

Abstract
Background and Aims
Growing evidence suggests that microRNAs coordinate various biological processes in the liver. We describe experiments to address the physiological roles of these new regulators of gene expression in the liver that are as of yet largely undefined.
Methods
We disrupted Dicer, an enzyme essential for the processing of microRNAs, in hepatocytes using a conditional knockout mouse model to elucidate the consequences of loss of microRNAs.
Results
The conditional knockout mouse livers showed the efficient disruption of Dicer1 at 3 weeks after birth. This resulted in prominent steatosis and the depletion of glycogen storage. Dicer1-deficient liver exhibited increased growth-promoting gene expression and the robust expression of fetal stage-specific genes. The consequence of Dicer elimination included both increased hepatocyte proliferation as well as overwhelming apoptosis. Over time, Dicer1-expressing wild-type hepatocytes that had escaped Cre-mediated recombination progressively repopulated the entire liver. Unexpectedly, however, two-third of the mutant mice spontaneously developed hepatocellular carcinomas derived from residual Dicer1-deficient hepatocytes at 1 year old.
Conclusions
Dicer and microRNAs have critical roles in hepatocyte survival, metabolism, developmental gene regulation and tumor suppression in the liver. Loss of Dicer primarily impairs hepatocyte survival, but can promote hepatocarcinogenesis in cooperation with additional oncogenic stimuli.
Introduction
Dicer, an endoribonuclease III type enzyme cleaves pre-microRNAs and double-stranded RNAs into mature microRNAs and short interfering RNAs. Previous studies have shown that the disruption of Dicer results in the loss of mature microRNAs, indicating that Dicer is necessary for microRNA processing 1-3. While the exact mechanisms are still under investigation, the involvement of microRNAs in the coordination of many biological processes via the regulation of mRNA expression and translation has been well established. Growing evidence suggests a physiological role of microRNAs in the liver. The downregulation of mir-122, the most abundant microRNA in hepatocytes, resulted in the suppression of cholesterol biosynthesis and the treated mice became resistant to high fat diet-induced steatosis4, 5. Furthermore, Grimm et al reported that the injection of a high-titer hairpin RNA-expression vector into mice resulted in fatal liver dysfunction and injury6. These mice showed impaired microRNA processing arising from oversaturation of exportin5-dependent microRNA transport and the results were interpreted to suggest that microRNAs are indispensable for proper liver function and hepatocyte survival.
In addition to the roles in the regulation of physiological functions of the liver, microRNAs are likely involved in hepatocarcinogenesis. Many of microRNAs show altered expression in hepatocellular carcinomas (HCCs)7, 8, and mir-122, which targets CCNG1, is frequently downregulated in hepatocellular carcinomas8. However, the mechanisms by which the altered microRNA expression contributes to hepatocarcinogenesis remains largely unclear.
To elucidate the role of Dicer and microRNAs in the liver, we generated hepatocyte-specific Dicer1 knockout mice. Since Dicer is encoded by a single locus within the mouse genome, the disruption of the single Dicer1 gene results in the loss of all microRNAs1–3, 9. Our findings revealed that Dicer is necessary for hepatocyte survival and proper metabolic regulation. Furthermore, a significant proportion of the knockout mice spontaneously developed hepatocellular carcinomas, providing evidence for the tumor suppressive activity of Dicer1.
Results
Efficient deletion of Dicer1 in young Albumin-Cre;Dicer1loxP/loxP mouse liver is followed by repopulation with Dicer1-expressing hepatocytes
Albumin-Cre transgenic mice and Dicer1loxP/loxP mice were crossed to achieve the hepatocyte-specific disruption of Dicer1 1, 10. Albumin-Cre;Dicer1loxP/loxP mice were born at the expected Mendelian ratio and survived to adulthood with no obvious growth phenotypes. An examination of Albumin-Cre;Dicer1loxP/loxP mice and their control littermates during young adulthood revealed apparent defects in liver morphology (Figure 1A). Three-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers were homogenously pale in color when compared with controls. At 6 weeks, small spots of normal color had appeared on a yellowish background. These normal-colored areas expanded and had largely replaced the liver at 12 weeks after birth.
Figure 1. Efficient deletion of Dicer1 in young Albumin-Cre;Dicer1loxP/loxP mouse liver and repopulation with Dicer1-expressing hepatocytes.
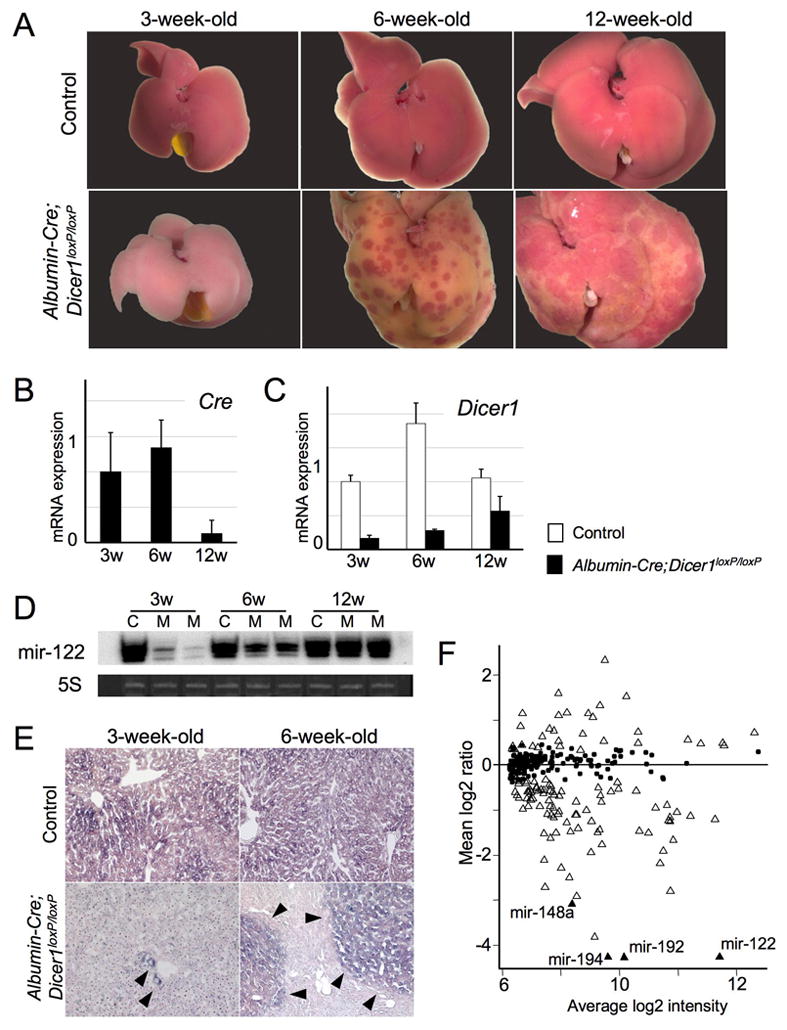
A: Gross morphology of control and Albumin-Cre;Dicer1loxP/loxP mouse livers at 3, 6 and 12 weeks after birth. At 3 weeks, the Albumin-Cre;Dicer1loxP/loxP mouse liver was pale compared with the control. At 6 weeks, the Albumin-Cre;Dicer1loxP/loxP liver had become yellowish, with the appearance of normal-colored spots. The normal-colored areas had expanded at 12 weeks. B: Quantitative PCR analysis of Cre transgene expression in Albumin-Cre;Dicer1loxP/loxP mouse liver. C: Quantitative PCR analysis of Dicer1 expression. Dicer1 expression recovered with age. D: Northern blotting for mir-122. Ethidium bromide staining of 5S RNA was used as a loading control. C: control; M: mutant (Albumin-Cre;Dicer1loxP/loxP). E: In situ hybridization for mir-122. Mir-122 expression was determined in control and Albumin-Cre;Dicer1loxP/loxP mouse livers at 3 and 6 weeks old. Livers from control mice showed diffuse expression of mir-122. Hepatocytes in the 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse liver did not express mir-122 except for a few cells (arrow heads). Nodules of mir-122-positive hepatocytes (arrowheads) appeared in the 6-week-old Albumin-Cre;Dicer1loxP/loxP mouse liver. F: Expression of microRNAs in 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers. Relative expression levels of mouse microRNAs were determined in comparison with control mouse livers. MicroRNAs with significantly altered expression (FDR<0.5) are represented by open and black triangles. Four previously reported liver-specific microRNAs are indicated by black triangles. Black dots indicate microRNAs without significant alteration.
