

Abstract
Direct methods for the trifluoromethylation of heteroaromatic systems are in extremely high demand in nearly every sector of chemical industry. Here we report the discovery of a general procedure using a benchtop stable trifluoromethyl radical source that functions broadly on a variety of electron deficient and rich heteroaromatic systems and demonstrates high functional group tolerance. This C-H trifluoromethylation protocol is operationally simple (avoids gaseous CF3I), scalable, proceeds at ambient temperature, can be used directly on unprotected molecules, and is demonstrated to proceed at the innately reactive positions of the substrate. The unique and orthogonal reactivity of the trifluoromethyl radical relative to aryl radicals has also been investigated on both a complex natural product and a pharmaceutical agent. Finally, preliminary data suggest that the regioselectivity of C-H trifluoromethylation can be fine-tuned simply by judicious solvent choice.
Keywords: medicinal chemistry, C-H functionalization, synthetic methodology
The trifluoromethyl group is becoming an increasingly common trait among molecules found in billion-dollar pharmaceuticals, agrochemicals, liquid crystals, dyes, and polymers (1–6). The inclusion of this motif and the unique properties its presence elicits is a testament to the success of chemical synthesis, as it is notably absent in Nature. Methodologies for the trifluoromethylation of arenes can be divided into two general categories (Fig. 1A): those that functionalize the inherently reactive positions of the substrate (“innate trifluoromethylation”) and those that utilize substrate prefunctionalization or a directing group (“programmed trifluoromethylation”). For most applications, “programmed” aryl trifluoromethylation holds a distinct advantage because it can selectively functionalize positions that are not naturally reactive. Indeed, incredibly powerful methods have recently emerged in this arena (10–18). On the other hand, methods that capitalize on innate reactivity avoid the complication of preparing prefunctionalized substrates. We became interested in exploring “innate” aryl trifluoromethylation during our recent studies in the area of direct C-H arylation of heterocycles (19) and quinones (20) using radicals derived from boronic acid precursors. Aromatic heterocycles containing the trifluoromethyl group represent an important subsection of molecules of practical interest, particularly for pharmaceuticals, and therefore previously undescribed methods for their rapid assembly are in high demand. Our goal was to identify a reagent that would circumvent the use of gaseous CF3I (21), a reagent that is often avoided in pharmaceutical settings. At the start of our work, the task of replacing this reagent for the innate trifluormethylation of nitrogen containing heterocycles remained an unmet challenge.
Fig. 1.

Discovery of a mild method for C-H trifluoromethylation (7–9).
As briefly illustrated in Fig. 1, we evaluated numerous reagents for the trifluoromethylation of 4-t-butylpyridine as a model compound, with the aim of exploring those that might proceed through electrophilic, nucleophilic, and radical conditions. Entries 1–10 are a sampling of the over 500 reactions that were screened using different oxidants, Lewis acids, solvents, and even phase-transfer catalysts. The only reagent that delivered appreciable amounts of the desired trifluoromethyl pyridine was sodium trifluoromethanesulfinate (CF3SO2Na, Langlois reagent, a benchtop stable, inexpensive solid). This result was surprising to us because pioneering studies by Langlois et al. have shown that the electrophilic trifluoromethyl radical generated using this reagent undergoes SERAr reactions with electron rich substrates such as phenol and aniline (22). Indeed, Langlois et al.’s original report specifically states that productive reactions were possible “provided that the substrates were substituted by mesomeric electron-releasing groups.” We were thus inspired to investigate how to reliably generate the putatively assigned CF3 radical and direct it toward the desired reaction in the context of medicinally relevant heterocycles (many of which are electron-withdrawing such as pyridines).
