

Abstract
Immobilizing a protein, that is fully compatible with the patient, on the surface of a biomedical device should make it possible to avoid adverse responses such as inflammation, rejection, or excessive fibrosis. A surface that strongly binds and does not denature the compatible protein is required. Hydrophilic surfaces do not induce denaturation of immobilized protein but exhibit a low binding affinity for protein. Here, we describe an energetic ion-assisted plasma process that can make any surface hydrophilic and at the same time enable it to covalently immobilize functional biological molecules. We show that the modification creates free radicals that migrate to the surface from a reservoir beneath. When they reach the surface, the radicals form covalent bonds with biomolecules. The kinetics and number densities of protein molecules in solution and free radicals in the reservoir control the time required to form a full protein monolayer that is covalently bound. The shelf life of the covalent binding capability is governed by the initial density of free radicals and the depth of the reservoir. We show that the high reactivity of the radicals renders the binding universal across all biological macromolecules. Because the free radical reservoir can be created on any solid material, this approach can be used in medical applications ranging from cardiovascular stents to heart-lung machines.
Keywords: unpaired electrons, protein immobilization, antibody array, biosensor, microarray
Unfavorable responses to biomedical devices necessitate intervention, prolong recovery after surgery, and often require the surgical removal or “revision” of implanted devices. Unfavorable responses include: inflammation; encapsulation in a thick layer of fibrotic tissue, known as the foreign-body response; and infection in the form of a colony of adherent bacterial cells or biofilm. The cost of intervention and prolonged recovery, including direct cost and that associated with further medical complications, is so high that any advances that reduce unfavorable responses are of great importance.
Exposed surfaces of medical devices that come into contact with body fluids typically promote nonspecific binding of molecules (mainly proteins) that results in a range of complications, such as the induction of clots and the activation of cellular immune responses. An inflammatory response, characterized by increases in the expression of at least ten leukocyte cluster of differentiation antigens (1), has been observed during cardiopulmonary bypass using heart-lung machines in patients undergoing heart transplantation. Attempts have been made to minimize these inflammatory responses to the surfaces that exchange the blood gases by cloaking them with proteins such as human plasma-derived albumin. These attempts have been largely unsuccessful (2), presumably because the albumin failed to bind to the surface during exposure to blood flow or was denatured.
Foreign-body responses to implantable devices result from the host’s identification of the implanted material as foreign. When attempts fail to break down and eliminate the material, a thick layer of fibrous scar tissue is formed around the implant to isolate it from the body. Such a layer of tissue may limit the effectiveness of the device, requiring its removal (3). For example, thick fibrous tissue at the electrodes of an implant used to stimulate neurons increases electrical impedance, resulting in prohibitive power consumption. Similarly, the formation of a fibrocellular coating inside a stent will reduce the flow rate through a coronary artery.
Serious infection results when invading bacteria or fungi attach to an implant surface and form a biofilm (4). The biofilm consists of the adherent cells embedded and proliferating in a self-synthesized extracellular matrix. Bacterial cells in a biofilm are often resistant to treatment with conventional antibiotics, so that in most instances the device must be surgically removed (5).
Inflammation of coronary vessel walls is the primary cause of coronary artery disease and involves increased extracellular protein (fibrin) deposits (6) and elevated monocytes, macrophages, and neutrophils, as well as platelet infiltration (7). There is strong evidence that bare stents, used to treat the condition, produce increased inflammation (7). A surface coating for stents that enables a covalent (nonreleasing) attachment of a patient’s own native plasma protein would be a valuable method of reducing the inflammatory potential of stents.
A surface that covalently binds but does not denature protein could effectively mask the original implant surface, preventing its recognition as foreign. Hydrophilic surfaces are well known to preserve the native conformations of proteins (8) by stabilizing polar amino acid side chains on the exterior of the molecule. However, hydrophilic surfaces typically do not retain adsorbed protein molecules (9). In fact, highly hydrophilic surfaces such as poly(ethylene glycol) are known for their protein-repellent properties (10). We achieve immobilization on a hydrophilic surface by creating radicals that form covalent bonds with amino side chains.
Free radicals have been implicated in aging (11) and in many diseases arising from the malfunctioning of proteins (12). However, we present evidence that shows that they can be used to immobilize proteins onto hydrophilic surfaces directly from a solution while retaining their function. An effective method of creating buried radicals is to treat an organic polymer with energetic ions (13, 14). After treatment with energetic ions, either postformation or during their deposition, these surfaces strongly immobilize proteins (15–29) and provide a means of cloaking biomaterial surfaces. What is required is a means of controlling the density of radicals to bind a full protein monolayer that is not compromised by excessive numbers of covalent bonds, while giving sufficient shelf life of the binding property for practical applications. Here, we develop a deep, quantitative understanding of how free radicals, embedded in a reservoir below the surface, interact with protein molecules to control the number of covalent bonds per molecule and the shelf life of the covalent immobilization capability. We demonstrate a hydrophilic surface that can be applied with robust adhesion to any implant and is capable of covalent immobilization of functional biological molecules directly from solution.
