


Abstract
We present and validate BlastR, a method for efficiently and accurately searching non-coding RNAs. Our approach relies on the comparison of di-nucleotides using BlosumR, a new log-odd substitution matrix. In order to use BlosumR for comparison, we recoded RNA sequences into protein-like sequences. We then showed that BlosumR can be used along with the BlastP algorithm in order to search non-coding RNA sequences. Using Rfam as a gold standard, we benchmarked this approach and show BlastR to be more sensitive than BlastN. We also show that BlastR is both faster and more sensitive than BlastP used with a single nucleotide log-odd substitution matrix. BlastR, when used in combination with WU-BlastP, is about 5% more accurate than WU-BlastN and about 50 times slower. The approach shown here is equally effective when combined with the NCBI-Blast package. The software is an open source freeware available from www.tcoffee.org/blastr.html.
INTRODUCTION
We describe in this work, a strategy to efficiently and accurately search databases for homologous non-coding RNAs (ncRNAs). This problem is growing in importance, mostly because so many new classes of ncRNAs have been recently reported (1–4), revealing ncRNA involvement at virtually all cellular levels, especially gene regulation. In broad terms, ncRNAs can be divided into two classes: those that are active via a well-defined and evolutionary constrained secondary and tertiary structure, and those for which no evidence is yet available to suggest a link between function and structure. The Rfam database is mostly made of RNAs with a well-defined and conserved secondary structure (5). The second group includes many of the recently discovered ncRNAs involved in gene regulation, such as Piwi-interacting RNAs (1) involved in gene silencing, lincRNAs (3) a novel class of regulated ncRNAs, epigenetic regulators-like HOTAIR (HOX antisense intergenic RNA) (6) or nuclear trafficking regulators-like NRON (ncRNA repressor of NFAT) (7).
Further characterizing these new classes of genes has become a prime target for biology, resulting in the development of an increasing number of in-silico methods for the prediction and comparison of RNA sequences. Analyzing ncRNAs is, however, a complex task. First of all, the genes are hard to discover, and although some of them, such as lincRNAs, exhibit the same properties as protein-coding genes, with introns/exon structures, similar chromatin markings (3) and capped poly-adenylated transcripts (8), they lack the equivalent of an open reading frame that could ease their detection. Homology-based analysis is not much easier since RNA sequences tend to evolve rapidly, either under the sole constraint of maintaining stable secondary structures (in the case of structured RNAs), or under unknown functional constraints whose effect on sequence variation is hard to anticipate. As a consequence, their level of sequence conservation is limited and often too weak to yield statistically meaningful alignments, even when considering moderate evolutionary distances such as mouse versus human. Using standard database search tools like Blast is, therefore, difficult and rarely as informative as in the case of protein-coding genes.
More sensitive searches are possible, using profiles for instance, but the highest level of sensitivity is only achieved when using algorithmic methods simultaneously taking into account sequence similarity and predicted (or experimentally known) secondary structures like the Sankoff algorithm does (9). These methods are computationally very expensive [Sankoff has a time complexity of O(L3N) where N is the number of sequences and L their length] and their practical usage requires the design of heuristic versions with lower complexity. These include Genetic Algorithms (10) or banded Sankoff implementations like Consan (11). In practice, however, the most popular applications to perform efficient database searches make use of stochastic context free grammars (SCFG) (12). These include Rsearch (13) and RaveNna (14) that are based on Infernal (15). Although their algorithmic details vary, the overall principle is similar: informing the database search with some secondary structure information.
A special form of SCFGs called covariance models (CMs) is widely used today in RNA homology search (16). Conceptually, CMs are similar to protein profiles, albeit computationally much more expensive to use. CM parameters are trained on high-quality (seed) alignments. In the Rfam framework (5), for example, seed alignments are taken from literature and are often manually refined to ensure high quality [e.g. using editors like RALEE (RNA alignment editor in Emacs) (17)]. These alignments are used to compute CMs, which are in turn used by Infernal to search databases. The whole setup is similar to HMM (Hidden Markov Model) searches using HMMER for Pfam (18,19) with the seed alignment quality being a critical parameter (20). Infernal searches are much more computationally demanding than HMMER-based searches, even when considering recent improvements (21). To reduce the search space a pre-filtering step is often necessary (22). This can be done by using tools especially developed for this task, e.g. (14) or (23) or by means of a Blast (24) search with ‘relaxed’ parameters (to avoid filtering out true positives). Several studies describe the optimization of BlastN parameters for searching RNA sequences (25,26). The method we describe here is precisely addressing this pre-filtering step and shall be seen as an attempt to improve database searches at a reasonable extra computational cost over a simple BlastN and without the need of profiles, SCFG or accurate secondary structure information.
