

Abstract
An insulin receptor-like signaling pathway regulates Caenorhabditis elegans metabolism, development, and longevity. Inactivation of the insulin receptor homolog DAF-2, the AGE-1 PI3K, or the AKT-1 and AKT-2 kinases causes a developmental arrest at the dauer stage. A null mutation in the daf-16 Fork head transcription factor alleviates the requirement for signaling through this pathway. We show here that a loss-of-function mutation in pdk-1, the C. elegans homolog of the mammalian Akt/PKB kinase PDK1, results in constitutive arrest at the dauer stage and increased life span; these phenotypes are suppressed by a loss of function mutation in daf-16. An activating mutation in pdk-1 or overexpression of wild-type pdk-1 relieves the requirement for AGE-1 PI3K signaling. Therefore, pdk-1 activity is both necessary and sufficient to propagate AGE-1 PI3K signals in the DAF-2 insulin receptor-like signaling pathway. The activating mutation in pdk-1 requires akt-1 and akt-2 gene activity in order to suppress the dauer arrest phenotype of age-1. This indicates that the major function of C. elegans PDK1 is to transduce signals from AGE-1 to AKT-1 and AKT-2. The activating pdk-1 mutation is located in a conserved region of the kinase domain; the equivalent amino acid substitution in human PDK1 activates its kinase activity toward mammalian Akt/PKB.
Keywords: Insulin signaling, dauer, PDK1 activation, PDK-1, life span
Insulin signaling in mammals causes a variety of cellular responses, including glucose uptake and glycogen synthesis in liver and muscle, fat storage in adipocytes, and changes in protein synthesis and gene expression (Kahn 1994). Activation of the insulin receptor leads to activation of other signaling molecules such as phosphoinositide-3-OH kinase (PI3K) and Ras and their downstream effectors, such as Akt/PKB and the MAP kinase cascade (Avruch 1998). The kinase cascades in turn regulate glucose transporter localization, metabolic enzymes, and the transcription and translation of these and other genes (Avruch 1998). In Caenorhabditis elegans, a signal transduction cascade from the DAF-2 insulin/IGF-1 receptor homolog (Kimura et al. 1997) to the DAF-16 Fork head transcription factor (Lin et al. 1997; Ogg et al. 1997) regulates metabolism, development, and longevity. Genetic analysis of this pathway has identified homologs of genes that had been implicated in mammalian insulin signaling by biochemical analyses (Morris et al. 1996; Avruch 1998; Paradis and Ruvkun 1998; this report), demonstrating the extensive conservation of this pathway between C. elegans and mammals. In addition, the C. elegans genetics has identified new signaling components, such as the DAF-16 Fork head transcription factor (Lin et al. 1997; Ogg et al. 1997) and the DAF-18 PTEN homolog (Ogg and Ruvkun 1998) not previously known to be coupled to insulin. For both the novel genes and the genes implicated previously in insulin signaling, genetic analysis in C. elegans allows specific components of the insulin-like signaling pathway to be disrupted so that the metabolic and developmental consequences can be monitored in the whole animal.
The C. elegans insulin/IGF-1 receptor pathway is required for reproductive growth and metabolism, as well as normal life span. Reduction of signaling through the DAF-2 insulin receptor-like signaling pathway causes the animals to arrest at a reversible diapause stage known as the dauer larval stage (Kimura et al. 1997). Dauer larvae do not feed or reproduce and their metabolism is shifted to energy storage (O’Riordan and Burnell 1989, 1990; Riddle and Albert 1997). Molecules that have been identified downstream of the DAF-2 insulin/IGF-1 receptor in C. elegans include the AGE-1 PI3K (Morris et al. 1996), the Akt/PKB homologs AKT-1 and AKT-2 (Paradis and Ruvkun 1998), the DAF-18 PTEN phosphatase (Ogg and Ruvkun 1998), and the DAF-16 Fork head/winged helix transcription factor family member (Lin et al. 1997; Ogg et al. 1997). Reducing the activity of daf-2 or genes that are positively regulated by daf-2 such as age-1, or akt-1 and akt-2, causes C. elegans to arrest at the dauer stage constitutively (Morris et al. 1996; Kimura et al. 1997; Paradis and Ruvkun 1998). Reduction-of-function mutations in genes that antagonize daf-2 and/or age-1 signaling, such as the daf-18 PTEN lipid phosphatase and the daf-16 transcription factor, suppress the dauer constitutive phenotype of daf-2 and/or age-1 mutants (Lin et al. 1997; Ogg et al. 1997; Ogg and Ruvkun 1998). Whereas a severe reduction in insulin receptor-like signaling induces arrest at the dauer stage, daf-2 and age-1 mutants that have been supplied with these gene activities until after the dauer arrest decision point show an increased life span that is dependent on daf-16 (Kenyon et al. 1993; Larsen et al. 1995; Morris et al. 1996).
In mammalian insulin signaling, insulin receptor activation leads to activation of PI3K and other signaling molecules (Avruch 1998). Activated PI3K generates 3-phosphoinositides, such as phosphatidylinositol-3,4-bisphosphate (PtdIns-3,4-P2), and phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3), which are thought to act as second messengers in signal transduction cascades because their levels rise rapidly in response to growth factor signaling (Toker and Cantley 1997). PtdIns-3,4-P2 and/or PtdIns-3,4,5-P3 bind to the pleckstrin homology domain of Akt/PKB and are required for its activation (Franke et al. 1997; Frech et al. 1997; Klippel et al. 1997). Phosphoinositide binding is thought to cause Akt/PKB to undergo a conformational change that makes two phosphorylation sites accessible to the pertinent kinases (Alessi et al. 1997b; Stokoe et al. 1997). Activation of Akt/PKB has been implicated in a variety of cellular responses to growth factor signaling, such as protection from apoptosis, glucose transporter translocation, and glycogen synthesis (Cross et al. 1995; Kohn et al. 1996; Dudek et al. 1997; Kauffmann-Zeh et al. 1997; Kulik et al.1997).
One of the kinases that phosphorylates Akt/PKB and is required for its activation is 3-phosphoinositide-dependent kinase-1 (PDK1) (Alessi et al. 1996; Alessi et al. 1997a,b). PDK1 phosphorylates the Thr-308 site on Akt/PKB in a phosphoinositide dependent manner (Allesi et al. 1997b). Although PDK1 binds PtdIns-3,4-P2 and PtdIns-3,4,5-P3 in vitro (Stephens et al. 1998), it is currently unclear whether the phosphoinositide dependence of Akt/PKB phosphorylation by PDK1 resides solely with the substrate, with PDK1, or with both. Because removal of the pleckstrin homology domain of Akt/PKB relieves the phosphoinositide dependence of PDK1 phosphorylation and PDK1 activity is not increased by growth factors that activate PI3K and Akt/PKB (Alessi et al. 1997a), it is possible that PDK1 activity is not PtdIns-3,4-P2 or PtdIns-3,4,5-P3 dependent. It has been suggested, however that phosphoinositide binding to PDK1 localizes the kinase to the plasma membrane, thereby co-localizing it with Akt/PKB (Andjelkovic et al. 1997) and increasing Akt/PKB activation (Anderson et al. 1998). The identity of the kinase responsible for the Ser-473 phosphorylation of Akt/PKB (so-called PDK2) is not well established although it has been reported that PDK2 is integrin-linked kinase (ILK) (Delcommenne et al. 1998). C. elegans AKT-1 has both the Thr-308 and Ser-473 equivalent phosphorylation sites whereas AKT-2 has only the Thr-308 equivalent site, raising the possibility that these proteins are differentially regulated (Paradis and Ruvkun 1998). PDK1 has also been shown to phosphorylate p70 S6 kinase in a phosphoinositide independent manner, thereby implicating PDK1 in translational control (Alessi et al. 1998; Pullen et al. 1998). More PDK1 substrates have been described recently, including the PKC isotypes ζ and δ (Chou et al. 1998; Le Good et al. 1998) and possibly PKA (Cheng et al. 1998).
Genetic screens for genes that control dauer development have revealed many molecules involved in the DAF-2 insulin receptor-like signal transduction cascade (Riddle et al. 1981; Riddle 1988; Paradis and Ruvkun 1998; this report). Here we report the identification of the C. elegans PDK1 homolog pdk-1. We establish the action of pdk-1 in the C. elegans insulin receptor-like signaling pathway by analysis of loss-of-function and gain-of-function mutations in pdk-1. pdk-1 activity is required for reproductive growth and metabolism and normal life span; a loss-of-function mutation in the daf-16 transcription factor bypasses this requirement. Activation of pdk-1 by substitution of a conserved residue in the kinase domain relieves the requirement for upstream AGE-1 PI3K signaling, but is dependent on akt-1 and akt-2 gene activity. The ability of this amino acid substitution to activate PDK1 is conserved across species. These studies of the first mutations in PDK1 in any animal show that C. elegans pdk-1 gene activity is both necessary and sufficient to propagate AGE-1 PI3K signals in the insulin receptor-like signaling pathway.
Results
Genetic identification of novel loss-of-function and gain-of-function mutations in the C. elegans DAF-2 insulin receptor-like signaling pathway
To identify new components regulating dauer formation, we performed a genetic screen for mutants that arrest at the dauer stage constitutively (Daf-c) at 27°C. Two alleles (sa680 and sa709) from this screen are recessive, map to the left arm of the X chromosome, and fail to complement for the Daf-c phenotype at 27°C, suggesting that they affect the same gene. Whereas both mutations cause a high percentage of dauer arrest at 27°C, the sa680 mutation also causes a high percentage of dauer arrest at 25°C (Table 1). The Daf-c phenotype of sa680 can be rescued maternally. An sa680/+ heterozygote produces no dauer progeny at 25°C, but ∼25% of its progeny (sa680 homozygotes) produce nearly all dauer progeny at 25°C. sa680 mutant dauers fail to recover at 15°C in the presence of plentiful food, whereas sa709 dauers recover readily. These phenotypes are similar to those of age-1 mutants. age-1 null mutants have a maternally rescued Daf-c phenotype at all temperatures and are defective in dauer recovery, whereas a weaker age-1 mutant has a Daf-c phenotype at 27°C and fails to maternally complement age-1 null mutants (Gottlieb and Ruvkun 1994; Malone et al. 1996; Morris et al. 1996).
