

Abstract
Upon ligand binding, the receptors of the TGFβ family phosphorylate Smad proteins, which then move into the nucleus where they activate transcription. To carry out this function, the receptor-activated Smads 1 and 2 require association with the product of deleted in pancreatic carcinoma, locus 4 (DPC4), Smad4. We investigated the step at which Smad4 is required for transcriptional activation. Smad4 is not required for nuclear translocation of Smads 1 or 2, or for association of Smad2 with a DNA binding partner, the winged helix protein FAST-1. Receptor-activated Smad2 takes Smad4 into the nucleus where they form a complex with FAST-1 that requires these three components to activate transcription. Smad4 contributes two functions: Through its amino-terminal domain, Smad4 promotes binding of the Smad2/Smad4/FAST-1 complex to DNA; through its carboxy-terminal domain, Smad4 provides an activation function required for Smad1 or Smad2 to stimulate transcription. The dual function of Smad4 in transcriptional activation underscores its central role in TGFβ signaling.
Keywords: TGFβ, activin, BMP, SMAD, signal transduction, transcriptional regulation, tumor suppressors
The transformation growth factor β (TGFβ) family signals a wide variety of biological responses through transcriptional regulation of genes encoding critical determinants of cell fate such as cell-cycle regulators (Pietenpol et al. 1990; Hannon and Beach 1994; Datto et al. 1995; Reynisdóttir et al. 1995; Iavarone and Massagué 1997), differentiation factors (Zentella and Massagué 1992), extracellular matrix proteins (Kerr et al. 1988; Rossi et al. 1988; Keeton et al. 1991; Inagaki et al. 1994) or homeobox gene products (Huang et al. 1995). Clues about the mechanism of transcriptional regulation by the TGFβ family have been provided by the recent discovery of the SMAD proteins as direct substrates of TGFβ family receptors and mediators of receptor signals to the nucleus. The founding member of the SMAD family is the product of the Drosophila gene Mad, which was identified as being required for signaling by the BMP homolog Decapentaplegic (Dpp) (Sekelsky et al. 1995). The discovery of Mad and the identification of nematode and vertebrate Mad-related gene products termed SMADs allowed the elucidation of a signaling pathway in which receptor phosphorylated SMADs move into the nucleus to activate transcription (for review, see Derynck and Zhang 1996; Wrana and Attisano 1996; Massagué et al. 1997).
The members of the SMAD family contain highly conserved amino- and carboxy-terminal domains (referred to as N and C domains, or MH1 and MH2 domains, respectively), separated by a more divergent linker region. On the basis of structural and functional criteria, the SMAD family can be divided into three subgroups. One group includes those SMADs that are direct receptor substrates. The second group includes co-SMADs, or SMADs that are not direct receptor substrates, but participate in signaling by associating with receptor-activated SMADs (for review, see Derynck and Zhang 1996; Wrana and Attisano 1996; Massagué et al. 1997). The third group includes proteins that interfere with SMAD activation and can be referred to as anti-SMADs (Hayashi et al. 1997; Topper et al. 1997).
Among the receptor-regulated SMADs, Smad1 and presumably its close homologs Smad5 and Smad9 are bone morphogenetic protein (BMP) receptor substrates and mediators of BMP signals in vertebrates (Graff et al. 1996; Hoodless et al. 1996; Lechleider et al. 1996; Liu et al. 1996; Thomsen 1996; Yingling et al. 1996; Kretzschmar et al. 1997a; Watanabe et al. 1997), whereas Mad in Drosophila (Newfeld et al. 1996; Wiersdorff et al. 1996) and Sma-2 and Sma-3 in Caenorhabditis elegans (Savage et al. 1996) mediate the actions of BMP-like factors in these organisms. Smad2 and Smad3 are substrates and mediators of related TGFβ and activin receptors in vertebrates (Baker and Harland 1996; Eppert et al. 1996; Graff et al. 1996; Macias-Silva et al. 1996; Zhang et al. 1996). Receptor-regulated SMADs are phosphorylated by the receptors at the carboxy-terminal end, which typically is an SSV/MS sequence (Macias-Silva et al. 1996; Kretzschmar et al. 1997a). The N and C domains of these SMADs interact with each other, causing auto inhibition (Baker and Harland 1996; Liu et al. 1996; Hata et al. 1997), and agonist-induced phosphorylation may relieve this inhibition.
On phosphorylation of the carboxy-terminal residues, SMADs move into the nucleus (Hoodless et al. 1996; Liu et al. 1996; Nakao et al. 1997a,b) where they participate in agonist-dependent transcriptional activation as originally inferred from studies showing that Smad1 fused to the GAL4 DNA-binding domain has transcriptional activity that is regulated by BMP (Liu et al. 1996). As further evidence for a direct role of SMADs in transcription, activin has been shown to activate the Mix.2 homeobox gene in Xenopus by inducing the association of Smad2 with FAST-1 (Chen et al. 1996), a nuclear protein that belongs to the winged helix transcription factor family and recognizes an activin responsive element (ARE) in the Mix.2 promoter (Huang et al. 1995). This has led to a model in which receptor-activated Smad2 translocates into the nucleus where it associates with a DNA-binding protein forming a transcriptional complex (Chen et al. 1996).
Signaling by receptor-regulated SMADs requires the participation of a co-SMAD. The only known member of this group in vertebrates is Smad4. Smad4 has the same overall structure as the receptor-regulated SMADs, but is more divergent and lacks the carboxy-terminal phosphorylation motif. Smad4 was originally identified as the product of the DPC4 tumor suppressor (Hahn et al. 1996) that is mutated or deleted in a high proportion of pancreatic cancers and in a smaller proportion of other cancers (Barrett et al. 1996; Hahn et al. 1996; Kim et al. 1996; Nagatake et al. 1996; Schutte et al. 1996). Inactivating missense mutations have been found both in the N and C domains in different tumor-derived DPC4 alleles. A similar distribution of inactivating mutations has been observed in another tumor suppressor in this family, Smad2 (Eppert et al. 1996; Riggins et al. 1996; Uchida et al. 1996). The C domain of SMADs has effector function in various biological assays (Baker and Harland 1996; Lagna et al. 1996; Liu et al. 1996) and mutations in this domain disrupt its activity (Shi et al. 1997). The crystal structure of the Smad4 C domain reveals that it is a trimer, and certain mutations disrupt the monomer interfaces of this trimer (Shi et al. 1997). Mutations in the N domain of Smad2 and Smad4 augment the autoinhibitory function of this domain (Hata et al. 1997) and may have additional effects (Kim et al. 1997).
A general requirement of Smad4 in TGFβ family signaling is suggested not only by the requirement of Smad4 for TGFβ responsiveness in mammalian cells (Lagna et al. 1996; Zhang et al. 1996) but also by its requirement for activin and BMP responses in Xenopus embryo (Lagna et al. 1996, Zhang et al. 1997). Thus, Smad4 is a shared co-Smad, participating in both TGFβ/activin and BMP signaling pathways. Smad4 associates with Smad1 or Smad2 when these SMADs are phosphorylated by specific receptors (Lagna et al. 1996) and is required for their signaling function (Lagna et al. 1996; Zhang et al. 1996, 1997). The interaction between the receptor-regulated SMADs and Smad4 is mediated by their C domains (Hata et al. 1997; Wu et al. 1997).