Results and Discussion
We first demonstrate a covalent immobilization capability that retains protein conformation on plasma-treated hydrophilic surfaces. Fig. 1 shows that there is no loss of protein from ion-treated polymer surfaces after washing with sodium dodecyl sulphate (SDS), a detergent capable of disrupting noncovalent interactions. Fig. 1A shows this using protein amide peak absorbances in the infrared, and Fig. 1B shows it using an enzyme-linked immunosorbent assay (ELISA) to detect the presence of the protein. This technique is well-established in the literature as a method for testing for the covalency of macromolecular attachment and has been reviewed (15). SDS is an ionic surfactant that unfolds proteins and disrupts the forces responsible for physisorption, while leaving the covalent bonds intact. The complete removal of physisorbed protein from a more hydrophobic control is used to ensure that steric hindrance does not prevent SDS from accessing all physisorption sites. Further discussion and references are given in Supporting Information. Fig. 1C shows a characteristic curve describing the resistance to elution by the SDS washing protocol used by Kiaei et al. (9) to remove albumin from a range of untreated polymers and plasma polymer surfaces. A clear trend (shown by the curve) with surface energy is apparent, with the strongest adsorption on the most hydrophobic (lowest energy) surfaces. Note that the room temperature SDS protocol employed by Kiaei et al. does not remove all of the physisorbed protein. Data from our plasma immersion ion implantation (PIII)-treated polymers (red squares) and untreated polymers (blue diamonds), where we employ a range of washing protocols (see Fig. S1 and Table S1), is also shown. Aggressive SDS protocols at 70–90 C completely elute protein from very hydrophobic surfaces such as polytetrafluor ethylene (PTFE). Our PIII-treated surfaces typically show 50–100% protein retention despite being hydrophilic. This indicates that physisorption cannot be responsible for the robust protein attachment observed on the ion-implanted surfaces and that a covalent linkage is formed. The ability to covalently immobilize onto a hydrophilic surface is a key advance that allows the retention of protein conformation (Fig. 1D) and bioactivity (Fig. 1E). The potential for cloaking of a biomaterial surface is demonstrated by the full coverage (Fig. 1F) of the ion-treated surface by protein. Ellipsometry analysis gave the thickness of the protein layer to be 9 nm, consistent with a monolayer of horseradish peroxidase (HRP). The refractive index was around 1.5 showing a normal dispersion with wavelength. This value agrees with refractive indices reported for proteins, which vary from 1.4 to 1.6. The thickness of the protein layer on the untreated surface was 8 nm, confirmed by both ellipsometry and atomic force microscopy (AFM), and the coverage of the protein on the surface was around 25–30%.
Fig. 1.

Attributes of protein immobilized on polymeric surfaces treated by energetic ions. (A) Infrared (ATR-FTIR) spectra of HRP enzyme immobilized on PMMA modified by nitrogen ions accelerated by a 20-kV bias at a fluence of 5 × 1015 ions/cm2 (PMMA background subtracted). Protein attached during incubation on the ion-treated surface (black solid spectrum, upward shifted to top) is retained after SDS washing (black dotted spectrum, upward shifted to second position) [2% SDS solution, 50 °C for 1 h]. Protein attached during incubation on the untreated surface (red solid spectrum, unshifted at third position) is completely removed after SDS washing (red dotted spectrum, unshifted bottom). (B) ELISA confirms that tropoelastin physisorbed from 20 μg/mL solution (gray bars) is retained only on the PIII-treated PTFE (Right) after SDS washing and not on the untreated PTFE (Left) (black bars). The white bars indicate the assay background signal without tropoelastin in solution. (C) Percent of protein retained after SDS washing (various solution strengths and temperatures) as a function of surface energy for various polymeric surfaces. Data is taken from Kiaei et al (9) (triangles) and this work (squares and diamonds). Points (squares and one triangle) lying above and to the right of the trend curve typical for physically adsorbed protein show exceptional protein retention given the hydrophilic nature of these surfaces. Untreated controls washed with various SDS protocols are shown as diamonds. (D) Conformation, as determined by the relative content of β-turns, α-helices, random coils, and β-sheets in the IR amide peak signal of surface-attached HRP, is closer to native on PIII-treated polystyrene than when adsorbed onto untreated polystyrene. (E) Amount (FTIR, solid symbols) and activity (TMB assay, open symbols) of HRP protein are both higher on PIII-treated than on untreated UHMWPE at all times of storage in buffer. (F) AFM images of HRP protein layers on polystyrene films spun onto silicon. The coverage of protein is incomplete on the untreated surface (Upper) and forms a densely packed monolayer on the nitrogen PIII-treated (2.5 × 1015 ions/cm2 at 20-keV bias) surface (Lower).
In order to show that cloaking in native protein could be expected to prevent an adverse response in vivo, we have performed a study using whole human blood under flow conditions that simulate circulation in arteries. The result is dramatically reduced thrombosis compared to stainless steel surfaces, a material commonly used in stents (Fig. 2C). Fig. 2 shows that platelets adhere to both the ion-treated plasma polymer film and to the bare stainless steel when incubated in the absence of blood plasma proteins (Fig. 2A) but that when the plasma proteins are present, the treated surface recruits a layer of protein from the blood that prevents the attachment of platelets while the adsorbed protein on the untreated surface initiates the formation of fibrinogen fibrils and the attachment of platelets, representing the initial stages of clot formation. Attenuated total reflection (ATR)-FTIR analysis (Fig. 2D) after SDS washing shows adsorbed protein is covalently attached to the ion-treated plasma polymer surface and only physically adsorbed on the bare stainless steel.
Fig. 2.