Table 1.
Effects of pdk-1(sa680) and pdk-1 (sa709) on dauer formation
|
Genotype
|
Phenotype at 25.4°C (%)
|
|||
|---|---|---|---|---|
|
L4 larvae and adult
|
dauer
|
dauer-like
|
no.a
|
|
| pdk-1(sa680) | 0 | 87 | 13 | 316 |
| osm-6(p811); pdk-1(sa680) | 0 | 99 | 1 | 320 |
| daf-5(e1385); pdk-1(sa680) | 0 | 92 | 8 | 205 |
| pdk-1(sa680) daf-12(m20) | 100 | 0 | 0 | 232 |
| daf-16(m27); pdk-1(sa680) | 99 | 1b | 0 | 275 |
| akt-1(mg144); pdk-1(sa680) | 63 | 37 | 1 | 572 |
| Phenotype at 26.8°C (%) | ||||
| Wild type | 96 | 4 | 0 | 251 |
| pdk-1(sa709) | 3 | 97 | 0 | 116 |
| daf-16(m27) | 98 | 2b | 0 | 258 |
| daf-16(m27); pdk-1(sa709) | 98 | 2b | 0 | 260 |
See Materials and Methods for description of categories.
Total number of animals scored.
Partial dauers as described in Vowels and Thomas (1992).
In addition to the DAF-2 insulin receptor-like signaling pathway, two other parallel pathways have been implicated in control of dauer arrest (Thomas et al 1993; Gottlieb and Ruvkun 1994). To determine in which of the genetic pathways sa680 functions, double mutants of sa680 with mutations that suppress the Daf-c phenotype of mutants in each pathway were analyzed. Mutations in osm-6 and daf-5 do not suppress the Daf-c phenotype of sa680 but mutations in daf-16 and daf-12 completely suppress the Daf-c phenotype (Table 1). The suppression of sa680 by a daf-16 mutation is consistent with placement of sa680 in the DAF-2 insulin receptor-like signaling pathway. The suppression by a daf-12 mutation is distinct from that observed for strong daf-2 alleles, but has been observed for weak daf-2 alleles, suggesting that sa680 only partially abolishes DAF-2 signaling (Gems et al. 1998). In support of the placement of sa680 in the insulin receptor-like signaling pathway, its Daf-c phenotype is partially suppressed by a gain-of-function mutation in the gene akt-1 (Table 1) that also suppresses the dauer arrest induced by loss of age-1 PI3K signaling (Paradis and Ruvkun 1998).
Further genetic mapping of sa680 placed it on the X chromosome to the right of unc-1. Also mapping to this genetic region is mg142, a dominant mutation that was isolated in a screen for suppression of the Daf-c phenotype of an age-1(mg44) null mutant. Reduction-of-function mutations in daf-16 and a dominant activating mutation in akt-1 have also been isolated in this screen (Paradis and Ruvkun 1998). The mg142 mutation suppresses the dauer constitutive phenotype of two nonsense mutations and one missense mutation in age-1 and is dominant (Table 2; data not shown). The mg142 dominant mutation suppresses the age-1 Daf-c phenotype, whereas the recessive sa680 and sa709 mutants phenocopy the age-1 Daf-c phenotype, suggesting that mg142 may activate the same gene that is inactivated by sa680 and sa709. The mg142 mutation does not have an obvious phenotype on its own (Table 2) and animals bearing the mutation arrest as dauer larvae on starved plates. The mg142 phenotypes are similar to that of the activating mutation in akt-1 (Paradis and Ruvkun 1998); both mutations activate the insulin signaling pathway enough to bypass the need for AGE-1 1PI3K signaling, but do not activate the pathway to the point that normal dauer arrest is affected strongly.
Table 2.
Effects of pdk-1(mg142) on dauer formation
|
Genotype of parent
|
Phenotype of progeny at 25°C at 48 hr post-egglay (%)
|
|||||
|---|---|---|---|---|---|---|
|
L4 larvae and adult
|
dauer
|
dauer-like
|
dead eggs
|
other
|
no.a
|
|
| Wild type | 100 | 0 | 0 | N.D. | 0 | 471 |
| pdk-1(mg142) | 99.7 | 0 | 0 | 0 | 0.3 | 329 |
| sqt-1(sc13) age-1(mg44)b | 0 | 86.6 | 0 | 0.5 | 12.9c | 187 |
| sqt-1(sc13) age-1(mg44); pdk-1(mg142)de | 92.6 | 0 | 0 | 0 | 7.4f | 149 |
| daf-16(m27); sqt-1(sc13) age-1(mg44)dg | 97.6 | 0 | 0 | 0 | 2.4 | 376 |
| daf-2(e1370) | 0 | 95.1 | 0 | 3.6 | 1.3 | 309 |
| daf-2(e1370); pdk-1(mg142)h | 0 | 0 | 94.6 | 4.6 | 0.8 | 240 |
| daf-16(m27); daf-2(e1370)i | 98.1 | 0 | 0 | 1.4 | 0.5 | 575 |
(N.D.) Not determined. See Materials and Methods for description of categories.
Total number of animals scored.
These animals are the homozygous progeny of age-1/balancer hermaphrodites.
Of 12 animals scored, 10 were dauers and 2 remained arrested at a younger larval stage by 96 hr post-egglay.
These animals are the homozygous age-1 progeny of homozygous age-1 hermaphrodites.
Rescoring of entire assay at 96 hr post-egglay: 50.3% of animals were gravid adults, 44.8% were sterile adults, 0.7% were dauer, and 4.2% were other (N = 143); sqt-1(sc13) age-1(mg44) animals remained essentially unchanged.
Six of seven animals scored were sterile adults and one of seven animals was a dauer by 96 hr post-egglay.
Rescoring of entire assay at 72 hr post-egglay: 97.1% of animals were gravid adults, 1.8% were sterile adults, and 1.1% were other (N = 379).
Rescoring of entire assay at 96 hr post-egglay: 79.9% of animals were sterile adults, 10.0% were dauer-like, 6.5% were dauers, and 3.5% were other (N = 229); daf-2(e1370) animals remained essentially unchanged.
Rescoring of entire assay at 72 hr post-egglay: 97.4% of animals were gravid adults, 2.6% were other (N = 575).
sa680, sa709, and mg142 are alleles of the C. elegans PDK1 homolog
We inspected the C. elegans genomic sequence of the region to which these alleles map for genes that have been implicated in receptor tyrosine kinase signaling. The C. elegans homolog of human PDK1 (Alessi et al. 1997a,b), which we named pdk-1, is located in this region and was an excellent candidate to be the gene defined by these alleles. We determined the pdk-1 DNA sequence in the sa680, sa709, and mg142 strains by PCR amplification and direct sequencing. This revealed a pdk-1 Gly-295–Arg substitution in the sa680 strain and a pdk-1 Ala-303–Val substitution in the mg142 strain, both conserved residues in the kinase domain (Fig. 1B). We did not detect a mutation in the pdk-1 coding region in the sa709 strain; this weak allele may have a mutation in a regulatory region of the gene. The Daf-c phenotypes of pdk-1(sa680) and pdk-1(sa709) were both efficiently rescued by a pdk-1(+) transgene, confirming their assignment as alleles of pdk-1 (data not shown; for description of transgene, see Materials and Methods).
Figure 1.
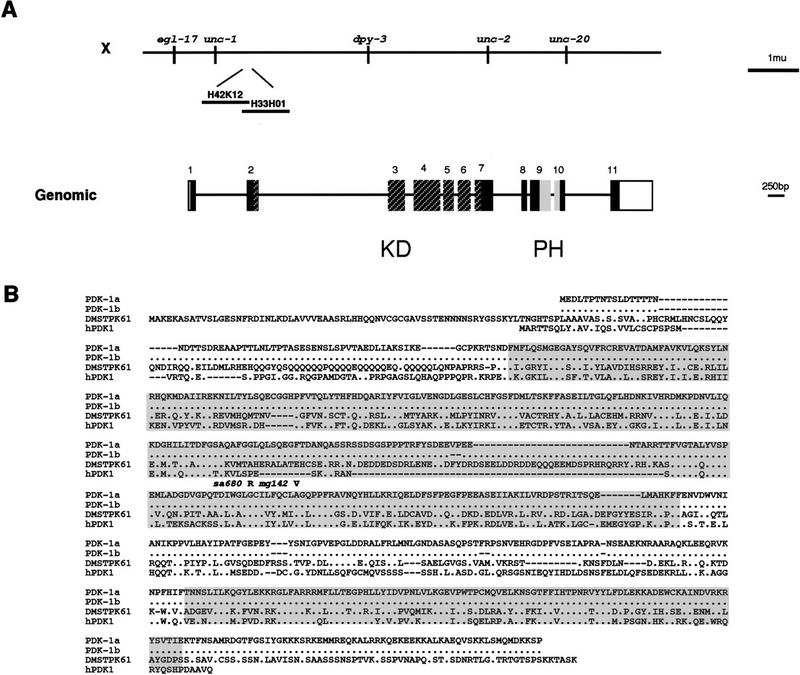
pdk-1 encodes a serine/threonine kinase. (A, top) Genetic and physical map of the pdk-1 region. pdk-1 is contained on fosmids H42K12 and H33H01. The starting methionine of PDK-1 is at 9848 bp on H42K12, the PDK-1 stop codon is at 3082 bp on H33H01; the fosmids overlap for 1201 bp (from 12897 to end on fosmid H42K12). (Bottom) Exon/intron structure of pdk-1. Coding regions are solid boxes; noncoding regions are open boxes; introns are lines. The kinase domain is indicated by hatched boxes (Hanks and Hunter 1995); the pleckstrin homology domain is indicated by the shaded boxes (Alessi et al. 1997a). (B) PDK-1a (GenBank accession no. AF130406); PDK1b (no. AF130407), DMSTPK61(EMBL Y07908), and human PDK1(GenBank accession no. AF017995) were aligned using PILEUP (GCG) (accession numbers for the proteins used in the PILEUP are indicated in parentheses). Identical residues are indicated by dots; gaps introduced to align the sequence are indicated by dashes. The kinase domain is indicated by the amino-terminal shaded area and the pleckstrin homology domain is indicated by the carboxy-terminal shaded area. The sa680 Gly-295–Arg substitution is indicated by an R above the PDK1a sequence. The mg142 Ala-303–Val substitution is indicated by a V above the PDK1a sequence.