Although Smad4 is recognized as a central mediator for TGFβ signaling, it was not clear which step of the SMAD-signaling pathway requires Smad4 function. In this study we made use of a Smad4-deficient cell line and the FAST-1/Mix.2 system reconstituted in mammalian cells to investigate this problem. We show that Smad4 is not required for nuclear translocation of a receptor-activated SMAD but its association with a receptor-activated SMAD promotes binding of the SMAD complex to DNA and, furthermore, it provides an essential transcriptional activation function.
Results
Nuclear translocation of Smad1 and Smad2 does not require Smad4
To investigate which step of the TGFβ and BMP signaling pathways requires Smad4, we first determined whether ligand-dependent nuclear translocation of Smad1 and Smad2 can occur in the absence of Smad4. When expressed as amino-terminal epitope-tagged constructs in SW480.7 human colon carcinoma cells (Goyette et al. 1992), which lack Smad4 (Zhang et al. 1996), Smad1 and Smad2 were mostly cytoplasmic under basal conditions (Fig. 1A). Upon cotransfection with constitutively active BMP or TGFβ receptors and treatment with BMP4 or TGFβ, respectively, Smad1 or Smad2 accumulated in the nucleus (Fig. 1A). No further increases in the nuclear localization of Smad1 or Smad2 were detected on cotransfection of Smad4 (data not shown). These results indicate that Smad4 is not required for nuclear translocation of Smad1 or Smad2.
Figure 1.


(A) Nuclear translocation of Smad1 and Smad2 in the absence of Smad4. SW480.7 cells were transfected with Flag–Smad1 or Flag–Smad2 and stimulated with BMP or TGFβ by cotransfection with activated BMP or TGFβ receptor plus treatment with BMP4 or TGFβ1. Immunofluorescence staining with anti–Flag antibody was then performed. After agonist stimulation, Flag–Smad1 staining was nuclear in up to 60% of the cells and Flag–Smad2 staining in up to 76% of the cells. (B) Smad4 requires a receptor-activated Smad for nuclear translocation in response to agonists. Amino-terminal Flag-tagged Smad4 was transfected alone or with the indicated SMADs and stimulated with BMP or TGFβ as in A. Shown are Flag immunostaining of cells transfected with Flag-Smad4 alone (control), with Smad1 and stimulated with BMP, or with Smad2 and stimulated with TGFβ. (C) A quantitation of Smad4 nuclear staining under various conditions in one representative experiment. BMP and TGFβ refers to BMP or TGFβ stimulation by cotransfection with activated BMP or TGFβ receptor and treatment with BMP4 or TGFβ1, respectively.
Smad4 nuclear translocation depends on receptor activated SMADs
Transfected Smad4 is localized predominantly in the cytoplasm in SW480.7 cells (Fig. 1B) or COS cells (data not shown), as visualized by immunostaining via an amino-terminal Flag epitope tag. The nuclear level of Flag–Smad4 was increased only slightly by BMP4 (Fig. 1C). Cotransfection with Smad1 or Smad2 had little effect on the cellular localization of Flag–Smad4 (Fig. 1C). Cotransfection of Smad1 or Smad2 (or Smad3, which is expressed at a higher level than Smad2 under these conditions), however, enabled Flag–Smad4 to accumulate in the nucleus in response to BMP4 or TGFβ, respectively (Fig. 1B,C). In the absence of cotransfected Smad1, Smad2, or Smad3, the endogenous level of these proteins in SW480.7 cells may be too low to carry a detectable proportion of the overexpressed Smad4 into the nucleus in response to ligand. Because agonist-induced activation of Smad1 and Smad2 leads to their association with Smad4 (Lagna et al. 1996; Kretzschmar et al. 1997a), we surmise that the activated Smads bind Smad4 in the cytoplasm and carry it into the nucleus.
Reconstitution of a Xenopus activin/TGFβ transcriptional response in mammalian cells
To investigate whether Smad4 might be essential for the formation of a transcriptional complex, we first reconstituted, in mammalian cells, the FAST-1-dependent transcriptional response from Xenopus, which is the only example to date of a natural transcriptional complex formed in response to a TGFβ family member and involving SMADs. Activin signaling in Xenopus early embryos induces the formation of an activin response factor (ARF) that contains Smad2 and FAST-1 (Chen et al. 1996), binds to the activin response element ARE in the Mix.2 promoter, and activates the reporter construct A3CAT that contains three copies of the ARE (Huang et al. 1995). As shown in Figure 2, A3CAT responded to activin when cotransfected with FAST-1 but not when transfected alone into the lung epithelial cell line R1B/L17. This response was not increased when the activin type I receptor ActR-IA was overexpressed. Overexpression of ActR-IB caused a high basal activation of A3CAT (Fig. 2), suggesting that ActR-IB can mediate this response. Because the ActR-IB kinase domain shares 97% sequence similarity with that of the TGFβ type I receptor TβR-I (Cárcamo et al. 1994), we tested whether A3CAT could be activated via TβR-I. R-1B/L17 cells, which lack TβR-I but become responsive to TGFβ on transfection of this receptor (Wrana et al. 1994; Weis-Garcia and Massagué 1996), showed a FAST-1- and TβR-I-dependent activation of A3CAT by TGFβ (Fig. 2). In a recent study, Hayashi et al. (1997) have also shown that the A3CAT reporter gene can be activated by TGFβ. Because of the limited availability of activin, the remaining work was carried out by use of TGFβ as the agonist.
Figure 2.

Reconstitution of the Mix2–FAST-1 transcriptional response in mammalian cells. R1B/L17 cells were cotransfected with the Mix2 reporter A3CAT and the indicated FAST-1 and receptor vectors, then treated with activin (A) or TGFβ (β). Reporter CAT activity was then determined. Data are the average ±s.d. of triplicates.
C domains mediate the Smad2/FAST-1 interaction
The presence of both Smad2 and FAST-1 in Xenopus ARF has been inferred from gel shift assays by use of the ARE probe (Chen et al. 1996). Consistent with the gel shift result, we were able to detect a TGFβ-induced Smad2–FAST-1 association in R-1B/L17 cells by coimmunoprecipitation assay (Fig. 3A). The interaction is specific because TGFβ did not induce association of Smad1 with FAST-1 (data not shown). The C domain in Smad2 has effector activity that is inhibited by the N domain (Baker and Harland 1996; Liu et al. 1996; Hata et al. 1997). Receptor-mediated phosphorylation relieves this repression and additionally enhances the signaling function of the isolated C domain (Hata et al. 1997). In agreement with this, the isolated C domain of Smad2 interacted constitutively, albeit weakly, with FAST-1, and this interaction was increased by TGFβ addition (Fig. 3A). Mutation of the receptor phosphorylation sites at the carboxyl terminus of Smad2 (Macias-Silva et al. 1996; Kretzschmar et al. 1997a) prevented the binding to FAST-1 (Fig. 3A, AAMA construct). A Smad2 nonsense mutant lacking the entire phosphorylation region [Smad2(1–429)] and a phosphorylation-defective mutant [Smad2(D450E) (Eppert et al. 1996)] were also unable to associate with FAST-1 (Fig. 3A). The various constructs were controlled to be expressed at comparable levels (data not shown). To map the binding region on FAST-1, we constructed a panel of Myc epitope-tagged FAST-1 deletion mutants (Fig. 3B), and found that Smad2 associates with the C domain but not the DNA-binding domain of FAST-1 (Fig. 3C). Thus, Smad2 can associate with FAST-1 in mammalian cells in an agonist-dependent and specific manner, and this interaction is mediated via the C domains of the two proteins.