Our approach takes advantage of the possibility of improving database search procedures by taking into account di-nucleotide content. This idea is not new and neighboring nucleotides have been known for a long time to exhibit some dependencies reflected in their evolutionary patterns (27–30). Durbin et al. (31) even proposed a Markov chain model able to take this phenomenon into account. While the reality of this phenomenon is relatively well-accepted at the genomic level and known to influence the stability of secondary structures at the transcript level (32,33), it remains debated whether the quantification of this effect can help improving gene finding methods (34,35). Di-nucleotides can also be used to improve multiple sequence alignments, as recently reported by Lu and Sze (36). Here we go further and show that in order to use di-nucleotide information, RNA sequences can be recoded using a 16 letters alphabet, thus making it possible to apply protein algorithms [BlastP, BlastClust (24)] to RNA sequences. This result is obtained by computing a suitable RNA substitution matrix (BlosumR) and using Rfam as a validation framework. While similar RNA-specific matrices had been previously estimated on ribosomal RNA (13,37), this work is, to our knowledge, the first reported attempt to use these models for efficient database searches in a Blast-like framework.
METHODS
Computation of the BlosumR matrices
In biology, the cost for aligning two symbols is often estimated using the log ratio between the observed frequency of substitutions and the expected ones, as measured on a collection of reference alignments (pair wise or multiple). This approach forms the basis of the PAM (point accepted mutation) (38) and BLOSUM (blocks of amino acid substitution matrix) matrices (39). We used a related approach in this work, to estimate a matrix that takes into account the cost for aligning two nucleotides to one another while taking into account the nature of their immediate neighbor. The cost for aligning two nucleotides Nx with Ny while taking into account Ny − 1 and Nx − 1 (their immediate neighbors) can therefore be expressed as follows:
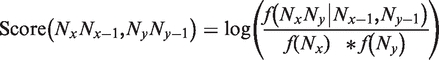 |
(1) |
where 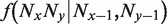 is the frequency of an alignment column NxNy given a neighboring column that contains Nx − 1, Ny − 1. The log-odd ratio in Equation (1) therefore reflects the influence of di-nucleotide content on the observed substitutions. In practice, this amounts to ‘enriching’ the substitution cost of each position with some information related to di-nucleotide content.
is the frequency of an alignment column NxNy given a neighboring column that contains Nx − 1, Ny − 1. The log-odd ratio in Equation (1) therefore reflects the influence of di-nucleotide content on the observed substitutions. In practice, this amounts to ‘enriching’ the substitution cost of each position with some information related to di-nucleotide content.
The counts used to compute our matrix were estimated on a data set containing 792 RNA families reported in Rfam release 9.1. These families were used because they are distinct from the ones used for validation (see next section). We named this matrix BlosumR (Figure 1B) because it relies on a procedure similar to that described for the BLOSUM matrix. For BlosumR, all pairwise projections yielding between 62% and 80% identity were extracted from the Rfam seed alignments. All positions containing gaps in the pairwise projection were excluded. The final matrix is scaled in half bits and contains entries reflecting the cost for the substitution of any pair of di-nucleotides. For validation purposes and in order to show the usefulness of di-nucleotide information, we also re-computed similar matrices from reference alignments with randomly reordered columns (i.e. reference alignments with randomized di-nucleotide content). The table used for the extension and the BlosumR matrix are displayed in Figure 1A and B.
Figure 1.

(A) Recoding scheme. The table shown represents the di-nucleotide to amino acid symbol recoding scheme. Rows correspond to the first letter of the di-nucleotide, while columns to the second. For example the di-nucleotide ‘AC’ is recoded to ‘E’. (B) BlosumR log-odd matrix. The matrix assigns a score for each possible substitution between di-nucleotides pairs. (C) BlosumN log-odd matrix. The matrix assigns a score for each possible substitution between nucleotides pairs. (D) BlastR pipeline. Rfam seed alignments are used to estimate a log-odd matrix (BlosumR). BlastP is then used along with BlosumR to search recoded RNA query sequences against a similarly recoded RNA database.
An interesting property of this formulation is its reliance on di-nucleotides. Since there are only 16 di-nucleotides, one can easily recode them using a 16 letters alphabet. Given a matrix like the one defined in Equation 1, it is therefore possible to recode the RNA sequences into pseudo-amino acid sequences and to align them with the BlosumR matrix used as a protein-like substitution matrix. This recoding involves replacing each nucleotide with a symbol coming from an extended 16-letters alphabet (Figure 1A and B). The choice of the 5′ or 3′ neighbor is arbitrary since it does not have any consequence on the amount and the quality of information eventually concentrated on every symbol after extension. The recoding is carried out by sliding a window of size 2 and replacing every nucleotide with one of the 16 letter alphabet symbol. Given an RNA sequence of size L, the recoding therefore results in a recoded sequence of size L-1. A neutral character is then added on the last position to preserve the original sequence length. We name such sequences recoded RNAs (reRNAs). Once recoded, the sequences can be fed to BlastP and searched against a database of extended sequences, as outlined in Figure 1D.