Comparison of hemocompatibility of ion-treated plasma polymers with bare stainless steel as used in stents. Scale bars, 20 μm. (A) Isolated platelets in buffer adhere to both surfaces. (B) The presence of protein in platelet-rich plasma prevents platelets adhering to the surface of the ion-treated plasma polymer while fibrinogen fibrils and platelets adhere to the stainless steel surface. (C) Acute thrombogenicity was measured under physiological flow conditions (80 mL/ min at 37 °C) in a modified Chandler loop. Thrombi formed in the presence of stainless steel strips (8 mm wide) after 30 minutes of flow, however, no thrombi formed in the presence of the ion-treated plasma polymer coated surfaces. (D) ATR-FTIR shows that protein from whole blood is covalently bound to the ion-treated polymer surface and not to the stainless steel. Protein attached during flow on the ion-treated plasma polymer surface (black solid spectrum, upward shifted to top) is retained after SDS washing (black dotted spectrum, upward shifted to second position) [2% SDS solution, 70 °C for 1 h, then 100 °C for 1 h]. Protein attached during flow on the stainless steel (red solid spectrum, upward to third position) is completely removed after SDS washing (red dotted spectrum, unshifted bottom). The sharp peaks in the top two spectra are due to characteristic vibrations of the ion-treated plasma polymer, which partially mask the amide II protein peak. Amide I is clearly visible in all spectra.
We now explore the mechanism for the recruitment of a full monolayer of conformationally stable protein on the ion-treated surfaces and demonstrate that our process creates free radicals, which react with the environment at the surface. Free radicals have an unpaired electron and therefore an associated electron spin. The electron spin density created by our ion treatment is quantified using electron spin resonance (ESR; Fig. 3). High concentrations of unpaired electrons in both ion-implanted (Fig. 3A) and plasma-deposited polymers (Fig. 3B) are measured for periods of many months after the ion treatment. Long-lived free radicals have previously been reported for irradiated polymers (30). Fig. 3C shows that surface energy and C═O IR adsorption bands are correlated with the changes in spin density. The concentration of C═O groups on the surface increases during exposure to atmosphere because of reactions with surface radicals (31). The surface energy measured at the first time point is significantly higher than that of an untreated surface and then progressively decreases as the radicals decay by recombination in the bulk and by reactions with the environment at the surface.
Fig. 3.

(A) ESR signal from PIII-treated PTFE showing the presence of free radicals. The red curve (largest amplitude) gives the signal for a freshly treated sample (20 min after treatment), the blue curve shows the signal after 28 mo of laboratory storage, and the black curve (smallest amplitude) is obtained from an untreated control sample. (B) ESR intensity as a function of pulse bias voltage applied during plasma polymer deposition. Measurements were taken 4 d after deposition (red circles) and 8 weeks after deposition both before (blue squares) and after (black triangles) reactivation by annealing. (C) Surface energy, normalized carbonyl group (C═O) absorbance from ATR-FTIR spectra of PIII-treated LDPE (nitrogen at 20 keV) and ESR intensity of free radicals (R∗) as a function of storage time. The surface energy decreases rapidly in the first few hours as free radicals are quenched at the surface by oxidation resulting in the appearance of oxygen-containing groups. Over longer time scales the oxygen group concentration reaches a plateau, and the surface energy stabilizes at a value much greater than that of the hydrophobic untreated LDPE surface. (D) Integrated ESR signal data (R∗ taken from C) and plotted on a logarithmic time axis to show more clearly the decay of free radicals in the treated LDPE sample. The curves are fits of Eq. 1 (blue curve) and to the same equation with an additive constant (432,000 ± 100,000 counts) to represent a residual density of free radicals remaining at long times (red curve, smaller curvature).
A quantitative understanding of the interaction of the free radicals with surface-contacting protein molecules is needed to give the control required to form implant surfaces cloaked in patient compatible proteins. Universality of protein attachment is a key requirement and is satisfied by the high reactivity of free radicals with amino acid residues (see Supporting Information). We now propose and confirm a model in which the covalent binding takes place via a reaction between an amino acid residue on the protein and a free radical on the ion-treated polymer surface that is created by the diffusion to the surface of an unpaired electron from a reservoir below the surface. This reservoir of unpaired electrons is created by the ion treatment. The number of unpaired electrons in the reservoir decreases with time as they migrate internally and to the surface and are quenched either in the bulk or by surface reactions with the environment. We use kinetic theory to derive (see Supporting Information) a quantitative description of the time evolution of unpaired electron (radical) number density, nr(t) in a reservoir of depth h:
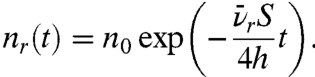 |
[1] |
The mean velocity of the unpaired electrons in the reservoir is 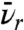 , S is the quenching probability upon reaching the surface, and A is the area of the surface. Eq. 1 does not include recombination in the bulk, which is shown to be insignificant compared to passivation at the surface in Supporting Information.
, S is the quenching probability upon reaching the surface, and A is the area of the surface. Eq. 1 does not include recombination in the bulk, which is shown to be insignificant compared to passivation at the surface in Supporting Information.