Figure 3.

FAST-1 interaction with Smad2. (A) TGFβ-induced FAST-1 association with Smad2. R1B/L17 cells were cotransfected with Myc–FAST-1, the indicated Flag–Smad2 derivatives, and TβR-I for TGFβ stimulation. After incubation with or without TGFβ, cell lysates were immunoprecipitated with anti-Flag antibody and the precipitates subjected to anti-Myc immunoblotting. Smad2(AAMA) contains Ser to Ala mutations in the carboxy-terminal SSMS sequence (Macias-Silva et al. 1996; Kretzschmar et al. 1997a). Smad2(C) refers to the Smad2 C domain (amino acids 248–467) (Hata et al. 1997). Smad2(1–429) and Smad2(D450E) are tumor-derived inactive Smad2 mutants (Eppert et al. 1996). (B) FAST-1 truncation constructs and their expression (as Myc-tagged constructs) in R-1B/L17 cells, as determined by anti-Myc immunoblotting analysis of cell lysates. (Solid rectangles) The winged helix DNA-binding domain (Chen et al. 1996). (C) Smad2 interaction with the FAST-1 C domain. R1B/L17 cells were cotransfected with Flag–Smad2, TβR-I, and the indicated Myc–FAST-1 derivatives. Cells were treated with TGFβ and analyzed by immunoprecipitation with anti-Myc antibody followed by anti-Flag immunoblotting.
The Smad2/FAST-1 interaction does not require Smad4
To determine whether FAST-1 and Smad2 can interact with each other in a Smad4-independent manner, we analyzed the association of Smad2 and FAST-1 in SW480.7 cells. As shown in Figure 4A, TGFβ induced the formation of the Smad2/FAST-1 complex in SW480.7 cells. The FAST-1 and Smad2 complex was unaffected when Smad4 was cotransfected (Fig. 4A). We also observed an interaction between Smad2 and FAST-1 in the yeast two-hybrid system and in vitro (data not shown). This indicates that the interaction between Smad2 and FAST-1 is direct and does not require Smad4.
Figure 4.
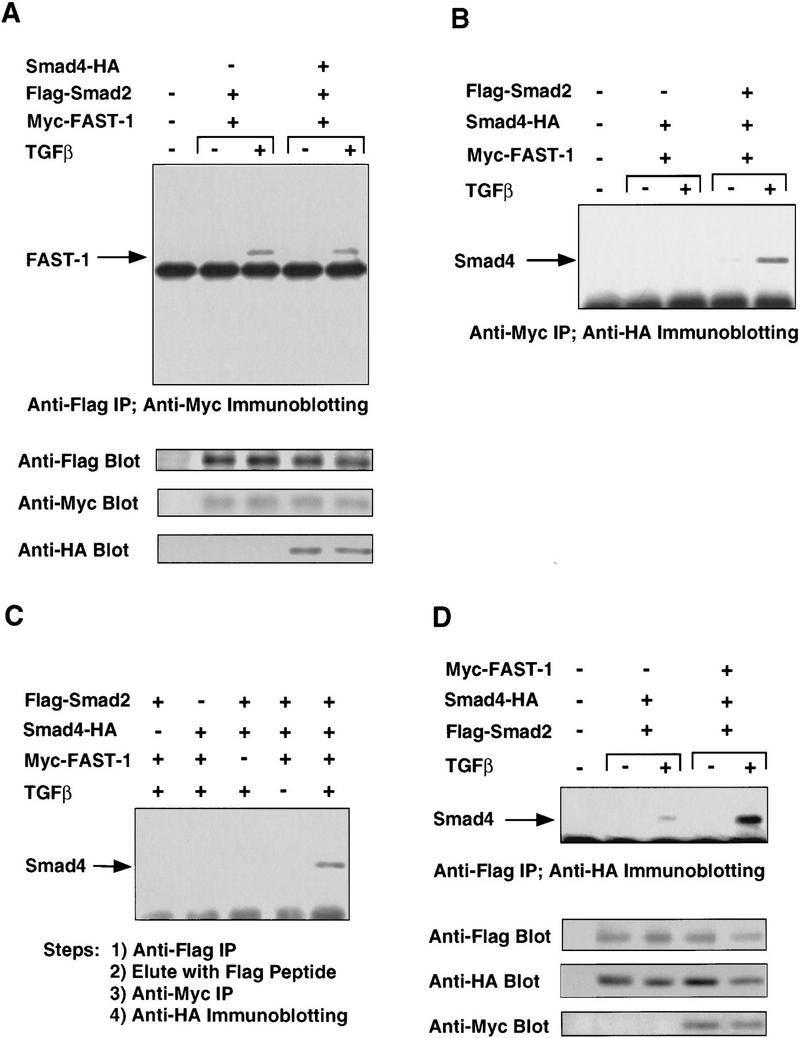
TGFβ-induced Smad2–Smad4–FAST-1 complex. (A) Smad2 interaction with FAST-1 does not require Smad4. SW480.7 cells were cotransfected with the indicated vectors and treated with TGFβ as indicated. Cell lysates were immunoprecipitated with anti-Flag antibody and the precipitates analyzed by anti-Myc immunoblotting. (B) Smad2-dependent interaction of Smad4 with FAST-1. SW480.7 cells were transfected with Myc–FAST-1, Flag–Smad2 and HA-tagged Smad4 vectors, and treated with TGFβ as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody and the precipitates analyzed by anti-HA immunoblotting. (C) Agonist-induced Smad2–Smad4–FAST-1 ternary complex. COS cells were cotransfected with the indicated vectors, and TβR-I(T204D) for TGF-β stimulation. Cells lysates were immunoprecipitated with agarose-immobilized anti-Flag antibody. Beads were eluted with Flag peptide, and the eluate precipitated by anti-myc antibody followed by anti-HA immunoblotting. (D) FAST-1 enhances the Smad2–Smad4 interaction. COS cells were cotransfected with the indicated vectors, and TβR-I(T204D) for TGFβ stimulation. Cell lysates were precipitated with anti-Flag antibody and the precipitates analyzed by anti-HA immunoblotting.
Smad4 forms a ternary complex with Smad2 and FAST-1
In contrast to Smad2, Smad4 did not form a stable complex with FAST-1 when cotransfected as epitope-tagged constructs in SW480.7 cells (Fig. 4B). TGFβ induced the association of Smad4 and FAST-1, however, when these constructs were cotransfected with Smad2 (Fig. 4B). To determine whether Smad2, Smad4, and FAST-1 are in the same complex, COS cells were cotransfected with Flag-tagged Smad2, HA-tagged Smad4, and Myc-tagged FAST-1. Cell lysates were immunoprecipitated with Flag antibody coupled to agarose beads. Immune complexes were eluted with Flag peptide and the eluate used in a second immunoprecipitation with Myc antibody. Finally, the precipitate was analyzed by immunoblotting with HA antibody. As shown in Figure 4C, a ternary complex was detected when cells were cotransfected with all three constructs and incubated with TGFβ, but not when any one construct was omitted or when TGFβ was not added. Furthermore, the TGFβ-induced association of Smad2 and Smad4 (Fig. 4D) was strongly enhanced when these constructs were cotransfected with FAST-1 (Fig. 4D). Similar results were obtained in R-1B/L17 cells (data not shown). FAST-1 therefore appears to stabilize the Smad2-Smad4 interaction.