The C. elegans pdk-1 gene has the hallmarks of the PDK1 family including an amino-terminal kinase domain and a carboxy-terminal pleckstrin homology domain (Alessi et al. 1997a) (Fig. 1A). In mammals, PDK1, in conjunction with the phospholipid products of PI3K, activates the Akt/PKB serine/threonine kinase via phosphorylation at the Thr-308 position in response to growth factor signaling (Alessi et al. 1996, 1997a). In C. elegans, akt-1 and akt-2, two homologs of Akt/PKB, transduce signals from AGE-1 PI3K to the DAF-16 transcription factor (Paradis and Ruvkun 1998). Simultaneous inactivation of akt-1 and akt-2 results in a Daf-c phenotype (Table 3; Paradis and Ruvkun 1998) and an activating mutation in akt-1 alleviates the need for AGE-1 PI3K signaling in C. elegans (Paradis and Ruvkun 1998). Therefore, a loss-of-function mutation in pdk-1 would be predicted to be unable to activate Akt/PKB and result in a Daf-c phenotype. An activating mutation in pdk-1 would be predicted to lead to increased akt-1 and/or akt-2 activity, thereby suppressing an age-1 null mutation. These predictions are supported by the observed phenotypes of the pdk-1 mutants.
Table 3.
Effects of pdk-1(RNAi), akt-1(RNAi), and akt-2(RNAi) on dauer formation
|
Strain
|
dsRNA injected
|
Phenotype of progeny at 20°C (%)
|
|||||
|---|---|---|---|---|---|---|---|
|
L4 larvae and adult
|
dauer
|
dauer-like
|
dead eggs
|
other
|
no.a
|
||
| age-1(mg44) | uninjected | 0 | 88.8 | 0 | 3.8 | 7.4 | 598 |
| age-1(mg44); pdk-1(mg142) | uninjected | 92.4 | 0 | 0 | 2.3 | 5.3 | 1325 |
| age-1(mg44); pdk-1(mg142) | pdk-1 | 9.8 | 85.1 | 2.3 | 1.2 | 1.6 | 686 |
| age-1(mg44); pdk-1(mg142) | akt-1 | 0 | 95.6 | 0 | 0.5 | 3.8 | 182 |
| age-1(mg44); pdk-1(mg142)b | akt-2 | 32.7 | 49.7 | 13.0 | 1.3 | 3.2 | 684 |
| age-1(mg44); pdk-1(mg142) | akt-1 + akt-2 | 0 | 96.1 | 0 | 1.1 | 2.8 | 280 |
| age-1(mg44); akt-1(mg144) | uninjected | 97.2 | 0 | 0 | 0.5 | 2.3 | 1485 |
| age-1(mg44); akt-1(mg144) | akt-1 | 0 | 91.3 | 0 | 0.3 | 8.4 | 311 |
| age-1(mg44); akt-1(mg144) | akt-2 | 62.8 | 24.8 | 11.7 | 0.4 | 0.4 | 537 |
| age-1(mg44); akt-1(mg144) | pdk-1 | 5.7 | 92.8 | 0.2 | 1.0 | 0.2 | 873 |
| Wild typec | pdk-1 | 86.4 | 4.9 | 4.1 | 4.6 | N.D. | 368 |
| Wild typed | akt-1 | 98.9 | 0 | 0 | 1.1 | N.D. | 542 |
| Wild typed | akt-2 | 97.3 | 0 | 0 | 2.7 | N.D. | 598 |
| Phenotype of progeny at 26°C (%) | |||||||
| Wild type | uninjected | 98.6 | 0 | 0 | 1.4 | N.D. | 497 |
| Wild typee | akt-1 + akt-2 | 22.9 | 64.8 | 8.1 | 0.5 | 3.6 | 580 |
| pdk-1(mg142) | uninjected | 100 | 0 | 0 | 0 | N.D. | 576 |
| pdk-1(mg142) | akt-1 + akt-2 | 15.9 | 77.4 | 2.2 | 1.2 | 3.3. | 674 |
(N.D.) Not determined. See Materials and Methods for description of categories. All age-1 strains also contained the linked sqt-1(sc13) mutation.
Total number of animals scored.
Overall, the dauer-inducing ability of this RNA in this strain background was variable between injected animals in a given trial and between trials. Animals that looked like dauer or dauer-like at 48 hr frequently became sterile or gravid adults by 72 hr.
This assay was performed at 27°C, a condition that is more dauer-inducing than 20°C. Uninjected wild-type control made 0.2% dauer at 27°C.
Data reproduced from Paradis and Ruvkun (1998) to facilitate comparison with new data. This assay was performed at 25°C, a condition that is more dauer-inducing than 20°C.
These data differs slightly from results published previously (Paradis and Ruvkun 1998). We believe this is due to differences in strain backgrounds and incubator temperatures (see Materials and Methods).
To confirm that the pdk-1(mg142) mutation causes the dominant suppression of the Daf-c phenotype of age-1 loss-of-function mutants, we performed a reversion experiment using RNA interference (RNAi) (Fire et al. 1998) to decrease pdk-1 gene activity in the age-1(mg44); pdk-1(mg142) strain. If a mutation in the pdk-1 locus is responsible for the suppression phenotype observed in this strain, RNAi of pdk-1 in this strain should revert the suppression phenotype and result in the age-1 Daf-c phenotype. Inhibition of pdk-1 activity by RNAi in the age-1(mg44); pdk-1(mg142) strain reverts the pdk-1 suppression phenotype (Table 3). We conclude that mg142 is a lesion in the pdk-1 locus. Reversion of the pdk-1(mg142) suppression phenotype also supports the conclusion that mg142 is a gain-of-function mutation in pdk-1 rather than a loss-of-function mutation.
Interestingly, reducing pdk-1 function by RNAi in wild-type animals (Table 3) does not cause a Daf-c phenotype similar to either pdk-1 loss of function allele. One possible explanation for this result is that RNAi of pdk-1 does not decrease pdk-1 gene activity sufficiently to cause a Daf-c phenotype in a wild-type background. However, RNAi of pdk-1(mg142) decreases gene activity enough to revert the ability of this gain-of-function allele to suppress the age-1 Daf-c phenotype (Table 3). Therefore, the possibility existed that RNAi of pdk-1 actually abolishes pdk-1 gene activity but pdk-1(sa680) was a novel, recessive interfering mutation rather than a loss-of-function mutation. If this were the case, inhibition of pdk-1 gene activity by RNAi would be expected to revert the Daf-c phenotype of pdk-1(sa680). We performed this experiment and did not observe any reversion of the pdk-1(sa680) Daf-c phenotype (data not shown). Therefore, we conclude that pdk-1(sa680) is a loss-of-function mutation and that RNAi of pdk-1 inhibits pdk-1 gene activity to a lesser extent than pdk-1(sa680). Inhibition of pdk-1 gene activity by RNAi also fails to enhance the pdk-1(sa680) Daf-c phenotype (data not shown). This implies that pdk-1(sa680) is a strong and possibly null allele, but because the sa680 mutation causes an amino acid substitution, pdk-1(sa680) could retain some activity.
Increased gene dosage of pdk-1(+) has a similar genetic activity to the pdk-1(mg142) mutation, suggesting that the phenotype of pdk-1(mg142) is attributable to an increase in gene activity. Increased gene dosage of pdk-1(+) suppresses the Daf-c phenotype of age-1(mg44) (Table 4). This suppression is dependent on a functional PDK1 kinase domain because a transgene bearing a substitution of a conserved lysine residue with asparagine (K98N in PDK1a) that inactivates the kinase activity of PDK1 is not able to suppress age-1(mg44) (Table 4). Similarly, increased akt-1 gene dosage was also sufficient to suppress age-1(mg44) (Table 4). Our conclusion is that increasing the gene dosage of pdk-1(+) is sufficient to compensate for the loss of AGE-1 PI3K signaling, presumably by increasing akt-1 and/or akt-2 activity. Importantly, the above genetic analyses of pdk-1 loss-of-function and gain-of-function mutations demonstrate that pdk-1(mg142) is a dominant activating mutation, as opposed to dominant negative or loss of function.
Table 4.
Effects of pdk-1 transgenes on dauer formation
|
Dauer constitutive mutation
|
Transgene
|
Phenotype at 25°C (%)
|
|||
|---|---|---|---|---|---|
|
L4 larvae and adult
|
dauer and dauerlike
|
other
|
no.a
|
||
| Wild type | none | 100 | 0 | N.D. | 218 |
| Wild type | pdk-1(+) | 100 | 0 | N.D. | 451 |
| Wild type | pdk-1(KD)b | 100 | 0 | N.D. | 539 |
| age-1(mg44) | none | 0 | 91.7 | 8.3 | 108 |
| age-1(mg44) | pdk-1(+) | 69.9 | 30.1 | N.D. | 322 |
| age-1(mg44) | pdk-1(KD)b | 0 | 98.1 | 1.9 | 207 |
See Materials and methods for description of categories. All age-1 strains also contained the linked sqt-1(sc13) mutation.
Total number of transgenic animals scored.
pdk-1(KD) is a kinase dead mutation that changes a conserved lysine to an asparagine (Chou et al. 1998).
