC, Saunders TL, Wood RI, Camper SA. Targeted disruption of the pituitary glycoprotein hormone α-subunit produces hypogonadal and hypothyroid mice. Genes & Dev. 1995;9:2007–2019. doi: 10.1101/gad.9.16.2007. [DOI] [PubMed] [Google Scholar]
- Koenig RJ, Lazar MA, Hodin RA, Brent GA, Larsen PR, Chin WW, Moore DD. Inhibition of thyroid hormone action by a non-hormone binding c-erbA protein generated by alternative mRNA splicing. Nature. 1989;337:659–661. doi: 10.1038/337659a0. [DOI] [PubMed] [Google Scholar]
- Lazar M. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev. 1993;14:184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- Lewinson D, Harel Z, Shenzer P, Silberman M, Hochberg Z. Effect of thyroid hormone and growth hormone on recovery from hypothyroidism of epiphyseal growth plate cartilage and its adjacent bone. Endocrinology. 1989;124:937–945. doi: 10.1210/endo-124-2-937. [DOI] [PubMed] [Google Scholar]
- Longcope C. The male and female reproductive systems in hypothyroidism. In: Braverman L, Utiger R, editors. Werner and Ingbar’s the thyroid. Philadelphia, PA: Lippincott-Raven; 1996. pp. 849–852. [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet. 1998;19:87–90. doi: 10.1038/ng0598-87. [DOI] [PubMed] [Google Scholar]
- Morreale de Escobar G, Obregon M, Ruiz de Oña C, Escobar del Ray F. Transfer of thyroxine from the mother to the rat fetus near term: effects on brain 3,5,3′-triiodothyronine deficiency. Endocrinology. 1989;122:1521–1531. doi: 10.1210/endo-122-4-1521. [DOI] [PubMed] [Google Scholar]
- Ng L, Forrest D, Haugen BR, Wood WM, Curran T. N-Terminal variants of thyroid hormone receptor β: Differential function and potential contribution to syndrome of resistance to thyroid hormone. Mol Endocrinol. 1995;9:1202–1213. doi: 10.1210/mend.9.9.7491112. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Nilsson A, Isaksson O, Bentham J, Lindahl A. Effects of tri-iodothyronine and insulin-like growth factor-I (IGF-I) on alkaline phosphatase activity, [3H]thymidine incorporation and IGF-I receptor mRNA in cultured rat epiphyseal chondrocytes. J Endocrinol. 1992;135:115–123. doi: 10.1677/joe.0.1350115. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Isgaard J, Tornell J, Nilsson A, Isaksson O, Lindahl A. Endocrine regulation of longitudinal bone growth. Acta Paediatr Suppl. 1993;391:33–40. doi: 10.1111/j.1651-2227.1993.tb12925.x. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Bengtsson B, Isaksson O, Andreassen T, Slootweg M. Growth hormone and bone. Endocr Rev. 1998;19:55–79. doi: 10.1210/edrv.19.1.0324. [DOI] [PubMed] [Google Scholar]
- Ray R, Asling C, Walker D, Simpson M, Li C, Evans H. Growth and differentiation of the skeleton in thyroidectomized-hypophysectomized rats treated with thyroxin, growth hormone or the combination. J Bone Joint Surg. 1954;36A:94–103. [PubMed] [Google Scholar]
- Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14:348–399. doi: 10.1210/edrv-14-3-348. [DOI] [PubMed] [Google Scholar]
- Reginato M, Zhang J, Lazar M. DNA-dependent and DNA-independent mechanisms regulate the differential heterodimerization of the isoforms of the thyroid hormone receptor with retinoid X receptor. J Biol Chem. 1996;271:28199–28205. doi: 10.1074/jbc.271.45.28199. [DOI] [PubMed] [Google Scholar]
- Rochette-Egly C, Lutz Y, Pfister V, Heyberger S, Scheuer I, Chambon P, Gaub M. Detection of retinoid X receptors using specific monoclonal and polyclonal antibodies. Biochem Biophys Res Comm. 1994;204:525–536. doi: 10.1006/bbrc.1994.2491. [DOI] [PubMed] [Google Scholar]