Histologically, both control and Albumin-Cre;Dicer1loxP/loxP mouse livers showed homogenous appearance at 3-week-old, even though hepatocytes of Albumin-Cre;Dicer1loxP/loxP mice showed abnormalities at cytological levels as described later (Supplementary Figure 1). At 6 weeks after birth, Albumin-Cre;Dicer1loxP/loxP mouse livers displayed round nodules consisting of enlarged, but otherwise normal hepatocytes. The nodules of normal-appearing hepatocytes further expanded and largely replaced the parenchyma of 12-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers.
We suspected that these progressive morphological changes represented the replacement of Dicer1-decifient hepatocytes with wild-type hepatocytes that had escaped Cre-mediated recombination. To test this hypothesis, we examined the expression of the Cre transgene and Dicer1. Quantitative PCR analysis revealed the wide-spread silencing of Cre expression and a concomitant recovery of Dicer1 expression between 6 and 12 weeks after birth (Figure 1B, C). Since Dicer is essential for the maturation of microRNAs, we also examined the expression of mir-122, a hepatocyte specific microRNA. Mir-122 expression was almost completely diminished at 3 weeks, but was re-established in older mice, indicating the recovery of proper microRNA processing (Figure 1D). In situ hybridization showed the extensive loss of mir-122 expression in 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers, ensuring the efficient disruption of Dicer1 at this stage (Figure 1E). However, at 6-week-old, we observed appearance of mir-122-positive hepatocyte nodules that corresponded to normal-colored spots observed in gross examination.
These findings indicate that Dicer1 allele is efficiently disrupted at 3 weeks in Albumin-Cre;Dicer1loxP/loxP mouse livers; however, the entire liver is gradually repopulated by Dicer1-positive hepatocytes over time, and this process is achieved by nodular growth of Dicer1-positive wild-type hepatocytes. We did not observe oval/stem cell marker expression, including cytokeratin 19, CD34 and A6 antigen, in Dicer1-positive hepatocyte nodules during this process (data not shown), suggesting that liver progenitor cells do not contribute to the repopulation process.
We also assessed microRNA expression in 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers using microarray (Figure 1F). The analysis identified 45 microRNAs downregulated more than 2-fold with FDR<0.05 in Dicer1-deficient livers (Supplementary Table 1, 2). Remarkably, all 4 previously reported liver-specific microRNAs, mir-122, -148a, -192 and -194, showed robust downregulation in Dicer1-deficient livers11. There were 9 microRNAs upregulated in Dicer1-deficient livers; they are likely expressed by non-parenchymal cells and reflect secondary effects.
Dicer1-deficient hepatocytes exhibit increased proliferative activity as well as overwhelming apoptosis
Immunohistochemistry for phosphohistone H3 revealed a modest increase in hepatocyte proliferation in 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers (Figure 2A, B). At the same time, Dicer1-deficient hepatocytes showed increased apoptosis as indicated by staining for cleaved caspase-3 (Figure 2A, C). Examination of 6-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers revealed that both Dicer1-deficient hepatocytes and Dicer1-positive hepatocytes located in expanding nodules showed similar increase in proliferation. In contrast, wild-type mouse livers displayed very low proliferative activity at this stage (Figure 2D, E). However, there was a marked difference in their apoptotic activity: apoptosis was almost exclusively observed in Dicer1-deficient hepatocytes (Figure 2D, F). These findings indicate that loss of Dicer1 in hepatocytes causes an increase in hepatocyte proliferation. Nonetheless, most of the mutant cells are lost to elevated apoptosis over time, allowing repopulation by wild-type hepatocytes that had escaped Cre-mediated recombination.
Figure 2. Increased hepatocyte proliferation and apoptosis in Dicer1-deficient hepatocytes.
