

Abstract
The induction of neurite outgrowth by NGF is a transcription-dependent process in PC12 cells, but the transcription factors that mediate this process are not known. Here we show that the bHLH transcriptional repressor HES-1 is a mediator of this process. Inactivation of endogenous HES-1 by forced expression of a dominant-negative protein induces neurite outgrowth in the absence of NGF and increases response to NGF. In contrast, expression of additional wild-type HES-1 protein represses and delays response to NGF. Endogenous HES-1 DNA-binding activity is post-translationally inhibited during NGF signaling in vivo, and phosphorylation of PKC consensus sites in the HES-1 DNA-binding domain inhibits DNA binding by purified HES-1 in vitro. Mutation of these sites generates a constitutively active protein that strongly and persistently blocks response to NGF. These results suggest that post-translational inhibition of HES-1 is both essential for and partially mediates the induction of neurite outgrowth by NGF signaling.
Keywords: bHLH transcription factors, neuronal differentiation, cell signaling, transcriptional repression, Hairy-related, PKC, neurotrophins
Basic helix–loop–helix (bHLH) genes control cell fate decisions, such as neurogenesis and myogenesis, in both vertebrates and invertebrates (Campuzano and Modolell 1992; Campos-Ortega 1993; Edmondson and Olson 1993; Jan and Jan 1993; Weintraub 1993). In Drosophila, neurogenesis is controlled by two distinct groups of genetically defined bHLH activator and repressor genes. The activators are the “proneural” genes such as the achaete and scute genes of the achaete–scute Complex (Jan and Jan 1990; Campuzano and Modolell 1992). The repressors are the “Hairy-related” genes such as hairy and Enhancer of Split (Rushlow et al. 1989; Delidakis and Artavanis-Tsakonas 1992; Knust et al. 1992). In addition to their distinct genetic functions as activators or repressors of neurogenesis, these two groups also have distinct DNA-binding and transcriptional regulation properties. The activators all bind to one class of DNA sites (class A sites) (Dang et al. 1992; Ohsako et al. 1994), whereas the repressors bind to different sites (class C or class B sites) (Sasai et al. 1992; Tietze et al. 1992; Ohsako et al. 1994; Van Doren et al. 1994). In addition, the genetic activators function as transcriptional activators in cellular transcription assays, whereas the genetic repressors function as transcriptional repressors (Oellers et al. 1994; Ohsako et al. 1994; Van Doren et al. 1994). These activator and repressor classes and their functional properties have been highly conserved. For example, MASH1, a mammalian achaete–scute homolog (Johnson et al. 1990; Guillemot et al. 1993) and HES-1, a Hairy and Enhancer of Split homolog, are conserved activator and repressor proteins, respectively (Sasai et al. 1992; Feder et al. 1993; Ishibashi et al. 1995).
Hairy-related proteins, such as HES-1, are defined by the presence of two distinct functional domains that confer transcriptional repressor activity to them. A repressor-specific class of basic region, containing a proline at a characteristic position, is a DNA-binding domain that binds to repressor-specific DNA sites (class C sites; Tietze et al. 1992; Ohsako et al. 1994; Van Doren et al. 1994). The other conserved feature is a 4-amino-acid domain, WRPW (Trp-Arg-Pro-Trp), located at the carboxyl terminus of each of these proteins, which functions as a transcriptional repression domain by recruiting a non-DNA-binding transcriptional corepressor called Groucho in Drosophila, or Transducin-like Enhancer of Split (TLE) in mammals (Paroush et al. 1994; Fisher et al. 1996; Grbavec and Stifani 1996). By virtue of these proline bHLH and WRPW domains, all Hairy-related proteins have the potential to function as DNA-binding, active transcriptional repressor proteins (Fisher et al. 1996) and function as repressors of differentiation (Ohsako et al. 1994). For example, HES-1 is a DNA-binding transcriptional repressor in mammalian cells (Sasai et al. 1992; Fisher et al. 1996), and it represses neuronal differentiation when expressed ectopically in the mammalian central nervous system (CNS) or chick retinal cells during development (Ishibashi et al. 1994; Tomita et al. 1996). Furthermore, HES-1 null mice have fewer than normal neuronal cells, attributable to premature differentiation of neuroblasts (Ishibashi et al. 1995).
HES-1, also called Rat Hairy-Like (RHL), is transcribed in the rat PC12 cell line before and during differentiation (Feder et al. 1993). PC12 cells are a pheochromocytoma tumor cell line that has been used widely as a model for neurotrophin signaling because they exhibit neurite outgrowth and sympathetic neuron-like differentiation after treatment with the neurotrophin nerve growth factor (NGF) (Greene and Tischler 1976). The NGF signaling pathway in PC12 cells has been studied extensively, and involves several kinase pathways including a Ras/mitogen-activated protein kinase (MAPK) pathway, a PI-3 kinase pathway, and a phospholipase C γ (PLCγ)/protein kinase C α (PKCα) pathway. These pathways are activated upon binding of NGF to the Trk A NGF receptor (for review, see Kaplan and Stephens 1994; Segal and Greenberg 1996). Recent analysis has shown that in addition to conventional cytoplasmic PKCs, a variety of conventional and nonconventional PKCs are activated or localized to the membrane or nucleus in PC12 cells during NGF signaling (Wooten 1992; Coleman and Wooten 1994; Borgatti et al. 1996). Other kinases, including pp90RSKs (Nakajima et al. 1996; Xing et al. 1996), also are activated during NGF signaling.
The induction by NGF of neurite outgrowth and neuronal marker gene expression in PC12 cells is known to be a transcription-dependent process and several nuclear targets for NGF have been identified. However, these have been general transcription factors such as Fos (Greenberg et al. 1985) and Nur77 (Yoon and Lau 1994), which do not appear to mediate the neurite outgrowth and differentiation response to NGF. Although expression of either activated Ras or activated MAPKK (MEK) is sufficient to induce neurite outgrowth in the absence of NGF (Bar-Sagi and Feramisco 1985; Cowley et al. 1994), such inductive properties have not been shown for the known transcription factor targets of NGF signaling.
Given that HES-1 is a defined repressor of neuronal differentiation, it is unclear why HES-1 mRNA, if translated into active protein, should be expressed during NGF signaling (Feder et al. 1993). We show here that endogenous HES-1 protein is expressed in uninduced PC12 cells, where it acts to inhibit differentiation, but that HES-1 DNA-binding activity is inhibited post-translationally during NGF signaling, thereby allowing differentiation to occur. Furthermore, increasing or decreasing the level of functional HES-1 protein by expression of additional wild-type HES-1 or dominant-negative HES-1 decreases or increases response to NGF, respectively, indicating that endogenous HES-1 is a component of the NGF signaling pathway. In addition, expression of a constitutively active HES-1 protein lacking consensus PKC phosphorylation sites in the DNA-binding domain persistently blocks NGF response, suggesting that inhibition of HES-1 DNA-binding activity is necessary for the induction of neurite outgrowth by NGF. Together, these results indicate that inhibition of endogenous HES-1 contributes to the induction of neurite outgrowth by NGF signaling.
Results
To determine whether endogenous HES-1 acts as a repressor in PC12 cells and how it functions during NGF signaling, we have expressed three different forms of HES-1 in stably and transiently transfected P12 cells: wild-type (WT HES-1), dominant-negative (DN HES-1), and a constitutively active serine mutant HES-1 (SM HES-1). Proteins were expressed from either a low-expression level promoter [uninduced, basal mouse mammary tumor virus (MMTV)] or high-expression level promoter cytomegalovirus (CMV), and all proteins carried an HA1/T7 double epitope tag fused at the amino terminus (see Materials and Methods). Except for the experiments noted for DN HES-1, all of the experiments described used cell lines transfected with MMTV expression constructs. Higher level expression of WT HES-1 (or SM HES-1) appears to be toxic, as transfection attempts repeatedly failed to generate any stably transfected CMV WT HES-1 cells. Low-level expression of proteins in clones stably transfected with MMTV vectors was further indicated by immunocytochemical analysis. It was necessary to use a combination of antibodies against both the HA and T7 epitopes to visualize protein expression in cells, because neither antibody proved sufficient to detect epitope-tagged protein when used individually (see Materials and Methods). Inadequate sensitivity of the epitope antibodies also prevented us from visualizing the epitope-tagged exogenously expressed protein by Western blot analysis, although we have also confirmed expression of stably transfected genes by reverse transcriptase–polymerase chain reaction (RT–PCR) (see Materials and Methods).