Eq. 1 describes an exponential decay with time constant 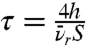 and initial radical density of n0. Fig. 3D shows that Eq. 1 gives a good fit to the decay of free radicals as measured by ESR for PIII-treated low-density polyethylene (LDPE). The PIII treatment was carried out in nitrogen plasma with pulsed bias of 20 kV. The curves show fits of Eq. 1 (R2 = 0.96; τ = 9 ± 1 d—blue curve) and of the same equation with an additive constant to represent a residual density of free radicals that remains at long times (R2 = 0.98; τ = 4 ± 1 d—red curve). The dependence of the radical decay time constant,τ, on the depth, h, of the reservoir shows that deep reservoirs retain the ability to covalently couple protein molecules to the surface for longer periods of time. When the reservoir is created in an already formed polymer, the reservoir depth h depends on the ion energy used for implantation and the type of ion used, whereas when the reservoir is a polymer deposited from a plasma containing monomeric precursors during ion bombardment, the depth h is the thickness of the deposited layer. We have observed high levels of covalent immobilization after more than a year of shelf storage.
and initial radical density of n0. Fig. 3D shows that Eq. 1 gives a good fit to the decay of free radicals as measured by ESR for PIII-treated low-density polyethylene (LDPE). The PIII treatment was carried out in nitrogen plasma with pulsed bias of 20 kV. The curves show fits of Eq. 1 (R2 = 0.96; τ = 9 ± 1 d—blue curve) and of the same equation with an additive constant to represent a residual density of free radicals that remains at long times (R2 = 0.98; τ = 4 ± 1 d—red curve). The dependence of the radical decay time constant,τ, on the depth, h, of the reservoir shows that deep reservoirs retain the ability to covalently couple protein molecules to the surface for longer periods of time. When the reservoir is created in an already formed polymer, the reservoir depth h depends on the ion energy used for implantation and the type of ion used, whereas when the reservoir is a polymer deposited from a plasma containing monomeric precursors during ion bombardment, the depth h is the thickness of the deposited layer. We have observed high levels of covalent immobilization after more than a year of shelf storage.
The covalent attachment process in which radicals diffuse to the surface and form covalent bonds with physisorbed proteins is illustrated schematically in the inset of Fig. 4A. The first step is the physisorption of a protein on the surface, and the second step is the formation of a covalent bond between a protein residue and a radical group. There are two more relevant time constants, one for the diffusion of proteins in solution to the surface, τ1, and the second for the diffusion of the unpaired electrons from the reservoir to the surface, τ2. The kinetic theory description of these two processes (see Supporting Information) leads to two coupled differential equations that are solved (see Supporting Information) to yield the following quantitative descriptions of the physisorption and covalent immobilization processes:
| [2] |
and
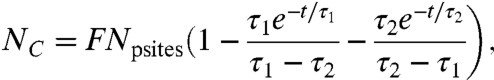 |
[3] |
where Np is the number of physisorbed protein molecules per unit area and Nc is the number of covalently immobilized protein molecules per unit area. The time constant τ1 depends linearly on the number density and diffusion coefficient of the molecules in solution, on the sticking coefficient of physisorbed molecules on the surface, and on the number of sites available for physisorption per unit area (see Eq. S9). The time constant τ2 depends linearly on the number density of unpaired electrons, on the diffusion coefficient of the unpaired electrons in the modified region of the polymer, and on the number of sites available for covalent immobilization per unit area (see Eq. S10). This number is FNpsites, where Npsites is the number of sites available for physisorption per unit area and F is the fraction of physisorption sites that is accessible to radicals diffusing from the interior reservoir.
Fig. 4.

Testing the predictions of the radical model against ELISA immobilization data. The model is illustrated schematically in the Inset. (A) The dependence of tropoelastin coverage on incubation time for PIII-treated PTFE. The lines show the fits of Eqs. 2 and 3 to the amounts adsorbed and covalently immobilized. (B) The time constant for covalent immobilization increases with sample age. (C) Tropoelastin physisorbed (open circles) and covalently bound (closed circles) to polystyrene treated for 10 s in nitrogen (2 mtorr) plasma discharges created at various rf plasma powers as indicated. The red line shows the fit of Eq. 4 with fitting parameter D = 0.0034 ± 10%. (D) Tropoelastin covalent binding to acetylene plasma-deposited layers of varying thickness on stainless steel sheet. The data are plotted against the independent variable time since deposition divided by layer thickness. The data, taken from two assays, is normalized to the average amount adsorbed. The red line shows the fit to Eq. 5 with fitted parameters B = 0.065 ± 0.019 d/nm and C = 976 ± 391 s.
Experiments were conducted to test the predictions of the model. We treated PTFE films with a PIII process. Voltage pulses of 20 kV were applied to a mesh over the films for 20 μs at a frequency of 50 Hz to provide the energetic ion bombardment from a nitrogen plasma. To examine protein adsorption, the samples were incubated in a 20 μg/mL solution of the extracellular matrix protein, tropoelastin, for a range of times. After removal from solution, they were washed in fresh buffer, and the amount of adsorbed tropoelastin was assayed using ELISA. Eq. 2 gives a good fit to the dependence of optical density obtained from ELISA (proportional to the amount of immobilized protein) on incubation time as shown in Fig. 4A. This gives a value of the adsorption time constant τ1 of 4.3 ± 1.2 min. The value is consistent with adsorption from a 500 μg/mL tropoelastin solution as measured in Yin et al. (figure 4 of ref. 20), who found a time constant 25 times shorter, as predicted by Eq. S9.