Smad4 promotes DNA binding and transcriptional activation by the ternary complex
To determine whether Smad4 is required for the formation of a TGFβ-inducible DNA–protein complex, we assayed SW480.7 cell extracts for TGFβ-inducible binding activity by use of the ARE oligonucleotide probe. Nuclear extracts from cells transfected with FAST-1 alone yielded a small amount of Myc–FAST-1–ARE complex, which comigrated with a background band but was revealed by supershift with anti-Myc antibody (Fig. 5A; data not shown). Importantly, no TGFβ-inducible ARE-binding complex was detected in cells transfected with vector alone or FAST-1 alone, or Smad2 and Smad4 (Fig. 5A, lanes 1–6). Although FAST-1 and Smad2 can form a stable complex as shown in the coimmunoprecipitation assay (Figs. 3A and 4A), little or no new ARE-binding complex was observed when Smad2 was cotransfected with FAST-1 in these cells (Fig. 5A, lanes 7,8). Cotransfection of Smad4 with FAST-1 yielded limited levels of binding (lanes 9,10). FAST-1, Smad2, and Smad4 transfected together, however, yielded a high basal level of binding activity that was further increased by TGFβ (lanes 11,12). We also performed the same experiment with whole cell extracts. In agreement with a previous report (Chen et al. 1996), whole cell extracts from cells transfected with FAST-1 alone yield high levels of FAST-1–ARE complex (data not shown). Smad4 as well as Smad2, however, were required for formation of a TGFβ-inducible ARE-binding complex (data not shown), in agreement with the results by use of nuclear extracts.
Figure 5.

Smad4 is essential for the formation of a TGFβ-inducible DNA-binding complex and transcriptional activation. (A) A TGFβ-inducible DNA-binding complex requiring FAST-1, Smad2, and Smad4. SW480.7 cells were cotransfected with Myc–FAST-1, Flag–Smad2, and Smad4–HA and treated with TGFβ as indicated. Nuclear extracts were used to perform gel mobility shift assays by use of the 50-bp ARE oligonucleotide as a probe. (B) Smad4 is essential for activation of the Mix2 reporter A3CAT. SW480.7 cells were cotransfected with the A3CAT reporter gene (Huang et al. 1995), FAST-1, Smad2, and Smad4, and treated with TGFβ (▪) or not (□), as indicated. CAT activity was then analyzed. (C) FAST-1, Smad2, and Smad4 bind together to DNA. SW480.7 cells were cotransfected with the indicated vectors and treated with TGFβ. Supershift assays were performed by use of nuclear extracts with the indicated antibodies individually or in various combinations.
The ability of Smad4 to associate with Smad2 and FAST-1 in response to TGFβ and the requirement of Smad4 for the generation of an ARE-binding complex correlated with an essential role of Smad4 in trancriptional activation of the A3CAT reporter gene (Fig. 5B). Cotransfection of FAST-1 and Smad2 led to a very low level of activation of the A3CAT reporter gene in the presence of TGFβ in SW480.7 cells. Transcriptional activation of the A3CAT reporter gene occurred when Smad-4 and FAST-1 were cotransfected (Fig. 5B), which is consistent with the notion that these cells contain low level of endogenous Smad2-like activity. Cotransfection of Smad2, Smad4, and FAST-1 together led to a strong basal activation of A3CAT that was further increased by TGFβ addition (Fig. 5B).
To establish that Myc–FAST-1, Flag–Smad2, and Smad4–HA are all in the same ARE-binding complex, we used antibodies against the epitope tags of each of these constructs to supershift the complex (Fig. 5C, lanes 7–9). Combinations of any two or all three antibodies yielded supershifts of progressively lower electrophoretic mobility (Fig. 5C, lanes 10–13). The Flag antibody did not lead to any detectable supershift in nuclear or whole cell extracts from cells transfected with FAST-1 and Smad2 (data not shown) further indicating that the Smad2/FAST-1 complex has little ARE-binding activity in these assays. The results indicate that the Smad2/Smad4/FAST-1 ternary complex binds DNA as one entity.
The N domain of Smad4 contributes to DNA binding
Because the isolated C domain of Smad4 can stably interact with Smad2 (Hata et al. 1997), we analyzed whether it can generate an ARE-binding complex together with Smad2 and FAST-1. As shown in Figure 6A, the Smad4 C domain transfected with Smad2 and FAST-1 led to an ARE-binding complex with 20-fold lower efficiency than the full-length Smad4. Furthermore, Smad4 constructs with partial or complete deletion of the N domain were inefficient at supporting A3CAT activation (Fig. 6B). Taken together, these observations imply that the N domain of Smad4 contributes to binding of the ternary complex to the ARE. In contrast, the N domain of Smad2 is not essential for activation of the A3CAT reporter gene (Fig. 6B).
Figure 6.

(A) The amino-terminal region of Smad4 is required for DNA binding by the ternary complex. SW480.7 cells were cotransfected with Myc-FAST-1, Flag-Smad2, and either Smad4 or Smad4(240–552), both tagged with the HA epitope. Cells were treated with TGFβ as indicated. Gel shift experiment was performed with whole cell extracts. The expression level of Flag-Smad2, Smad4, and Smad4(240–552) was analyzed by immunoblotting with antibodies against the Flag or HA epitopes. (B) Both the N and the C domains of Smad4 are required to activate the A3CAT reporter gene. SW480.7 cells were cotransfected with A3CAT, FAST-1, the indicated full length or truncated Smad4 and Smad2 constructs. Cells were treated with TGFβ, and CAT activity was analyzed.
The C domain of Smad4 is required for transcriptional activation by receptor-regulated Smads
To determine whether Smad4 has additional roles in the transcriptional complex besides promoting binding to DNA, we used a transcription assay in which Smad binding to a GAL4 reporter gene is ensured by a DNA-binding domain of GAL4 fused amino-terminally to Smads. In this assay, GAL4–Smad1 (Liu et al. 1996) and GAL4–Smad2 (F. Liu et al., unpubl.) activate transcription in response to BMP and TGFβ/activin, respectively, in R1B/L17 cells that contain wild-type Smad4. To determine whether the transcription activities of GAL4–Smad1 and GAL4–Smad2 are dependent on Smad4, we performed the same assay in SW480.7 cells that lack endogenous Smad4. Figure 7A shows that GAL4–Smad1 and GAL4–Smad2 had very low activity in SW480.7 cells even in the presence of ligand stimulation, but were greatly stimulated by BMP or TGFβ when cotransfected with wild-type Smad4. Importantly, the isolated C domain of Smad4 [Smad4(240–552) construct] was as effective as the full-length Smad4 in restoring agonist-induced transcriptional activation by GAL4–Smad2 (Fig. 7B) or GAL4–Smad1 (data not shown). In contrast, a Smad4 construct with a small carboxy-terminal truncation [Smad4(1–514)] was unable to rescue these responses (Fig. 7B). Taken together, these results indicated that the transcriptional activity of Smad1 and Smad2 is dependent on Smad4, and the C domain of Smad4 provides this activity.
Figure 7.