Induction of neurite outgrowth in the absence of NGF by expression of a DN HES-1 protein
To generate a DNA-binding defective, dominant-negative form of HES-1 (DN HES-1), 3 amino acids in the basic region were mutated such that the mutant protein cannot bind to DNA, although it can still dimerize with endogenous WT HES-1 to form a non-DNA-binding heterodimer complex (data not shown). Expression of DN HES-1 in stably transfected cells resulted in a flatter cell morphology with the induction of numerous short neuritic projections (1- to 2-cell diameters), together with a greatly potentiated response to NGF. In addition, ∼10% of the clones isolated from each of four separate transfections exhibited a marked extension of neurites (2- to 10-cell diameters in length) in the absence of NGF (Fig. 1A). This more extreme phenotype was stable for several months of continuous culture, although it generally diminished over time. In contrast, cells transfected with a control expression plasmid were never observed to extend neurites (Fig. 2A,D). The DN HES-1 transfected cells also showed an increased expression of the endogenous neuronal marker genes neurofilament 160 (NF 160), peripherin, and GAP-43 (Fig. 1B,C,D), compared to controls. Thus, the DN HES-1 cells exhibited neurite outgrowth and neuronal marker gene expression in the absence of NGF, suggesting that inactivation of HES-1 results in at least partial differentiation of PC12 cells.
Figure 1.

Induction of neurite outgrowth in the absence of NGF by cells expressing DN HES-1. (A) Phase-contrast micrograph of cells from a stably transfected DN HES-1 clone. In contrast to control cells (Fig. 2D) many DN HES-1-expressing cells extend neurites in the absence of NGF. (B–D) Western blot analysis comparing neuronal marker protein expression in equal amounts of total protein from control and DN HES-1 expressing cells; (B) neurofilament 160 kD (NF 160), (C) peripherin, (D) GAP-43. (E) Gel-retardation assay comparing the level of endogenous repressor-specific (class C) DNA-binding activity in nuclear extracts from control cells to DN HES-1 cells, normalized to total protein. (F) Confirmation that the endogenous class C-binding complex contains HES-1. Class C binding proteins in PC12 nuclear extracts (N.E.) were affinity purified using a biotinylated Class C DNA probe; Western blot analysis of the DNA-purified protein using an anti-HES-1 antibody (kindly provided by Drs. J. Feder and Y.N. Jan) revealed a band (indicated by lower arrow on left side, lane 1) that comigrated with the anti-HES-1-labeled band present in nuclear extracts (indicated by arrow, lane 3). A DNA-purification positive control, using bacterially expressed GST HES-1 fusion protein in a parallel reaction, is evident as a higher molecular mass band (indicated by upper arrow on left side, lane 2) with characteristically observed degradation products. The upper band in lanes 1 and 2 (indicated by asterix) and not present in lane 3, is probably streptavidin protein eluted from the streptavidin beads during the purification.
Figure 2.

Increased response to NGF in DN HES-1-expressing cells. (A) Three stable DN HES-1 clones (1–3) show increased response to low amounts of NGF (1 or 5 ng/ml for 24 hr) relative to control cells. This is a representative graph from a triplicate experiment that was repeated five times. The error is the standard deviation of the mean. (B,C) DN HES-1-expressing cells (C) also showed a much greater length of neurites than did the control cells (B) when exposed to NGF. (D–F) Clones from both the DN HES-1 (E) and WT HES-1 (F) transfected cells showed HA/T7 epitope-tagged protein expression and nuclear localization, whereas no staining was evident in control cells (D).
To confirm the functional activity of the expressed DN HES-1 protein, nuclear extracts from DN HES-1 cells were tested for the inhibition of binding of endogenous HES-1 protein to a labeled repressor-specific class C site in a gel-retardation assay. Nuclear extracts from control cells contained a single endogenous class C binding complex (Fig. 1E, lane 1). In contrast, nuclear extracts from cells expressing DN HES-1 exhibited a significantly reduced level of class C binding activity relative to the control cells in a gel-retardation assay (Fig. 1E, lane 2). To verify that the endogenous class C binding complex contains HES-1, nuclear extracts were incubated with a biotin-conjugated class C DNA site and the DNA-bound complex was purified with streptavidin-conjugated beads. Western blot analysis of the purified protein complex using affinity purified anti-HES-1 antibodies (Chen et al. 1997) showed that it contained an anti-HES-1 reactive band that comigrated with endogenous HES-1 protein from nuclear extracts (Fig. 1F, lanes 1,3). The same anti-HES-1 antibodies, but not control antibodies, also partially disrupted DNA binding by the endogenous class C binding complex, further indicating that it contains HES-1 (data not shown).
Modulation of NGF signaling by expression of DN HES-1 or WT HES-1 proteins
To test whether DN HES-1 protein expression increased response to NGF, DN HES-1 and WT HES-1 stably transfected clones were isolated and specific expression and nuclear localization of epitope-tagged protein was confirmed (Fig. 2D–F). DN HES-1-transfected cells were treated with relatively low amounts of NGF (1 or 5 ng/ml, where 10 ng/ml is normally necessary for full response). DN HES-1 cells showed a significant quantitative increase in the percentage of cells with neurites relative to control cells (Fig. 2A). Moreover, the response of these cells was also greatly increased with respect to the length of neurites compared to the control cells (Fig. 2B,C). Thus, inactivation of endogenous HES-1 by DN HES-1 results in a significantly enhanced response to NGF both with respect to the number and the length of neurites.
In contrast, two stable clones expressing (WT HES-1) exhibited almost no morphological response to NGF after 48 hr of treatment (Fig. 3A,E), and a third clone showed an intermediate phenotype (not shown). A control clone exhibited a normal response to NGF, showing increased neurite outgrowth with increasing NGF, up to a saturation point of ∼10 ng/ml of NGF (Fig. 3A,C). Although WT HES-1 expression strongly represses the neurite outgrowth response after 48 hr of NGF treatment, when these cells are cultured in NGF for longer periods they do respond; however, that response is greatly inhibited and delayed relative to control cells (shown in Fig. 9, below). Thus, WT HES-1 expression is sufficient to strongly inhibit and delay NGF signaling.
Figure 3.

Decreased response to NGF in WT HES-1-expressing cells. (A) NGF response is greatly repressed in cells expressing WT HES-1 (▵ and □, two WT HES-1 stable clones) compared to stable control cells (•) after 48 hr of treatment with NGF 100 ng/ml. This is a representative graph from triplicate experiments that were repeated four times. The error is the standard deviation of the mean. (B) Control PC12 stable cells without NGF. (C) Control PC12 stable cells treated with 100 ng/ml of NGF for 48 hr extend neurites. (D) HES-1 expressing stable PC12 cells without NGF. (E) HES-1 expressing stable PC12 cells treated with 100 ng/ml of NGF for 48 hr do not significantly respond to NGF.
Figure 9.

Persistent block of NGF response in SM HES-1-, but not WT HES-1-expressing cells. (A) The neurite outgrowth response of control, WT HES-1, and SM HES-1 cells was analyzed over 5 days of exposure to NGF. WT HES-1 stable cells have essentially no neurite outgrowth response to 100 ng/ml of NGF at 2 days of exposure (also see Fig. 2), but by 5 days they have regained a similar response in terms of percent cells with neurites. In contrast, three stable SM HES-1 PC12 clones do not respond substantially to NGF after 5 days of exposure. This graph shows data from two independent experiments performed in triplicate, the error bars are the standard deviation of the mean. (B–D) Although the percentage of cells bearing neurites after 5 days of NGF treatment is similar to that of control cells (B), the extent of the neurites in WT HES-1 cells (C) is substantially reduced. The SM HES-1 cells exhibit essentially no neurite outgrowth (D). (E) Semiquantitative RT–PCR of glycerol-3-phosphate dehydrogenase (G3PDH) (input control) and exogenous HES-1 reveals an equivalent level of expression between the WT and SM HES-1 cell lines, indicating that the difference in response is attributable to mutation of the basic region serines. The numbers above the bands correspond to the clones analyzed in panel A, the plus sign is a cDNA control. (F,G) Immunohistochemical staining with antibodies against the T7 and HA epitopes on the exogenous SM HES-1 proteins reveals consistent nuclear expression of these proteins in the SM HES-1 stable PC12 lines (G) and an absence of staining in the control cells (F).
Together the WT HES-1 and DN HES-1 results show that increased or decreased levels of functional HES-1 protein levels decrease or increase response to NGF, respectively, suggesting that endogenous HES-1 is a functional component of the NGF signaling pathway.
Post-translational inhibition of endogenous HES-1 DNA-binding activity during NGF signaling
To determine whether DNA-binding activity of endogenous HES-1 protein is inhibited during NGF signaling in vivo, we compared the level of HES-specific class C DNA-binding activity present in nuclear extracts from untreated control cells with that in extracts from cells treated for 24 hr with NGF. We observed that the level of endogenous binding activity decreased significantly after 24 hr of NGF treatment relative to the level in untreated cells (Fig. 4A, lanes 1,2). In contrast to the decrease in endogenous HES-1 DNA-binding activity, Western blot analysis of HES-1 protein levels in the same nuclear extracts showed no decrease in level of protein between control and 24-hr NGF-treated cells (Fig. 4B, lanes 1,2), thereby indicating that endogenous HES-1 is inhibited post-translationally with respect to DNA-binding activity during NGF signaling.
Figure 4.