Computation of the BlosumN matrices
The single nucleotide matrix BlosumN (Figure 1C) was estimated on the same Rfam reference multiple sequence alignments, using the same interval of 62–80% for the selection of pairwise projections. The following formula was used to estimate the matrix:
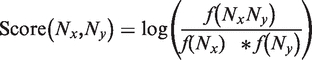 |
(2) |
Where f(NxNy) is the frequency of an alignment column NxNy.
Validation data set
In order to establish the relative merits of the di-nucleotide-based searches with respect to simpler methods such as BlastN, one requires a reference data set that can be used to measure sensitivity and specificity. For this purpose we used Rfam which is a collection of RNA alignments. For each family a high-quality seed alignment and a corresponding full alignment are provided. Full alignments are created by searching databases for homologues of the seed sequences using Infernal. It is appropriate to use Rfam as a reference since our goal is not to over-perform Infernal in accuracy, but rather to show how di-nucleotides based searches can produce results close enough to Infernal so that they might be used for efficient and sensitive pre-filtering. Validation was done on a subset of Rfam that we named Rfam-G because it contains all the sequences of the 603 families of Rfam 9.0 that can be mapped onto a reference genome. Rfam-G contains a total of 10 409 sequences (591 families).
It is important to point out that the Rfam-G families are distinct from the 792 Rfam families used for estimating the BlosumR and BlosumN matrices. Although the genomic location is not used here, the rationale behind the development of Rfam-G is to create a framework in which both homology search and homology-based RNA gene discovery methods can be evaluated with a comparable data set (manuscript in preparation).
True positive/false positive curves
One can validate database search procedures by producing receiver operator curves (ROC) or a similar representation that involves plotting the number of false positives (FPs) versus true positives (TPs) while varying some meta-parameter affecting the trade-off between specificity and sensitivity (typically a threshold). When doing so, the method's capacity to separate TPs and FPs is globally estimated without any dependence on a meta-parameter (threshold). The relative performances of two methods can then be compared by measuring the area under the curve (AUC) or by comparing the number of identified TPs for a given number of accepted FPs.
In order to produce these graphs, Rfam-G families were combined. For each family, we selected as a representative sequence the most closely related member (as estimated by measuring the average pairwise percent identity on the corresponding Rfam-G seed alignment). Each representative was then searched against the full Rfam-G database and in the resulting output, family members of that same representative sequence were labeled as Proven Positives while non-members were labeled as Proven Negatives. This search/labeling procedure was carried out with the representative sequences of all Rfam-G families, resulting in 591 distinct Blast outputs (one per family). These outputs were concatenated and sorted by E-values. FPs versus TPs graphs were produced by scanning the sorted output line by line, and counting for each line the number of TPs and the number of FPs (respectively, number of Proven Positives and Proven Negatives having a score better or equal to that of the considered line). This strategy is fairly standard and similar to the one recently used by Biegert and Söding (40). As pointed out by these authors, large families (Ribosomal RNA or RNAse-P in our case) tend to dominate these FP/TP curves, a drawback that can be corrected by weighting the contribution of each sequence with the inverse of its family size (i.e. down-weighting the contribution of big families). We used this approach to estimate the quantities referred to as weighted TPs and weighted FPs in Figure 2 and 3.
Figure 2.

Receiver–operator-like curve (ROC) for WU-Blast. The horizontal axis indicates the weighted number of reported TPs, while the vertical indicates the corresponding weighted number of FPs. Going from left to right on the top of the graph, the blue curve shows WU-BlastN, the orange curve corresponds to WU-BlastP/BlosumN and the green one shows WU-BlastR.
Figure 3.

ROC for optimized Blast. Similar layout as Figure 2. Going from left to right on the top of the graph, the blue curve corresponds to default WU-BlastN, the pink curve shows NCBI-BlastN optimized by Roshan et al. (26), the brown curve corresponds to WU-BlastN optimized by Freyhult et al. (25) and the green one shows WU-BlastR.