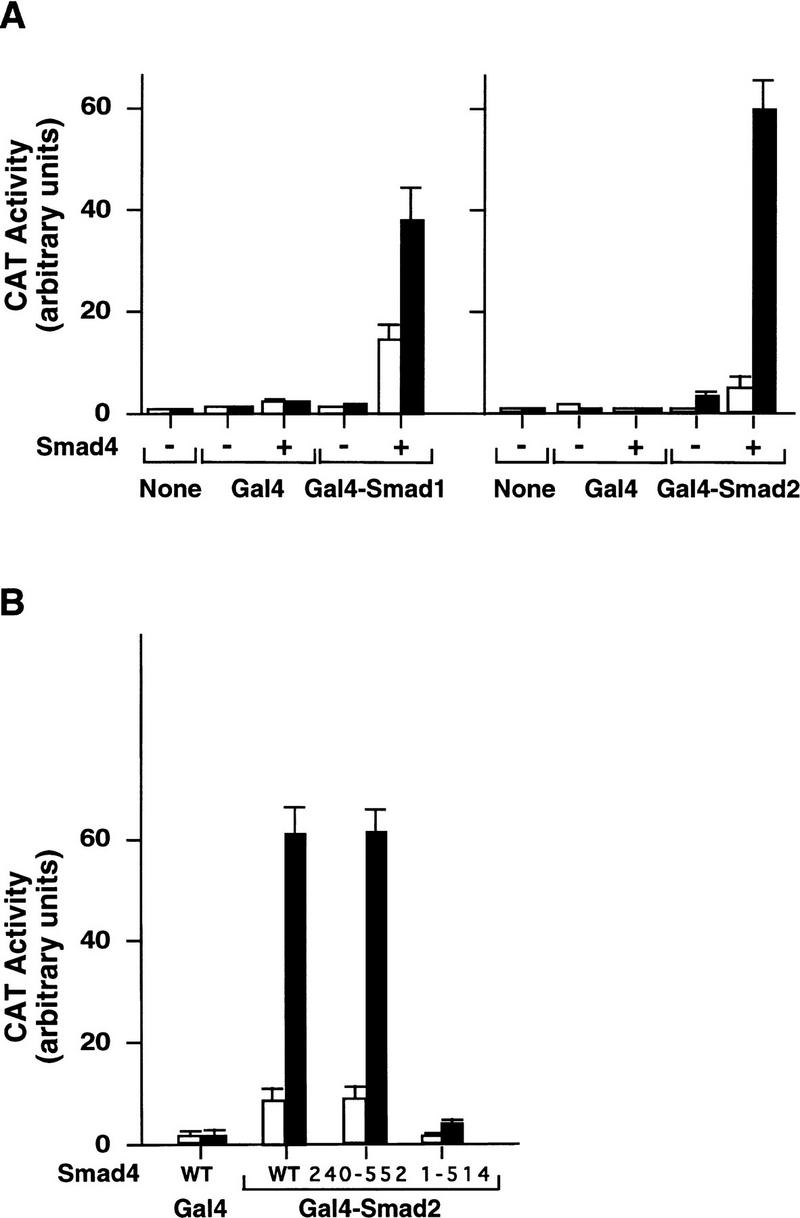
Ligand-induced transcriptional activity of GAL4–Smad1 and GAL4–Smad2 depends on the C domain of Smad4. SW480.7 cells were cotransfected with GAL4–Smad1, GAL4–Smad2, wild-type Smad4, Smad4(240–552), or Smad4(1–514) along with the GAL4 reporter gene G1E1BCAT (Lillie and Green 1989). Cells were incubated with 2 nm BMP4 (▪) in A (left) or without (□), or with 500 pm TGFβ in A (right) and B, as indicated, and CAT activity was then analyzed.
Discussion
Previous work has shown that receptor-activated SMADs move to the nucleus to activate transcription (Chen et al. 1996; Hoodless et al. 1996; Liu et al. 1996; Nakao et al. 1997a,b), and their signaling function somehow requires Smad4 (Lagna et al. 1996; Zhang et al. 1996, 1997) with which the receptor-activated SMADs form a complex (Lagna et al. 1996). By examining the requirement of Smad4 in three basic steps of the SMAD pathway, namely, nuclear translocation, binding to DNA, and transcriptional activation, the present study sheds light into the role of Smad4 in this process. The evidence presented here suggests that Smad4 is dispensable for nuclear translocation of receptor-activated SMADs, but it plays a role in their binding to DNA and is essential for their ability to activate transcription.
Nuclear translocation of Smad4 by receptor-activated SMADs
Translocation of receptor-regulated SMADs into the nucleus is a highly controlled process. Phosphorylation of Smad1 at carboxy-terminal sites by the BMP receptor (Kretzschmar et al. 1997a) and phosphorylation of Smad2 at similar sites by the TGFβ receptor (Macias-Silva et al. 1996) mediate translocation of these SMADs into the nucleus, whereas phosphorylation of Smad1 by mitogen-activated protein (MAP) kinase in response to mitogenic factors inhibits nuclear translocation (Kretzschmar et al. 1997b). Phosphorylation by TGFβ family receptors also endows SMADs with the ability to associate with Smad4 (Kretzschmar et al. 1997a). Because Smad4 is required for signal transduction by diverse SMAD pathways, it was possible that the association with Smad4 might mediate nuclear translocation of receptor-activated SMADs. Investigation of this question in the present studies by use of the Smad4-deficient cell line SW480.7 clearly indicates that Smad4 is not required for the nuclear translocation of Smad1 or Smad2 in response to their respective agonists. Nuclear translocation of receptor-activated SMADs is clearly observed under these conditions and is not further enhanced by transfection of Smad4. Furthermore, a Smad4 construct containing an amino-terminal epitope tag is cytoplasmic and remains in the cytoplasm upon BMP or TGFβ stimulation when transfected alone. When cotransfected with receptor-regulated SMADs, this Smad4 construct is translocated into the nucleus. These results suggest that receptor-activated SMADs can translocate into the nucleus independently of Smad4 and can take Smad4 into the nucleus after associating with it in the cytoplasm.
Formation of a ternary complex
Is Smad4 part of a transcriptional complex with receptor-regulated SMADs? To address this question, we have reconstituted in mammalian cells the FAST-1-dependent transcriptional response described by Whitman and colleagues in Xenopus (Chen et al. 1996). By use of gel mobility shift assays, it was inferred previously that upon activation by the receptor, Smad2 associates with FAST-1 in the nucleus (Chen et al. 1996). By transfecting FAST-1 and Smad2 into TGFβ responsive lung epithelial cells, we provide evidence for this interaction on the basis of coimmunoprecipitation of a Smad2/FAST-1 complex. Using a panel of Smad2 and FAST-1 deletion mutants, we have determined that the C domains of the two proteins mediate this interaction. In FAST-1, this Smad2-binding domain is separate from the previously identified DNA-binding domain (Chen et al. 1996). In Smad2, the C domain is also involved in the formation of homo-oligomers and in the interaction with Smad4 (Hata et al. 1997; Wu et al. 1997). The C domain is highly conserved among SMADs and its tertiary structure is known in Smad4 (Shi et al. 1997). The structure of the Smad4 C domain has several solvent-exposed regions that are conserved in Smad2 and may be involved in interactions with other proteins. One of these proteins, in the case of Smad2, may be FAST-1.