Activated pdk-1 requires akt-1 and akt-2 gene activities
The biochemical studies of Akt/PKB activation by PDK1 (Alessi et al. 1997b; Stokoe et al. 1997) predict that pdk-1(mg142) suppression of the Daf-c phenotype of age-1 mutants requires akt-1 and akt-2 gene activities. Reduction of akt-1 gene activity alone by RNAi was sufficient to interfere with pdk-1(mg142) suppression of the age-1 Daf-c phenotype (Table 3). Reduction of only akt-2 gene activity partially impaired the ability of pdk-1(mg142) to suppress age-1(mg44) (Table 3). Simultaneous inactivation of akt-1 and akt-2 by RNAi causes a Daf-c phenotype (Table 3; Paradis and Ruvkun 1998) that is epistatic to the activating mutation pdk-1(mg142) (Table 3), suggesting that akt-1 and akt-2 act downstream of pdk-1. These results are consistent with the model that pdk-1(mg142) activates akt-1 and akt-2 signaling in the absence of upstream AGE-1 PI3K inputs. These results show that akt-1 and akt-2 are the major outputs of pdk-1 signaling.
Biochemical experiments have shown that PDK1 phosphorylation of Thr-308 on Akt/PKB is required for Akt/PKB activity (Alessi et al. 1996, 1997b). We tested whether the activating mutation in akt-1, akt-1(mg144), relieves the requirement for the PDK1 phosphorylation event. The akt-1(mg144) mutation is able to partially suppress the dauer constitutive phenotype of the pdk-1(sa680) loss-of-function mutation (Table 1), showing that akt-1(mg144) activity does not require full pdk-1 activity. This experiment is consistent with the model that akt-1 acts downstream of pdk-1. However, pdk-1 activity is necessary for activated akt-1 to signal in the absence of AGE-1 generated phosphoinositide signaling. Inactivation of pdk-1 by RNAi in an age-1 (mg44); akt-1(mg144) strain abolishes the ability of akt-1(mg144) to suppress the Daf-c phenotype of age-1(mg44) (Table 3). This observation contrasts with the ability of activated akt-1(mg144) to bypass the requirement for pdk-1 signaling in a genetic background with normal AGE-1 phosphoinositide signaling (Table 1). One interpretation of this result is that AKT-1 has both non-PDK-1 phosphoinositide dependent and PDK-1 dependent inputs to its normal activation and that the akt-1(mg144) mutation relieves the requirement for PDK-1 inputs only if AGE-1 PI3K-generated phosphoinositides are present.
As would be expected for a mutation that is predicted to increase akt-1 and possibly akt-2 activity, pdk-1(mg142) behaves similarly to the activating mutation in akt-1, akt-1(mg144) (Paradis and Ruvkun 1998). Like akt-1(mg144), pdk-1(mg142) suppresses a null mutation in age-1 more efficiently than it suppresses a loss-of-function mutation in the DAF-2 insulin receptor-like protein (Table 2). This result supports the idea that a bifurcation in the signaling pathway occurs downstream of the DAF-2 insulin receptor-like homolog, and that the AGE-1 PI3K and its downstream signaling molecules represent one branch of these parallel pathways. All signaling from the DAF-2 insulin receptor-like homolog must converge on daf-16, because daf-16(m27) completely suppresses the Daf-c phenotypes of both daf-2 and age-1 mutants (Table 2) (Vowels and Thomas 1992; Gottlieb and Ruvkun 1994; Larsen et al. 1995).
A human PDK1 Ala-277–Val mutant that is equivalent to pdk-1(mg142) activates PDK1 kinase activity toward Akt/PKB
The genetic evidence that the pdk-1(mg142) mutation activates PDK1 kinase activity toward Akt/PKB was biochemically verified using the mammalian homologs of these kinases. The Ala-303–Val substitution in pdk-1(mg142) is in a region of the kinase domain that is conserved in human PDK1 (hPDK1) (Fig. 1B). The equivalent substitution mutation was constructed in hPDK1 (hPDK1.A277V) and its kinase activity on mammalian Akt/PKB substrate was compared with wild-type hPDK1. Human embryonic 293T kidney cells were transfected transiently with wild-type hPDK1, a kinase inactive mutant hPDK1.K110N (Chou et al. 1998) and the hPDK1.A277V mutant, all tagged with a Myc epitope. Transfected cells were lysed under native conditions, and the hPDK1 proteins were immunoprecipitated with a monoclonal anti-Myc antibody. The activity of each hPDK1 protein derivative was determined in an in vitro protein kinase assay using recombinant His-tagged Akt/PKB substrate in the presence of PtdIns-3,4,5-P3, as described previously (Chou et al. 1998) (Fig. 2A). The hPDK1.A277V mutant has a significantly higher protein kinase activity (2.9-fold) toward the Akt/PKB substrate than wild-type hPDK1 (Fig. 2A). As expected, the kinase-inactive mutant poorly phosphorylates the substrate (Fig. 2A). This confirms biochemically the genetic evidence that an Ala-to-Val substitution at this conserved position increases PDK1 protein kinase activity. In addition, we noted that the hPDK1.A277V mutation induces a gel-mobility shift reminiscent of the hyperphosphorylation found in other signaling protein kinases, such as p70 S6 kinase (Romanelli et al. 1999; Fig. 2B). This presumed phosphorylation of hPDK1 may be indicative of activation of the enzyme. It remains to be established, however, whether this modification applies only to the Ala-277–Val-activating mutation or is a more general mechanism of hPDK1 activation by upstream signaling proteins.
Figure 2.

Substitution of A277V in human PDK1 leads to activation of the enzyme. Wild-type human PDK1 (hPDK1), kinase inactive hPDK1 (hPDK1.K110N), and the activating A277V mutation in hPDK1 (hPDK1.A277V) expression clones were transfected transiently in duplicate into 293T cells and after the transfection protocol, the protein kinase activity of each mutant toward Akt/PKB was assayed in an in vitro protein kinase assay (see Material and Methods). The data are representative of four independent experiments. (A) Relative amounts of hPDK1 proteins (hPDK1, hPDK1.K110N, and hPDK1.A277V) were normalized for hPDK1 protein concentration from B using anti-Myc antibody and ECL detection (Amersham) on a Bio-Rad Molecular Imager, and the adjusted amounts of lysate used for immunoprecipitation with anti-Myc antibody. The immunoprecipitates were washed and an in vitro kinase assay using His–Akt as substrate was carried out. The phosphorylation of Akt/PKB (His–Akt) was assessed by autoradiography and quantitated on a Bio-Rad Molecular Imager. The data are normalized to wild-type hPDK1 activity (1) and represent the average of the two lanes for each hPDK1 construct. In the hPDK1.A277V lanes, only the lower species (comigrating with His–Akt in the wild-type hPDK1 lanes) were used for quantitation as the identity of the higher migrating species is unclear. (B) Total cell lysate was assayed for total protein content (Bio-Rad Protein Assay) and equal amounts of protein loaded on a 7.5% SDS–polyacrylamide gel, and resolved proteins transferred to nitrocellulose and immunoblotted with anti-Myc antibody. Additional anti-Myc immunoreactive bands are observed with the A277V mutant when compared with either wild-type or kinase-inactive proteins.
pdk-1(sa680) extends C. elegans life span
Mutations in age-1 increase C. elegans life span greater than twofold (Klass 1983; Larsen et al. 1995; Morris et al. 1996). Mutations in daf-16 suppress this life span increase (Kenyon et al. 1993; Larsen et al. 1995). A loss-of-function mutation in pdk-1 increases C. elegans life span almost twofold, similarly to a mutation in age-1 (Fig. 3). daf-16(m27) suppresses the longevity phenotype of pdk-1(sa680) (Fig. 3). These results show that longevity regulation signals from the DAF-2 and AGE-1 signaling pathway are propagated by PKD-1, presumably via AKT-1 and AKT-2, to the DAF-16 transcription factor. Interestingly, both daf-16(m27); pdk-1(sa680) and the activating mutation in pdk-1 on its own have a slightly shortened life span relative to wild type (Fig. 3). This result agrees with previously published results showing decreased longevity in a daf-16(m26) mutant strain (Larsen et al. 1995). The significance of a slightly decreased life span of a particular strain, however, is difficult to interpret because strain-to-strain variation has been observed even between different wild-type isolates (Kenyon 1997). Therefore, a more rigorous analysis would have to be performed to conclude that the small decrease in life span observed in these strains is caused by these particular mutations.
Figure 3.

Adult life span of pdk-1(sa680) mutants. pdk-1(sa680) extends adult life span and a mutation in daf-16 suppresses the life span increase at 25°C. Mean life spans were 17 days for wild type (□, n = 50), 15 days for pdk-1(mg142) (*, n = 50), 15 days for daf-16(m27); pdk-1(sa680) (●, n = 49), 27 days for pdk-1(sa680) (⋄, n = 50), 27 days for sqt-1(sc13) age-1(hx546) ( , n = 25). Mean life spans are significantly different (P≤2e−9; see Materials and Methods) from each other except pdk-1(sa680) compared with sqt-1(sc13) age-1(hx546), and daf-16(m27); pdk-1(sa680) compared with pdk-1(mg142). Life spans were determined in parallel for all strains; data is from one representative experiment that has been performed at least one other time.