Validation of the clustering capacity
A methodology was designed in order to estimate the clustering capacities of BlastR. This methodology estimates the capacity of a clustering method (BlastClust, a component of the NCBI-Blast package) to reconstruct a known clustering (Rfam-G) when using a comparison method to estimate pair-wise sequence similarity. The clustering was applied onto a data set made of all Rfam-G sequences. BlastClust clusters were then evaluated using the following strategy: given the N Rfam-G sequences, an N × N matrix is defined. In the reference matrix (the one built on the original Rfam-G families), a vertex is set to 1 and considered a Proven Positive whenever the two corresponding sequences belong to the same family, the other vertices are set to 0 (Proven Negative). Given the output of BlastClust, a similar matrix is built, where new-found clusters are used to mark the vertices. The two matrices are then compared. TPs can be estimated by counting the number of vertices set to 1 in the reference matrix and to 1 in the new matrix; similarly for FPs (0 and 1), False Negatives (1 and 0) and True Negatives (0 and 0). Counts can be weighted as described above and used to estimate sensitivity and specificity using standard formulas:
| (3) |
| (4) |
We used this method to benchmark BlastClust (Figure 5), varying the inclusion threshold in order to obtain a range of values for Sn and Sp. These values were used to plot a graph Sn versus (1 − Sp). The areas under the resulting curves were integrated. By default, BlastClust uses MegaBlast (41) as a comparison engine for nucleotide sequences and BlastP for proteins. In the context of this work, we only used BlastP as a comparison engine since MegaBlast is meant to be used for closely related sequences and is therefore not sensitive enough for the kind of clustering considered here.
Figure 5.

ROC analysis of clustering capacities. Horizontal axis: 1-specificity. Vertical axis: sensitivity. Each curve shows the trade-off between these two quantities when varying the inclusion threshold of BlastClust. Starting from the bottom right corner to the top left, the bottom curve corresponds to NCBI-BlastP/BlosumN, while the top curve corresponds to NCBI-BlastR.
Database search algorithms
In order to validate the relevance of the sequence recoding and the use of BlosumR matrices, we used the BlastP algorithm to search reRNAs against their corresponding reRNA databases. We named this approach BlastR. For comparison purposes, BlastR was designed to run both with WU-BlastP (version 2.0 MP, Gish, unpublished) and with NCBI-BlastP (standalone NCBI Package version 2.2.18). BlastR was also compared against several different flavors of BlastN and BlastP packages (Table 1). Considering the nucleic nature of the reRNA sequences, we used for BlastP the BlastN default gap penalties while keeping all other parameters to their default BlastP values. In order to further emulate BlastN behavior, sequences were reverse complemented before extension (i.e. the recoded databases contain both the direct and the reverse sequence).
Table 1.
Blast flavors used in the validation and associated parameters
| Name | GOP | GEP | Word Size | Matrix | Sequence tType |
|---|---|---|---|---|---|
| NCBI-BlastN | 5 | 2 | 11 | +1/−3 | Nuc |
| NCBI-BlastN Optimized | 8 | 6 | 4 | +5/−4 | Nuc |
| NCBI-BlastP/BlosumN | 5 | 2 | 3 | BlosumN | Nuc |
| NCBI-BlastR | 5 | 2 | 3 | BlosumR | Recoded Nuc |
| WU-BlastN | 10 | 10 | 11 | +5/−4 | Nuc |
| WU-BlastN Optimized | 20 | 10 | 7 | +5/−4 | Nuc |
| WU-BlastP/BlosumN | 10 | 10 | 3 | BlosumN | Nuc |
| WU-BlastR | 10 | 10 | 3 | BlosumR | Recoded Nuc |
‘Name’ indicates the Blast flavor name, ‘GOP’ is the gap opening penalty, ‘GEP’ the gap extension penalty. ‘Word size’ indicates the word size for seeding alignment. ‘Matrix’ indicates the scoring scheme that has been used. ‘Sequence type’ indicates whether the considered sequences were nucleotides or recoded nucleotide sequences (respectively ‘Nuc’ and ‘Recoded Nuc’). BlastP can be used with any Blosum like scoring scheme (see text).
The substitution matrices used were the default NCBI and WU-Blast matrices (+1/−3 or +5/−4, respectively) when searching RNA sequences with BlastN, and the BlosumN or BlosumR matrices when using BlastP. The analyses presented in the main body of the text were all carried out using WU-Blast, although similar results were obtained using NCBI-Blast (Figure S1 in Supplementary Data). WU-Blast is now distributed under the name AB-Blast but the behavior of the AB-Blast package was identical to WU-Blast in our benchmarks.
Implementation/distribution
BlastR is a Perl wrapper for BlastP. Three wrappers are distributed, one for AB-Blast (Gish, unpublished data), one for WU-Blast, and one for NCBI-Blast. The package has been designed to behave like these three implementations of Blast, supporting exactly the same command line. BlastR is part of the standard T-Coffee distribution, an open-source freeware available from www.tcoffee.org/blastr.html.
RESULTS
In this work, we describe a procedure for searching ncRNAs in databases. Our approach relies on three distinct components: the use of a 16 letters alphabet reflecting di-nucleotide content (recoded sequences), the use of the BlastP algorithm to align the nucleotide sequences, and the use of new substitution matrices trained on Rfam seed alignments (BlosumR and BlosumN). Our first task was to determine the net contribution of each component when doing database searches.