Formation of the Smad2/FAST-1 complex in response to TGFβ does not require Smad4 and is not enhanced by overexpression of Smad4, as determined in Smad4-deficient cells. When Smad4 is present, however, it forms a ternary complex with Smad2 and FAST-1, as determined by coimmunoprecipitation of the three proteins. The interaction of Smad4 with FAST-1 requires Smad2. The evidence, therefore, suggests that upon phosphorylation by the TGFβ receptor, Smad2 associates with Smad4 forming a complex that moves into the nucleus where it binds FAST-1. Recently, Chen et al. (1997) also observed that Smad4 is in a complex with FAST-1 and Smad2 from injected Xenopus embryos.
Interaction with DNA: Involvement of the Smad4 N domain
Our gel mobility shift assays and A3CAT reporter gene assays indicate that formation of the Smad2/Smad4/FAST-1 complex is essential for optimal binding to the ARE and transcriptional activation of A3CAT. The DNA-bound complex detected in the presence of Smad2, Smad4, and FAST-1 contains these three proteins, as determined by gel mobility supershift assays with the appropriate antibodies. Little or no TGFβ-inducible ARE-binding complex or A3CAT activation were observed in cells expressing Smad2 and FAST-1 but devoid of Smad4. Smad4 is therefore required for optimal binding of the ternary complex to the ARE.
Previous work has shown that the C domain of Smad4 is sufficient for an interaction with receptor-activated Smad2 (Hata et al. 1997). The present results, however, indicate that the C domain of Smad4 is not sufficient to promote binding to the ARE and activation of A3CAT. The amino-terminal region of Smad4 is required for optimal binding of the ternary complex to DNA. The inability of the Smad4 C domain to activate transcription in SW480.7 cells was observed not only with the A3CAT reporter but also with the TGFβ-responsive p3TP–luciferase reporter (F. Liu et al., unpubl.). A previous study showed that transfection of a Smad4 C domain can lead to transcriptional activation of p3TP-luciferase in R1B/L17 cells that contain wild-type of Smad4 (Hata et al. 1997). It should be noted that these results are not incompatible because the Smad4 C domain could act in R-1B/L17 cells by associating with the endogenous Smad4.
The mechanism by which Smad4 promotes interaction of the ternary complex with DNA remains to be elucidated. FAST-1 can bind to ARE directly in yeast (Chen et al. 1996), and FAST-1/ARE complexes have been detected with whole cell extracts from FAST-1-transfected cells (Chen et al. 1996; F. Liu et al., unpubl.). Therefore, Smad4 might act by enhancing the intrinsic DNA-binding activity of FAST-1 in the ternary complex. Although Smad4 does not appear to stabilize the interaction between FAST-1 and Smad2 in solution, we cannot exclude the possibility that the Smad4 N domain stabilizes the interaction between FAST-1 and Smad2 on DNA. Alternatively, one attractive possibility is that the Smad4 N domain has affinity for DNA. This possibility is supported by the recent observation that the homologous amino-terminal region of Drosophila Mad can bind directly to a specific DNA sequence in the promoter of the Dpp target gene vestigial (Kim et al. 1997). Although we were unable to detect DNA binding by Smad4 alone, this could be as a result of a low affinity of Smad4 for DNA. Smad4 might contact DNA only after being recruited into the ternary complex with Smad2 and FAST-1. These possibilities warrant further investigation.
The Smad4 N domain has been shown to inhibit the ability of the C domain to associate with Smad2 (Hata et al. 1997). The Smad4 N domain, however, has been shown recently to enhance agonist-dependent signaling (de Caestecker et al. 1997). In light of this and the present results, it is possible that Smad4 N and C domains interact with each other in the basal state in a reciprocally inhibitory fashion.
Trancriptional activation requiring the Smad4 C domain
If the DNA-binding requirement of a SMAD complex is bypassed, is Smad4 still required for transcriptional activation? We investigated this question by fusing the GAL4 DNA-binding domain to receptor regulated SMADs to provide DNA-binding activity independent of FAST-1 or other cofactors. In cells containing endogenous Smad4, these GAL4–Smad1 and GAL4–Smad2 fusions can mediate agonist-dependent activation of a GAL4 reporter gene (Liu et al. 1996; Hayashi et al. 1997). In Smad4 deficient SW480.7 cells, however, these constructs are inactive and do not mediate transcriptional activation in response to BMP4 or TGFβ. Transcriptional activation under these conditions is rescued by cotransfection of full-length Smad4 and, importantly, by cotransfection of the Smad4 C domain. Thus, when the DNA-binding function is supplied by an ectopic DNA-binding domain, receptor-activated SMADs still require Smad4 for transcriptional activation. In this case, the C domain of Smad4 is sufficient to provide this rate-limiting function. It is possible that the Smad4 C domain, either alone or jointly with the associated C domain of Smads 1 or 2, activates basal transcription machinery.
In conclusion, the present results shed light on the role of Smad4 in the SMAD-signaling pathway. Collectively, the evidence suggests a model (Fig. 8) in which a receptor-regulated SMAD, such as Smad1 or Smad2, moves into the nucleus where it can associate with a sequence-specific DNA-binding protein such as FAST-1 without requiring Smad4. Association of Smad2 with Smad4, however, presumably in the cytoplasm, and formation of a ternary complex with FAST-1 in the nucleus are required for optimal binding of this complex to DNA and for transcriptional activation. The contributions to DNA binding and transcriptional activation appear to be made through distinct regions of Smad4. The dual function of Smad4 in transcriptional regulation highlights its central role in TGFβ signaling.
Figure 8.

A model depicting the participation of Smad4 in a transcriptional complex with Smad2 and FAST-1, extending the previous model of Chen et al. (1996). Upon phosphorylation by the activated receptor, Smad2 translocates to the nucleus. Smad4 associates with Smad2 through their C domains but is not required for this translocation. In the nucleus, the Smad2/Smad4 complex associates with FAST-1. In this ternary complex, the N domain of Smad4 promotes binding to DNA, whereas the C domain of Smad4 is essential for activation of transcription.
Materials and methods
Constructs
Smad2 cDNA (GenBank accession no. AF027964) was obtained by sequencing and ligation of human EST cDNA clones. GAL4–Smad2 was constructed by inserting the human Smad2 cDNA into the pSG424 vector (Sadowski and Ptashne 1989). Smad3 cDNA and the various Myc tagged FAST-1 derivatives were constructed in the CS2 vector (Turner and Weintraub 1994). All other constructs have been described (Liu et al. 1996; Hata et al. 1997; Kretzschmar et al. 1997a).
Immunofluorescence
SW480.7 cells were transfected with lipofectin reagent (GIBCO BRL) for 20 hr and then plated into chamber slides. BMP or TGFβ stimulation was provided by cotransfecting the activated BMP type I receptor, BMPR-IA(Q233D) (Hoodless et al. 1996) or the activated TGFβ type I receptor, TβR-I(T204D) (Wieser et al. 1995), and incubated with 2 nm BMP4 or 1 nm TGFβ1 for 1 hr. Cells were then fixed by methanol/acetone. Immunostaining was performed by incubation with the M2 Flag antibody (Eastman Kodak) at 1 μg/ml for 1 hr followed by incubation with the FITC-conjugated goat anti-mouse antibody (1 : 100) (Jackson Immunologicals) for 1 hr.