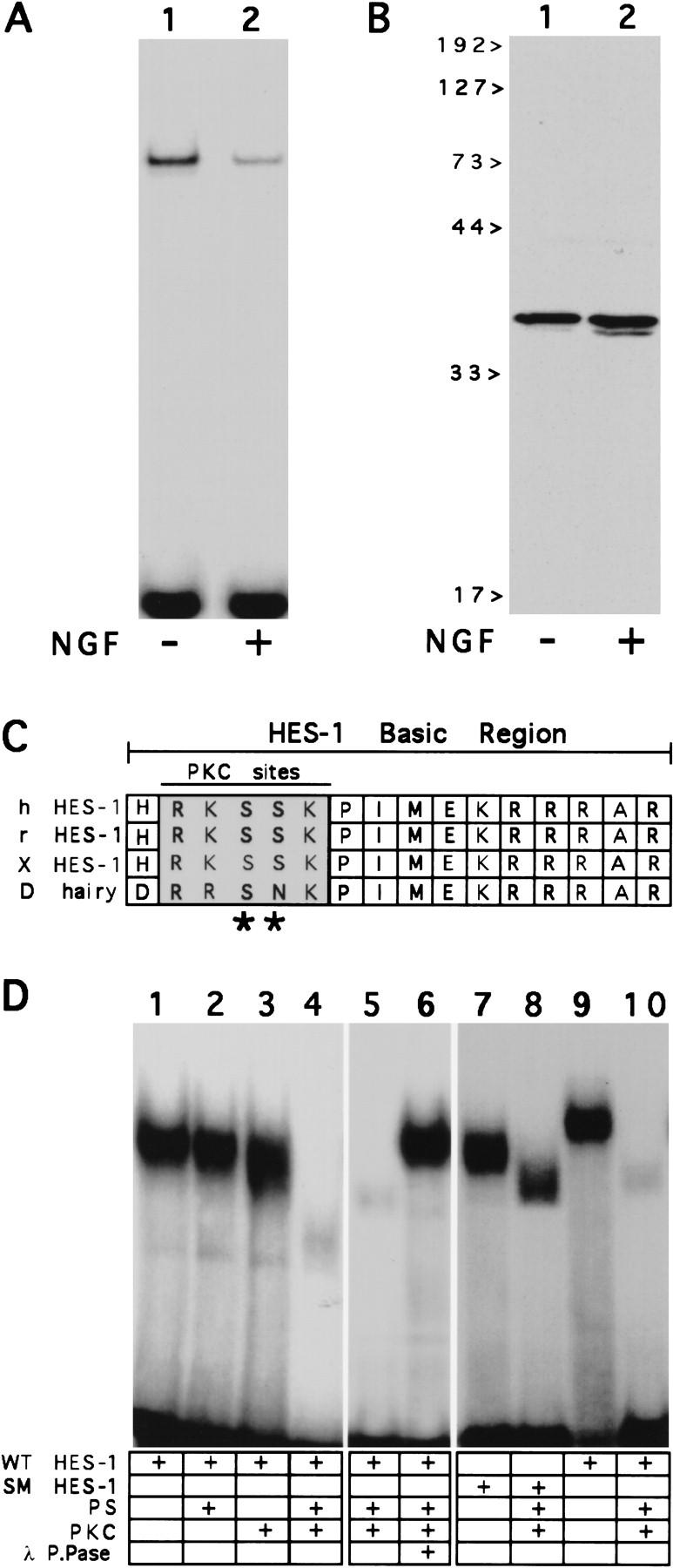
Post-translational inhibition of HES-1 DNA binding activity in vivo and in vitro. (A,B) Post-translational inhibition of endogenous HES-1 DNA binding during NGF signaling. (A) HES-1 DNA-binding activity in nuclear extracts from control PC12 cells (lane 1) and PC12 cells treated with 100 ng/ml of NGF for 24 hr (lane 2). (B) Anti-HES-1 Western blot analysis of the same PC12 nuclear extracts, showing that the level of endogenous HES-1 protein in PC12 cell nuclear extracts (lane 1) is not decreased after 24 hr of NGF treatment (lane 2). (C) Sequence alignment of the DNA-binding domains (basic regions) from the human, rat and Xenopus HES-1 homologs, together with Drosophila hairy. The HES-1 homologs each contain two adjacent serines within PKC consensus phosphorylation sites (*) in the basic region. (D) Inhibition of purified HES-1 protein DNA-binding activity by PKC in vitro. (Lanes 1–4) Incubation of purified, bacterially expressed and HES-1 bHLH domain with PKC results in inactivation of HES-1 DN- binding activity in a phosphotidyl serine (PS)-dependent manner. (Lanes 5,6) Subsequent addition of lambda protein phosphatase (lane 6) restores DNA-binding activity to PKC-treated HES-1 (lane 5). (Lanes 7–10) Treatment of SM HES-1 with PKC does not inhibit DNA-binding activity (cf. lane 7 with lane 8), whereas PKC does inhibit WT HES-1 DNA-binding activity (cf. lane 9 with lane 10). SM HES-1 protein migrates differently than WT HES-1 on native gels (lanes 7,9), despite being identical to the WT HES-1 with the exception of the two basic region serine residues; both migrate identically on SDS-PAGE. Also, phosphorylation of an additional PKC site present in the loop region of HES-1 apparently makes SM HES-1 protein run faster than unphosphorylated protein (lane 8).
Inhibition of HES-1 DNA-binding activity in vitro by phosphorylation by PKC
We have observed that there are two adjacent serine residues in the HES-1 DNA-binding domain, which are both consensus phosphorylation sites for PKC and that these sites are conserved in the Xenopus, rat, and human HES-1 homologs (Fig. 4C). To test whether HES-1 can be phosphorylated by PKC and if so, whether this inhibits DNA-binding activity in vitro, the WT HES-1 bHLH domain (bHLH HES-1) was expressed in bacteria as a fusion protein containing a poly-histidine leader and purified by metal chelate chromatography. Bacterially expressed HES-1, which is not phosphorylated, binds well to a class C DNA site as a homodimer (Fig. 4D, lanes 1,9). However, incubation with purified PKC completely inhibited the HES-1 DNA-binding activity in a phosphatidyl serine (PS) (Fig. 4D, lanes 1–4) and ATP-dependent manner (not shown). This inhibition was reversible by the subsequent addition of λ protein phosphatase (Fig. 4D, lanes 5,6). To test whether the inhibition of binding was attributable to phosphorylation of the two serine residues in the basic region, the codons for those residues were mutated to encode nonphosphorylatable residues and the serine mutant protein SM HES-1 was expressed in bacteria and purified as above. In contrast to the wild-type bHLH HES-1 protein, which is inhibited with respect to DNA-binding activity, the DNA-binding activity of the SM HES-1 protein was not inhibited by treatment with PKC. This protein bound as well after PKC treatment as did untreated protein (Fig. 4D, lanes 7,8), although phosphorylation of an additional site, probably a PKC consensus site present in the loop region, caused the protein–DNA complex to run faster in the gel than the untreated protein.
Together these results indicate that phosphorylation of HES-1 by PKC inhibits HES-1 DNA-binding activity, and that this is attributable to phosphorylation of the PKC consensus sites in the HES-1 DNA-binding domain.
TPA induced restoration of NGF response to WT HES-1-expressing cells
Previous studies have shown that various cytoplasmic and nuclear PKCs are activated or localized during NGF signaling in PC12 cells (Wooten 1992; Coleman and Wooten 1994; Borgatti et al. 1996). To determine whether activation of endogenous PKCs had an effect on the repression of neurite outgrowth observed in the WT HES-1-expressing cells, these cells were exposed to the phorbol ester PKC activator, TPA. Remarkably, treatment of WT HES-1 cells with TPA (150 nm TPA for 24 hr) completely restored the response of WT HES-1-expressing cells to NGF (Fig. 5C,D), although the cells showed essentially no response to NGF or TPA alone (Fig. 5A,B). (Control experiments showed that the WT HES-1 protein levels do not decrease after NGF, TPA, or NGF + TPA treatment; see Fig. 6B, below.) These results show that activation of endogenous PKCs by TPA is sufficient to substantially inhibit the functional repression activity of exogenously expressed WT HES-1 protein. Interestingly, previous studies have shown that exposure of wild-type PC12 cells to TPA modestly increases the response to NGF (Burstein et al. 1982).
Figure 5.