Results
Several of the seminal reports on the use of CF3SO2Na by Langlois and others employed catalytic metal salts (22, 23), whereas one study on the trifluoromethylation of disulfides showed that the reaction could still be productive in their absence (24). Further investigations of the reaction parameters were carried out with 4-acetylpyridine (Fig. 2). It quickly became apparent that metal additives were not required for a productive reaction, although trace metals found in the CF3SO2Na could be responsible for reaction initiation. (Trace metal analysis performed at the Scripps Institution of Oceanography revealed ppm levels of several metal salts present in commercially available CF3SO2Na. See SI Appendix.) Excess reagents were required for appreciable conversion to product (entries 4–6), although the reason for this requirement was unclear. Additionally, stirring had a counterintuitive effect on the biphasic reaction: Conversion appeared to be inversely correlated with the rate of stirring (entries 6, 9–10). The sensitivity of yield to reaction parameters encouraged us to probe the mechanistic subtleties of the reaction in more detail.
Fig. 2.

Initial investigations into reaction parameters: a GC yield with tetradecane as an internal standard. b tBuOOH added as a single portion. c 0.25 μL/s syringe drive addition of tBuOOH. d Isolated yield.
Reaction calorimetric monitoring of the trifluoromethylation of 4-acetylpyridine in CH2Cl2/H2O reveals that the mode of addition of reagents is critical to achieving high yield. When all reagents are mixed simultaneously, a large heat flow signal is observed immediately after mixing (Fig. 3A). However, conversion of the arene substrate to product is minimal during the period of this heat flow, and a similarly large heat flow signal was observed when the oxidant and Langlois reagent are mixed in the absence of the arene substrate (Fig. 3B). Similar results are observed for the system with water alone as solvent. Thus the exothermic signal may be attributed to unproductive reactions between the peroxide and sulfinate rather than to the desired reaction chemistry, suggesting that under well-mixed reaction conditions a significant fraction of the reagents is diverted away from the desired reaction pathway. This result helps to explain both variable yields and the requirement for large excesses of tBuOOH and CF3SO2Na.
Fig. 3.

Observed heat flow in the two-phase system: DCM∶H2O 2.5∶1, with 0.18 M arene, 5 equiv. tBuOOH, 3 equiv. CF3SO2Na, (B) stirred at 600 rpm; (A) in the absence of arene, stirred at 600 rpm; (C) with arene, unstirred with separate organic and aqueous layers.
Fig. 3C shows that the unproductive heat flow signal is minimized in reactions where the CH2Cl2 and H2O phases are deliberately kept separate, with the sulfinate residing in the aqueous layer and the arene and tBuOOH initially solvated in the organic layer. Fig. 4 compares fraction conversion of the arene to product for reactions carried out at two different stirring speeds and for an unstirred reaction in which separate organic and aqueous layers are maintained. At high stirring speeds, product formation initially proceeds more rapidly but essentially halts at ca. 60% conversion. Reaction with the phases segregated proceeds in a steady manner and ultimately provides the highest yield, albeit with the requirement for longer reaction times.
Fig. 4.

Fraction conversion to product over time as a function of stirring speed in the two-phase reaction shown in Fig. 2 using 0.18 M arene, 5.0 equiv. tBuOOH, 3.0 equiv. CF3SO2Na in (DCM∶H2O 2.5∶1). Final reaction conversions were measured at 1,500 min.
The combination of previous studies (22–25) with these data allowed for a putative mechanism to be proposed (Fig. 5). The tert-butoxy radical, presumably generated from trace metal or another initiator, reacts with 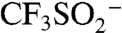 to provide CF3SO2•. This transient intermediate disproportionates, releasing SO2 and CF3•. It should be noted that in situ EPR studies of CF3SO2Na and tBuOOH in aqueous solution indicated the presence of radical intermediates. The fate of the inherently reactive CF3• after its generation can follow many pathways, three of which are shown in Fig. 5. The productive pathway involves capture of the trifluoromethyl radical with Ar-H, followed by reoxidation to Ar-CF3, concomitantly generating another molecule of tBuO•. Two other competitive reaction pathways lead to undesired by-products. Abstraction of a hydrogen atom yields CF3H (observable by 19F-NMR). Alternatively, reaction with isobutene generated from tBuOOH (26) followed by reaction with an arene substrate leads to alkyltrifluoromethyl by-products such as (26). To confirm this pathway, the reaction of 4-cyanopyridine was run with added 2-methyl-2-butene and provided moderate yield of 3 (along with the isobutene by-product), suppressing the direct trifluoromethylation.