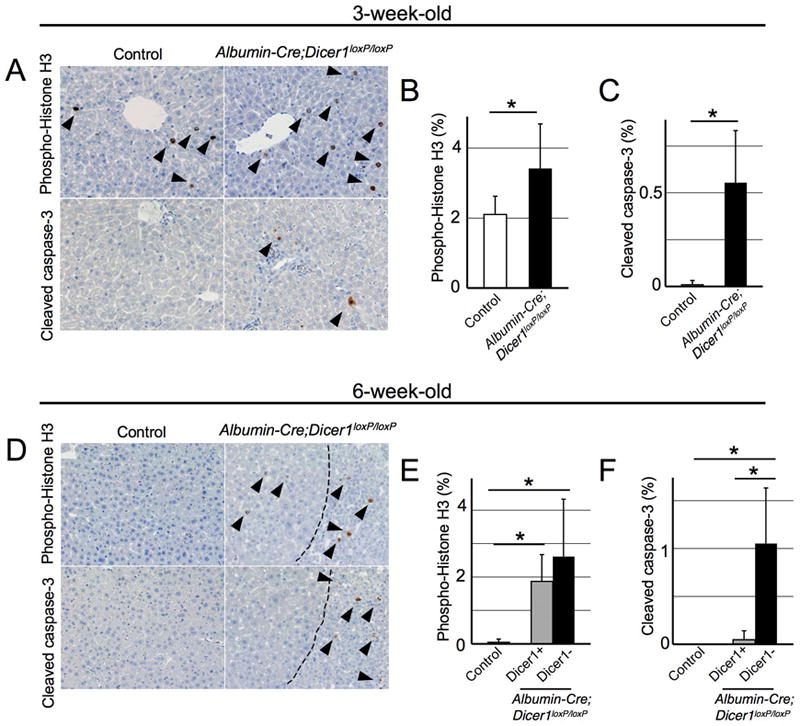
A, D: Immunohistochemistry for phospho-histone H3 and cleaved caspase-3 in 3- (A) and 6-week-old (D) mouse livers. Dotted lines in 6-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers indicate borders between Dicer1-positive hepatocyte nodules (left side) and Dicer1-negative areas (right side). B, C, E, F: Quantification of hepatocyte proliferation and apoptosis in Albumin-Cre;Dicer1loxP/loxP mouse livers (n=4-7, each group). At least 1000 cells were counted for quantification of phospho-histone H3 and cleaved caspase-3-positive hepatocytes. For 6-week-old Albumin-Cre;Dicer1loxP/loxP mice, hepatocytes in Dicer1-positive (grey columns) and –negative (black columns) areas were separately examined. *, P<0.05
Loss of Dicer1 impairs lipid and glucose metabolism
To further address the consequences of Dicer loss, we characterized 3-week-old mice, a time point at which Dicer1 is efficiently eliminated. Except for some alterations in lipid levels, serum analyses did not reveal significant changes that would indicate liver dysfunction (Table 1). Histological analysis confirmed that the normal liver architecture was preserved (Figure 3A). However, Dicer1-deficient hepatocytes had small vacuoles in their cytoplasm; these vacuoles were identified as lipid droplets using electron microscopy. The prominent lipid accumulation is likely responsible for the discoloration of the mutant livers. In contrast, glycogen granules normally present in hepatocytes were barely detectable in Dicer1-deficient hepatocytes. These fin dings were confirmed using histochemical analysis: oil red O staining confirmed the presence of numerous lipid droplets and periodic acid-Schiff staining showed the depletion of glycogen in Dicer1-deficient hepatocytes. A detailed analysis of the lipid composition revealed a remarkable elevation of cholesterol ester and triglyceride, whereas the free cholesterol and free fatty acid levels were mildly increased (Figure 3B-E). Our studies also revealed that the depletion of glycogen storage led to impaired blood glucose maintenance. Albumin-Cre;Dicer1loxP/loxP mice kept on normal feeding showed only a mild decrease in their glucose levels; however, the mutant mice became severely hypoglycemic after 6 hours of fasting (Figure 3F). Thus, Dicer function is critical for the regulation of liver lipid and glucose metabolism.
Table 1.
Metabolic measurements of 3-week-old Albumin-Cre;Dicer1loxP/loxP mice
| Control | Albumin-Cre; Dicer1loxP/loxP | P | |
|---|---|---|---|
| Alanine aminotransferase (U/L) | 40.0±17.0 | 45.6±24.8 | 0.70 |
| Aspartate aminotransferase (U/L) | 104.5±60.2 | 136.6±82.8 | 0.52 |
| γ-Glutamyl transpeptidase (U/L) | 3.0±3.8 | 3.4±1.5 | 0.85 |
| Albumin (g/dL) | 2.7±0.4 | 2.6±0.4 | 0.83 |
| Total protein (g/dL) | 4.8±0.3 | 4.0±0.9 | 0.14 |
| Direct bilirubin (mg/dL) | 0.1±0.1 | 0.1±0.1 | 0.65 |
| Indirect bilirubin (mg/dL) | 0.0±0.0 | 0.2±0.1 | 0.053 |
| Triglyceride (mg/dL) | 46.3±11.8 | 32.6±19.0 | 0.22 |
| Free fatty acid (μPEq/L) | 553.8±156.3 | 1065±333.2 | 0.023 |
| Cholesterol ester (mg/dL) | 56.3±7.0 | 41.6±3.4 | 0.017 |
| Free cholesterol (mg/dL)_ | 18.3±1.0 | 58.6±13.6 | 0.0026 |
n=4-5, each group.
Figure 3. Impaired lipid and glucose metabolism in Albumin-Cre;Dicer1loxP/loxP mice.
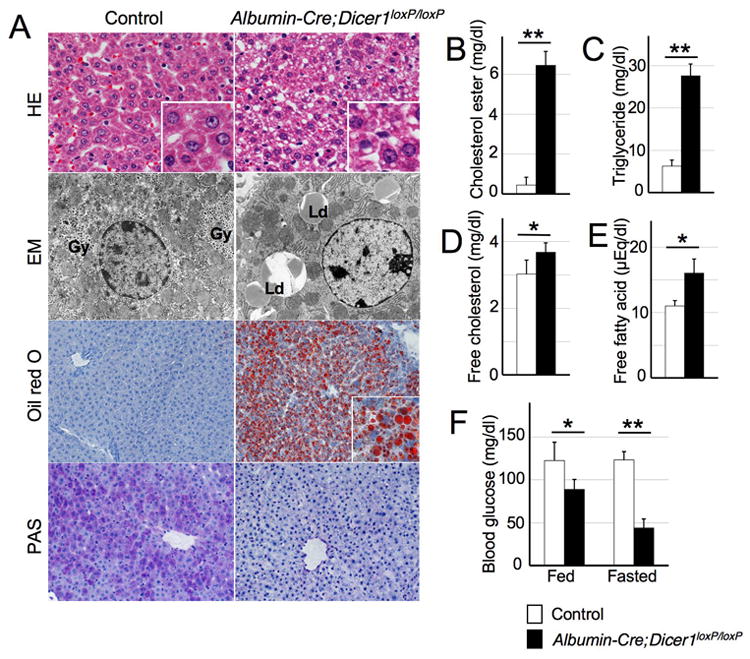
A: Histology and ultrastructure of control and Albumin-Cre;Dicer1loxP/loxP mouse livers. Hepatocytes of control mice displayed uniform eosinophilic cytoplasm and were arranged in a trabecular pattern. The liver architecture was preserved in the Albumin-Cre;Dicer1loxP/loxP mice, but the hepatocytes exhibited small cytoplasmic vacuoles. Electron microscopy analysis of Dicer1-deficient hepatocytes revealed a lack of glycogen granules that are readily observed in control hepatocytes (Gy). Instead, the Dicer1-deficient hepatocytes contained an abundance of lipid droplets (Ld). Lipid accumulation was barely detectable in the control liver, whereas numerous lipid droplets were visible in Dicer1-deficient liver by oil red O staining. PAS staining highlighted glycogen in the control liver, but not in Dicer1-deficient liver. B-E: Lipid analysis of the liver. The lipid composition of the liver tissue was determined following methanol/chloroform extraction. F: Blood glucose analysis. Tail vein blood glucose was measured in a fed condition or after 6 hours of starvation. n=4-5, each group. *, P<0.05; **, P<0.0001
We also examined changes in non-parenchymal cell population. Sinusoidal endothelial cells and portal tracts, including bile ducts, were not significantly altered (Supplementary Figure 2A). Some hematopoietic cell colonization was observed in Dicer1-deficient livers, and the presence of megakaryocytes and erythroblasts was confirmed by immunohistochemistry. (Supplementary Figure 2B). In contrast, hematopoietic cells are completely absent at 3-week-old in control littermates. While the liver is a place for hematopoiesis during fetal stages, liver normally loses the capacity to support hematopoiesis around the perinatal stage. The persistent presence of hematopoietic cells may represent an immature differentiation state of Dicer1-deficient liver.