In parallel, a group of samples was subjected to rigorous SDS washing prior to ELISA detection of the tropoelastin to quantify the proportion of the protein covalently bonded at each stage. Because the protein detected in this case is covalently immobilized, it would be expected to show the time dependence predicted by Eq. 3. A fit of this data by Eq. 3 is shown in Fig. 4A. The parameters determined in the physisorption experiments (Npsites and τ1) were used in the fit of Eq. 3 with F = 1, leaving only one free parameter, the time constant for covalent binding τ2, which was found to be 35 ± 9 min. All fitting parameters are shown in Table 1. The shape of the experimental curve for covalent attachment is distinctly different from that for physisorption, especially in its behavior at short incubation times, and is well reproduced by the model. The presence of two different time constants in the binding process, one for physisorption and one for radical diffusion and covalent binding, is confirmed.
Table 1.
Results of model fitting to data for binding of tropoelastin to PTFE
| Fitting equation | Npsites*, m-2 | τ1, s | τ2, s | χ2 | R2 |
| Eq. 2 | 1.48 ± 0.07 | 258 ± 74 | not applicable | 0.01666 | 0.94358 |
| Eq. 3 | 1.48 | 258 | 2,122 ± 564 | 0.00478 | 0.98568 |
*Npsites has units of (area)-1 and is scaled according to the optical density of the ELISA for the purposes of fitting the ELISA data.
To test the predicted dependence of the covalent binding capability on sample age due to the reduction in the number density of free radicals in the reservoir over time, we compare in Fig. 4B the dependence of the amount of protein covalently attached on incubation time for new and aged (448 d) PIII-treated PTFE films. The time constant, τ2 , for free radical binding found by fitting Eqs. 2 and 3 to the data for the old sample was 3 ± 1 d, two orders of magnitude greater than for the new sample (approximately 30 min, as found above) a result consistent with the ESR-determined decay constant for radicals (see discussion after Eq. S10).
In Fig. 4C we test for the effect of changing the initial number density of free radicals in the reservoir, and in Fig. 4D we test for the effect of the depth of the reservoir, on the amount of protein covalently bound. This is achieved by plasma treating polymer surfaces at a range of plasma powers (Fig. 4C) to vary the density of free radicals and depositing plasma polymer layers containing a constant density of radicals to a range of thicknesses (Fig. 4D). Fig. 4C shows that the amount of protein covalently bound after 1 h of incubation in 10 μg/mL tropoelastin solution increases with the power used in the plasma treatment. In this case, the polymer was polystyrene, and it was treated for 10 s in a 2-mtorr (0.27-Pa) nitrogen plasma at rf powers of 20, 40, 60, 80, and 100 W. Increasing the power is expected to increase the plasma density and thus the density of unpaired electrons created below the surface of the treated polymer. Making the assumption that the relationship between the power and unpaired electron density is linear (14) (i.e. nr = Kp, where p is the power and K is a constant), the expected dependence of the amount of covalently bound protein Nc (from Eqs. S8 and S10) is given by
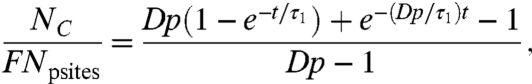 |
[4] |
where p is power and τ1 is a constant for this experiment. 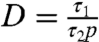 is also constant as τ2 is proportional to 1/p (see Eq. S10). The fit (R2 = 0.9) shown in Fig. 3C is that produced by Eq. 4 with t = 1 h (the incubation time in protein solution) and τ1 = 8 min (adjusted for 10 μg/mL protein solution). The fitted value of D gives τ2 = 24 ± 8 min at the highest power of 100 W, similar to that observed for fresh PIII-treated polymers above, implying a similar number density of free radicals. For the lowest free radical density at 20 W, the time constant (τ2 = 118 ± 41 min) is significantly longer, as would be expected, because of the reduced number density. The quality of the fit achieved with D as the only fitted parameter verifies the model.
is also constant as τ2 is proportional to 1/p (see Eq. S10). The fit (R2 = 0.9) shown in Fig. 3C is that produced by Eq. 4 with t = 1 h (the incubation time in protein solution) and τ1 = 8 min (adjusted for 10 μg/mL protein solution). The fitted value of D gives τ2 = 24 ± 8 min at the highest power of 100 W, similar to that observed for fresh PIII-treated polymers above, implying a similar number density of free radicals. For the lowest free radical density at 20 W, the time constant (τ2 = 118 ± 41 min) is significantly longer, as would be expected, because of the reduced number density. The quality of the fit achieved with D as the only fitted parameter verifies the model.
We now test the prediction of the model for the dependence of τ2 on the thickness h and the time of storage after treatment and prior to incubation with protein, ts. The model predicts the following from Eqs. S4 and S10 (see Eq. S11) :
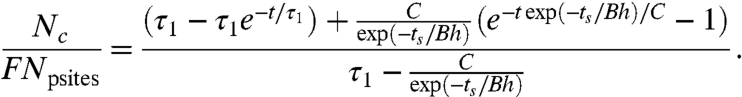 |
[5] |
Fig. 4D shows the amount of tropoelastin covalently attached as a function of ts/h for fixed incubation time, t = 1 h and τ1 = 8 min (adsorption time constant expected for a 10 μg/mL tropoelastin solution). Eq. 5 is fitted (R2 = 0.88) to the data with independent variable ts/h. The fitted values of B and C give τ2 = 20 ± 14 min at the lowest value of ts/h, which is close to that observed for fresh PIII-treated polymers. For the highest value of ts/h, the covalent binding time (τ2 = 5.3 ± 3.7 d) is higher as predicted.