to provide CF3SO2•. This transient intermediate disproportionates, releasing SO2 and CF3•. It should be noted that in situ EPR studies of CF3SO2Na and tBuOOH in aqueous solution indicated the presence of radical intermediates. The fate of the inherently reactive CF3• after its generation can follow many pathways, three of which are shown in Fig. 5. The productive pathway involves capture of the trifluoromethyl radical with Ar-H, followed by reoxidation to Ar-CF3, concomitantly generating another molecule of tBuO•. Two other competitive reaction pathways lead to undesired by-products. Abstraction of a hydrogen atom yields CF3H (observable by 19F-NMR). Alternatively, reaction with isobutene generated from tBuOOH (26) followed by reaction with an arene substrate leads to alkyltrifluoromethyl by-products such as (26). To confirm this pathway, the reaction of 4-cyanopyridine was run with added 2-methyl-2-butene and provided moderate yield of 3 (along with the isobutene by-product), suppressing the direct trifluoromethylation.
Fig. 5.

Putative mechanism and radical alkene capture.
With progress toward a general set of optimized conditions, the scope of this reaction was evaluated on a broad cross-section of heterocyclic space (Fig. 6). For the sake of experimental consistency, these reactions were carried out with standard stirring, although this resulted in incomplete conversion for select substrates. For cases where conversion was exceptionally low, a second portion of tBuOOH and CF3SO2Na could be added after 24 h to drive the reaction toward completion. Pyridines (1–7, 14), pyrroles (8–9), indoles (10), pyrimidines (12), pyrazines (13, 15), phthalazines (16), quinoxalines (17, 18), deazapurine (24), thiadiazoles (11), uracils (19, 25), xanthines (20–22), and pyrazolinopyrimidines (23) can all be directly trifluoromethylated with this simple procedure. It should be noted that several of the compounds in Fig. 6 are previously undescribed chemical entities (see SI Appendix), whereas others are drugs (25) or trifluoromethylated derivatives thereof (7, 10, 20–23). In the case of trifluridine (25), this compound has been previously prepared either with an elaborate procedure involving gaseous CF3I as a slowly added radical source (at an elevated temperature under an inert atmosphere in the presence of a metal catalyst) (27), or through a three-step procedure involving protection, XeF2-mediated trifluoromethylation, and deprotection in 40% overall yield (28). Many functional groups are tolerated such as unprotected alcohols, amines, ketones, esters, halides, and nitriles. For cases where more than one reactive site is present, products may be formed as a single compound (7–10, 13, 19, 23–25) or as mixture of regioisomers (1, 4–6, 12, 14, 16–18). Although in some cases selectivity appeared to be governed by the electronics of the heterocycle, a general predictive model for site selectivity remains a focus of study. This reaction has proven to be scalable with uracil (19) and caffeine (21) being trifluoromethylated on a gram-scale. For highly reactive substrates (e.g., caffeine, to yield 21) or those that are not soluble in dichloromethane (e.g., deoxyuridine, to yield 25), the reaction could even be carried out under purely aqueous conditions without an organic workup.
Fig. 6.

Scope of heterocycle trifluoromethylation. a Heterocycle (1.0 equiv), sodium trifluoromethanesulfinate (3.0 equiv), tButyl-hydroperoxide (5.0 equiv), 23 ºC, 3–24 h; isolated yields of chromatographically and spectroscopically pure products yields displayed, unless otherwise noted. b Reaction showed incomplete conversion after 24 h, and a second addition of sodium trifluoromethanesulfinate (3.0 equiv) and tButyl-hydroperoxide (5.0 equiv) was added. c Reaction run without organic solvent.