Dysregulated expression of fetal stage-specific genes in Dicer1-deficient livers
We next sought to determine whether Dicer1-deficient hepatocytes retain their terminally differentiated mature phenotypes. Quantitative PCR analysis revealed that the expression of liver-enriched transcription factors was maintained, with Onecut1 and its direct target gene Hnf1b upregulated significantly in Dicer1-deficient livers12 (Figure 4A). Hnf1a and Cebpb also showed minor changes in expression levels. To further determine the differentiation status of Dicer1-deficient hepatocytes, we examined the expression of a battery of developmentally regulated genes. This analysis revealed dysregulated expression of genes that mark immature hepatocytes, whereas the expression pattern of genes normally found in mature hepatocytes was preserved (Figure 4B). The latter genes included Sdh and Tdo that are activated during the terminal stages of liver development. Thus, with some quantitative alterations, the fundamental hepatocyte transcriptional programs of mature hepatocytes were maintained in the absence of Dicer1. However, Dicer function is required to repress the fetal gene expression program in adult liver.
Figure 4. Expression of liver-enriched transcription factors, developmentally regulated genes and mir-122 target genes in Dicer1-deficient livers.
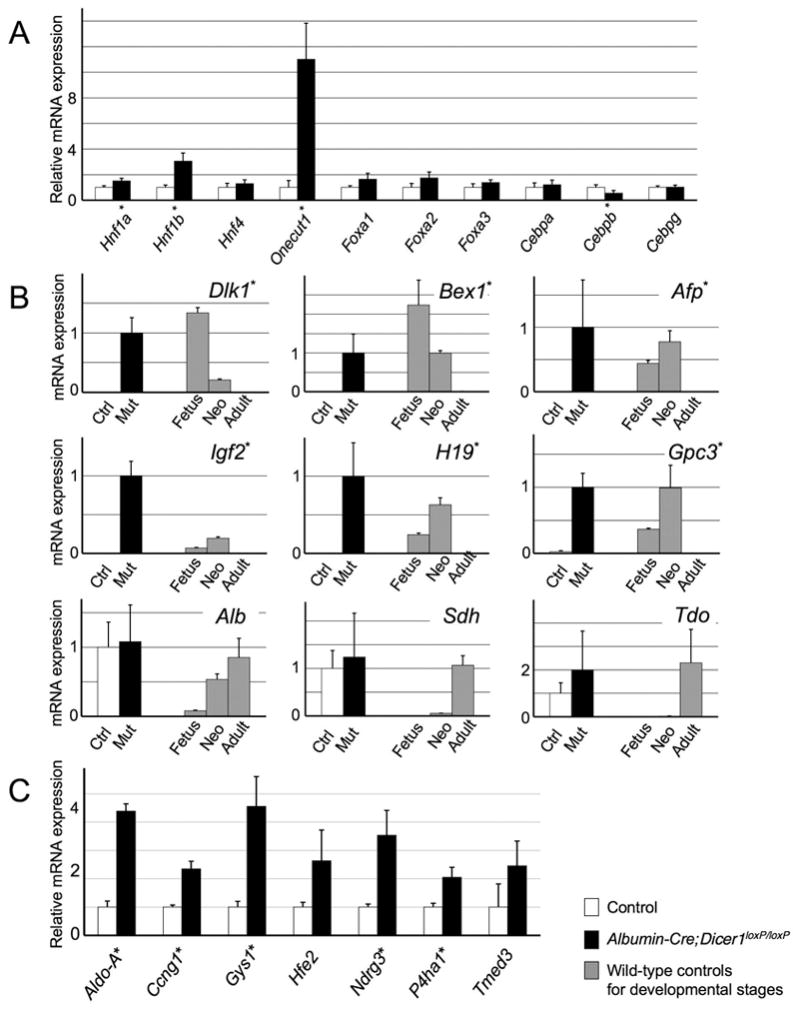
A: Relative expression levels of liver-enriched transcription factors. Quantitative PCR analysis of liver samples from Albumin-Cre;Dicer1loxP/loxP and control littermates at 3 weeks after birth (n=4, each group). B: The expression of developmentally regulated genes in control and Albumin-Cre;Dicer1loxP/loxP mice at 3 weeks (n=5, each group). The expression levels of wild-type mouse livers at different developmental stages were used as references (n=3, each group). C: Expression of putative direct targets of mir-122 in Albumin-Cre;Dicer1loxP/loxP mouse livers (n=4, each group). Values are shown as mean ± S.D. *, P<0.05. Ctrl: Control; Mut: mutant (Albumin-Cre;Dicer1loxP/loxP mice); Fetus: embryonic day 14 fetus; Neo: postnatal day 0 neonate; Adult: postnatal day 42 adult
Dicer1-deficient livers show increased expression of cell cycle-promoting genes and suppression of genes for steroid biosynthesis
To define global changes in gene expression, we conducted a cDNA microarray analysis. The analysis generated a list of genes with more than 2-fold changes, including 1415 upregulated and 944 downregulated genes, that was analyzed for the enrichment in Gene Ontology (GO) categories to identify biological pathways regulated by Dicer1. The most enriched “GO term” among the overexpressed genes was “cell division” (P=8.2×10−13). A list of growth promoting genes, including those encoding cyclins and aurora kinases, was found to be upregulated in the Dicer1-deficient livers (Table 2). The “GO term” most overrepresented among downregulated genes was “steroid biosynthesis” (P=1.6×10−15: Table 2). Of note, two previous studies using antisense oligonucleotide have shown that genes for cholesterol synthesis were downregulated in the absence of mir-1224, 5. In addition, we observed that previously identified direct targets of mir-122 were upregulated modestly in Dicer1-deficient liver (Figure 4C).