Immunoprecipitation and immunoblot assay
R1B/L17 and COS cells were transfected with DEAE–dextran, and SW480.7 cells were transfected with lipofectin. Cells were induced with 500 pm TGFβ1 for 1 hr and then lysed in 1 ml of TNE buffer [10 mm Tris (pH 7.8), 150 mm NaCl, 1 mm EDTA, 1% NP-40] in the presence of protease inhibitors. Immunoprecipitation was performed by incubation with the M2 Flag monoclonal antibody (Eastman Kodak) or with 9E10 Myc monoclonal antibody (Santa Cruz Biotechnology) for 1 hr. Immunoprecipitates were separated in an 8% SDS-PAGE (except for Fig. 3B, in a 14% SDS-PAGE) and transferred to a PVDF membrane. Immunoblotting was performed by use of antibodies against the epitopes Flag, Myc, or HA (12CA5 antibody, Boehringer Mannheim), followed by incubation with the HRP-conjugated goat anti-mouse antibody and detected by chemiluminescence (Amersham).
To detect the ternary complex, transfected COS cells were lysed in 1 ml of TNE buffer and immunoprecipitated with agarose-coupled M2 Flag antibody for 3.5 hr. The precipitates were eluted twice with 250 μg/ml of Flag peptide (Eastman Kodak) and the eluate diluted with 0.8 ml of TNE buffer, immunoprecipitated with anti-Myc antibody for 6 hr, and subjected to anti-HA immunoblotting.
Gel mobility shift and supershift assay
SW480.7 cells were transfected with lipofectin and treated with TGFβ for 18 hr. Nuclear extracts were prepared by resuspending cells in a hypotonic buffer containing 10% glycerol, 20 mm HEPES (pH 7.9), 0.1 mm EDTA, 50 mm KCl, 2 mm DTT, 0.15 mm spermine, 0.5 mm spermidine, and protease and phosphatase inhibitors. Cell suspensions were frozen in liquid nitrogen and then thawed on ice. The nuclei fraction was recovered by centrifugation and then incubated on ice with a hypertonic buffer containing 20% glycerol, 20 mm HEPES (pH 7.9), 0.1 mm EDTA, 600 mm KCl, and 2 mm DTT with protease and phosphatase inhibitors. After centrifugation, the supernatant was recovered as nuclear extract. Whole cell extract was prepared by freezing cell pellet in liquid nitrogen and lysis of the frozen cell pellet in a buffer containing 10 mm HEPES (pH 7.9), 300 mm NaCl, 0.1 mm EGTA, 20% glycerol, and 0.2% NP-40 with protease inhibitors and phosphatase inhibitors. DNA-binding assays were performed essentially as described (Huang et al. 1995) with 1 ng of radiolabeled ARE probe. For antibody supershift assays, extracts were incubated for 10 min in binding buffer, then 15 min with the probe, and 10 min with antibodies. DNA–protein complexes were resolved on a 4% (40 : 1) polyacrylamide gels containing 1% glycerol.
CAT assays
SW480.7 cells or R1B/L17 cells were transfected with DEAE–dextran and treated with 2 nm BMP4 (Genetics Institute), 2.5 nm activin A (National Hormone and Pituitary Program, National Institute of Diabetes and Digestive Kidney Diseases), or 500 pm TGFβ1 (R&D Systems) for 18–22 hr. CAT activity was quantitated by scintillation counting or PhosphorImager analysis.
Acknowledgments
We gratefully acknowledge M. Whitman for the FAST-1 cDNA and the A3CAT reporter gene, Y. Zhang and R. Derynck for the Smad3 cDNA, H. Ge, C. Fasching, and E. Stanbridge for SW480.7 cells, Genetics Institute for BMP4, the National Hormone and Pituitary Program for activin, K. Manova for assistance with cell imaging, A. Hata and R.S. Lo for constructs, and J. Doody, R.S. Lo, D. Wotton, Y.G. Shi, C. Zhang, and Y.-G. Chen for helpful discussions and assistance. This work was supported by grants from the National Institutes of Health to J.M. and to the Memorial Sloan-Kettering Cancer Center. F.L. and C.P. are recipients of postdoctoral fellowships from the Jane Coffin Childs Memorial Fund for Medical Research and the International Agency for Research on Cancer, respectively. J.M. is an investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL j-massague@ski.mskcc.org; FAX (212) 717-3298.
References
- Baker J, Harland RM. A novel mesoderm inducer, mMadr-2, functions in the activin signal transduction pathway. Genes & Dev. 1996;10:1880–1889. doi: 10.1101/gad.10.15.1880. [DOI] [PubMed] [Google Scholar]
- Barrett MT, Schutte M, Kern SE, Reid BJ. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res. 1996;56:4351–4353. [PubMed] [Google Scholar]
- Cárcamo J, Weis FMB, Ventura F, Wieser R, Wrana JL, Attisano L, Massagué J. Type I receptors specify growth inhibitory and transcriptional responses to TGF-β and activin. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner of MAD proteins in TGF-β signaling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Datto MB, Yu Y, Wang X-F. Functional analysis of the transforming growth factor β responsive elements in WAF1/Cip1/p21 promoter. J Biol Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- de Caestecker MP, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts AB, Lechleider RJ. Characterization of functional domains within Smad4/DPC4. J Biol Chem. 1997;272:13690–13696. doi: 10.1074/jbc.272.21.13690. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y. Intracellular signaling: The Mad way to do it. Curr Biol. 1996;6:1226–1229. doi: 10.1016/s0960-9822(96)00702-6. [DOI] [PubMed] [Google Scholar]
- Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui L-C, Bapat B, Gallinger S, Andrulis IL, Thomsen GH, Wrana JL, Attisano L. MADR2 maps to 18q21 and encodes a TGFβ-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Goyette MC, Cho K, Fasching CL, Levy DB, Kinzler KW, Paraskeva C, Vogelstein B, Stanbridge EJ. Progression of colorectal cancer is associated with multiple tumor suppressor gene defects but inhibition of tumorigenicity is accomplished by correction of any single defect via chromosome transfer. Mol Cell Biol. 1992;12:1387–1395. doi: 10.1128/mcb.12.3.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JM, Bansal A, Melton DA. Xenopus Mad proteins transduce distinct subsets of signals for the TGFβ superfamily. Cell. 1996;85:479–487. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate the tumour suppressors Smad2 and Smad4. Nature. 1997;388:82–86. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Huang H-C, Murtaugh LC, Vize PD, Whitman M. Identification of a potential regulator of early transcriptional responses to mesoderm inducers in the frog embryo. EMBO J. 1995;14:5965–5973. doi: 10.1002/j.1460-2075.1995.tb00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A, Massagué J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Truter S, Ramirez F. TGF-β stimulates a2(I) collagen gene expression through a cis-acting element that contains an Sp1-binding site. J Biol Chem. 1994;269:14828–14834. [PubMed] [Google Scholar]
- Keeton MR, Curriden SA, van Zonneveld A-J, Loskutoff D. Identification of regulatory sequences in the type 1 plasminogen activator inhibitor gene responsive to transforming growth factor β. J Biol Chem. 1991;266:23048–23052. [PubMed] [Google Scholar]
- Kerr LD, Olashaw NE, Matrisian LM. Transforming growth factor β1 and cAMP inhibit transcription of epidermal growth factor- and oncogene-induced transin RNA. J Biol Chem. 1988;263:16999–17005. [PubMed] [Google Scholar]