One of the most intriguing aspects of this methodology stems from its functional group compatibility, predictable positional selectivity, and orthogonality to other types of innate C-H functionalization. As illustrated in Fig. 7, three types of heterocycles were exposed to both nucleophilic and electrophilic modes of direct functionalization. Caffeine contains a C-H bond that can be viewed as having both electrophilic and nucleophilic properties (ambiphilic). Therefore, this position reacts smoothly with both a nucleophilic aryl radical species (derived from the corresponding aryl boronic acid) (19) and the electrophilic trifluoromethyl radical to furnish 26 and 21, respectively. Arylation of this position has recently been reported by Knochel using a metallation/Negishi sequence (29) and 21 has only been prepared by ring synthesis (30).
Fig. 7.

Diverging regioselectivity of reactive radicals on complex substrates: “Nucleophilic” versus “electrophilic” radical regioselectivity. Isolated yields of purified products after silica gel chromatography (21 and 26) or preparative HPLC (27–30).
Position selectivity in systems with more than one functionalizable C-H bond is also predictable. In the case of dihydroquinine, the C-7 position is considered the most sterically accessible and electron-rich, whereas the C-2 position is most electrophilic. In accord with the predicted outcome, direct arylation (to yield 27) and trifluoromethylation (to yield 29) proceed exclusively at C-2 and C-7, respectively. Finally, in the case of the blockbuster drug Varenicline (31, 32), the C-5 position is deemed most nucleophilic whereas the C-2 position is highly electrophilic. Thus, direct arylation proceeds exclusively at C-2 (to yield 28) and direct trifluoromethylation (to yield 30) proceeds with 4∶1 selectivity at C-5. In the case of quinine and Chantix derived substrates, access to these functionalized molecules would require multistep routes (total synthesis).
Aside from the useful levels of selectivity observed, it is worth noting that these functionalization methods take place on unprotected systems in water at room temperature and without any need for solvent purification or an inert atmosphere. This methodology therefore holds promise for use in both combinatorial diversification and parallel divergent synthesis.
Although this method clearly avoids substrate prefunctionalization and is operationally simple, it is not without limitations. Several substrates deliver only moderate amounts of product, although some of the lower yielding cases from Fig. 6 are the result of product volatility (1, 4, and 5 cannot be subjected to rotary evaporation, whereas 6 and 15 quickly vanish under high vacuum) or from difficulties recovering starting material due to incomplete conversion. If one desires a single isomer when multiple C-H bonds are present, the standard conditions reported in Fig. 6 cannot override the innate reactivity dictated by the substrate. Of the 23 examples in Fig. 6, three of the substrates give equimolar mixtures of regioisomers (6, 12, and 14), and compound 17 delivers a mixture of four products.
Although many of the regioisomeric mixtures listed in Fig. 6 may be chromatographically separated, efforts were taken to identify experimental conditions that could override innate substrate bias. As shown in Fig. 8, preliminary results suggest that solvent effects play a significant role in the regioselectivity of the trifluoromethylation of 4-acetylpyridine. The standard, aqueous reaction conditions are selective for C-2 in a ratio of 2.4–2.6∶1. However, when substituting dimethylsulfoxide for dichloromethane, a complete reversal in selectivity is observed, providing the C-3 product with greater than 5∶1 selectivity. Investigation into the origin of solvent-mediated regiocontrol predicated on mechanistic understanding is ongoing and will be reported in due course.
Fig. 8.

Preliminary results on the effect of solvent on regioselectivity in the trifluoromethylation of 4-acetylpyridine. (Top arrow) DCM∶H2O 2.5∶1, with 0.18 M arene, 5 equiv. tBuOOH, 3 equiv. CF3SO2Na. (Bottom arrow) DMSO∶H2O 2.5∶1, with 0.18 M arene, 5 equiv. tBuOOH, 3 equiv. CF3SO2Na. GC yield with tetradecane as an internal standard. Both products are volatile and are lost upon rotary evaporation (see SI Appendix for details).