Table 2.
Altered gene expression in Albumin-Cre;Dicer1loxP/loxP mouse liver
| Genes related to Cell division | Genes related to Steroid synthesis | ||||
|---|---|---|---|---|---|
| Gene name | Microarray | qPCR | Gene name | Microarray | qPCR |
| Aurka | 2.8* | 4.0* | Dhcr7# | 0.30* | 0.29* |
| Aurkb | 2.2, 2.5* | 3.5* | Fdft1# | 0.21*, 0.28* | 0.37* |
| Ccna2 | 2.4*, 2.6* | 3.7* | Fdps# | 0.066* | 0.09* |
| Ccnb1 | 2.8*, 3.0* | 4.0* | Hmgcr | 0,36, 0.39 0.18*, 0.22* |
0.29 |
| Ccnb2 | 3.0* | 4.0* | Hmgcs1# | 0.22*, 0.29* | 0.37* |
| Ccnd1 | 2.8*, 3.1*, 3.1* | 3.8* | Hsd17b7 | 0.35* | 0.51 |
| Ccng1# | 2.2* | 2.3* | Mvk# | 0.14*, 0.34* | 0.34 |
| E2f5 | 0.7, 1.5, 2.0* | 2.4* | Pmvk | 0.14* | 0.25* |
| Plk1 | 3.6* | 5.2* | Tm7sf2 | 0.28* | 0.27* |
Gene expression levels of 3-week-old Albumin-Cre;Dicer1loxP/loxP mouse livers analyzed by cDNA microarray (n=3, each group), and the results were further confirmed by quantitative PCR (qPCR; n=4, each group). Values are indicated as fold changes compared with control mouse livers.
: Genes with significantly altered expression. FDR<0.05 for microarray and P<0.05 for qPCR.
Development of hepatocellular carcinoma in Albumin-Cre;Dicer1loxP/loxP mice
Given that Dicer1-deficient hepatocytes were mostly replaced by wild-type cells at 12 weeks after birth, we were surprised to observe that one-third of Albumin-Cre;Dicer1loxP/loxP mice developed HCCs after 6 months (Figure 5A-C). Furthermore, 12-month-old mice showed increased tumor incidence and progression of the disease, including 3 mice that succumbed to tumors at 9–11 months. However, none of the mice developed metastatic lesions. Similarly to human HCCs, these tumors exhibited considerable histological variations, including trabecular, pseudoglandular and solid arrangements with variable degree of steatosis (Figure 5B).
Figure 5. Development of hepatocellular carcinoma in Albumin-Cre;Dicer1loxP/loxP mice.
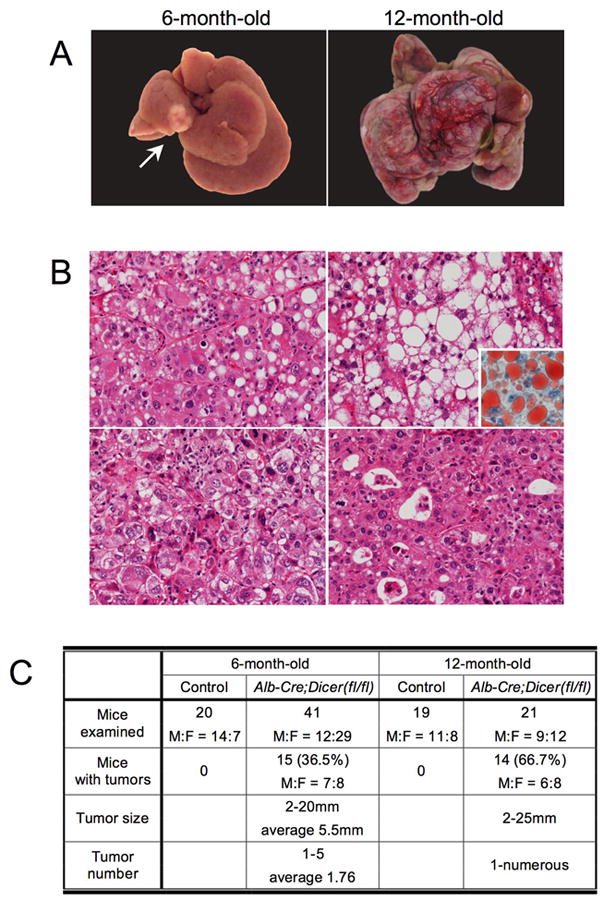
A: Hepatocellular carcinomas in Albumin-Cre;Dicer1loxP/loxP mice at 6 and 12 months after birth. A single tumor was observed in a 6-month-old Albumin-Cre;Dicer1loxP/loxP mouse liver (left, arrow). A 12-month-old Albumin-Cre;Dicer1loxP/loxP mouse liver carried multiple tumors that had replaced the liver parenchyma (right). B: Histological spectrum of hepatocellular carcinomas included thick trabecular arrangement with mild steatosis (top, left), prominent steatosis (top, right) as highlighted by oil red O staining (inset), poorly differentiated tumors with a solid growth pattern (bottom, left) and a pseudoglandular pattern (bottom, right). C: Summary of tumor incidence in Albumin-Cre;Dicer1loxP/loxP mice. Note that 12-month-old mice with tumors include 3 mice that had succumbed to HCCs at 9-11 months of age.
Remarkably, the tumors exhibited a decreased expression of Dicer1, persistent Cre transgene expression, a lack of mir-122 expression, and high expression levels of fetal liver genes (Figure 6A-D). Non-neoplastic areas consisted exclusively of histologically normal hepatocytes without evidence of preneoplastic or dysplastic changes (data not shown). In addition, non-neoplastic liver tissues obtained from tumor-bearing mice had lost Cre expression and were positive for Dicer1 and mir-122 at levels comparable to those in the control littermates (Figure 6A-C). These fin dings indicate that the HCCs were derived from residual Dicer1-deficient hepatocytes, whereas the non-neoplastic areas were entirely repopulated by wild-type hepatocytes.
Figure 6. Hepatocellular carcinomas in Albumin-Cre;Dicer1loxP/loxP mice are derived from Dicer1-deficient hepatocytes.