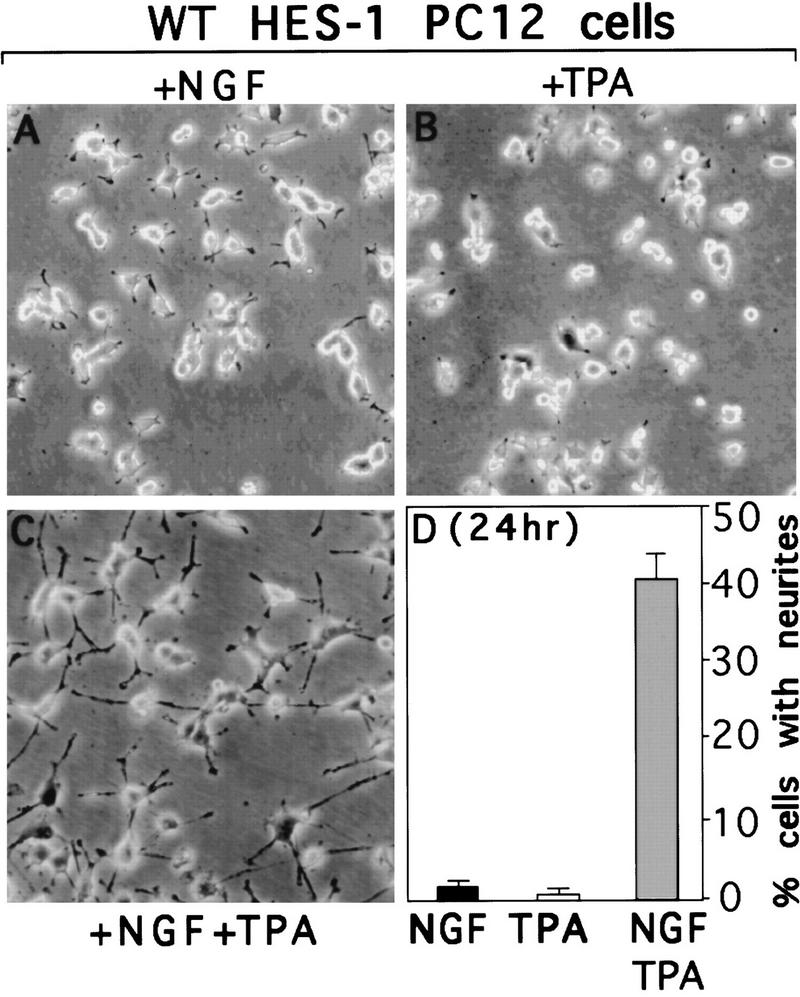
TPA induced restoration of NGF response to WT HES-1-expressing cells by activation of endogenous PKCs. (A) WT HES-1-expressing cells do not extend neurites after 24 hr of exposure to 100 ng/ml of NGF. (B) WT HES-1-expressing cells do not extend neurites after 24 hr of exposure to TPA. (C) TPA cotreatment for 24 hr restores a normal NGF response to WT HES-1-expressing cells. (D) Quantitation of the degree of neurite outgrowth observed in the NGF, TPA, and NGF+TPA treated WT HES-1-expressing cells. This is a representative graph of a triplicate experiment that was repeated five times. The error is the standard deviation of the mean. Although not shown, the percentage of HES-1 cells with neurites at the 24-hr +NGF time point was 3.6% ± 0.9%, significantly less than the control cell value of 23.1% ± 1.6%. Also, the control cells exhibited a potentiated response to TPA plus NGF, which can be seen in Figure 8, A and B.
Figure 6.

Inhibition of endogenous HES-1 DNA binding during NGF- and PKC-dependent signaling. Gel-shift analysis of PC12 nuclear extracts, using a C class DNA probe, reveals that endogenous HES-1 DNA-binding activity is strongly inhibited after a 24-hr treatment with the PKC activator TPA (A, lane 3), relative to equal amounts of untreated control extract (A, lane 1). The NGF-induced decrease in HES-1 DNA binding (A, lane 2), is blocked by coaddition of the PKC inhibitor bisindolylmaleimide (A, lane 4). Western blot analysis of PC12 nuclear extracts blotted with anti-HES-1 antibody (B) reveals that, as in the case for NGF (lane 2, also see Fig. 4B), the loss of HES-1 DNA binding is not attributable to a reduction in protein levels after treatment with TPA (lane 3), or TPA plus NGF (lane 4).
Inhibition of endogenous HES-1 DNA-binding activity by a PKC-like kinase during NGF signaling
To further determine whether the decrease in endogenous HES-1 DNA-binding activity observed after NGF treatment is attributable to phosphorylation by an endogenous PKC or PKC-like kinase, we treated control PC12 cells with the classic PKC activator TPA. After TPA treatment for 24 hr in the absence of NGF, extracts from treated cells showed a strong decrease in endogenous HES-1-binding activity (Fig. 6, lanes 1,3) analogous to that observed during NGF signaling (Fig. 6A, lane 2), although protein levels did not decrease after treatment with NGF and/or TPA (Fig. 6B, lanes 1–4). This decrease in DNA-binding activity occurred in the absence of NGF (Fig. 6, lane 3), indicating that activation of endogenous PKCs or PKC-like kinases can mimic the effect of NGF signaling on HES-1 activity. Moreover, treatment of cells with the general PKC inhibitor bisindolylmaleimide, which competes with ATP for the ATP-binding site in the various PKCs, blocked the NGF-induced decrease completely (Fig. 6A, lane 4).
Thus, the NGF-induced decrease in HES-1 DNA binding activity in vivo is dependent on activation of an endogenous PKC or PKC-like kinase, which may phosphorylate directly the PKC sites present in HES-1 DNA-binding domain.
DN HES-1 expression restores NGF response to cells inhibited by the PKC inhibitor bisindolylmaleimide
Given that the PKC inhibitor bisindolylmaleimide blocked the post-translational inhibition of endogenous HES-1 DNA-binding activity observed during NGF signaling (Fig. 6A, lane 4), we then asked whether this PKC inhibitor blocks NGF-induced neurite outgrowth, and if so, whether expression of DN HES-1 could bypass this blockage. Although the NGF-induced neurite outgrowth response of control PC12 cells is strongly reduced by bisindolylmalmeimide (Fig. 7A–C), the neurite outgrowth response of a DN HES-1-expressing clone is still robust in the presence of an equivalent amount of the PKC inhibitor (Fig. 7A,D,E). Thus, DN HES-1 expression makes the NGF-induced neurite outgrowth response of the cells independent of PKC activation, suggesting that HES-1 functions downstream of PKC in the NGF signaling pathway. These results are quantitated in the graph shown in Figure 7A.
Figure 7.

NGF response is not inhibited by the PKC inhibitor bisindolylmaleimide (bis) in DN HES-1-expressing PC12 cells. Response of control cells (B) to 48 hr of NGF (50 ng/ml) is greatly reduced by addition of bisindolylmaleimide (4 μm) (C), whereas the NGF response in DN HES-1 cells in the presence of bisindolylmaleimide (E) is only slightly reduced compared to that in NGF alone (D). This effect is quantitated in the graph shown in A, which is the average of two independent experiments with the error as the standard deviation of the mean.
Requirement of the basic region serine residues for TPA-induced restoration of NGF response in HES-1 overexpressing cells
We then wished to determine whether the TPA-induced restoration of NGF response in WT HES-1-expressing cells (see Fig. 5) is attributable to phosphorylation of the PKC consensus sites in the HES-1 DNA-binding domain. Cells stably transfected with expression plasmids encoding full-length WT HES-1 or SM HES-1 were compared for the ability of TPA to restore NGF responsiveness (Fig. 8A,C,D). The wild-type and mutant plasmids were identical except for the 2-amino-acid codons encoding the PKC phosphorylation sites in the basic region. Whereas WT HES-1-expressing cells responded to NGF nearly as well as control cells after TPA treatment (Fig. 8A–C), cells expressing SM HES-1 showed little response to NGF even after TPA treatment (Fig. 8A,D). Thus, mutation of the PKC sites in the HES-1 basic region generates a protein that is insensitive to inhibition by TPA-induced activation of PKC.
Figure 8.

TPA restores the NGF response to WT HES-1 but not SM HES-1-overexpressing cells. (A) Quantitation of the percentage of cells from the control, WT HES-1, and SM HES-1 stably transfected clones that have neurites after 24 hr of NGF + TPA treatment. This is a representative graph from triplicate experiments repeated three times. (B) Response of control stably transfected cells to treatment with NGF + TPA for 24 hr. (C) Response of WT HES-1-expressing cells to NGF + TPA is similar to that of control cells. (D) SM HES-1-expressing cells do not extend neurites in response to NGF + TPA treatment. (E) Neurite outgrowth response of PC12 cells transfected transiently with equal amounts of the same expression plasmids as above, plus a β-galactosidase reporter vector, in the presence of NGF alone or NGF plus TPA. The percentage of transfected cells (identified by β-galactosidase activity) with neurites was determined, from which the percent repression of neurite outgrowth for the WT HES-1 and SM HES-1 transfected cells was calculated relative to the corresponding control value (taken as 0% repression). The data shown are from two independent sets of triplicate experiments, the error is the standard deviation of the mean. WT HES-1-transfected cells exhibited a significantly lower repression of NGF-induced neurites in the presence of TPA (∼27% repression without TPA, ∼12% with TPA). SM HES-1-transfected cells, in contrast, exhibited nearly identical percent repression of neurites in both NGF (50%) and NGF + TPA (46%), indicating that SM HES-1 repression activity is insensitive to TPA induced activation of endogenous PKCs. The SM HES-1 transfected cells also exhibited higher repression of neurites than WT HES-1-transfected cells in either NGF (50% vs. 27%) or NGF + TPA (46% vs. 12%). The observation that transiently transfected cells are not all completely repressed at this time point, unlike WT HES-1 stable cells, may relate to the degree and timing of HES-1 expression; in particular the relationship between the timing of transient transfection to the start of NGF treatment and the cell cycle stages of these cells.