These results enable fundamental properties of unpaired electrons in the reservoir to be estimated and checked for consistency with known data. A value of 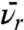 of (7 ± 2) × 10-13 m/s is obtained from B assuming that Sr is unity. The fitted values of B and C give n0 through the relation n0 = BFNpsites/C, which can be derived from the definitions of B and C below Eq. S11. Assuming that all physisorption sites are available for covalent binding (F = 1) and that the number of physisorption sites can be estimated by assuming close packing of protein molecules of 10-nm diameter, we find that the free radical density immediately after treatement is predicted to be n0 = 6 × 1025 m-3. This value is consistent with measured values of nr determined by ESR of 5 × 1023 m-3 in our plasma polymer 1 d after deposition and of 2.7 × 1026 m-3 in a similar amorphous carbon material deposited from a plasma (14). An upper limit for n0 is given by the case of 1 spin per carbon atom, which would give approximately 1 × 1029 m-3. All the parameters determined from the model are therefore self consistent and consistent with known values.
of (7 ± 2) × 10-13 m/s is obtained from B assuming that Sr is unity. The fitted values of B and C give n0 through the relation n0 = BFNpsites/C, which can be derived from the definitions of B and C below Eq. S11. Assuming that all physisorption sites are available for covalent binding (F = 1) and that the number of physisorption sites can be estimated by assuming close packing of protein molecules of 10-nm diameter, we find that the free radical density immediately after treatement is predicted to be n0 = 6 × 1025 m-3. This value is consistent with measured values of nr determined by ESR of 5 × 1023 m-3 in our plasma polymer 1 d after deposition and of 2.7 × 1026 m-3 in a similar amorphous carbon material deposited from a plasma (14). An upper limit for n0 is given by the case of 1 spin per carbon atom, which would give approximately 1 × 1029 m-3. All the parameters determined from the model are therefore self consistent and consistent with known values.
The model explains in a natural way what happens when the connectivity of the polymer is changed so that there are regions in which unpaired electrons are mobile and regions where they are not. Carbon-based systems containing extended states, such as π conjugated carbon structures, have mobile unpaired electrons that can move freely throughout the conjugated region and can hop across small gaps between the regions (32). The inclusion of elements that discourage the π conjugation reduces the mobility of the unpaired electrons. This is observed in plasma polymer samples with added hydrogen, oxygen, or stainless steel inclusions (see Fig. S2). In all cases, the covalent binding capability is dramatically reduced.
Some polymers, such as polydimethylsiloxane (PDMS) and Elast-Eon (a PDMS-polyurethane copolymer), which have high concentrations of silicon, show no and reduced covalent binding, respectively. The reduced covalent binding correlates with the reduced concentration of mobile unpaired electrons on carbon sites. In PDMS, the unpaired electrons are principally on Si-O or Si sites and are distinguished from those on carbon sites by a different g value of the electron spin (see Fig. S3). The unpaired electrons on Si or Si-O are immobile and do not facilitate covalent binding at the surface. In Elast-Eon there are both immobile and mobile spins. PIII treatment up to a critical fluence increases the density of mobile spins and then decreases it again as carbon is selectively etched from the structure and immobile spins associated with silicon become dominant. The covalent bonding capability reaches a maximum and then declines with further treatment (see Fig. S4).
We have shown that ion treatments create layers containing unpaired electrons that provide a universal protein binding platform, given sufficient electron mobility within the layer. The free radicals can covalently link a wide range of amino acid residues (see Table S2), showing that this technique of covalent immobilization is universal with respect to biomolecules. Because these layers can be created on any material, they are universal in that sense also and are versatile interfaces for covalent coupling of functional biomolecules without the need for specific linker chemistry. The role of free radicals is underlined by the high reactivity immediately after treatment (Fig. S5) and the action of radical blockers (Fig. S6 and Table S3).
We have developed a quantitative understanding of how the unpaired electrons are effective in covalent surface immobilization of protein molecules from solution. The dependences of the rate of covalent immobilization on the age of the sample and the density of free radicals show that the irreversible protein immobilization observed is associated with the free radicals. The dependence of the rate of covalent immobilization on the depth of the free radical reservoir and the connectivity of subsurface structures in which electrons are mobile, such as regions of π conjugated carbon, show that the unpaired electrons come from the bulk and diffuse to the surface.
There is a need for such surfaces in biomimetic surface coatings for medical implants (33, 34), environmental biosensors (35), and antibody arrays for early and precise disease diagnosis (36), all of which require functional (33, 37) immobilized proteins. The capability of immobilizing the whole range of proteins expressed in a cell would enable “reverse phase” microarrays (38) to monitor disease progression through changes in protein expression. “Cloaking” a prosthetic implant with a conformal coverage of selected bioactive proteins or peptide segments could be used to elicit an optimal local host response such as adherence of a target cell type (28, 33, 39, 40). Used on implantable biomedical devices, such cloaked surfaces would make truly biomimetic implants that elicit optimum local cellular responses by means of a covalently immobilized functional protein layer derived from the patient’s protein present at the site of the implant.