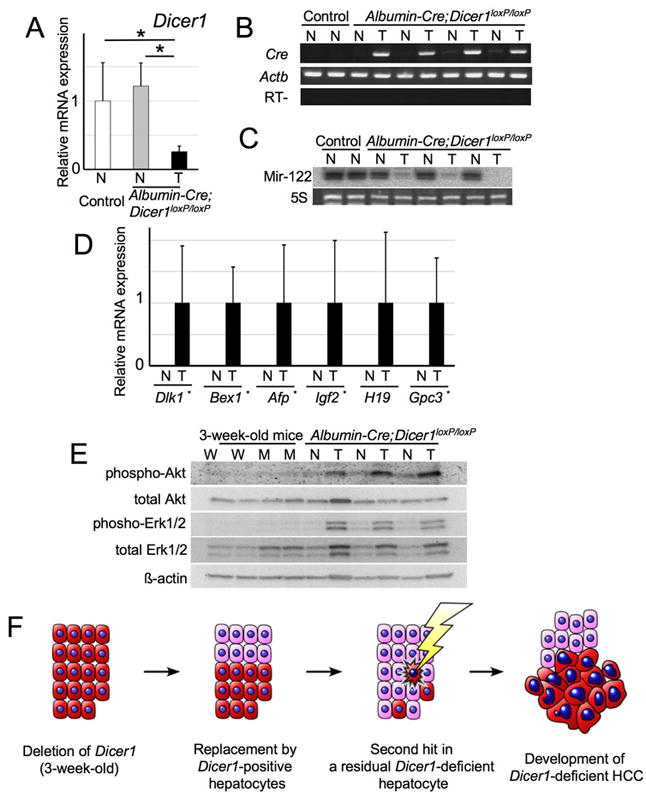
A: Expression of Dicer1 in non-neoplastic and tumor samples of control and Albumin-Cre;Dicer1loxP/loxP mice (n=5-6, each group). mRNA expression was determined by quantitative PCR. Values are mean ± S.D. *, P<0.05. T: Tumor; N: Non-neoplastic liver tissue. B: RT-PCR analysis of Cre transgene expression. Actb served as a positive control. C: Northern blot for mir-122. Ethidium bromide staining of 5S RNA served as a loading control. D: Quantitative PCR analysis of oncofetal genes (n=5-6, each group). E: Expression of phospho-Erk and phospho-Akt in HCCs and Dicer1-deficient livers. Protein samples from HCCs, corresponding non-neoplastic liver, 3-week-old Dicer1-deficient and control livers were treated with the indicated antibodies. F: Model of hepatocarcinogenesis in Albumin-Cre;Dicer1loxP/loxP mice. Albumin-Cre;Dicer1loxP/loxP mice exhibit the efficient deletion of Dicer1 at 3 weeks after birth. However, Dicer1-deficient hepatocytes (red cells) gradually undergo apoptosis and Dicer1-expressing wild-type hepatocytes (pink cells) that have escaped Cre-mediated recombination repopulate the entire liver over time. When a Dicer1-deficient hepatocyte acquires secondary oncogenic stimuli presumably through additional genetic alterations, the absence of Dicer1 cooperatively promotes the development of hepatocellular carcinoma. spectrum of hepatocellular carcinomas included thick trabecular arrangement with mild steatosis (top, left), prominent steatosis (top, right) as highlighted by oil red O staining (inset), poorly differentiated tumors with a solid growth pattern (bottom, left) and a pseudoglandular pattern (bottom, right). C: Summary of tumor incidence in Albumin-Cre;Dicer1loxP/loxP mice. Note that 12-month-old mice with tumors include 3 mice that had succumbed to HCCs at 9-11 months of age.
As disruption of Dicer1 results in impaired survival of hepatocytes, we suspected that Dicer1-deficient HCCs harbor additional molecular alterations that protect from apoptosis and transform the Dicer1-deficient hepatocytes. Western blotting showed the increased expression of Erk1/2 and the enhanced phosphorylation of Erk1/2 and Akt in Dicer1-deficient HCCs, but not in Dicer1-deficient non-neoplastic livers (Figure 6E). Expression analysis of a panel of tumor-related genes did not identify robust and consistent alterations of particular oncogenes in HCCs, but showed overexpression of Mycn and Bcl2 in a subset of tumors (Supplementary Figure 3). This may suggest that a variety of oncogenic signals can transform Dicer1-deficient hepatocytes. Sequencing analysis did not reveal any activating mutations in major oncogenes involved in mouse hepatocarcinogenesis, including Ctnnb1, Hras and Braf (data not shown).
Discussion
Our findings indicate that the loss of Dicer1 compromises hepatocyte survival in vivo. As indicated by the almost complete loss of mir-122, the Alb promoter mediated expression of Cre recombinase in hepatocytes achieved the efficient disruption of Dicer1 in the liver at 3 weeks after birth. However, Dicer1-deficient hepatocytes exhibited increased apoptosis and wild-type hepatocytes that had escaped the Cre-mediated recombination of Dicer1 gradually repopulated the entire liver in the absence of progenitor cell expansion. Previous studies have noted that wild-type hepatocytes can display a selective growth advantage over hepatocytes with metabolic defects in vivo13, 14. A small number of donor wild-type hepatocytes can repopulate a metabolically defective liver in this setting, a process known as therapeutic liver repopulation15. The progressive replacement of Dicer1-deficient hepatocytes by Dicer1-positive wild-type hepatocytes in the Albumin-Cre;Dicer1loxP/loxP mouse livers closely simulates this process and might be mediated by a common mechanism.
Examination of the Dicer1-deficient livers revealed the requirement of Dicer for proper lipid and glucose metabolism. Of note, the suppression of mir-122, the most abundant microRNA in the liver, has been previously described to result in the downregulation of genes involved in cholesterol biosynthesis4, 5. These mice became resistant to high fat diet-induced steatosis, while glucose metabolism was not affected4. Indeed, Dicer1-deficient livers also showed alterations in gene expression that are consistent with the consequences of mir-122 loss, including the upregulation of mir-122 targets and the decreased expression of genes involved in cholesterol biosynthesis. Since mir-122 constitutes 70% of the entire microRNA pool in the liver16, it is not surprising that the effects of mir-122 loss are prominent in Dicer1-deficient liver tissues where all microRNA processing is impaired. However, in contrast to the findings obtained in the mir-122 knockdown study, the disruption of Dicer1 resulted in marked steatosis, indicating that Dicer regulates lipid metabolism through a mir-122-dependent as well as a mir-122-independent pathway. This suggests the presence of microRNAs other than mir-122 that play critical roles in metabolic regulation in the liver. Since loss of Dicer impacts expression of a large number of genes, it is difficult to identify a specific pathway responsible for the steatosis in Dicer1-deficient livers. Further studies, including modulation of single microRNAs, would be required to clarify the mir-122-independent mechanisms for the regulation of lipid and glucose metabolism in liver.