To control against artifacts that might arise during generation and selection of stable cell lines, transient transfection experiments were performed with the same WT HES-1 and SM HES-1 expression plasmids and the results shown in Figure 8E. The percentage of the transfected cells (which were β-galactosidase positive) that extended neurites was determined and the percent neurite repression in WT HES-1 versus SM HES-1-expressing cells, relative to the corresponding control cell response, was then calculated (Fig. 8E, also see legend). The percent repression of neurite outgrowth by SM HES-1 was unchanged by the addition of TPA to the medium (Fig. 8E, column 3 versus column 4). In contrast, for WT HES-1 the level of repression was reduced significantly by the addition of TPA (Fig. 8E, column 1 versus column 2).
Thus, the results of both the transient and stable transfection experiments show that WT HES-1 neurite repression activity is inhibited upon activation of endogenous PKC by TPA, and this inhibition is at least partially dependent on the PKC phosphorylation sites in the HES-1 DNA-binding domain. Mutation of these sites makes SM HES-1 insensitive to inhibition by TPA, suggesting that this mutant protein functions as a constitutively active DNA-binding repressor.
Requirement of the basic region serine residues for NGF response in HES-1-expressing cells
The transient transfection results shown in Figure 8E also suggest that the neurite repression activity of the WT HES-1 protein is inhibited during normal NGF signaling (i.e., in the absence of TPA). The reduced repression by WT HES-1 relative to SM HES-1 (27% vs. 50%, respectively; Fig. 8E, columns 1,3) may be attributable to post-translational inhibition of the HES-1 DNA-binding domain by endogenous PKC activated during normal NGF signaling.
To further address whether HES-1 repression activity is inhibited through the basic region PKC sites during normal NGF signaling, the neurite repression activities of WT HES-1 and SM HES-1 were also compared in the stably transfected cells treated with NGF for extended periods of time. As mentioned above (see Fig. 3) and shown in Figure 9, the response of WT HES-1-expressing cells to NGF is greatly repressed for the first 48 hr of NGF treatment (Fig. 9A, day 2). However, if these cells are cultured for longer periods in NGF they do extend neurites (Fig. 9A, “WT HES #1”), although the response is still inhibited (Fig. 9B,C). For example, after 5 days of NGF treatment, the percentage of cells with neurites is nearly normal; however, those neurites that are present are markedly reduced in length relative to the neurites extended by control cells (Fig. 9B,C).
To test whether phosphorylation of the basic region serines allows WT HES-1-overexpressing cells to respond eventually to NGF, WT HES-1, and SM HES-1 stably transfected cells were compared for neurite outgrowth response to NGF during 5 days of NGF treatment. As for DN HES-1 and WT HES-1 (see Fig. 2E,F), SM HES-1 was expressed specifically and localized to the nucleus (Fig. 9F,G). RT–PCR analysis revealed that the stably transfected SM HES-1 and WT HES-1 expression plasmids were expressed at equivalent levels (Fig. 9E). In contrast to WT HES-1-expressing cells, three clones of SM HES-1-expressing cells examined showed essentially no response to NGF even after a 5-day period of exposure (Fig. 9A). In addition, these cells showed a nearly complete absence of even short processes as compared to the WT HES-1-expressing cells (Fig. 9C,D).
Thus, mutation of the PKC sites in the HES-1 DNA-binding domain generates a constitutively active repressor protein that persistently blocks the response to NGF. These results also suggest that post-translational inhibition of HES-1 by phosphorylation of PKC sites in its DNA-binding domain is essential for the induction of neurite outgrowth by NGF.
Discussion
Post-translational inhibition of HES-1 DNA-binding activity during NGF signaling
Our results show that endogenous HES-1 is expressed in uninduced PC12 cells, but that its DNA-binding activity is inhibited post-translationally during NGF signaling. This inhibition is dependent on activation of an endogenous PKC or PKC-like kinase, but the exact identity of this kinase and the pathway leading to its activation have not yet been defined. HES-1 DNA-binding activity is inhibited in vitro by phosphorylation by PKC of serine residues in the HES-1 DNA-binding domain. In contrast, a mutated HES-1 protein (SM HES-1) lacking the serine residues in the basic region is resistant to post-translational inhibition by PKC in vitro. Moreover, as discussed further below, this SM HES-1 protein also behaves in vivo as a constitutively active repressor protein that is insensitive to post-translational inhibition during NGF signaling. Although we have not shown directly that phosphorylation of these serine residues occurs in endogenous HES-1 during NGF signaling, we have shown that these serines are essential for NGF induction of neurite outgrowth in a functional assay. Although indirect, this functional assay demonstrates the importance of these residues in HES-1 for NGF signaling in vivo and suggests that these residues are targets for phosphorylation during NGF signaling. The simplest interpretation of our results is that endogenous HES-1 binds to DNA sites in neuronal target genes and represses their transcription until it is inhibited post-translationally during NGF signaling by phosphorylation of its DNA-binding domain. This model for the induction of neurite outgrowth by derepression of target gene transcription is depicted in Figure 10.
Figure 10.

Model for the induction of neurite outgrowth through NGF through post-translational inhibition of endogenous HES-1. (A) In the absence of NGF (−NGF), endogenous HES-1 binds to class C sites in target neuronal differentiation genes and actively represses transcription. DNA-bound HES-1 actively represses target gene transcription by recruiting a TLE corepressor, as shown previously (see Discussion). (B) During NGF signaling, post-translational inhibition of HES-1 DNA-binding activity by phosphorylation of the HES-1 basic region results in derepression of target gene transcription, thus mediating the induction of neurite outgrowth and differentiation. The phosphorylation of HES-1 during NGF signaling requires the activation of one or more endogenous PKC or PKC-like kinase (“PKCs”), which phosphorylates directly or indirectly the consensus PKC sites in the HES-1 basic region.
Previous studies have demonstrated that NGF signaling induces the activation or localization of both cytoplasmic and nuclear PKC isoforms in PC12 and other cells (Leach et al. 1989; Borgatti et al. 1996), some in a time period similar to that observed for post-translational inhibition of endogenous HES-1 (Borgatti et al. 1996). Thus, the “HES kinase” that phosphorylates the HES-1 basic region directly may be a PKC or PKC-like kinase possibly localized to, or activated in, the nucleus. However, it could also be some other kinase dependent on the activation of an upstream PKC kinase. It also is possible that the kinase that directly phosphorylates and inhibits HES-1 is constitutively active, and that regulated inhibition of a phosphatase during NGF signaling results in increased phosphorylation of HES-1 and inhibition of DNA binding. In addition, one of the serine residues in the basic region is not only a canonical PKC site, but also is a potential phosphorylation site for several other kinases including PKA, Cam kinases, and ribosomal S6 kinase (RSKs). Preliminary results have shown that phosphorylation of this serine by PKA can inhibit, at least partially, HES-1 DNA-binding activity (P. Castella and M. Caudy, unpubl.). Given that both PKCs and at least some RSKs are activated during NGF signaling (Xing et al. 1996), HES-1 may be a target for multiple kinases activated or functioning during NGF signaling.
The expression of WT HES-1 protein in addition to endogenous HES-1 results in a substantial repression of response to NGF. This repression is overcome by the activation of endogenous PKC by TPA, which restores NGF response to WT HES-1-expressing cells. The delayed response observed during normal NGF signaling (i.e., in the absence of TPA), may be attributable to the additional time needed to phosphorylate and inhibit the additional wild-type HES-1 protein. In contrast, SM HES-1 protein appears to function as a constitutively active repressor in vivo. The TPA-induced restoration of NGF response observed in WT HES-1-overexpressing cells does not occur in cells expressing the SM HES-1 protein. Moreover, the persistent lack of response to NGF observed in SM HES-1-expressing cells also suggests that this protein is insensitive to post-translational inhibition during normal NGF signaling. This constitutive block of NGF response also suggests that post-translational inhibition of HES-1 is essential for NGF signaling to induce neurite outgrowth in PC12 cells.
An alternative mechanism that could explain some of our results is that NGF signaling might increase the expression or activity of a protein inhibitor of HES-1 DNA-binding activity. For example, a dominant-negative protein analogous to the Id repressor for bHLH activator proteins (Benezra et al. 1990) could inhibit endogenous HES-1 binding. However, if there were such a protein inhibitor it would seem likely to also inhibit the SM HES-1 protein, as it has the normal HLH dimerization domain. This, however, has not been observed.
HES-1 functions as a DNA-binding, active repressor of neuronal differentiation
Our results are consistent with previous studies that have shown that HES-1 functions as a repressor of neuronal differentiation during vertebrate embryogenesis (Ishibashi et al. 1994, 1995; Tomita et al. 1996). Previous studies have also shown that HES-1 and other Hairy-related proteins are DNA-binding, transcriptional repressor proteins that repress target gene transcription (Sasai et al. 1992; Ohsako et al. 1994; Van Doren et al. 1994; Fisher et al. 1996) by recruiting a TLE (Groucho homolog) corepressor through the WRPW domain at their carboxyl termini (Paroush et al. 1994; Fisher et al. 1996; Grbavec and Stifani 1996; Fisher and Caudy 1998). These results suggest that the endogenous HES-1 protein, which is present and active with respect to DNA binding in uninduced cells, should actively repress target genes until it is post-translationally inhibited during NGF signaling (Fig. 10).