We started by estimating the contribution of the BlastP algorithm. It is important to realize that BlastP and BlastN do not use the same algorithm. BlastP takes advantage of k-tuple similarity, while BlastN relies on perfect k-tuple identity. Furthermore, the two algorithms rely on a different low-level parameterization (different word size, gap opening and gap extension penalties, HSP (high-scoring segment pairs) extension thresholds, two-hit/single-hit algorithms, multiple-hit window size, drop-off value for gapped alignment, drop-off value for un-gapped extensions) and the BlastP algorithm supports the use of user defined log-odd matrices. It was, therefore, necessary to quantify how these variations may affect subsequent analysis. We did so by searching nucleotide sequences with BlastP while treating them as protein sequences made of a four letters alphabet and using as a substitution matrix BlosumN (BlastP/BlosumN), a nucleotide log-odd substitution matrix estimated on the Rfam seed alignments. We estimated the behavior of WU-BlastP/BlosumN by running one selected query from each Rfam family, concatenating the resulting outputs and sorting them by E-value. The resulting list was used to produce the FP versus TP plots in Figure 2 (‘Material and Methods’ section). As a reference we also included WU-BlastN with default parameters.
The results clearly show that using WU-BlastP/BlosumN as a substitution matrix yields an improvement over BlastN. It is hard to determine whether the improvement is an effect of the matrix, the algorithm or a combination, since neither WU-BlastN nor NCBI-BlastN support the use of ad hoc matrices. We further quantified the difference in behavior between BlastN and BlastP by precisely estimating the number of TPs returned while accepting a specified number of FPs. Results (Table 2), suggest that when accepting up to 10 FPs, WU-BlastP/BlosumN reports 535 TPs, that is to say about >4% of WU-BlastN. We did not see a similar improvement when feeding NCBI-BlastP the BlosumN matrix (Figure S1 in Supplementary Data). In WU-Blast, the increased sensitivity comes at a significant computational cost and we show in Table 2 that on the same benchmark data set, WU-BlastP takes about 150 times longer to produce a result than WU-BlastN.
Table 2.
Comparison between various Blast configurations
| Blast Flavor | Mat | #TP10 | #TP20 | #HSPs | CPU (s) |
|---|---|---|---|---|---|
| WU-BlastN | +5/−4 | 514 | 538 | – | 3 |
| WU-BlastP/BlosumN | BlosumN | 535 | 553 | – | 440 |
| WU-BlastN Opt | +5/−4 | 533 | 551 | – | 6 |
| WU-BlastR | BlosumR | 541 | 557 | – | 169 |
| NCBI-BlastN | +1/−3 | 523 | 530 | 51565 | 6 |
| NCBI-BlastP/BlosumN | BlosumN | 492 | 521 | 553738 | 156 |
| NCBI-BlastN Opt | +1/−3 | 499 | 525 | 149065 | 103 |
| NCBI-BlastR | BlosumR | 509 | 539 | 171891 | 89 |
‘Blast Flavor’ indicates which Blast was used, ‘Mat’ indicates which matrix, ‘#TP10′ and ‘#TP20′ indicate how many weighted TPs were reported for a total of, respectively, 10 and 20 accepted weighted FPs. ‘#HSPs’ indicates how many HSPs were reported (only for NCBI). ‘CPU’ indicates the CPU time in seconds.
We then replaced BlosumN with BlosumR, the di-nucleotide matrix and ran the benchmark on recoded RNA sequences. This flavor of Blast is named BlastR and its benchmark behavior is shown on Figures 2 and 3. The improvement of WU-BlastR over WU-BlastP is modest (557 TPs versus 553 when accepting 20 FPs) but more significant when considering the NCBI package (539 versus 521). Yet in both cases, this improvement comes along with a significant increase in efficiency. WU-BlastR requires nearly 60% less CPU (Central Processing Unit) time than WU-BlastP/BlosumN (Table 2) while NCBI-BlastR requires about 40% less CPU time than NCBI-BlastP/BlosumN. The lower CPU requirement is consistent with a significant lower number of reported HSPs (at least in NCBI BlastP) as shown on Table 2. Albeit modest, the improvement remains consistent over the whole TP versus FP graph. We finally compared BlastR with two optimized versions of BlastN, one from Freyhult et al. (25) and one from Roshan et al. (26). The results on Figure 3 and Table 2 suggest a moderate but consistent improvement of BlastR over these optimized flavors of BlastN.