Recent studies have established that microRNAs are frequently deregulated in many types of cancers including HCCs17, 18. Some evidence suggests that the dominant role of microRNA processing during transformation may be tumor suppression. Lu and colleagues reported that microRNAs are predominantly down-regulated in human tumors and Kumar and colleagues showed that the simultaneous deletion of Dicer1 and expression of activated Kras enhances lung tumorigenesis17, 19. The dysregulated expression of fetal liver genes in Dicer1-deficient hepatocytes is intriguing considering the notion that cancers mimic corresponding fetal tissue, a hypothesis that is supported by the common expression of a group of genes referred to as oncofetal genes20, 21 . The reactivation of fetal stage-specific genes is also a common finding in human HCCs. Alpha-fetoprotein, encoded by AFP, is the most widely used serum marker of HCCs in clinical practice22, 23. Glypican-3, which is a gene product of GPC3, is another increasingly recognized serum and histological marker of human HCCs24, 25. Not only are these oncofetal genes known as tumor markers, several of their gene products, including delta-like 1, glypican-3 and insulin-like growth factor 2, have been implicated in the malignant potential of HCCs26–28. For instance, insulin-like growth factor 2 is a highly potent mitogen, and the suppression of this gene alone has therapeutic benefits in a mouse implantation model of HCC26.
It is also notable that Dicer1-deficient liver showed increased expression of cell cycle promoting genes, that is another common feature of hepatocellular carcinomas. Even though we cannot exclude that these changes represent secondary effects such as a consequence of dysregulated fetal gene expression, it is tempting to speculate that these growth-promoting genes are controlled by a microRNA-mediated machinery. Of note, Ccng1, which is upregulated also in Dicer1-deficient liver, has been identified as a direct target of mir-1224, 8. Thus, Dicer1-deficient hepatocytes exhibit some characteristics consistent with transformed hepatocytes, including dysregulation of fetal liver genes and increased expression of cell cycle-promoting genes. However, the fact that Dicer1-deficient hepatocytes und ergo apoptosis indicates that these changes are not sufficient to initiate tumorigenesis by themselves.
Our observations indicate that the disruption of Dicer1 primarily impairs hepatocyte survival, however, it also promotes hepatocarcinogenesis in cells that escape the initial wave of cell death. Although these opposing effects of Dicer1-loss on non-neoplastic hepatocytes and HCCs appear paradoxical, our findings are consistent with previous experimental observations. Impaired microRNA processing have resulted in reduced proliferative activity, tissue degeneration or senescence in variety types of primary cells1–3, 19, 29–31. On the other hand, deletion of Dicer1 promoted cellular transformation in the presence of activated Kras both in vitro and in vivo19. Interestingly, it is known that the introduction of prototypical oncogenes into normal cells can induce apoptosis32, 33. In these situations, the presence of appropriate survival signals can circumvent the apoptotic reaction and transform the cells33–35. Thus, the opposing effects of Dicer1-loss on cell survival and transformation may somehow simulate those of oncogenes.
The fact that only a minor subset of Dicer1-deficient hepatocytes gives rise to HCCs suggests the requirement of a “second-hit” that promotes hepatocarcinogenesis in Dicer1-deficient hepatocytes. We found that Dicer1-deficient HCCs consistently exhibited phosphorylation of Erk1/2 and Akt, processes that are also common in human HCCs36, 37. Since non-neoplastic Dicer1-deficient livers did not show the activation of Erk1/2 or Akt, these events are not an immediate consequence of Dicer1 loss but are acquired during tumorigenesis. Some secondary events leading to the activation of these pathways might be involved in the transformation of Dicer1-deficient hepatocytes, and may promote hepatocarcinogenesis synergistically with the loss of Dicer1 (Figure 6F).
The present study revealed novel and pivotal roles of Dicer1 in hepatocyte survival, metabolism, developmental gene regulation and tumor suppression in the liver. Reactivation of the fetal gene expression program might be a key mechanism of hepatocarcinogenesis induced by the loss of Dicer1. Since Dicer1 is essential for microRNA processing, the phenotypes observed here would reflect the regulatory roles of microRNAs in the liver. Further studies will examine the functions of individual microRNAs to elucidate the precise mechanisms for the regulation of liver function by Dicer.
Materials and Methods
Mice
Alb promoter-driven Cre recombinase transgenic mice (Albumin-Cre mice) and mice carrying the floxed allele of Dicer1 (Dicer1loxP/loxP mice) have been previously described1, 10. Albumin-Cre and Dicer1loxP/loxP mice were crossed to obtain hepatocyte-specific Dicer1 knockout mice (Albumin-Cre;Dicer1loxP/loxP mice). Dicer1loxP/loxP littermates were used as controls throughout the experiment. The mice used in the present study were maintained in barrier facilities according to the protocols approved by the Committee on Animal Research of the University of California-San Francisco and the Committee for Ethics in Animal Experimentation at the National Cancer Center, Japan.
Histological Analysis
Formalin-fixed and paraffin-embedded sections were subjected to hematoxylin and eosin. Immunohisochemistry was performed either on paraffin-embedded or frozen sections using the avidin-biotin complex method as previously described38. Primary antibodies used are listed on Supplementary Table 3. Oil red O staining was performed on frozen sections fixed with formalin. Periodic acid-Schiff staining was performed on frozen sections fixed with Carnoy’s fixative. The electron microscopy analysis was performed using standard procedures.
In situ hybridization against mir-122 was performed on frozen sections using locked nucleic acid probe labeled with digoxigenin (Exiqon, Denmark) as previously described39. The signal was visualized using NBT/BCIP as a chromogen.
Serum, blood glucose and liver lipid analysis
Blood samples were collected from the inferior vena cava at necropsy. The serum was then separated and subjected to analysis (IDEXX, MA and SRL, Tokyo, Japan). Mouse tail vein blood glucose levels were measured using a standard glucometer. A liver lipid analysis was performed using commercially available kits (Wako, Tokyo, Japan and Eiken, Tokyo, Japan) following methanol-chloroform extraction.
Reverse transcription-PCR
Reverse-transcription reaction and conventional PCR were performed using standard protocols. Quantitative PCR reactions were performed using SYBR Green PCR master mix (Applied Biosystems, CA). The expression of liver genes was compared with the expression level of Gusb, as previously described38. The primer sequences are available upon request.
Northern blotting
Total RNA was fractioned on a 15% polyacrylamide gel, transferred to nylon membrane and hybridized to a radiolabeled oligonucleotide complementary to mir-1225. Ethidium bromide staining of 5S RNA served as a control.