The ability of DN HES-1 expression to induce neurite outgrowth in the absence of NGF demonstrates that inhibition of endogenous HES-1 DNA binding activity can be sufficient to induce differentiation, at least partially. It is unclear why activation of endogenous PKC by TPA, which strongly inhibits the DNA-binding activity of endogenous HES-1 protein, does not also induce neurite outgrowth although it greatly potentiates the response to NGF. It may be that broad activation of PKCs by TPA has additional actions beyond inhibition of HES-1 that repress rather than promote neurite outgrowth. In addition, DN HES-1 expression may result in the titration or sequestration of additional corepressor factors that normally interact with endogenous HES-1, such as the TLE corepressor. Such sequestration, or “squelching” of cofactors involved in repression could lead to further activation of the differentiation pathway than does only the inactivation of endogenous HES-1.
HES-1 as a mediator of NGF signaling
Three lines of evidence indicate that HES-1 mediates the induction of neurite outgrowth functionally by NGF signaling. First, the observation that DN HES-1 expression increases neurite outgrowth in response to NGF, whereas WT HES-1 expression decreases it, suggests that endogenous HES-1 is a functional component of the NGF signaling pathway. Second, the observation that expression of SM HES-1 blocks NGF response constitutively suggests that post-translational inhibition of overexpressed WT HES-1 is necessary before NGF signaling can induce neurite outgrowth. Third, the finding that DN HES-1 expression can induce neurite outgrowth in the absence of NGF indicates that endogenous HES-1 is actively repressing neurite outgrowth before NGF signaling. These results suggest that the post-translational inhibition of endogenous HES-1 actively mediates neurite outgrowth and differentiation.
The functional targets for repression by HES-1 in PC12 cells and in vivo are not known, but the MASH1 bHLH activator gene is a likely target for direct repression by HES-1. Gene disruption studies have shown that MASH1 is necessary for sympathoadrenal precursor differentiation in mice (Guillemot et al. 1993). MASH1 transcription is activated in PC12 cells 2 days after NGF treatment (Johnson et al. 1990) and a recent study of regulation of the HASH1 (human MASH1) promoter in cultured cells has shown that HES-1 directly binds to the promoter of HASH1 and represses HASH1 transcription (Chen et al. 1997). In addition, MASH1 is up-regulated transcriptionally in HES-1 null mice (Ishibashi et al. 1995). Direct transcriptional repression of MASH1 by HES-1 would be analogous to the direct transcriptional repression of the achaete proneural gene by the Hairy repressor protein in Drosophila (Ohsako et al. 1994; Van Doren et al. 1994), suggesting that this particular regulatory pathway has been conserved.
Regulation of expression and function of HES-1 by opposing Notch and NGF cell signaling pathways
The finding that HES-1 mediates NGF signaling is consistent with the emerging view that transcriptional repressor proteins are as important in regulating cell differentiation as are transcriptional activator proteins (Chong et al. 1995; Schoenherr and Anderson 1995). HES-1 is widely transcribed during embryogenesis (Sasai et al. 1992), but also is activated transcriptionally by the Notch signaling pathway during Notch-mediated lateral inhibition (Jarriault et al. 1995). The post-translational inhibition of HES-1 described in this study mediates a different cell signaling pathway, NGF signaling in the PC12 model cell system. In Notch signaling, the transcriptional activation of HES-1 inhibits neuronal differentiation, whereas in NGF signaling the post-translational inhibition of HES-1 induces neuronal differentiation by a derepression mechanism.
It is interesting to note that NGF signaling is potentially dominant over Notch signaling during neuronal differentiation. Although speculation, it may be that one function of NGF during neuronal differentiation is to protect a committed precursor from lateral inhibition by the Notch pathway.
Implications for bHLH proteins as common targets for cell signaling pathways
The present results have implications both for roles of HES-1 in additional cell signaling processes and for a general role of bHLH activator and repressor proteins as targets for regulation by cell signaling pathways. It seems likely that post-translational inhibition of HES-1 is a general mechanism for mediating many or all neurotrophin-mediated differentiation pathways, because the various other neurotrophins, BDNF, NT-3, and NT4/5, are known to activate similar downstream pathways as NGF (Kaplan and Stephens 1994; Segal and Greenberg 1996). It is also possible that HES-1 mediates other types of transcription-dependent signaling events regulated by NGF or other neurotrophins, such as synaptic plasticity (Lo 1995).
More generally, HES-1 is one of several members of a “functional cassette” of bHLH activator and repressor proteins that work together to provide a precise regulatory switch mechanism for controlling cell fate decisions in Drosophila and mammals (Jan and Jan 1993). Although the known pathways for regulating HLH factors in Drosophila work at the transcriptional level, it is plausible that post-translational regulation of the same key regulatory factors by cell signaling pathways has been added to transcriptional regulation. Moreover, although post-translational inhibition of HES-1 DNA-binding activity is the first example of post-translational inhibition of any of the neuronal bHLH proteins, a similar regulation of myogenin by PKC activated by fibroblast growth factor (FGF) signaling has been previously reported (Li et al. 1992). Our working hypothesis is that given their common role in regulating neuronal and other cell differentiation pathways in Drosophila and vertebrates, the cassette or module of bHLH activator and repressor transcription factors is likely to be the target for many cell signaling pathways controlling cell differentiation.
Materials and methods
Expression constructs
The cDNA for HES-1, kindly provided by Dr. John Feder, has been described previously (Feder et al. 1993). The HES-1 PvuII–XhoI fragment (amino acids 4–282) was cloned into pcDNA3 containing a 5′ HA/T7 epitope tag to generate pcDNA3–WT HES-1. The Sculptor site-directed mutagenesis system (Amersham) was used to mutate the WT HES-1 construct to generate pcDNA3–DN HES-1 (where amino acids E43, K44, and R47 were each mutated to A) and pcDNA3–SM HES-1 (where amino acids S37 and S38 were mutated to V37 and M38, derived from the corresponding position in the Enhancer of split m7 basic region). The PKC sites were identified using the MacPattern program (Fuchs 1994). Versions of each construct with the CMV promoter replaced by an MMTV promoter derived from pMSG (Pharmacia) were also made. Details of restriction maps, cloning sites, and primers are available upon request.
Cell cultures, stable transfections, and neurite analysis
PC12 cells, kindly provided by Moses V. Chao (Cornell University Medical College), were plated on tissue culture dishes (Corning) and maintained in Dulbecco’s modified essential medium (DMEM) (Cellgro), 5% fetal calf serum (Hyclone), 10% horse serum (Hyclone), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Sigma) at 37°C in 100% humidity and 10% CO2. Transfections were carried out using DOTAP (Boehringer Mannheim) or Lipofectamine (GIBCO BRL) reagents according to the manufacturers’ instructions. Transfectants were selected using G418 (GIBCO BRL) at 500 μg/ml of G418 (net) and isolated clones maintained with 250 μg/ml of G418. WT HES-1 and SM HES-1 stable clones were generated using the MMTV promoter construct and DN HES-1 lines with either the MMTV or CMV promoter construct. Expression was confirmed by RT–PCR or cell immunostaining (described below), with transgene-expressing clones representing between 10% and 30% of all stable lines generated.
Treatment with various reagents was carried out after the cells were plated out and left to attach for 24 hr. NGF (2.5s murine NGF, Promega) was used at 100 ng/ml final concentration except where indicated in the figure legends, TPA (Sigma) was used at a final concentration of 150 nm, Bisindolylmaleimide (CalBiochem) at a final concentration of 4 μm in dimethylsulfoxide (DMSO). A neurite was defined as a phase dark process with a clearly defined growth cone that was at least 1.5-cell diameters in length. Experiments were carried out in triplicate and repeated at least once in all cases. Error bars are the standard deviation of the mean, with n = 340 to 1420 cells counted per data point.
Transient transfections
Transient transfections were performed using Lipofectamine reagent (GIBCO BRL) in 35-mm dishes with 0.1 μg of pcDNA3 (CMV)-β-gal and 2 μg of either pcDNA3(MMTV)–WT HES-1, pcDNA3(MMTV)–SM HES-1, or pcDNA3(MMTV) (control) according to the manufacturer’s instructions. Twelve hours after transfection, the cells were replated on 100-mm dishes and 50 ng/ml of NGF (2.5s murine NGF, Promega) was added to cells for a further 48 hr, after which the cells were fixed and stained for β-galactosidase activity with X-gal according to standard protocols (Maniatis et al. 1982). The experiments were carried out in triplicate and repeated at least once, with n = 200 to 725 positive cells counted per data point. The errors are the standard deviation of the mean.