The activating mutation pdk-1(mg142) does not suppress the increased longevity phenotype of age-1 (data not shown). The activating mutation in akt-1 also does not suppress the life span increase of age-1 mutants (Paradis and Ruvkun 1998). One possible interpretation of this data is that the degree of pathway activation by pdk-1(mg142) or akt-1(mg144) in the absence of normal PtdIns-3,4-P2 and PtdIns-3,4,5-P3 signaling is enough to bypass the need for AGE-1 PI3K signaling in reproductive development but not in life span. Animals carrying only a maternal contribution of age-1 activity are long lived but do not arrest at the dauer stage (Morris et al. 1996), which is consistent with the model that a wild-type life span demands a higher level of pathway activation.
pdk-1 loss-of-function mutants have pleiotropic phenotypes
pdk-1(sa680) and pdk-1(sa709) mutants have several other pleiotropic phenotypes in addition to the Daf-c and aging phenotypes (Table 5). pdk-1(sa680) mutant animals grown at 20°C are defective in egg-laying (Egl), have a longer body than wild type (Lon), form clumps of animals rather than being dispersed on the bacterial lawn (Cpy) (Thomas et al. 1993), and have low fertility. Like the Daf-c phenotype, the Lon, Cpy, and fertility phenotypes of pdk-1(sa680) are rescued maternally. The pdk-1(sa680) Egl phenotype, however, does not show complete maternal rescue. A mutation in daf-16 suppresses all of the phenotypes and the akt-1(mg144) mutation partially suppresses the Daf-c and fertility phenotypes, but does not suppress the Egl, Lon, and Cpy phenotypes (though partial suppression of these qualitative phenotypes might be difficult to score accurately). Both the weaker suppression of the Daf-c phenotype and the lack of suppression of the Egl, Lon, and Cpy phenotypes by the akt-1(mg144) activating mutation could reflect weaker activation of the pathway by akt-1(mg144) than by daf-16(m27). Alternatively, pdk-1(sa680) could have outputs in parallel to AKT-1 that are not activated by akt-1(mg144) but that converge on DAF-16.
Table 5.
Summary of pdk-1 loss-of-function phenotypes
|
Genotype
|
Daf-c
|
Egl
|
Lon
|
Cpy
|
Brood size at 20°Ca
|
|---|---|---|---|---|---|
| Wild type | + | + | + | + | 291 ± 69 (5) |
| pdk-1(sa709) | +/−b | +/−b | − | +/−b | N.D.c |
| pdk-1(sa680) | − | − | − | − | 24 ± 20 (4) |
| osm-6(p811); pdk-1(sa680) | − | − | − | − | 38 ± 17 (5) |
| daf-5(e1385); pdk-1(sa680) | − | − | − | − | 36 ± 23 (5) |
| pdk-1(sa680) daf-12(m20) | + | − | − | − | 17 ± 16 (5) |
| daf-16(m27); pdk-1(sa680) | + | + | + | + | 230 ± 43 (5) |
| akt-1(mg144); pdk-1(sa680) | +/−d | − | − | − | 135 ± 61 (3) |
Mean brood size ± s.d. with the number of animals assayed in parantheses.
Indicates that pdk-1(sa709) has defective phenotypes that are weaker than those of pdk-1(sa680).
Not determined. However, casual observation suggests that pdk-1(sa709) does not have an obvious fertility defect.
Partial suppression of the Daf-c phenotype.
PDK-1/GFP is widely expressed
The expression pattern of pdk-1 was examined in transgenic animals containing a translational fusion of the genomic pdk-1 locus to green fluorescent protein (GFP) (Chalfie et al. 1994). This construct contains the entire genomic coding region from pdk-1, including 5′ upstream regulatory sequences, fused in frame at the carboxyl terminus to GFP. The PDK-1/GFP fusion efficiently rescues the Daf-c phenotype of pdk-1(sa680), indicating that the fusion protein is functional (data not shown). PDK-1/GFP expression is observed in late stage embryos and throughout the life of the animal. In post-embryonic animals, PDK-1/GFP expression is observed in the cell bodies and processes of the majority of neurons in the head and tail (Fig. 4A), in the motor neurons of the ventral nerve cord (Fig. 4B) and neuronal processes along the body of the animal, in the cells and neurons of the pharynx (Fig. 4A), in intestinal cells (Fig. 4B), and in hypodermal cells (Fig. 4A). In L4s and adults, PDK-1/GFP is also expressed in the somatic gonad (data not shown).
Figure 4.

PDK-1/GFP expression. (A) PDK-1/GFP expression in an L1 animal. Expression in the procorpus and anterior bulb of the pharynx (ant.ph.) is shown; expression in the entire pharynx is also observed. PDK-1/GFP expression in the cell bodies of many neurons in the head and tail is visible; occasional expression in neuronal nuclei is observed. Expression of PDK-1/GFP is also observed in the hypodermal cells (hyp) of the tail. (B) PDK-1/GFP expression in the intestine (int.) and ventral nerve cord (VNC) of an L1 animal. Anterior is to the left, ventral is down; the animal is slightly twisted because of co-injection with the rol-6 marker. Cell bodies and axons of the VNC neurons (arrowheads) clearly express PDK-1/GFP as do the cell bodies of the intestinal cells (arrows); VNC and intestinal nuclei do not appear to express PDK-1/GFP.
The PDK-1/GFP expression pattern is similar to the expression patterns of AKT-1/GFP and AKT-2/GFP (Paradis and Ruvkun 1998). This result is consistent with a role for PDK-1 in activating AKT-1 and AKT-2. The broad expression patterns of these genes are consistent with a function in the sensory neurons that regulate dauer formation (Bargmann and Horvitz 1991) or in the target tissues that are remodeled during dauer development, such as the pharynx, hypodermis, and intestine (Riddle and Albert 1997).
Discussion
The C. elegans PDK1 homolog is required to prevent developmental arrest at the dauer larval stage. Loss-of-function mutations in pdk-1 cause a dauer constitutive phenotype and life span increase that are suppressed by loss-of-function mutations in the daf-16 transcription factor. This confirms the placement of pdk-1 in the DAF-2 insulin receptor-like signaling pathway and indicates that the DAF-16 transcription factor is a major output of PDK1 signaling in C. elegans. An activating mutation in pdk-1 is sufficient to efficiently bypass the need for AGE-1 PI3K signaling in reproductive development. In agreement with the model that PDK1 activates Akt/PKB signaling, the ability of the activating mutation in pdk-1 to suppress the dauer constitutive phenotype of a null age-1 PI3K mutant is dependent on akt-1 and partially dependent on akt-2. This shows that pdk-1 functions between AGE-1 PI3K and AKT-1/AKT-2 to transduce insulin receptor-like signals and promote reproductive growth and metabolism. PDK-1 probably does not directly phosphorylate DAF-16 because DAF-16 does not have PDK1 consensus phosphorylation sites (TFCGT; Chou et al. 1998). The direct PDK-1 targets are more likely to be AKT-1 and AKT-2 that bear this consensus phosphorylation site and act downstream of PDK-1. DAF-16 does have Akt/PKB consensus phosphorylation sites suggesting that it is directly regulated by Akt/PKB phosphorylation (Paradis and Ruvkun 1998). In support of this model, it was shown recently that Akt/PKB phosphorylation of a mammalian homolog of DAF-16 inhibits its nuclear localization and transcriptional activity and promotes cell survival (Brunet et al. 1999).
PDK1 is required for Akt/PKB activation in other systems (Alessi et al. 1996, 1997b). It is known that simultaneous reduction of akt-1 and akt-2 gene activity causes dauer arrest in C. elegans (Paradis and Ruvkun 1998). Our genetic analyses also predict that the reduction of function mutations in pdk-1 impair its ability to activate akt-1 and akt-2, thereby resulting in a dauer constitutive phenotype. Because reduction of akt-1 and akt-2 gene activity is epistatic to the activating mutation in pdk-1, it appears that akt-1 and akt-2 are the relevant outputs of pdk-1 in regulating the growth arrest and metabolic shift associated with dauer arrest. PDK1 also has other known outputs in mammals, such as p70 S6 kinase (Alessi et al. 1998; Pullen et al. 1998) and PKC isotypes ζ and δ (Chou et al. 1998; Le Good et al. 1998). The suppression of the activated pdk-1(mg142) phenotype by inhibition of akt-1 and akt-2 gene activities strongly argues that these other possible PDK1 outputs are not relevant to the regulation of dauer arrest by pdk-1. So far, none of these other molecules has been implicated by genetic analysis in the DAF-2 insulin receptor-like signaling pathway.
The pdk-1 loss-of-function and gain-of-function mutations that map to conserved residues in the kinase domain of the protein can be interpreted in light of the known crystal structure of the homologous mammalian PKA kinase (Zheng et al. 1993). The Gly-295 residue changed to Arg in pdk-1(sa680) (Fig. 1B) is located in subdomain IX (Hanks and Hunter 1995) of the kinase domain and is conserved completely in all PDK1 family members and well conserved in serine/threonine kinases in general. This conserved glycine is located in α-helix F buried deep within the kinase domain (Zheng et al. 1993). Therefore a Gly-to-Arg substitution at this position would be predicted to interfere with kinase domain tertiary structure and reduce kinase activity. Our genetic analysis is consistent with sa680 being a severe loss-of-function mutation in pdk-1.
The Ala-303 residue changed to Val in the dominant-activating pdk-1(mg142) allele is also a conserved residue in subdomain IX of the kinase domain of PDK-1 (Fig. 1B) predicted to lie in α-helix F only eight amino acids away from Gly-295. The alanine residue is conserved in all PDK1 family members but is not conserved in all serine/threonine kinase domains. Other PKA and PKC family members have a valine at that position, as do other serine/threonine kinase family members, suggesting that this mutation does not interfere with kinase activity. In fact, the equivalent amino acid substitution in human PDK1 (Ala-277–Val) increases human PDK1 kinase activity relative to hPDK1 wild-type kinase activity. This result agrees with our genetic analysis, which shows that pdk-1(mg142) is an activating mutation and implies that the function of alanine at this position is conserved across species.
The fact that an Ala-to-Val substitution at this position (C. elegans 303/human 277) results in an activated kinase indicates that Ala-303/277 may normally limit PDK1 activity. Ala-303/277 may function simply to reduce PDK1 activity at all times, or more interestingly, it may down-regulate PDK1 activity only in the absence of PtdIns-3,4-P2 and PtdIns-3,4,5-P3 signals. Isolation of the activating Ala-303–Val substitution in C. elegans PDK-1, and the demonstration that the equivalent substitution activates human PDK1, shows that PDK1 can be activated above its basal level. So far, PDK1 had been refractive to additional activation on treatment with stimuli that activate Akt/PKB (Alessi et al. 1997a).