In order to further investigate the source of the improvement obtained when replacing BlosumN with BlosumR, we re-estimated BlosumR matrices on Rfam seed alignments with shuffled columns. The alignments thus produced retain the same level of identity, the same single nucleotide mutation patterns, but lose their di-nucleotide dependencies. We produced 1000 such replicates based on the 792 Rfam family set on which the BlosumR matrix was estimated, and we used these 1000 replicates to search Rfam-G with BlastR. The results (Figure 4) show the distribution of reported TPs (for 10 accepted FPs) obtained with these replicates. Interestingly, the behavior of BlastR when used with shuffled BlosumR matrices is similar to the behavior of BlastP/BlosumN. This observation suggests that the differences observed on Figure 2 results from the use of di-nucleotide information. This result measured on a large number of replicates confirms the existence of a modest but real signal resulting from di-nucleotide dependencies and susceptible to improve database searches. It is possible to estimate these dependencies by measuring the log-odd ratio between the observed and expected frequency of every di-nucleotide given the background frequencies of the four nucleotides. Results (Figure S2 in Supplementary Data) confirm that the strongest dependency is between a nucleotide and its immediate neighbor. The second peak of dependency occurs when considering the dependency between a given nucleotide and its +4 neighbor; this distal dependency is, however, significantly lower.
Figure 4.

Score distribution with shuffled matrices. Thousand shuffled matrices were used to produce the corresponding number of replicate benchmarks. For each replicate, the weighted number of TPs for a weighted total of 10 accepted FPs was measured and used as a score for the replicate. The graph shows the distribution of these scores, with the vertical line on the right indicating the value associated with BlastR.
One of the main objectives of BlastR is to allow the identification of new ncRNA families. In order to estimate the relative merits of BlastR for that purpose, we designed a simple benchmark test. It involves re-clustering Rfam-G ab initio and using an estimate of the similarity between the new clusters and the original one as a measure of success. Results are displayed on Figure 5. This test was used to compare BlastR and BlastP/BlosumN. The results are in agreement with those reported on Figure 2 and suggest that BlastR consistently provides a better trade-off between sensitivity and specificity.
It is important to point out that with the exception of its substitution matrix BlastR has not been tuned in any way for RNA. It is merely the off-the-shelf BlastP package used along with a di-nucleotide substitution matrix and a BlastN gap penalty scheme. We therefore did not make any specific attempt to properly estimate the parameters of the Extreme Value Distribution (K and λ) used to model the E-value estimations. The problem of addressing the effect of unsuitable values for K and λ is not new (40), and it is well-known that inaccurate values result in under or over estimated E-values for the reported hits. To assess the effect on BlastR we systematically compared the E-values reported by BlastN with the E-Values reported by BlastR when considering the same hit (Figure 6A and B). The correlation between the log (E-value) is linear and on that same scale BlastR values are roughly twice lower than BlastN. Interestingly, the same linear correlation and the same doubling effect occur when comparing BlastN E-values with BlastP/BlosumN E-values (Figure S3 in Supplementary Data). This observation rules out our di-nucleotide matrix (BlosumR) as the main explanation for the doubling effect. It is, therefore, most likely that in BlastR, the E-value underestimation results from inappropriate values of K and λ in BlastP. Fortunately, the consequences on the present analysis are limited. The log-shift does not affect hit ranking, and therefore does not challenge any of the results reported here. In practice, to benefit from the increased sensitivity of BlastR (or BlastP/BlosumN), users simply need to use an E-Value of 10−6 as a threshold for significance, rather than the value of 10−3 that appear to be most suitable for BlastN on this data set. Aside from this E-value shift the results on Figure 6 confirm the capacity of BlastR to discriminate very accurately between proven positives (blue dots) and proven negatives (red dots).
Figure 6.

(A) E-values correlation between WU-BlastN and WU-BlastR. The horizontal axis indicates the ten based logarithm of the E-values recorded on WU-BlastN output. The vertical axis corresponds to that same quantity measured on WU-BlastR outputs. Each dot on the graph corresponds to the same hit observed on two outputs. Blue dots are hits labeled as proven positives, red dots correspond to proven negatives. (B) Close up of the previous graph centered on the high E-value area.
DISCUSSION
In this work, we describe and validate a database search method specific for non-coding RNA sequences. We show that through simple sequence recoding and estimation of an ad hoc substitution model, one can improve the accuracy of standard tools such as Blast. Sequence recoding is achieved by switching from the nucleotide four-letters alphabet to a protein-like 16-letters alphabet. Because they are recoded using an amino acid-like alphabet, the resulting sequences can be dealt with using protein analysis tools.