RT–PCR
To compare transgene expression, total RNA was purified from cells grown on 100-mm dishes using Tri-Reagent (Molecular Research Corp.) according to the manufacturer’s protocol. Two micrograms of total RNA was reversed transcribed with 1 μg oligonucleotide dT21 (Integrated DNA Technologies, IDT) and Superscript II reverse transcriptase (GIBCO BRL) according to the manufacturer’s protocols in the presence of 20 units of RNasin RNase inhibitor (Promega). Twenty percent of the RT reaction was PCR amplified with either glycerol-3-phosphate dehydrogenase (G3PDH) primers (Clontech) or epitope tag 5′/HES-1 3′ transgene-specific primers (IDT, sequences upon request) for 35 (G3PDH) or 40 (exogenous HES-1) cycles using TAQ gold enzyme and buffers (Perkin Elmer) according to the manufacturer’s protocol. PCR products were visualized with Sybr Green (Molecular Probes) on 3% agarose gels.
Immunocytochemistry
Cells were cultured on tissue culture slides (VWR SuperCell, Erie Scientific) and fixed and immunostained according to standard protocols (Harlow and Lane 1988). Anti-HA antibody was used at 1:80 (2 μg/ml) (Boehringer Mannheim), anti-T7 antibody was used at 1:3000 (Novagen), secondary antibody (goat anti-mouse biotin conjugated, Jackson Immunological) was used at 1:1000 and streptavidin–HRP (Jackson Immunological) at 1:500 (1 μg/ml). Staining was visualized with 0.6 mg/ml of diaminobenzidine (DAB) (Sigma)/0.03% H2O2.
Western blot analysis
Neuronal markers
Cells were lysed in RIPA buffer [150 mm NaCl, 1% NP40, 0.5% DOC, 0.1% SDS in 50 mm Tris (pH 8.0), all reagents from Sigma], the cell extracts were spun in a microfuge for 15 min at 4°C and the supernatant concentrated in a Centricon 3000 (Amicon) for 2 hr at 6700g. Protein concentrations were determined by Bradford assay (Bio-Rad). Western blot analysis was performed according to standard protocols (Harlow and Lane 1988) using 10% SDS-PAGE gels for GAP-43 and peripherin, 8% SDS-PAGE gels for NF 160. Anti-GAP-43 was used at 1:2000, anti-NF 160 was used at 1:400, anti-peripherin (rabbit IgG, kindly provided by Dr. Carol Troy, College of Physicians and Surgeons, Columbia University, New York, NY) was used at 1:2000, goat anti-mouse (or goat anti-rabbit) IgG/biotin secondary antibody was used at 1:50,000 (Jackson Immunological) and streptavidin/HRP (Jackson Immunological) tertiary reagent was used at 1:1000. Antibody binding was visualized using 0.03% (wt/vol) chloronapthol/0.03% H2O2.
HES-1
PC12 cell nuclear extracts were fractionated on 12% SDS–polyacrylamide gels and blotted onto nitrocellulose (Bio-Rad) as above. The anti-HES-1 antibodies (kindly provided by Drs. John Feder, Mercator Genetics Inc., Menlo Park, CA, and Yuh-Nung Jan, University of California, San Francisco), were used as an equal mixture of two affinity-purified rabbit polyclonals raised to an amino- and a carboxy-terminal peptide sequence, respectively at a 1:400 (1:800 each) dilution and visualized with a goat anti-rabbit HRP conjugated secondary antibody (Jackson Immunological) at 1:50,000, followed by chemiluminescence (Rennaisance, New England Nuclear). All Western data shown are from autoradiographs (BioMax film, Kodak) in the linear range of exposure.
Nuclear extracts
Nuclear extracts were prepared basically as to the protocol of Schreiber et al. (1989) except that PBS was used instead of TBS and the following inhibitors were added: aprotinin at 10 μg/ml, leupeptin at 1 μg/ml, 100 μm Na3VO4, 1 mm NaPPi, 1 mm NaF, 100 μm Na2MO4 (all from Sigma). Nuclear extracts were prepared from 100-mm tissue culture dishes at about 60%–70% confluency and total protein content was determined by Bradford assay (BioRad). All nuclear extracts for a particular experiment, including controls, were prepared simultaneously.
Electrophoretic mobility-shift assay
For gel-shift assays with nuclear extracts, equal amounts of extract (typically 5 μg of total protein at ∼2 mg/ml) were added to a 29-μl binding reaction containing 3.3 mm DTT, 20 mm HEPES (pH 7.6), 50 mm NaCl, 0.5 mm EDTA, 5% glycerol, 300 μg/ml BSA (final concentrations), and 0.25–1.0 μg poly[d(I-C)]. The poly[d(I-C)] concentration was optimized for each batch of extracts to minimize nonspecific binding. The gel retardation protocol was as described previously (Ohsako et al. 1994). The C class oligonucleotides contain the site CACGCG and are derived either from the Drosophila achaete promoter (Ohsako et al. 1994) or from the mouse HES-1 promoter (Takebayashi et al. 1994). The sequence for the HES-1 oligonucleotide is 5′-AgCggT gCCgCG TgTCTTggAgCT-3′. All experiments shown are with the mammalian probe but indistinguishable results have been attained with either version.
For gel shifts with purified HES-1 protein, ∼25 ng of bacterially expressed and purified HES-1 protein was incubated in the binding buffer described above. For the PKC phosphorylation reactions, the protein was preincubated in 1× PKC buffer [20 mm HEPES (pH 7.4), 0.34 mm EDTA, 1.67 mm CaCl2, 1 mm DTT, 10 mm MgCl2) in a 10-μl volume at room temperature for 50 min with 1 μm ATP (Sigma), 0.6 mg/ml of phosphatidyl serine (ICN), and 0.02 units of PKC (Promega) as indicated. The reaction mixture was diluted to 30 μl in the standard binding reaction and further incubated with radiolabeled class C DNA probe for 20 min before fractionating on a 5% native gel. For phosphatase treatment, MnCl2 (2 mm final) and 400 units of λ protein phosphatase (New England Biolabs) are added to the PKC reaction mixture for the final 10 min before initiating the binding reaction.
All gel shifts shown are from autoradiographs (BioMax film, Kodak) exposed in the linear range of sensitivity.
Recombinant protein
The HES-1 proteins used for in vitro DNA binding are PCR-generated bHLH constructs (primer sequences available upon request) coding for residues E33 through A98, cloned into pET-14b (Novagen) and purified from DE3pLysS cells (Novagen) using the histidine leader according to the manufacturer’s protocols. All constructs were sequenced (Cornell central sequencing facility) and purification determined to be >90% by Coomassie staining of SDS-PAGE gels (not shown). Protein quantification was by Bradford assay (BioRad) and Coomassie staining against a protein standard titration on SDS-PAGE.
The amino terminal glutathionine S-transferase (GST) full-length HES-1 fusion protein was generated from pcDNA3–WT HES-1 (minus the His/HA/T7 tags), with the HES-1 fragment cloned in-frame into pGEX4T-1 (Pharmacia) and expressed and purified as described previously (Fisher et al. 1996).
Affinity purification of DNA-binding protein
The C class DNA-binding complex was purified from PC12 cell nuclear extracts using a top strand 5′-biotin-labeled version of the C class probe (IDT). The depletion protocol is as described in Ausubel et al. (1992), using 1 mg of nuclear extract, 100 ng of biotin-labeled oligonucleotide/50 μl of streptavidin, and 4% agarose beads (Sigma) in a standard, scaled-up DNA-binding assay followed by Western blot analysis of the pellet. In parallel, ∼10 ng of GST–HES-1 bacterially expressed fusion protein was used as a positive binding control. Radiolabeled (bottom strand) biotinylated probe, which binds identically to the unlabeled oligonucleotide (not shown) was used to determine the efficiency of depletion (∼90%+, not shown) by gel shift comparison of the depleted extract to a nondepleted control.
Acknowledgments
We thank Al Fisher, Dr. Moses Chao, Dr. Lorraine Gudas, and Dr. Ulrike Gaul for critical comments on the manuscript. We also thank the members of the Caudy, Chao, and Wagner laboratories for valuable discussion of this work. M.C. also gives special thanks to Dr. Moses Chao for ongoing advice and support, and to Drs. John Feder and Yuh-Nung Jan for the gift of the anti-HES-1 antibodies used in this study. A.S. was supported by a postdoctoral fellowship from the Swedish Cancer Society. P.C. was supported by the Graduate Program in Cell Biology and Genetics at C.U.M.C. J.R. was supported by National Institutes of Health (NIH) postdoctoral training grant (T32NS07384). J.W. was funded by grants from the NIH (EY06454; NS31728). M.C. was funded by grants from the NIH (NS 28652), Alfred P. Sloan Foundation, the Pew Scholars in Biomedical Sciences Program, the Mather Foundation, the Cornell Scholars Program, and by an institutional grant from the Markey Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mcaudy@mail.med.cornell.edu; FAX (212) 746-8175.