Based on analogy to the known PKA kinase domain crystal structure, Ala-303/277 is solvent exposed and on the same face as the pseudosubstrate binding region of PKA (1CMK; Zheng et al. 1993). There are two simple models for how the Ala-303/277–Val substitution activates PDK1. One possibility is that Val-303/277, either directly or indirectly perhaps via α-helix F movement, increases recognition and/or phosphorylation of substrates by PDK1. A related idea is that Val-303/277 increases recognition of the phosphoinositides or other upstream regulatory molecules for PDK1 itself. Another possibility is that Val-303/277 interferes with an inhibitory interaction, for example with a pseudosubstrate, that normally decreases PDK1 phosphorylation of substrates. The mobility shift observed with hPDK1.A277V that is suggestive of hyperphosphorylation of the protein supports either model.
An activating mutation in pdk-1 or overexpressing pdk-1(+) bypasses the need for AGE-1 PI3K signaling. The current model for Akt/PKB activation is that PtdIns-3,4-P2 and/or PtdIns-3,4,5-P3 bind to the pleckstrin homology domain of Akt/PKB, thereby making the Thr-308 site accessible to phosphorylation by PDK1, and possibly also serving to localize the protein to the cell membrane (Alessi et al. 1997b; Andjelkovic et al. 1997; Stokoe et al. 1997; Anderson et al. 1998). Our results are consistent with both possibilities. The role of the phosphoinositides in activating PDK1 has not been established at present; our genetic analysis shows that pdk-1 is a downstream target of AGE-1 PI3K. Whereas the pdk-1 gain-of-function mutation bypasses the requirement for AGE-1-mediated PtdIns-3,4-P2 and PtdIns-3,4,5-P3 production, it is unlikely that AGE-1 PI3K is the only source for PtdIns-3,4-P2 and PtdIns-3,4,5-P3 in the cell. The fact that inactivation of the DAF-18 PTEN lipid phosphatase can suppress null mutations in AGE-1 PI3K (Ogg and Ruvkun 1998) suggests that there is another source of PtdIns-3,4-P2 and PtdIns-3,4,5-P3 in the absence of age-1 activity. This indicates that the activating mutations in akt-1 and pdk-1 do not necessarily activate the proteins in the absence of the phosphoinositides; it is possible that these mutations sensitize the proteins to levels of phosphoinositides that are subthreshold for wild-type AKT-1 and PDK1.
The increased life span observed in animals carrying a loss-of-function pdk-1 mutation shows that pdk-1 functions in a major C. elegans longevity regulating pathway that includes the DAF-2 insulin receptor-like protein and downstream AGE-1 PI3K (Klass 1983; Larsen et al. 1995; Morris et al. 1996). The dependence of the pdk-1(sa680) life span increase on daf-16 also maps pdk-1 to this pathway (Kenyon et al. 1993; Larsen et al. 1995). One popular theory is that aging results from reactive oxygen species inflicting damage on cellular proteins and nucleic acids; therefore molecules that neutralize reactive oxygen species would be protective against aging (Sohal and Weindruch 1996). It is not clear how mis-regulated daf-16 activity results in increased life span. One possibility is that daf-16 activates transcription of genes that promote longevity in the organism. In support of this idea, age-1 mutant animals (which have mis-regulated daf-16 activity similar to pdk-1(sa680) animals) have increased activity of superoxide dismutase and catalase (Larsen 1993), two proteins implicated in scavenging reactive oxygen species. daf-16 could promote longevity by activating the transcription of superoxide dismutase and catalase genes, thereby increasing the ability of C. elegans to protect against oxidative damage.
The pleiotropy of the pdk-1 loss-of-function alleles indicates that pdk-1 also regulates the signaling pathways that control body size, egg laying, and social behavior. A TGF-β related signaling pathway functions in parallel to the DAF-2 insulin receptor-like signaling pathway to regulate dauer formation (Thomas et al. 1993; Gottlieb and Ruvkun 1994). Mutations in the TGF-β ligand, receptor, and downstream effectors also result in Daf-c, Egl, and Cpy phenotypes (Estevez et al. 1993; Thomas et al. 1993; Ren et al. 1996). pdk-1 mutants are the first in the DAF-2 insulin receptor-like signaling pathway to display the Egl and Cpy phenotypes (although age-1 mutants also appear slightly Egl; data not shown). This suggests that pdk-1 has outputs to the parallel TGF-β signaling pathway or to targets in common with those regulated by the TGF-β pathway. The fact that a mutation in the daf-16 transcription factor suppresses all the phenotypes of pdk-1(sa680), however, suggests that these other outputs depend on daf-16. Furthermore, the interaction between these pathways may be complex because daf-5(e1385) suppresses the pleiotropic phenotypes of mutants in the TGF-β pathway (Thomas et al. 1993) but does not suppress pdk-1(sa680) pleiotropies, whereas daf-16(m27) suppresses the pleiotropies of pdk-1(sa680) but not those of TGF-β pathway mutants (M. Ailion, T. Inoue, and J.H. Thomas, pers. comm.).
The role of pdk-1 in regulating akt-1 and akt-2 activity in the DAF-2 insulin receptor-like signal transduction cascade suggests the following model (Fig. 5). Under reproductive growth conditions an insulin-like ligand binds to and activates the DAF-2 insulin/IGF-I receptor. Activation of DAF-2 recruits and activates the AGE-1 PI3K and other signaling pathways emanating from DAF-2. Our genetic experiments place PDK1 downstream of and positively regulated by AGE-1 PI3K, and upstream of and negatively regulating DAF-16. Although it is a formal possibility that PDK-1 signals in parallel to AKT-1/AKT-2 to antagonize DAF-16, our analysis suggests that PDK-1 is dependent on AKT-1 and AKT-2 to exert its function. Therefore, we favor the model that PDK-1 antagonizes DAF-16 by activating AKT-1 and AKT-2. The phosphoinositides generated by AGE-1 activate PDK1 and, in cooperation with phosphorylation by PDK1 (and the as yet unidentified PDK2), activate AKT-1 and AKT-2. AKT-1 and AKT-2, and presumably inputs from other DAF-2 activated pathways, negatively regulate the DAF-16 transcription factor, possibly via direct phosphorylation of DAF-16. The nuclear localization of the mammalian DAF-16 homolog FKHRL1 is antagonized by Akt/PKB signaling (Brunet et al. 1999), consistent with the genetic evidence that C. elegans DAF-16 is active when insulin receptor-like signaling is decreased. Unphosphorylated DAF-16 could function to activate genes necessary for dauer arrest, metabolism, and increased life span or could repress genes necessary for reproductive growth.
Figure 5.

Model for regulation of reproductive growth and metabolism by the DAF-2 insulin receptor-like signaling pathway. On DAF-2 activation, AGE-1 generates PtdIns-3,4-P2 and PtdIns-3,4,5-P3 that are required for PDK-1 and AKT-1/AKT-2 activation. Other parallel pathways from DAF-2 are also activated and all signaling converges on DAF-16. See text for details.
Biochemical analysis of the insulin signaling pathway suggests that PI3K, PDK1, and Akt/PKB act in the same cell to transduce insulin signals (Kahn 1994; Alessi et al. 1997b; Avruch 1998). Our model is that the DAF-2 insulin receptor-like protein, AGE-1 PI3K, PDK-1, AKT-1/AKT-2, the DAF-18 PTEN phosphatase, and the DAF-16 transcription factor act in the same cell to transduce signals from an as yet unidentified insulin-like ligand to transcriptional outputs regulated by DAF-16 in the nucleus. It has not yet been determined if these genes in fact act in the same cells to regulate dauer formation. Interestingly, DAF-2 has been shown to function cell nonautonomously to regulate dauer formation and longevity (Apfeld and Kenyon 1998). One possible interpretation of this result is that the DAF-2 mediated signal transduction cascade itself generates another signal that controls reproductive growth, metabolism, and life span. This result, however, does not clarify if AGE-1, PDK-1, AKT-1/AKT-2, DAF-18, and DAF-16 function in the same cells as DAF-2 and further functional analysis of these genes is required to address that question. pdk-1, akt-1, akt-2, and daf-16 are all broadly expressed (Ogg et al. 1997; Paradis and Ruvkun 1998). It is still not understood whether these molecules function in secretory neurons to generate the dauer-inducing signal, or function in the target tissues that are remodeled during dauer formation to receive and respond to the dauer-inducing signal.
Mammalian homologs of many of the genes (daf-2, age-1, akt-1, akt-2, pdk-1) that regulate C. elegans dauer arrest had been shown biochemically to act in the insulin signal transduction cascade (Avruch 1998). Our genetic analysis of pdk-1 strongly endorses the placement of pdk-1 in the insulin pathway and also reveals important regulatory regions on the protein. Also, our molecular and genetic analysis of dauer formation suggests that much of the insulin signaling pathway was in place in the common ancestor of nematodes and vertebrates. The fact that the nematode and mammalian pathways share so many conserved molecules suggests that, as in the case of the DAF-16 transcription factor and DAF-18 phosphatase, a combination of C. elegans genetics and the nearly complete genome sequence can reveal new molecules involved in insulin signal transduction.
Materials and methods
Strains and transgenic lines
The following strains were used: Wild type: N2 Bristol, GR1318 pdk-1(mg142), JT9609 pdk-1(sa680), JT9604 osm-6(p811); pdk-1(sa680), JT9606 daf-5(e1385); pdk-1(sa680), JT9605 pdk-1(sa680) daf-12(m20), JT9607 daf-16(m27); pdk-1(sa680), JT10108 akt-1(mg144); pdk-1(sa680), JT709 pdk-1(sa709), JT6064 daf-16(m27), JT9902 daf-16(m27); pdk-1(sa709), GR1188 sqt-1(sc13) age-1(mg44)/mnC1, GR1316 sqt-1(sc13) age-1(mg44); pdk-1(mg142), GR1196 daf-16(m27); sqt-1(sc13) age-1(mg44), GR1306 sqt-1(sc13) age-1(mg44); akt-1(mg144), HT211 sqt-1(sc13) age-1(hx546), GR1122 daf-2(e1370), SP122 daf-2(e1370); pdk-1(mg142), GR1105 daf-16(m27); daf-2(e1370), GR1319 unc-4(e120) age-1(mg44)/mnC1.