We examined the effect of using the BlastP algorithm to analyze recoded RNA sequences. This recoding amounts to increasing the information density within sequences by overloading each position with information related to its 3′ neighbor. While in theory, a nucleotide could be informed with the nature of any other nucleotide in its vicinity (i.e. one, two, three or more nucleotides apart), we showed here that the strongest dependency occurs between immediate neighbors. Interestingly, we also found an increased signal at position +4 (Figure S2 in Supplementary Data). This means that if one was ready to use a slightly larger alphabet, it might be worthwhile exploring the effect of combining these three nucleotides (NN××N) into a unique symbol. As shown in Figures 2 and 3, most of the improvement reported here is achieved by replacing the standard identity matrix in BlastN with a log-odd matrix estimated on Rfam RNA alignments (BlosumN) and feeding this matrix to the BlastP algorithm. We then show how a more discriminative matrix can be estimated by considering di-nucleotide substitutions to search nucleotide sequences recoded accordingly. We name the resulting procedure BlastR and show how its increased database search performances also result in improved clustering capacities when evaluating the re-clustering of Rfam-G. The modifications we propose here are simple and amount to a database pre-processing procedure (reverse complementation, sequence extension) combined with the use of di-nucleotide-specific substitution matrix (BlosumR). As such, they can be added on the top of any Blast-like package (PSI-Blast, Smith and Waterman, HMMER, etc.). We are currently distributing three such adaptations: one for the NCBI-Blast, one for WU-Blast and one for AB-Blast. We report comparable results when using these three packages.
An important effect of the recoding is to create sequences with a lateral dependency. In the recoded sequences, some symbol combinations are not allowed anymore because they are incompatible with the underlying recoding. For instance the symbol ‘D’ that corresponds to the ‘AA’ di-nucleotide cannot be followed by the symbol ‘I’ that correspond to ‘CA’. In order to be compatible, two extended symbols must have identical nucleotides on their 5′ and 3′. This property results from the fact that the di-nucleotides do not define an arbitrary reading frame, but merely correspond to a sliding window of size 2. Such a high lateral dependency would dramatically decrease the estimated E-values if Blast were considering dependencies between adjacent positions. Blast, however, like Smith and Waterman or the most common string matching algorithms, ignores such dependencies. It relies on the simplifying assumption that all positions are independent from one another and sums up the contribution of each position to estimate the final score. As a consequence, the privileged association between two symbols does not affect the score.
Overall, the observation that taking into account di-nucleotide composition when searching databases results in an improvement should not come as a surprise. Nucleotides forming dinucleotides have been known for a long time not to be fully independent. The question is, therefore, not whether the di-nucleotide signal exists, but whether it is strong enough to make a difference when added on the top of any analysis. In this work, we designed a simple protocol to enrich standard database search methods with di-nucleotide signal. We show here that the effect is strong enough to influence sensitivity. Although our results suggest that the best combination between CPU requirement and sensitivity is obtained when using recoded sequences, it is worth noting that we also report a significant increase in accuracy by the mere fact of using a nucleotide log-odd matrix (BlastP/BlosumN). Of course, one would have liked to test these nucleotide matrices within BlastN. Unfortunately, this is not possible as the currently available BlastN implementations have not been designed to use ad hoc scoring schemes.
Considering the importance of RNA analysis and the exponential growth of this research field, any small improvement in detection capacities could have major consequences. It must be stressed here that BlastR is not an attempt to improve over structure-aware, CPU intensive methods, like Infernal. These packages use RNA information in a way that is much closer to optimality. Our goal is not to outperform them but rather to ask how close one can get to their accuracy while using CPU inexpensive methods such as Blast. For this reason, we used the output of these methods (namely Infernal/Rfam) as a standard of truth and asked what fraction of the signal generated by accurate methods can be recovered using BlastR. A niche exists for methods like BlastR since today even the most sophisticated procedures designed for the assembly of ncRNA databases like Rfam rely in their early stage on BlastN-based analyses. Whenever BlastN is used, BlastR could be used as a drop-in replacement. BlastR has roughly the same CPU requirements as BlastP. That makes it about 10–100 times slower than BlastN but 2–5 orders of magnitude faster than any other non-Blast based alternatives such as Infernal (21).