References
- Ausubel FM, Brent R, Kingsston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. second ed. New York, NY: Greene Publishing Associates and John Wiley & Sons; 1988. [Google Scholar]
- Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell. 1985;42:841–848. doi: 10.1016/0092-8674(85)90280-6. [DOI] [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: A negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- Borgatti P, Mazzoni M, Carini C, Neri LM, Marchisio M, Bertolaso L, Previati M, Zauli G, Capitani S. Changes of nuclear protein kinase C activity and isotype composition in PC12 cell proliferation and differentiation. Exp Cell Res. 1996;224:72–78. doi: 10.1006/excr.1996.0112. [DOI] [PubMed] [Google Scholar]
- Burstein DE, Blumberg PM, Greene LA. Nerve growth factor-induced neuronal differentiation of PC12 pheochromocytoma cells: Lack of inhibition by a tumor promoter. Brain Res. 1982;247:115–119. doi: 10.1016/0006-8993(82)91033-2. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA. Early neurogenesis in Drosophila melanogaster. In: Bate M, Martinez Arias A, editors. The development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 1091–1129. [Google Scholar]
- Campuzano S, Modolell J. Patterning of the Drosophila nervous system: The achaete-scute gene complex. Trends Genet. 1992;8:202–208. doi: 10.1016/0168-9525(92)90234-u. [DOI] [PubMed] [Google Scholar]
- Chen H, Thiagalingam A, Chopra H, Borges MW, Feder JN, Nelkin BD, Baylin SB, Ball DW. Conservation of the Drosophila lateral inhibition pathway in human lung cancer: A hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc Natl Acad Sci. 1997;94:5355–5360. doi: 10.1073/pnas.94.10.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Coleman ES, Wooten MW. Nerve growth factor-induced differentiation of PC12 cells employs the PMA-insensitive protein kinase C-zeta isoform. J Mol Neurosci. 1994;5:39–57. doi: 10.1007/BF02736693. [DOI] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- Dang CV, Dolde C, Gillison ML, Kato G. Discrimination between DNA sites by a single amino acid residue of Myc-related basic-helix-loop-helix proteins. Proc Natl Acad Sci. 1992;89:599–602. doi: 10.1073/pnas.89.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delidakis C, Artavanis-Tsakonas S. The Enhancer of split [E(spl)] locus of Drosophila encodes seven independent helix-loop-helix proteins. Proc Natl Acad Sci. 1992;89:8731–8735. doi: 10.1073/pnas.89.18.8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson DG, Olson EN. Helix-loop-helix proteins as regulators of muscle-specific transcription. J Biol Chem. 1993;268:755–758. [PubMed] [Google Scholar]
- Feder J, Sheng M, Jan LY, Jan YN. A rat gene with sequence homology to the Drosophila gene hairy is rapidly induced by growth factors known to influence neuronal differentiation. Mol Cell Biol. 1993;13:105–113. doi: 10.1128/mcb.13.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, A. and M. Caudy. 1998. The function of Hairy-related bHLH repressor proteins in cell fate decisions. BioEssays (in press). [DOI] [PubMed]
- Fisher A, Ohsako S, Caudy M. The WRPW motif of Hairy-related bHLH repressor proteins acts as a four amino acid transcription repression and protein-protein interaction domain. Mol Cell Biol. 1996;16:2670–2677. doi: 10.1128/mcb.16.6.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs R. Predicting protein function: A versatile tool for the Apple Macintosh. Comput Appl Biosci. 1994;10:171–178. doi: 10.1093/bioinformatics/10.2.171. [DOI] [PubMed] [Google Scholar]
- Grbavec D, Stifani S. Molecular interaction between TLE1 and the carboxyl-terminal domain of HES-1 containing the WRPW motif. Biochem Biophys Res Commun. 1996;223:701–705. doi: 10.1006/bbrc.1996.0959. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Greene LA, Ziff EB. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- Greene LA, Tischler SA. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Cell. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner AL. Mammalian achaete-scute homolog 1 is required for for the early development of olfactory and autonomic neurons. Cell. 1993;75:463–476. doi: 10.1016/0092-8674(93)90381-y. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- Ishibashi M, Moriyoshi K, Sasai Y, Shiota K, Nakanishi S, Kageyama R. Persistent expression of helix-loop-helix HES-1 prevents mammalian neural differentiation in the central nervous system. EMBO J. 1994;13:1799–1805. doi: 10.1002/j.1460-2075.1994.tb06448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M, Ang SL, Shiota K, Nakanishi S, Kageyama R, Guillemot F. Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix–loop–helix factors, premature neurogenesis, and severe neural tube defects. Genes & Dev. 1995;9:3136–3148. doi: 10.1101/gad.9.24.3136. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Genes required for specifying cell fates in Drosophila embryonic sensory nervous system. Trends Neurosci. 1990;13:493–498. doi: 10.1016/0166-2236(90)90083-m. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Functional gene cassettes in development. Proc Natl Acad Sci. 1993;90:8305–8307. doi: 10.1073/pnas.90.18.8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch [see comments] Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Birren SJ, Anderson DJ. Two rat homologues of Drosophila achaete-scute specifically expressed in neuronal precursors. Nature. 1990;346:858–861. doi: 10.1038/346858a0. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- Knust E, Schrons H, Grawe F, Campos-Ortega JA. Seven genes of the Enhancer of split complex of Drosophila melanogaster encode helix-loop-helix proteins. Genetics. 1992;132:505–518. doi: 10.1093/genetics/132.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach KL, Powers EA, Ruff VA, Jaken S, Kaufmann S. Type 3 protein kinase C localization to the nuclear envelope of phorbol ester-treated NIH 3T3 cells. J Cell Biol. 1989;109:685–695. doi: 10.1083/jcb.109.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhou J, James G, Heller HR, Czech MP, Olson EN. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell. 1992;71:1181–1194. doi: 10.1016/s0092-8674(05)80066-2. [DOI] [PubMed] [Google Scholar]
- Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1982. [Google Scholar]
- Nakajima T, Fukamizu A, Takahashi J, Gage FH, Fisher T, Blenis J, Montminy MR. The signal-dependent coactivator CBP is a nuclear target for pp90RSK. Cell. 1996;86:465–474. doi: 10.1016/s0092-8674(00)80119-1. [DOI] [PubMed] [Google Scholar]
- Oellers N, Dehio M, Knust E. bHLH proteins encoded by the Enhancer of split complex of Drosophila negatively interfere with transcriptional activation mediated by proneural genes. Mol Gen Genet. 1994;244:465–473. doi: 10.1007/BF00583897. [DOI] [PubMed] [Google Scholar]
- Ohsako S, Hyer J, Panganiban G, Oliver I, Caudy M. hairy function as a DNA binding HLH repressor of Drosophila sensory organ formation. Genes & Dev. 1994;8:2743–2755. doi: 10.1101/gad.8.22.2743. [DOI] [PubMed] [Google Scholar]
- Paroush Z, Finley R, Kidd T, Wainwright SM, Ingham P, Brent R, Ish-Horowicz D. Groucho is required for Drosophila neurogenesis, segmentation, and sex determination and interacts directly with hairy-related bHLH proteins. Cell. 1994;79:805–815. doi: 10.1016/0092-8674(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Rushlow CA, Hogan A, Pinchin SM, Howe KM, Lardelli M, Ish-Horowicz D. The Drosophila hairy protein acts in both segmentation and bristle patterning and shows homology to N-myc. EMBO J. 1989;8:3095–3103. doi: 10.1002/j.1460-2075.1989.tb08461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai Y, Kageyama R, Tagawa Y, Shigemoto R, Nakanishi S. Two mammalian helix–loop–helix factors structurally related to Drosophila hairy and Enhancer of split. Genes & Dev. 1992;6:2620–2634. doi: 10.1101/gad.6.12b.2620. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. Silencing is golden: Negative regulation in the control of neuronal gene transcription. Curr Opin Neurobiol. 1995;5:566–571. doi: 10.1016/0959-4388(95)80060-3. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Ann Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosome locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. J Biol Chem. 1994;269:5150–5156. [PubMed] [Google Scholar]
- Tietze K, Oellers N, Knust E. Enhancer of splitD, a dominant mutation of Drosophila, and its use in the study of functional domains of a helix-loop-helix protein. Proc Natl Acad Sci. 1992;89:6152–6156. doi: 10.1073/pnas.89.13.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Ishibashi M, Nakahara K, Ang SL, Nakanishi S, Guillemot F, Kageyama R. Mammalian hairy and Enhancer of split homolog 1 regulates differentiation of retinal neurons and is essential for eye morphogenesis. Neuron. 1996;16:723–734. doi: 10.1016/s0896-6273(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Van Doren M, Bailey AM, Esnayra J, Ede K, Posakony JW. Negative regulation of proneural gene activity: hairy is a direct transcriptional repressor of achaete. Genes & Dev. 1994;8:2729–2742. doi: 10.1101/gad.8.22.2729. [DOI] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: Redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wooten MW. Differential expression of PKC isoforms and PC12 cell differentiation. Exp Cell Res. 1992;199:111–119. doi: 10.1016/0014-4827(92)90468-n. [DOI] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- Yoon JK, Lau LF. Involvement of JunD in transcriptional activation of the orphan receptor gene nur77 by nerve growth factor and membrane depolarization in PC12 cells. Mol Cell Biol. 1994;14:7731–7743. doi: 10.1128/mcb.14.12.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]