The following transgenic lines were used: SGP300 mgEx464, SGP301 mgEx465, SGP303 mgEx467, SGP304 mgEx468, SGP305 mgEx469, SGP320 sqt-1(sc13) age-1(mg44)/+; mgEx464, SGP319 sqt-1(sc13) age-1(mg44)/+; mgEx465, SGP317 sqt-1(sc13) age-1(mg44); mgEx467, SGP316 sqt-1(sc13) age-1(mg44); mgEx468, SGP318 sqt-1(sc13) age-1(mg44); mgEx469, SGP294 mgEx479, SGP295 mgEx480, SGP296 mgEx481, SGP306 pdk-1(sa680); mgEx470, SGP307 pdk-1(sa680); mgEx471, SGP308 pdk-1(sa709); mgEx472, SGP312 pdk-1(sa709); mgEx476, SGP313 pdk-1(sa709); mgEx477.
Isolation and mapping of pdk-1(sa680) and pdk-1(sa709)
pdk-1(sa680) and pdk-1(sa709) were isolated in a screen for Daf-c mutants at 27°C. sa680 was outcrossed four times and sa709 twice before characterizing the mutant phenotypes in detail. At all temperatures, sa680 males all arrest as dauers or die before the dauer stage. Therefore, mapping markers were always crossed into the sa680 mutant, often using tra-2(q276) to obtain XX males heterozygous for X chromosome markers. Both sa680 and sa709 were mapped to the left arm of the X chromosome between unc-1 and dpy-3. Pooling results from both Unc non-Dpy and Dpy non-Unc recombinants picked from either sa680/unc-1(e719) dpy-3(e27), or sa709/unc-1(e719) dpy-3(e27), we get the following map data: unc-1 (13) sa680 (46) dpy-3, and unc-1 (2) sa709 (8) dpy-3. This is very close to the position of pdk-1 on the physical map.
pdk-1(mg142) isolation and mapping
pdk-1(mg142) was isolated as a suppressor of age-1(mg44) in a screen described previously (Paradis and Ruvkun 1998). All mapping experiments were done in a sqt-1(sc13) age-1(mg44) mutant background. age-1(mg44) causes a Daf-c phenotype that can be rescued maternally (Riddle 1988). Therefore, in each mapping experiment, we first homozygosed sqt-1 age-1, then genotyped the other loci by scoring phenotypes segregated in the subsequent generation. For technical reasons, we did not score the subsequent generation of animals that had homozygosed a mapping marker in the same generation as sqt-1 age-1 (i.e., we did not score the broods of sqt-1 age-1; dpy-3, for example). During backcrossing, we found pdk-1(mg142) to be X-linked. Subsequent two-factor crosses showed that mg142 is linked to dpy-3(e27) by roughly 11 m.u. Three-factor crosses between mg142 and dpy-6(e14) unc-3(e151) suggested that mg142 is to the left of dpy-6. Further, three-factor crosses between mg142 and unc-1(e719) dpy-7(e88) showed that mg142 is either close to the right, or possibly to the left, of unc-1. These results are as follows. First, we recovered 42 homozygous mg142 animals and 19 heterozygous mg142/unc-1(e719) dpy-7(e88) animals, representing the nonrecombinant classes. In the recombinant classes, we found that 15 out of 15 Dpy non-Unc recombinants carried mg142 (although we might have overlooked recombinants that did not carry mg142 because it is difficult to score Unc in Dpy dauers). This result places mg142 far from dpy-7. Only one out of 15 Unc non-Dpy recombinants carried mg142. This result places mg142 right of and close to unc-1, an interpretation that is consistent with the pdk-1 molecular cloning result. We also isolated two recombinants that carried mg142 unc-1(e719) dpy-7(e88) chromosomes, however, suggesting that mg142 is to the left of unc-1. We suspect this class to be the result of double-recombination events. Extrapolation of this data to account for all recombinant and nonrecombinant classes gives a two factor map distance between mg142 and unc-1 of 3.3 m.u. or a three-factor map distance for mg142 of 1.7 m.u. right of unc-1.
Test for pdk-1(mg142) dominance
unc-4(e120) age-1(mg44)/+ males were crossed to sqt-1(sc13) age-1(mg44); pdk-1(mg142) hermaphrodites. In the F1 we recovered the following cross progeny: 249 wild-type nondauer males; 242 wild-type nondauer hermaphrodites; 10 wild-type dauers; and 13 other (totals are from two trials). The broods of wild-type nondauer hermaphrodites that segregated Sqt, Unc, and wild type were scored for dauer versus non-dauer and gave the following results: 70.6% non-dauer and 29.4% dauer (n = 422, one trial). We noticed that the dauers seemed to recover 24–48 hr after scoring in contrast with age-1(mg44) dauers. We believe that this is evidence for a maternal effect of pdk-1(mg142) suppression of age-1.
pdk-1(sa680) and pdk-1(sa709) dauer formation assays
Table 1 combines the results from two independent experiments, each with similar results. For pdk-1(sa680) dauer formation assays, in one experiment parents were allowed to lay eggs at room temperature for 8 hr and plates were then shifted to 25.4°C. In the second, a 17-hr overnight egg lay was performed at 25.4°C before parents were removed (in this assay, some sa680 parents had ‘bagged’ from internal hatching of progeny). In both cases, the plates were counted 48 hr post-egglay. Care was taken to examine the sides of the plate, as sa680 dauers have a high propensity to climb the walls of the plate. Tight synchrony was not needed because of lack of recovery of sa680 dauers, and the assays were performed as described because sa680 is defective in egg-laying. In addition to forming dauers, sa680 mutants form a low percentage of ‘dauer-like’ animals that arrest development following the L2 stage and have dark, dauer-like intestines, but are not radially constricted like dauers and do not cease movement or pumping. Dauer-like animals such as these have also been seen in mutants of daf-2 (Gems et al. 1998) and age-1 (Gottlieb and Ruvkun 1994). For pdk-1(sa709) dauer formation assays, parents were allowed to lay eggs at room temperature for 5.5 hr and plates were then shifted to 26.8°C. Progeny were counted 41 hr post-egglay.
pdk-1(mg142) dauer formation assays
Gravid adults were allowed to lay eggs at 25°C for 3 hr. Progeny were scored for all classes at 48 hr post-egglay and some strains were scored at 72 or 96 hr post-egglay. For all assays performed in this study, numbers represent the summary of at least two trials of each genotype in at least two experiments performed on different days. In all assays, the following scoring categories were used. Dauer-like refers to animals that were arrested and had a dark intestine like age-1 or daf-2 dauers but were not as fully constricted as these dauers, or that were arrested and constricted like age-1 and daf-2 dauers but did not have as dark an intestine as age-1 or daf-2 dauers. The dauer-like category is equivalent to the partial dauer category in Paradis and Ruvkun (1998). ‘Other’ refers to animals that could not be classified as dauer because the animal was young, had a grossly aberrant morphology, or was dead.
Allele sequencing
Genomic DNA from pdk-1(mg142), pdk-1(sa680), and pdk-1(sa709) strains was PCR-amplified and directly sequenced. At least two different outcrossed pdk-1(mg142) and pdk-1(sa680) strains were sequenced.
cDNA characterization
pdk-1a gene structure was confirmed by sequencing of a cDNA (yk216b6 provided by Y. Kohara, National Institute of Genetics, Mishima, Japan). pdk-1b gene structure was confirmed by sequencing of cDNAs (yk478b12, yk499g8, yk551e8 provided by Y. Kohara). The 5′ end of the gene was determined by 5′ RACE (GIBCO) using a gene-specific primer to exon 4 and is assumed to be the same for both pdk-1a and pdk-1b. The pdk-1 message analyzed by 5′ RACE was trans-spliced with a SL1 leader sequence 10 bp upstream of the first methionine.
RNAi
The pdk-1 coding region was amplified from yk478b12 (Y. Kohara) and RNA prepared as described (Paradis and Ruvkun 1998). Preparation of akt-1 and akt-2 RNAs and injections were performed as described (Paradis and Ruvkun 1998). After injection, the worms were allowed to recover at 20°C for 24 hr, then were moved to fresh plates and allowed to lay eggs at the appropriate temperature for 24 hr. Broods were scored for dead eggs 24-hr post-egglay and for dauers or L4 larvae and adults 48 hr post-egglay. For assays performed at 20°C only, a second consecutive 24-hr egglay at 20°C was performed and scored. We found slight differences between the dauer-inducing ability of akt-1 + akt-2 RNAi in a fresh stock of N2 wild-type strain received from the C. elegans Genetic Center and in N2 that had been cultured in our lab [ccf. Table 3 of this report with Table 2 of Paradis and Ruvkun (1998)]. We performed the akt-1 + akt-2 RNAi assay at 26°C for greater consistency between assays in the new N2 background. We suspect that either the presence of modifier mutations or fluctuations of temperature within incubators have affected penetrance of the Daf-c phenotype of akt-1 + akt-2 RNAi.