In this analysis, we have focused our interest on evaluating how BlastR can be used to identify homologs among a set of well-defined RNA sequences. This exercise supposes the existence of data sets made of mature RNA transcripts (as opposed to genes embedded within a genome). This situation is realistic, given the large number of RNAseq experiments being performed in an increasing number of projects that deliver fully assembled transcriptomes. Of course BlastR could also be used to scan genomes for embedded multiexonic genes but the performance of our method in this precise context remains to be evaluated. This work is currently underway, taking advantage of the Rfam-G data set described here. Last but not least, it is interesting to note that the use of protein-like sequences makes it possible to use all available flavors of Blast including the most sophisticated, such as PSI-Blast. Unfortunately, our attempts at doing so have been hampered by the presence of many hard coded protein-dependent parameters within PSI-Blast. The development of PSI-BlastR would therefore require a more invasive software engineering approach, but it is certainly an avenue worth exploring.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: The Centre de Regulacio Genomica (CRG) (to C.N.); the Plan Nacional (BFU2008-00419) (to I.E. and E.R.); ‘La Caixa’ international PhD program fellowships (to G.B.); grant 205321-103973 from the Swiss National Research Foundation (at early stages of this work, to E.B.). This work was also co-financed by the European Commission, within the 7th Framework Programme (Grant Agreement KBBE-2A-222664) (‘Quantomics’) and by the Plan Nacional (BFU2008-00419).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- 2.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 8.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 9.Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J. Appl. Math. 1985;45:810–825. [Google Scholar]
- 10.Notredame C, O'Brien EA, Higgins DG. RAGA: RNA sequence alignment by genetic algorithm. Nucleic Acids Res. 1997;25:4570–4580. doi: 10.1093/nar/25.22.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowell RD, Eddy SR. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinformatics. 2006;7:400. doi: 10.1186/1471-2105-7-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein RJ, Eddy SR. RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinformatics. 2003;4:44. doi: 10.1186/1471-2105-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberg Z, Ruzzo WL. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- 15.Eddy SR. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics. 2002;3:18. doi: 10.1186/1471-2105-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardner PP. The use of covariance models to annotate RNAs in whole genomes. Brief Funct. Genomic. Proteomic. 2009;8:444–450. doi: 10.1093/bfgp/elp042. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- 18.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 20.Menzel P, Gorodkin J, Stadler PF. The tedious task of finding homologous noncoding RNA genes. RNA. 2009;15:2075–2082. doi: 10.1261/rna.1556009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Borovok I, Aharonowitz Y, Sharan R, Bafna V. A sequence-based filtering method for ncRNA identification and its application to searching for riboswitch elements. Bioinformatics. 2006;22:e557–565. doi: 10.1093/bioinformatics/btl232. [DOI] [PubMed] [Google Scholar]
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Freyhult EK, Bollback JP, Gardner PP. Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA. Genome Res. 2007;17:117–125. doi: 10.1101/gr.5890907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roshan U, Chikkagoudar S, Livesay DR. Searching for evolutionary distant RNA homologs within genomic sequences using partition function posterior probabilities. BMC Bioinformatics. 2008;9:61. doi: 10.1186/1471-2105-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park L. Relative mutation rates of each nucleotide for another estimated from allele frequency spectra at human gene loci. Genet Res. 2009;91:293–303. doi: 10.1017/S0016672309990164. [DOI] [PubMed] [Google Scholar]
- 28.O'Toole AS, Miller S, Haines N, Zink MC, Serra MJ. Comprehensive thermodynamic analysis of 3′ double-nucleotide overhangs neighboring Watson-Crick terminal base pairs. Nucleic Acids Res. 2006;34:3338–3344. doi: 10.1093/nar/gkl428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F, Zhao Z. The influence of neighboring-nucleotide composition on single nucleotide polymorphisms (SNPs) in the mouse genome and its comparison with human SNPs. Genomics. 2004;84:785–795. doi: 10.1016/j.ygeno.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Wolfe KH, Sharp PM. Mammalian gene evolution: nucleotide sequence divergence between mouse and rat. J. Mol. Evol. 1993;37:441–456. doi: 10.1007/BF00178874. [DOI] [PubMed] [Google Scholar]
- 31.Durbin R, Eddy SR, Krogh A, Mitchison GJ. Models of Proteins and Nucleic Acids. Cambridge UK: Cambridge University Press; 1998. Biological sequence analysis: probabilistic; p. 72. [Google Scholar]
- 32.Clote P, Ferre F, Kranakis E, Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005;11:578–591. doi: 10.1261/rna.7220505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babak T, Blencowe BJ, Hughes TR. Considerations in the identification of functional RNA structural elements in genomic alignments. BMC Bioinformatics. 2007;8:33. doi: 10.1186/1471-2105-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Workman C, Krogh A. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 1999;27:4816–4822. doi: 10.1093/nar/27.24.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rivas E, Eddy SR. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, Sze SH. Improving accuracy of multiple sequence alignment algorithms based on alignment of neighboring residues. Nucleic Acids Res. 2009;37:463–472. doi: 10.1093/nar/gkn945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith AD, Lui TW, Tillier ER. Empirical models for substitution in ribosomal RNA. Mol. Biol. Evol. 2004;21:419–427. doi: 10.1093/molbev/msh029. [DOI] [PubMed] [Google Scholar]
- 38.Dayhoff MO, Schwartz RM, Orcutt BC. Atlas of Protein Sequence and Structure. Vol. 5. Washington, DC, USA: National Biomedical Research Foundation; 1978. pp. 345–352. [Google Scholar]
- 39.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biegert A, Soding J. Sequence context-specific profiles for homology searching. Proc. Natl Acad. Sci. USA. 2009;106:3770–3775. doi: 10.1073/pnas.0810767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.