- Kim SK, Fan Y, Papadimitrakopoulou V, Clayman G, Hittelman WN, Hong WK, Lotan R, Mao L. DPC4, a candidate tumor suppressor gene, is altered infrequently in head and neck squamous cell carcinoma. Cancer Res. 1996;56:2519–2521. [PubMed] [Google Scholar]
- Kim J, Johnson K, Chen HJ, Carroll S, Laughon A. Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature. 1997;388:304–308. doi: 10.1038/40906. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Liu F, Hata A, Doody J, Massagué J. The TGF-β mediator Smad1 is directly phosphorylated and functionally activated by the BMP receptor kinase. Genes & Dev. 1997a;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signaling pathways converge on the TGFβ family mediator Smad1. Nature. 1997b;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGFβ signaling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Lechleider RJ, de Caestecker MP, Dehejia A, Polymeropoulos MH, Roberts AB. Serine, phosphorylation, chromosomal localization and TGFβ signal transduction by human bsp-1. J Biol Chem. 1996;271:17617–17620. doi: 10.1074/jbc.271.30.17617. [DOI] [PubMed] [Google Scholar]
- Lillie JW, Green MR. Transcription activation by the adenovirus E1A protein. Nature. 1989;338:39–44. doi: 10.1038/338039a0. [DOI] [PubMed] [Google Scholar]
- Liu F, Hata A, Baker J, Doody J, Cárcamo J, Harland R, Massagué J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFβ receptor and phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massagué J, Hata A, Liu F. TGFβ signaling through the Smad pathway. Trends Cell Biol. 1997;7:187–192. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- Nagatake M, Takagi Y, Osada H, Uchida K, Mitsudomi T, Saji S, Shimokata K, Takahashi T, Takahashi T. Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:2718–2720. [PubMed] [Google Scholar]
- Nakao A, Röijer E, Imamura T, Souchelnytskyi S, Stenman G, Heldin C-H, ten Dijke P. Identification of Smad2, a human Mad-related protein in the transforming growth factor-β signaling pathway. J Biol Chem. 1997;272:2896–2900. doi: 10.1074/jbc.272.5.2896. [DOI] [PubMed] [Google Scholar]
- Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J-i, Heldin C-H, Miyazono K, ten Dijke P. TGF-β receptor-mediated signaling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newfeld SJ, Chartoff EH, Graff JM, Melton DM, Gelbart WM. Mothers against dpp encodes a conserved cytoplasmic protein required in DPP/TGFβ responsive cells. Development. 1996;122:2099–2108. doi: 10.1242/dev.122.7.2099. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Stein RW, Moran E, Yacuik P, Schlegel R, Lyons RM, Pittelkow RM, Münger K, Howley PM, Moses HL. TGF-β1 Inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming protein with pRB binding domains. Cell. 1990;61:777–785. doi: 10.1016/0092-8674(90)90188-k. [DOI] [PubMed] [Google Scholar]
- Reynisdóttir I, Polyak K, Iavarone A, Massagué J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arest in response to TGF-β. Genes & Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, Willson JKV, Markowitz SD, Kinzler KW, Vogelstein B. Mad-related genes in the human. Nature Genet. 1996;13:347–349. doi: 10.1038/ng0796-347. [DOI] [PubMed] [Google Scholar]
- Rossi P, Karsenty G, Roberts AB, Roche NS, Sporn MB, de Crombrugghe B. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-β. Cell. 1988;5:405–414. doi: 10.1016/s0092-8674(88)80033-3. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Ptashne M. A vector for expressing GAL4(1–147) fusion in mammalian cells. Nucleic Acid Res. 1989;17:7539. doi: 10.1093/nar/17.18.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage C, Das P, Finelli A, Townsend SR, Sun C-Y, Baird SE, Padgett RW. Caenorhabditis elegans genes sma-2, sma-3 and sma-4 define a conserved family of transforming growth factor β pathway components. Proc Natl Acad Sci. 1996;93:790–794. doi: 10.1073/pnas.93.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Hedrik L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero CA, Meltzer PS, Hahn SA, Kern SE. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Sekelsky JJ, Newfeld SJ, Raftery LA, Chartoff EH, Gelbart WM. Genetic characterization and cloning of Mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics. 1995;139:1347–1358. doi: 10.1093/genetics/139.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. doi: 10.1038/40431. [DOI] [PubMed] [Google Scholar]
- Thomsen G. Xenopus mothers against decapentaplegic is an embryonic ventralizing agent that acts downstream of the BMP-2/4 receptor. Development. 1996;122:2359–2366. doi: 10.1242/dev.122.8.2359. [DOI] [PubMed] [Google Scholar]
- Topper JN, Cai J, Qiu Y, Anderson KR, Xu YY, Deeds JD, Feeley R, Gimeno CJ, Woolf EA, Tayber O, Mays GG, Sampson BA, Schoen FJ, Gimbrone MA, Falb D. Vascular MADs: Two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc Natl Acad Sci. 1997;94:9314–9319. doi: 10.1073/pnas.94.17.9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Weintraub H. Expression of achaete–scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes & Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- Uchida K, Nagatake M, Osada H, Yatabe Y, Kondo M, Mitsudomi T, Masuda A, Takahashi T, Takahashi T. Somatic in vivo alterations of the JV18-1 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:5583–5585. [PubMed] [Google Scholar]
- Watanabe TK, Suzuki M, Omori Y, Hishigaki H, Horie M, Kanemoto N, Fujiwara T, Nakamura Y, Takahashi E. Cloning and characterization of a novel member of the human Mad gene family (MADH6) Genomics. 1997;42:446–451. doi: 10.1006/geno.1997.4753. [DOI] [PubMed] [Google Scholar]
- Weis-Garcia F, Massagué J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 1996;15:276–289. [PMC free article] [PubMed] [Google Scholar]
- Wiersdorff V, Lecuit T, Cohen SM, Mlodzik M. Mad acts downstream of Dpp receptors, revealing differential requirement for dpp signaling in initiation and propagation of morphogenesis in the Drosophila eye. Development. 1996;122:2153–2162. doi: 10.1242/dev.122.7.2153. [DOI] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massagué J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L. MAD-related proteins in TGFβ signaling. Trends Genet. 1996;12:493–496. doi: 10.1016/s0168-9525(96)30109-1. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Wu R-Y, Zhang Y, Feng X-H, Derynck R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol Cell Biol. 1997;17:2521–2528. doi: 10.1128/mcb.17.5.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling JM, Das P, Savage C, Zhang C, Padgett RW, Wang X-F. Mammalian Dwarfins are phosphorylated in response to TGF-β and are implicated in control of cell growth. Proc Natl Acad Sci. 1996;93:8940–8944. doi: 10.1073/pnas.93.17.8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentella A, Massagué J. TGF-β induces myoblast differentiation in the presence of mitogens. Proc Natl Acad Sci. 1992;89:5176–5180. doi: 10.1073/pnas.89.11.5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng X-H, Wu R-Y, Derynck R. Receptor-associated Mad homologs synergize as effectors of the TGFβ response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Musci T, Derynck R. The tumor suppressor Smad4/DPC4 as a central mediator of Smad function. Curr Biol. 1997;7:270–276. doi: 10.1016/s0960-9822(06)00123-0. [DOI] [PubMed] [Google Scholar]