Materials and Methods
Polymer sheets were from Goodfellow, and protein, polyamino acids, blockers, and reagents were from Sigma Aldrich. PIII was carried out at 20 kV in rf plasma. Plasma polymers were deposited from rf plasma containing hydrocarbon precursors. ESR spectra were recorded on a Bruker Elexsys E500 EPR spectrometer; contact angles were measured using Kruss DS10. FTIR spectra were recorded using a Digilab FTS7000 fitted with Harrick Ge ATR. AFM images were acquired in tapping mode using a PicoSPM with WSxM software (Nanotec Electronica). Ellipsometry was used to determine spun polymer and protein thickness and optical constants (M2000V, JA Woollam). Assays of HRP activity were performed on 25-μL aliquots using the optical density produced by tetramethylbenzidene (TMB) at 450 nm, measured with a Beckman DU530. ELISA was used to complement the FTIR assessments of protein coverage. The primary antibody was mouse anti-elastin antibody (BA-4), and the secondary antibody was goat anti-mouse IgG-HRP conjugated. Static thrombogenicity assays were carried out by incubation of surfaces with isolated platelet suspension or platelet-rich plasma in 24 or 48 well plates blocked with 3% BSA. For assessing thrombogenesis under flow, whole human blood was circulated over the surfaces being tested in a modified Chandler loop (41). Further details of all materials and methods are contained in Supporting Information.
Supplementary Material
Acknowledgments.
We thank Stacey Hirsh for comments on the manuscript and Jennifer Tilley, Robert Thompson, Pourandohkt Naseri, and Susan Graham for collecting some of the data shown in Supporting Information. We acknowledge the Australian Research Council for funding and the Wellcome Trust Equipment Fund for provision of the ESR. We acknowledge industry partners Cochlear Ltd. and SpineCell Pty Ltd. for financial and in-kind research support.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103277108/-/DCSupplemental.
References
- 1.Lal S, et al. Increases in leukocyte CD antigen expression during cardiopulmonary bypass in patients undergoing heart transplantation. Proteomics. 2004;4:1918–1926. doi: 10.1002/pmic.200300756. [DOI] [PubMed] [Google Scholar]
- 2.Ohqvist G, Settergren G, Lundburg S. Pulmonary oxygenation, central haemodynamics and glomerular filtration following cardiopulmonary bypass with colloid or non-colloid priming solution. Scand J Thorac Cardiovasc Surg. 1981;15:257–262. doi: 10.3109/14017438109100583. [DOI] [PubMed] [Google Scholar]
- 3.Higgins DM, et al. Localized immunosuppressive environment in the foreign body response to implanted biomaterials. Am J Pathol. 2009;175:161–170. doi: 10.2353/ajpath.2009.080962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson JM, Marchant RE. Biomaterials: Factors favoring colonization and infection. In: Waldvogel FA, Bisno AL, editors. Infections Associated with Indwelling Medical Devices. 3rd Ed. Washington, DC: ASM Press; 2000. pp. 89–109. [Google Scholar]
- 5.Wolcott RD, Rhoads DD. A study of biofilm-based wound management in subjects with critical limb ischaemia. J Wound Care. 2008;17:145–155. doi: 10.12968/jowc.2008.17.4.28835. [DOI] [PubMed] [Google Scholar]
- 6.Wilson GJ, et al. Comparison of inflammatory response after implantation of Sirolimus- and Paclitaxel-eluting stents in porcine coronary arteries. Circulation. 2011;120:141–149. doi: 10.1161/CIRCULATIONAHA.107.730010. [DOI] [PubMed] [Google Scholar]
- 7.Inoue T, et al. Comparison of activation process of platelets and neutrophils after coronary stent implantation versus balloon angioplasty. Am J Cardiol. 2000;86:1057–1062. doi: 10.1016/s0002-9149(00)01159-0. [DOI] [PubMed] [Google Scholar]
- 8.Karlsson M, Ekeroth J, Elwing H, Carlsson U. Reduction of irreversible protein adsorption on solid surfaces by protein engineering for increased stability. J Biol Chem. 2005;280:25558–25564. doi: 10.1074/jbc.M503665200. [DOI] [PubMed] [Google Scholar]
- 9.Kiaei D, Hoffman AS, Horbett TA. Tight binding of albumin to glow discharge treated polymers. J Biomater Sci Polym Ed. 1992;4:35–44. [PubMed] [Google Scholar]
- 10.Lopez GP, et al. Glow discharge plasma deposition of tetraethylene glycol dimethyl ether for fouling-resistant biomaterial surfaces. J Biomed Mater Res. 1992;26:415–439. doi: 10.1002/jbm.820260402. [DOI] [PubMed] [Google Scholar]
- 11.Harman D. Aging: A theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 12.Giunta S, Ronchi P, Valli B, Franceschi C, Galeazzi L. Transformation of beta-amyloid (AP) (1–42) tyrosine to L-Dopa as the result of in vitro hydroxyl radical attack. Amvloid. 2000;7:189–193. doi: 10.3109/13506120009146833. [DOI] [PubMed] [Google Scholar]
- 13.Popok VN, Azarkob II, Odzhaevb VB, Tóthc A, Khaibullin RI. High fluence ion beam modification of polymer surfaces: EPR and XPS studies. Nucl Instrum Methods Phys Res B. 2001;178:305–310. [Google Scholar]
- 14.Jones BJ, Barklie RCK, Khan RUA, Carey JD, Silva SRP. Electron paramagnetic resonance study of ion implantation induced defects in amorphous hydrogenated carbon. Diam Relat Mater. 2001;10:993–997. [Google Scholar]