Conclusion
In summary, a practical method for the trifluoromethylation of medicinally relevant heterocycles has been explored predicated on the idea of “innate C-H functionalization.” A series of mechanistic studies on the nuances of this unique reactivity was performed and a robust protocol was developed that displays broad substrate scope and functional group compatibility. Additionally, differences in selectivity observed with different solvent systems suggest promise for effective tuning of the innate substrate reactivity. The operational ease of this transformation coupled with its predictable reactivity in complex settings bodes well for its widespread application.
Supplementary Material
Acknowledgments.
We are grateful to Dr. Jotham Coe (Pfizer Inc.) for valuable discussions and a generous sample of Chantix and to Professor Arnold Rheingold for X-ray crystallographic analysis. Special thanks go to R. Rodriguez for early findings and suggestions. Financial support for this work was provided by the National Institutes of Health/National Institute of General Medical Sciences (GM-073949), Ministerio de Eduacion y Ciencia (to Y.J.), the Deutscher Akademischer Austauschdienst postdoctoral fellowship (to T.B.), National Science Foundation predoctoral fellowship (to I.B.S.), Japan Society for the Promotion of Science postdoctoral fellowship (to Y.F.), Amgen, and Bristol-Myers Squibb (unrestricted research support).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109059108/-/DCSupplemental.
References
- 1.Smart BE. Introduction: Fluorine chemistry. Chem Rev. 1996;96:1555–1556. doi: 10.1021/cr960075e. [DOI] [PubMed] [Google Scholar]
- 2.Filler R, Kobayashi Y, Yagupolskii LM, editors. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Amsterdam: Elsevier; 1993. [Google Scholar]
- 3.Welch JT. Advances in the preparation of biologically-active organofluorine compounds. Tetrahedron. 1987;43:3123–3197. [Google Scholar]
- 4.Ma JA, Cahard D. Strategies for nucleophilic, electrophilic, and radical trifluoromethylations. J Fluorine Chem. 2007;128:975–996. [Google Scholar]
- 5.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 6.Furuya T, Kamlet AS, Ritter T. Catalysis for fluorination and trifluoromethylation. Nature. 2011;473:470–477. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiehn MS, Vinogradova EV, Togni A. Electrophilic trifluoromethylation of arenes and N-heteroarenes using hypervalent iodine reagents. J Fluorine Chem. 2010;131:951–957. [Google Scholar]
- 8.Umemoto T, Ishihara S. Power-variable electrophilic trifluoromethylating agents. S-, Se-, and Te- (trifluoromethyl)dibenzothio-, -seleno-, and -tellurophenium salt system. J Am Chem Soc. 1993;115:2156–2164. [Google Scholar]
- 9.Prakash GK, Yudin AK. Perfluoroalkylation with organosilicon reagents. Chem Rev. 1997;97:757–786. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]
- 10.Roy S, Gregg BT, Gribble GW, Le VD, Roy S. Trifluoromethylation of aryl and heteroaryl halides. Tetrahedron. 2011;67:2161–2195. [Google Scholar]
- 11.Tomashenko OA, Grushin VV. Aromatic trifluoromethylation with metal complexes. Chem Rev. 2011 doi: 10.1021/cr1004293. 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]
- 12.Oishi M, Kondo H, Amii H. Aromatic trifluoromethylation catalytic in copper. Chem Commun. 2009;14:1909–1911. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]
- 13.Knauber T, Arikan F, Roschenthaler GV, Goossen LJ. Copper-catalyzed trifluoromethylation of aryl iodides with potassium (trifluoromethyl)trimethoxyborate. Chemistry. 2011;17:2689–2697. doi: 10.1002/chem.201002749. [DOI] [PubMed] [Google Scholar]