Microarray analysis
Total RNA was extracted from 3 control and 3 Albumin-Cre; Dicer1loxP/loxP mice. cRNA synthesis and labeling was performed using GeneChip Two-cycle target labeling and control reagent (Affymetrix, CA). Gene expression profile was assessed by GeneChip genome 430 2.0 array (Affymetrix) following manufacture’s protocol. Data analysis was performed on the NIA array analysis website40. Genes that showed more than 2-fold changes with the false discovery rate <0.05 were considered as significantly altered. Gene ontology analysis was performed using the DAVID Functional annotation tool41. Overrepresented GO terms for biological pathways among significantly altered genes were analyzed with default settings.
MicroRNA expression was also determined for the same set of RNA samples. Samples were labeled using miRCURY LNA microRNA Power Hy3/Hy5 Labeling kit (Exiqon) and microRNA expression profiles were assessed by miRCURY LNA v.10.0 - hsa, mmu & rno (Exiqon). MicroRNAs that showed more than 2-fold changes with the false discovery rate <0.05 were considered as significantly altered.
Western blotting
Western blotting was performed as previously described38. Primary antibodies used are listed on Supplementary Table 3.
Statistical Analysis
The results are presented as the mean ± standard deviation. Statistical significance was determined by Student two-tailed t test, with a P value of <0.05 was considered significant.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The authors thank Shigeru Tamura for photographic assistance, Fumio Hasegawa for electron microscopy and John P Morris IV for critical reading of the manuscript.
Grant support: This work was supported by a Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cance r Control and a Grant-in-Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan, and a program for promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedic al Innovation (NiBio), Japan. Work in M.H.’s laboratory was supported by a NIH grant (CA112537).
Abbreviations
- HCC
hepatocellular caricinoma
- HPF
high power field
- GO
gene ontology
Footnotes
Conflict of interest: There are no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci U S A. 2005;102:10898–903. doi: 10.1073/pnas.0504834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci U S A. 2005;102:12135–40. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 6.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 7.Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–63. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 8.Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L, Negrini M. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–9. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 9.Calabrese JM, Seila AC, Yeo GW, Sharp PA. RNA sequence analysis defines Dicer’s role in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:18097–102. doi: 10.1073/pnas.0709193104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–15. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 11.Barad O, Meiri E, Avniel A, Aharonov R, Barzilai A, Bentwich I, Einav U, Gilad S, Hurban P, Karov Y, Lobenhofer EK, Sharon E, Shiboleth YM, Shtutman M, Bentwich Z, Einat P. MicroRNA expression detected by oligonucleotide microarrays: system establishment and expression profiling in human tissues. Genome Res. 2004;14:2486–94. doi: 10.1101/gr.2845604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clotman F, Lannoy VJ, Reber M, Cereghini S, Cassiman D, Jacquemin P, Roskams T, Rousseau GG, Lemaigre FP. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–28. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 13.Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–56. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- 14.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, Grompe M. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–73. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 15.Grompe M, Laconi E, Shafritz DA. Principles of therapeutic liver repopulation. Semin Liver Dis. 1999;19:7–14. doi: 10.1055/s-2007-1007093. [DOI] [PubMed] [Google Scholar]
- 16.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–9. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 17.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 18.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 19.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–7. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 20.Uriel J. Cancer, retrodifferentiation, and the myth of Faust. Cancer Res. 1976;36:4269–75. [PubMed] [Google Scholar]
- 21.Alexander P. Foetal "antigens" in cancer. Nature. 1972;235:137–40. doi: 10.1038/235137a0. [DOI] [PubMed] [Google Scholar]
- 22.Taketa K. Alpha-fetoprotein: reevaluation in hepatology. Hepatology. 1990;12:1420–32. doi: 10.1002/hep.1840120625. [DOI] [PubMed] [Google Scholar]
- 23.Johnson PJ. The role of serum alpha-fetoprotein estimation in the diagnosis and management of hepatocellular carcinoma. Clin Liver Dis. 2001;5:145–59. doi: 10.1016/s1089-3261(05)70158-6. [DOI] [PubMed] [Google Scholar]
- 24.Zhu ZW, Friess H, Wang L, Abou-Shady M, Zimmermann A, Lander AD, Korc M, Kleeff J, Buchler MW. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut. 2001;48:558–64. doi: 10.1136/gut.48.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, Filmus J. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. doi: 10.1016/s0016-5085(03)00689-9. [DOI] [PubMed] [Google Scholar]
- 26.Yao X, Hu JF, Daniels M, Shiran H, Zhou X, Yan H, Lu H, Zeng Z, Wang Q, Li T, Hoffman AR. A methylated oligonucleotide inhibits IGF2 expression and enhances survival in a model of hepatocellular carcinoma. J Clin Invest. 2003;111:265–73. doi: 10.1172/JCI15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Zhang X, Zhang M, Zhu JD, Zhang YL, Lin Y, Wang KS, Qi XF, Zhang Q, Liu GZ, Yu J, Cui Y, Yang PY, Wang ZQ, Han ZG. Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis. 2007;28:1094–103. doi: 10.1093/carcin/bgl215. [DOI] [PubMed] [Google Scholar]
- 28.Capurro MI, Xiang YY, Lobe C, Filmus J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005;65:6245–54. doi: 10.1158/0008-5472.CAN-04-4244. [DOI] [PubMed] [Google Scholar]
- 29.Damiani D, Alexander JJ, O’Rourke JR, McManus M, Jadhav AP, Cepko CL, Hauswirth WW, Harfe BD, Strettoi E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008;28:4878–87. doi: 10.1523/JNEUROSCI.0828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–5. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–74. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 32.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 33.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109:321–34. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 34.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 35.Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–90. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakanishi K, Sakamoto M, Yamasaki S, Todo S, Hirohashi S. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer. 2005;103:307–12. doi: 10.1002/cncr.20774. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. Increased MAPK expression and activity in primary human hepatocellular carcinoma. Biochem Biophys Res Commun. 1997;236:54–8. doi: 10.1006/bbrc.1997.6840. [DOI] [PubMed] [Google Scholar]
- 38.Sekine S, Gutierrez P, Lan B, Feng S, Hebrok M. Liver specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. 2007;45:361–8. doi: 10.1002/hep.21523. [DOI] [PubMed] [Google Scholar]
- 39.Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc. 2007;2:1508–14. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- 40.Sharov AA, Dudekula DB, Ko MS. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics. 2005;21:2548–9. doi: 10.1093/bioinformatics/bti343. [DOI] [PubMed] [Google Scholar]
- 41.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.