- 15.Bilek M, McKenize DR. Plasma modified surfaces for covalent immobilization of functional biomolecules in the absence of chemical linkers: Towards better biosensors and a new generation of medical implants. Biophys Rev. 2010;2:55–65. doi: 10.1007/s12551-010-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin YB, Bax D, McKenzie DR, Bilek MMM. Protein immobilization capacity and covalent binding coverage of pulsed plasma polymer surfaces. Appl Surf Sci. 2010;256:4984–4989. [Google Scholar]
- 17.Yin YB, et al. Acetylene plasma polymerized surfaces for covalent immobilization of dense bioactive protein monolayers. Surf Coat Technol. 2009;203:1310–1316. [Google Scholar]
- 18.Yin YB, et al. Covalently bound biomimetic layers on plasma polymers with graded metallic interfaces for in vivo implants. Plasma Process Polym. 2009;6:658–666. [Google Scholar]
- 19.Yin YB, et al. Plasma polymer surfaces compatible with a CMOS process for direct covalent enzyme immobilization. Plasma Process Polym. 2009;6:68–75. [Google Scholar]
- 20.Yin YB, et al. Covalent immobilisation of tropoelastin on a plasma deposited interface for enhancement of endothelialisation on metal surfaces. Biomaterials. 2009;30:1675–1681. doi: 10.1016/j.biomaterials.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Yin Y, et al. Acetylene plasma coated surfaces for covalent immobilization of proteins. Thin Solid Films. 2009;517:5343–5346. [Google Scholar]
- 22.Nosworthy NJ, Ho JPY, Kondyurin A, McKenzie DR, Bilek MMM. The attachment of catalase and poly-L-lysine to plasma immersion ion implantation-treated polyethylene. Acta Biomater. 2007;3:695–704. doi: 10.1016/j.actbio.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 23.MacDonald C, Morrow R, Weiss AS, Bilek MMM. Covalent attachment of functional protein to polymer surfaces: A novel one-step dry process. J R Soc Interface. 2008;5:663–669. doi: 10.1098/rsif.2007.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondyurin A, Nosworthy NJ, Bilek MMM. Attachment of horseradish peroxidase to polytetrafluorethylene (teflon) after plasma immersion ion implantation. Acta Biomater. 2008;4:1218–1225. doi: 10.1016/j.actbio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Kondyurin A, et al. Covalent attachment and bioactivity of horseradish peroxidase on plasma-polymerized hexane coatings. Plasma Process Polym. 2008;5:727–736. [Google Scholar]
- 26.Ho JPY, et al. Plasma-treated polyethylene surfaces for improved binding of active protein. Plasma Process Polym. 2007;4:583–590. [Google Scholar]
- 27.Bax DV, McKenzie DR, Weiss AS, Bilek MMM. Linker-free covalent attachment of the extracellular matrix protein tropoelastin to a polymer surface for directed cell spreading. Acta Biomater. 2009;5:3371–3381. doi: 10.1016/j.actbio.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Bax DV, McKenzie DR, Weiss AS, Bilek MMM. The linker-free covalent attachment of collagen to plasma immersion ion implantation treated polytetrafluoroethylene and subsequent cell-binding activity. Biomaterials. 2010;31:2526–2534. doi: 10.1016/j.biomaterials.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Hirsh SL, et al. A comparison of covalent immobilization and physical adsorption of a cellulase enzyme mixture. Langmuir. 2010;26:14380–14388. doi: 10.1021/la1019845. [DOI] [PubMed] [Google Scholar]
- 30.Arakl K. ESR study of free radicals formed by gamma-irradiation of poly(ethylene terephthalate) J Polym Sci B. 1965;3:993–996. [Google Scholar]
- 31.Jiang H, Grant JT, Enlow J, Su W, Bunning TJ. Surface oxygen in plasma polymerized films. J Mater Chem. 2009;19:2234–2239. [Google Scholar]
- 32.Ambegaokar V, Halperin BI, Langer JS. Hopping conductivity in disordered systems. Phys Rev B Condens Matter Mater Phys. 1971;4:2612–2620. [Google Scholar]
- 33.Olsson P, Sanchez J, Mollnes TE, Riesenfeld J. On the blood compatibility of end-point immobilized heparin. J Biomater Sci Polym Ed. 2000;11:1261–1273. doi: 10.1163/156856200744192. [DOI] [PubMed] [Google Scholar]
- 34.Werner C, Maitz MF, Sperling C. Current strategies towards hemocompatible coatings. J Mater Chem. 2007;17:3376–3384. [Google Scholar]
- 35.DeGrado WF. Computational biology—Biosensor design. Nature. 2003;423:132–133. doi: 10.1038/423132a. [DOI] [PubMed] [Google Scholar]
- 36.Hanash S. Disease proteomics. Nature. 2003;422:226–232. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 37.Nojiri C, Senshu K, Okano T. Nonthrombogenic polymer vascular prosthesis. Artif Organs. 1995;19:32–38. doi: 10.1111/j.1525-1594.1995.tb02241.x. [DOI] [PubMed] [Google Scholar]
- 38.Paweletz CP, et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981–1989. doi: 10.1038/sj.onc.1204265. [DOI] [PubMed] [Google Scholar]
- 39.Pompe W, et al. Functionally graded materials for biomedical applications. Mater Sci Eng A Struct Mater. 2003;362:40–60. [Google Scholar]
- 40.Uchida M, Oyane A, Kim HM, Kokubo T, Ito A. Biomimetic coating of laminin-apatite composite on titanium metal and its excellent cell-adhesive properties. Adv Mater. 2004;16:1071–1074. [Google Scholar]
- 41.Waterhouse A, et al. The immobilization of recombinant human tropoelastin on metals using a plasma-activated coating to improve the biocompatibility of coronary stents. Biomaterials. 2010;31:8332–8340. doi: 10.1016/j.biomaterials.2010.07.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.