- 14.Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. A broadly applicable copper reagent for trifluoromethylations and perfluoroalkylations of aryl iodides and bromides. Angew Chem Int Ed Engl. 2011;50:3793–3798. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu L, Qing FL. Copper-mediated oxidative trifluoromethylation of boronic acids. Org Lett. 2010;12:5060–5063. doi: 10.1021/ol1023135. [DOI] [PubMed] [Google Scholar]
- 16.Senecal T, Parsons AT, Buchwald ST. Room temperature aryl trifluoromethylation via copper-mediated oxidative cross-coupling. J Org Chem. 2011;76:1174–1176. doi: 10.1021/jo1023377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho EJ, et al. The palladium-catalyzed trifluoromethylation of aryl chlorides. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Truesdale L, Yu JQ. Pd(II)-catalyzed ortho-trifluoromethylation of arenes using TFA as a promoter. J Am Chem Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]
- 19.Seiple IB, et al. Direct C-H arylation of electron-deficient heterocycles with arylboronic acids. J Am Chem Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujiwara Y, et al. Practical C-H functionalization of quinones with boronic acids. J Am Chem Soc. 2011;133:3292–3295. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kino T, et al. Trifluoromethylation of various aromatic compounds by CF3I in the presence of Fe(III) compound, H2O2 and dimethylsulfoxide. J Fluorine Chem. 2010;131:98–105. [Google Scholar]
- 22.Langlois BR, Laurent E, Roidot N. Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]
- 23.Langlois BR, Laurent E, Roidot N. “Pseudo-cationic” trifluoromethylation of enol esters with sodium trifluoromethanesulfinate. Tetrahedron Lett. 1992;33:1291–1294. [Google Scholar]
- 24.Langlois BR, Montegre D, Roidot N. Synthesis of S-trifluoromethyl-containing a-amino acids from sodium trifluoromethanesulfinate and dithio-amino acids. J Fluorine Chem. 1994:63–66. [Google Scholar]
- 25.Billard T, Roques N, Langlois BR. Synthetic uses of thio- and selenoesters of trifluoromethylated acids. 1. Preparation of trifluoromethyl sulfides and selenides. J Org Chem. 1999;64:3813–3820. [Google Scholar]
- 26.Chen CJ, Bozzelli JW. Analysis of tertiary butyl radical + O2, iobutene + HO2, isobutene + OH, and isobutene-OH adducts + O2: A detailed tertiary butyl oxidation mechanism. J Phys Chem. 1999;103:9731–9769. [Google Scholar]
- 27.Uraguchi D, Yamamoto K, Ohtsuka Y, Tokuhisa K, Yamakawaa Y. Catalytic trifluoromethylation of uracil to 5-trifluoromethyluracil by use of CF3I and its industrial applications. Appl Cat A General. 2008;342:137–143. [Google Scholar]
- 28.Kumar V, Malhotra SV. Synthesis of nucleoside-based antiviral drugs in ionic liquids. Bioorg Med Chem Lett. 2008;18:5640–5642. doi: 10.1016/j.bmcl.2008.08.090. [DOI] [PubMed] [Google Scholar]
- 29.Bresser T, Monzon G, Mosrin M, Knochel P. Scaleable preparation of sensitive functionalized aromatics and heteroaromatics via directed metalation using tmpZnCl-LiCl. Org Process Res Dev. 2010;14:1299–1130. [Google Scholar]
- 30.Hirota K, Sako MH. A high chemical reativity of 5-azidouracils and its synthetic application: novel synthesis of 8-substituted 1,3-dimethylxanthine derivatives. Heterocycles. 1997;46:547–554. [Google Scholar]
- 31.Coe JW, et al. Varenicline: An alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- 32.Coe JW, Watson HA, Singer RA. Varenicline: Discovery synthesis and process chemistry developments. In: Gadamasetti K, Braish T, editors. Process Chemistry in the Pharmaceutical Industry. Vol 2. Boca Raton, FL: CRC Press; 2008. pp. 23–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.