Construction of pdk-1 transgenic lines and scoring of transgene effects on dauer formation
A 9.2-kb PCR product of genomic DNA from the pdk-1(+) genomic region containing 2.7 kb of 5′ upstream regulatory sequence, 6.1 kb of coding sequencing containing introns and exons, and 0.4 kb of pdk-1 3′ UTR was purified using QIAquick (Qiagen) and injected at 10 ng/μl with ttx-3::GFP at 50 ng/μl (pPD95.75-C40H5-GFP O. Hobert, MGH and Harvard Medical School) as a co-injection marker (Mello et al. 1991). pdk-1(KD) was constructed and injected in the same manner except PCR primers were used to introduce the K98N mutation. Once the arrays were established in wild type, they were crossed into an age-1(mg44) background. To score arrays in a wild-type background, gravid adults were allowed to lay eggs at 25°C for 3 hr. All animals were scored 72 hr post-egglay. To score arrays in an age-1 background, gravid adults were allowed to lay eggs at 25°C for 5 hr. All animals were scored 72 hr post-egglay. For a description of the categories used in Table 4, see the ‘pdk-1(mg142) dauer formation assays’ section of Materials and Methods. The numbers in Table 4 represent the tally of two independent transgenic lines of pdk-1(KD) (mgEx464 and mgEx465) and three independent transgenic lines of pdk-1(+) (mgEx467, mgEx468, and mgEx469) for each genotype. This same pdk-1 construct was injected with ttx-3::GFP into sa680 and sa709 strains to assay for rescue of the Daf-c phenotype (1/2 lines rescued sa680 and 3/3 lines rescued sa709).
Life span assays
Animals were grown at 20°C until the L4 larval stage and then transferred to plates (10 animals/plate) containing 400 μm fluorodeoxyuridine (Sigma) at 25°C. Animals were scored every 1–3 days subsequently and moved periodically to keep growth conditions mold free. Animals were scored as dead if they failed to respond to a gentle tap on the head and tail with a platinum wire. Life span is defined as the day animals were at the L4 larval stage (t = 0) to the day the animal was scored as dead. A t-test was performed to compare mean life spans of each strain pairwise.
PDK-1/GFP expression
The PDK-1/GFP translational fusion was constructed as follows. A 9-kb PCR product of genomic DNA from the pdk-1 genomic region comprising 2.9 kb of 5′ upstream regulatory region and 6.1 kb of coding region including exons and introns was fused by PCR in-frame to a GFP with unc-54 3′UTR PCR product from pPD95.75 (A. Fire, Carnegie Institute of Washington, Baltimore, MD). PCR products were purified using QIAquick (Qiagen) and injected with rol-6 (pRF4, 100 ng/μl) as the coinjection marker (Mello et al. 1991). For UV microscopy, worms were anesthetized in 5 μm NaN3 M9 buffer and mounted on a 2% agarose pad. Three independent transgenic lines (mgEx479, mgEx480, and mgEx481) were scored to determine the expression pattern of PDK-1/GFP.
PDK1 kinase activity assays
To generate the Ala-277–Val substitution in hPDK1, site-directed mutagenesis (Quickchange, Stratagene) was performed on the Myc.hPDK1 cDNA (Chou et al. 1998) using the following primers: SP170 5′-CCAGCTTGTGGTAGGACTCCCAC and SP171 5′-GTGGGAGTCCTACCACAAGCTGG. To determine the protein kinase activity of the hPDK1 proteins, the cDNAs encoding Myc.hPDK1, Myc.hPDK1.K110N, and Myc.hPDK1.A277V were transiently transfected into 293T cells using the calcium phosphate method (Ausubel et al. 1998). Following the transfection protocol, cells were recovered for 48 hr in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum at 37°C in a 5% humidified CO2 atmosphere. Cell monolayers were washed twice with ice-cold phosphate buffered saline, then lysed in a 1% NP-40 lysis buffer as described previously (Chou et al. 1998). HPDK1 was immunoprecipitated using the monoclonal anti-Myc antibody and immunoprecipitates washed stringently as described (Chou et al. 1998). The kinase assay was performed using 2 mg of a recombinant His-tagged full-length Akt/PKB purified to homogeneity from baculovirus-infected cells, in the presence of 10 μm PtdIns-3,4,5-P3/100 μm phosphatidylserine and 100 μm phosphatidylcholine (Chou et al. 1998). The kinase assay was stopped by addition of SDS sample buffer, and proteins resolved by SDS-polyacrylamide. Phosphorylated Akt/PKB was detected by autoradiography and on a Bio-Rad Molecular Imager for quantitation. Expression of hPDK1 was detected on a 7.5% SDS–polyacrylamide gel, transferred to nitro-cellulose and visualized using the anti-Myc antibody and ECL (Amersham). hPDK1 protein concentration was quantitated using a Bio-Rad Molecular Imager, and equal amounts of wild-type hPDK1, hPDK1. K11ON, and hPDK1. A277Val were used in the in vitro kinase assay.
Acknowledgments
We thank P. Delerme and Y. Liu for expert technical assistance; Y. Kohara for providing cDNAs; O. Hobert and B. Reinhart for help with cell identifications; J. Avruch for discussions on PDK1 in particular and insulin signaling in general; J. Hogan for help with the PDK1-PKA crystal structure comparison; and T. Inoue and members of the Ruvkun laboratory for helpful suggestions and critical reading of the manuscript. Some of the strains used in this study were provided by the C. elegans Genetics Center, which is supported by the National Center for Research Resources. M.A. is a Howard Hughes Medical Institute predoctoral fellow. This work was supported by the National Institutes of Health grant RO1AG14161 to G.R. and RO1CA75134 to A.T.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Ruvkun@frodo.mgh.harvard.edu; FAX (617) 726-6893.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997a;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Ba. Curr Biol. 1997b;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Kozlowski MT, Weng QP, Morrice N, Avruch J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr Biol. 1998;8:69–81. doi: 10.1016/s0960-9822(98)70037-5. [DOI] [PubMed] [Google Scholar]
- Anderson KE, Coadwell J, Stephens LR, Hawkins PT. Translocation of PDK1 to the plasma membrane is important in allowing PDK1 to activate protein kinase B. Curr Biol. 1998;8:684–691. doi: 10.1016/s0960-9822(98)70274-x. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J. Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell. 1998;95:199–210. doi: 10.1016/s0092-8674(00)81751-1. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York, NY: John Wiley and Sons; 1998. [Google Scholar]
- Avruch J. Insulin signal transduction through protein kinase cascades. Mol Cell Biochem. 1998;182:31–48. [PubMed] [Google Scholar]
- Bargmann CI, Horvitz HR. Control of larval development by chemosensory neurons in Caenorhabditis elegans. Science. 1991;251:1243–1246. doi: 10.1126/science.2006412. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Cheng X, Ma Y, Moore M, Hemmings BA, Taylor SS. Phosphorylation and activation of cAMP-dependent protein kinase by phosphoinositide-dependent protein kinase. Proc Natl Acad Sci. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Estevez M, Attisano L, Wrana JL, Albert PS, Massague J, Riddle DL. The daf-4 gene encodes a bone morphogenetic protein receptor controlling C. elegans dauer larva development. Nature. 1993;365:644–649. doi: 10.1038/365644a0. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phoshatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474–8481. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL. Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics. 1998;150:129–155. doi: 10.1093/genetics/150.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S, Ruvkun G. daf-2, daf-16, and daf-23: Genetically interacting genes controlling dauer formation in Caenorhabditis elegans. Genetics. 1994;137:107–120. doi: 10.1093/genetics/137.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. The eukaryotic protein kinase superfamily. In: Hardie G, Hanks S, editors. The protein kinase facts book protein–serine kinases. San Diego, CA: Academic Press, Inc.; 1995. pp. 7–47. [Google Scholar]
- Kahn CR. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kenyon C. Environmental factors and gene activities that influence life span. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 791–813. [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtlang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura K, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Klass M. A method for the isolation of longevity mutants in the nematode C. elegans and initial results. Mech Ageing Dev. 1983;22:279–286. doi: 10.1016/0047-6374(83)90082-9. [DOI] [PubMed] [Google Scholar]
- Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PL. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc Natl Acad Sci. 1993;90:8905–8909. doi: 10.1073/pnas.90.19.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Malone EA, Inoue T, Thomas JH. Genetic analysis of the roles of daf-28 and age-1 in regulating dauer formation. Genetics. 1996;143:1193–1205. doi: 10.1093/genetics/143.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration transforming sequences. EMBO J. 1991;10:3959–5970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhaditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- O’Riordan VB, Burnell AM. Intermediary metabolism in the dauer larva of the nematode Caenorhabditis elegans-1. Glycolysis, gluconeogenesis, oxidative phosphorylation and the tricarboxylic acid cycle. Comp Biochem Biophys. 1989;92B:223–238. [Google Scholar]
- ————— Intermediary metabolism in the dauer larva of the nematode Caenorhabditis elegans-II. The glyoxylate cycle and fatty acid oxidation. Comp Biochem Physiol. 1990;95B:125–130. [Google Scholar]
- Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes & Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Ren P, Lim C-S, Johnsen R, Albert PS, Pilgrim D, Riddle DL. Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science. 1996;274:1389–1392. doi: 10.1126/science.274.5291.1389. [DOI] [PubMed] [Google Scholar]
- Riddle DL. The dauer larva. In: Wood WB, editor. The nematode Caenorhabditis elegans. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1988. pp. 393–412. [Google Scholar]
- Riddle DL, Albert PS. Genetic and environmental regulation of dauer larva development. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 739–768. [PubMed] [Google Scholar]
- Riddle DL, Swanson MM, Albert PS. Interacting genes in nematode dauer larva formation. Nature. 1981;290:668–671. doi: 10.1038/290668a0. [DOI] [PubMed] [Google Scholar]
- Romanelli A, Martin KA, Toker A, Blenis J. p70 S6 kinase is regulated by protein kinase czeta and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5- trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PRJ, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Thomas JH, Birnby DA, Vowels JJ. Evidence for parallel processing of sensory information controlling dauer formation in Caenorhabditis elegans. Genetics. 1993;134:1005–1117. doi: 10.1093/genetics/134.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A, Cantley LC. Signalling through the lipid products of phophoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Vowels JJ, Thomas JH. Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics. 1992;130:105–123. doi: 10.1093/genetics/130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Knighton DR, Xuong NH, Taylor SS, Sowadski JM, Ten Eyck LF. Crystal structures of the myristylated catalytic subunit of cAMP- dependent protein kinase reveal open and closed conformations. Protein Sci. 1993;2:1559–1573. doi: 10.1002/pro.5560021003. [DOI] [PMC free article] [PubMed] [Google Scholar]