- Rüsch A, Erway L, Oliver D, Vennström B, Forrest D. Thyroid hormone receptor β-dependent expression of a potassium conductance in inner hair cells at the onset of hearing. Proc Natl Acad Sci. 1998;95:15758–15762. doi: 10.1073/pnas.95.26.15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sap J, Muñoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennström B. The c-erbA protein is a high affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- Sap J, Muñoz A, Schmitt J, Stunnenberg H, Vennström B. Repression of transcription mediated at a thyroid hormone response element by the v-erbA oncogene product. Nature. 1989;340:242–244. doi: 10.1038/340242a0. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Müller M, Schaffner W. Rapid detection of octamer binding proteins with mini-extracts prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupnik M, Chin W, Habener J, Ridgway E. Transcriptional regulation of the thyrotropin subunit genes by thyroid hormone. J Biol Chem. 1985;260:2900–2903. [PubMed] [Google Scholar]
- Sjöberg M, Vennström B. Ligand-dependent and -independent transactivation by thyroid hormone receptor β2 is determined by the structure of the hormone response element. Mol Cell Biol. 1995;15:4718–4726. doi: 10.1128/mcb.15.9.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder P. The pituitary in hypothyroidism. In: Braverman L, Utiger R, editors. Werner and Ingbar’s the thyroid. Philadelphia, PA: Lippincott-Raven; 1996. pp. 836–840. [Google Scholar]
- Solé E, Calvo R, Obregón M, Meseguer A. Effects of thyroid hormone on the androgenic expression of KAP gene in mouse kidney. Mol Cell Endocrinol. 1996;119:147–159. doi: 10.1016/0303-7207(96)03603-9. [DOI] [PubMed] [Google Scholar]
- Torresani J, DeGroot L. Triiodothyronine binding to liver nuclear extracts in vitro. Endocrinology. 1975;96:1201–1209. doi: 10.1210/endo-96-5-1201. [DOI] [PubMed] [Google Scholar]
- Vivanco Ruiz M, Bugge T, Hirschmann P, Stunnenberg H. Functional characterization of a natural retinoic acid response element. EMBO J. 1991;10:3829–3838. doi: 10.1002/j.1460-2075.1991.tb04952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- Wikström L, Johansson C, Saltó C, Barlow C, Campos Barros A, Baas F, Forrest D, Thorén P, Vennström B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor α1. EMBO J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G, Robson H, Shalet S. Thyroid hormone actions on cartilage and bone: interactions with other hormones at the epiphyseal plate and effects on linear growth. J Endocrinol. 1998;157:391–403. doi: 10.1677/joe.0.1570391. [DOI] [PubMed] [Google Scholar]
- Wondisford F, Magner J, Weintraub B. Thyrotropin. In: Braverman L, Utiger R, editors. Werner and Ingbar’s the thyroid. Philadelphia, PA: Lippincott-Raven; 1996. pp. 190–207. [Google Scholar]
- Xue-Yi C, Xin-Min J, Zhi-Hong D, Rakeman M, Ming-Li Z, O’Donnell K, Tai M, Amette K, DeLong N, DeLong G. Timing of vulnerability of the brain to iodine deficiency in endemic cretinism. N Engl J Med. 1994;331:1739–1744. doi: 10.1056/NEJM199412293312603. [DOI] [PubMed] [Google Scholar]
- Zhang W, Behringer R, Olson E. Inactivation of the myogenic bHLH gene MRF4 results in up-regulation of myogenin and rib abnormalities. Genes & Dev. 1995;9:1388–1399. doi: 10.1101/gad.9.11.1388. [DOI] [PubMed] [Google Scholar]
- Zhu X-G, McPhie P, Lin K-H, Cheng S-Y. The differential hormone-dependent transcriptional activation of thyroid hormone receptor isoforms is mediated by interplay of their domains. J Biol Chem. 1997;272:9048–9054. doi: 10.1074/jbc.272.14.9048. [DOI] [PubMed] [Google Scholar]