


Abstract
Ataxia telangiectasia (ATM) mutated and Artemis, the proteins defective in ataxia telangiectasia and a class of Radiosensitive-Severe Combined Immunodeficiency (RS-SCID), respectively, function in the repair of DNA double strand breaks (DSBs), which arise in heterochromatic DNA (HC-DSBs) following exposure to ionizing radiation (IR). Here, we examine whether they have protective roles against oxidative damage induced and/or endogenously induced DSBs. We show that DSBs generated following acute exposure of G0/G1 cells to the oxidative damaging agent, tert-butyl hydroperoxide (TBH), are repaired with fast and slow components of similar magnitude to IR-induced DSBs and have a similar requirement for ATM and Artemis. Strikingly, DSBs accumulate in ATM−/− mouse embryo fibroblasts (MEFs) and in ATM or Artemis-defective human primary fibroblasts maintained for prolonged periods under confluence arrest. The accumulated DSBs localize to HC-DNA regions. Collectively, the results provide strong evidence that oxidatively induced DSBs arise in HC as well as euchromatic DNA and that Artemis and ATM function in their repair. Additionally, we show that Artemis functions downstream of ATM and is dispensable for HC-relaxation and for pKAP-1 foci formation. These findings are important for evaluating the impact of endogenously arising DNA DSBs in ATM and Artemis-deficient patients.
INTRODUCTION
Oxidative DNA damage caused by reactive oxygen species (ROS) generated during metabolism makes a significant contribution to genomic instability, carcinogenesis and cellular ageing. ROS predominantly introduces DNA base or sugar damage leading to single-strand break (SSB) formation (1). However, DNA double strand breaks (DSBs) can also arise following replication past ROS-induced lesions, their encounter with the transcription machinery or when they arise in close proximity. Although DSBs arise at a lower frequency than SSBs, they are biologically significant if unrepaired or misrepaired. While there are multiple, overlapping pathways for base excision repair (BER) and SSB repair, such that loss of proteins involved in the repair pathways tends to slow but not abolish repair, DSBs are repaired more slowly and loss of the major DSB rejoining pathway is highly significant.
DNA non-homologous end-joining (NHEJ) is the major DSB repair pathway in mammalian cells (2). Homologous recombination (HR) functions primarily at the replication fork and as a second pathway in late S/G2 phase. Most DSB repair studies have examined DSBs generated by ionizing radiation (IR) (X- or γ-rays). Studies using pulsed field gel electrophoresis (PFGE) have demonstrated fast and slow repair components (3). Enumerating γ-H2AX as a DSB marker have confirmed these findings and have shown that the slow DSB repair process represents the repair of DSBs that arise in heterochromatic DNA (HC-DSBs) (4). Further, distinct genetic requirements were observed for the repair of HC versus euchromatic (EU) DSBs; while the fast DSB repair process requires the core NHEJ proteins, Ku70, Ku80, DNA-PKcs, XRCC4, DNA ligase IV and XLF/Cernunnos, the slow process in G0/G1 phase additionally requires ataxia telangiectasia mutated protein (ATM), Artemis, γH2AX, MDC1, RNF8, RNF168, the MRN complex and 53BP1 (5,6). A role for ATM in the repair of HC DSBs is further supported by the finding that KAP-1, a heterochromatic building factor, is an ATM substrate and KAP-1 siRNA relieves the requirement for ATM for DSB repair (4,7). It is proposed that HC poses a barrier to DSB repair that is relieved by ATM-dependent KAP-1 phosphorylation, that pKAP-1 foci at HC-DSBs are required for HC-DSB repair and that their formation requires the damage response mediator proteins described above.
ROS-induced DNA damage is similar to, but distinct from, IR-induced damage. Although a substantial contribution to IR-induced damage represents oxidative damage from secondary electrons, multiple lesions can arise in close proximity generating complex DSBs. This is especially true of high linear energy transfer (LET) radiation but is also a feature of low LET (X- or γ-ray) radiation (8). Two types of ‘complex’ lesions can arise: those with non-ligatable termini due to associated base or sugar damage (dirty DSBs) or multiply damaged sites where multiple DSBs, DSBs/SSBs and/or base damage arise in close proximity. ROS-induced DSBs represent dirty DSBs but do not have multiple damages in close proximity. The impact of damage complexity on DSB repair is evident by the slow rejoining kinetics of DSBs induced by high LET radiation (9). However, the in vivo impact of complex lesions following low LET radiation is less clear. Additional questions in considering endogenous versus IR-induced DSBs are whether higher order chromatin structure confers protection against endogenously induced DNA damage and whether differences exist between mouse and human cells. Mouse cells appear to sustain a greater level of oxidative damage and are more sensitive to oxygen tension than human cells, either because they generate high levels of ROS or because they have reduced scavenging capacity (10). Thus, while mice and mouse cells defective in NHEJ proteins show elevated endogenous genomic instability, it is unclear if this is fully replicated in human patients or cell lines (11).
The elevated genetic instability in mouse cells lacking NHEJ proteins strongly suggests that NHEJ plays a role in repairing DSBs arising from oxidative damage. Whether these DSBs arise following replication or in non-replicating cells is currently unclear (11,12). Whether ATM and Artemis have roles in repairing endogenously arising DSBs has not been shown to date. A-T cells are hypersensitive to the killing and clastogenic effects of a range of oxidative stress-inducing agents (e.g. hydrogen peroxide and t-butyl hyrdoperoxide) and fail to activate cell cycle checkpoint arrest after oxidative damage (13–15). However, whether this sensitivity can be entirely attributed to ATM’s checkpoint function and/or whether ATM functions in the repair of endogenously induced DSBs is unknown. Mouse cells lacking Artemis also show endogenous genomic instability but this has not been reported in human Artemis defective cells (16). The effect of endogenously arising DSBs in Artemis cells is an important question since most Artemis patients have null mutations. While their immunodeficiency can be controlled following bone marrow transplantation, the majority of patient cells still lack Artemis activity.
One difficulty in examining the impact of oxidatively induced DSBs is the low frequency at which they arise, most particularly in non-replicating cells. The use of oxidative DNA damaging agents has hitherto been difficult due to the high ratio of SSB: DSB damage induced by ROS, which has restricted the use of insensitive physical methods such as PFGE for DSB analysis. γ-H2AX foci analysis represents a sensitive technique allowing these questions to be addressed; further it can be used to examine DSB repair at sites of differing chromatin complexity. Damage response signalling is carried out predominantly by two phosphoinositol 3-kinase like kinases (PIKKs), ATM and ataxia telangiectasia and Rad3 related (ATR) (although DNA-PK can provide some overlapping function with ATM) (17,18). ATR is activated by single-stranded regions of DNA, which can arise during replication but is not activated by single-strand nicks or base damage. ATM, in contrast, is predominantly activated by DSBs. Thus, similar to the response to IR, the majority of oxidative damage in non-replicating cells fails to activate ATR, and ATM is only activated when DSBs arise. γ-H2AX analysis has thus provided a sensitive assay to monitor DSB formation and repair following exposure to IR in non-replicating cells (5,19,20).
Here, we use this sensitive technique to assess endogenously induced DSB formation and repair or DSBs induced by an agent generating oxidative damage. We examine the repair of ROS-induced DSBs with a focus on the function of ATM or Artemis. We show that DSBs can arise endogenously in non-replicating cell cultures in EU and HC regions and that Artemis and ATM function in the repair of such endogenously induced HC-DSBs. Further, we show that ATM and Artemis function in the repair of DSBs induced by tert-butyl hydroperoxide (TBH), an agent that induces oxidative DNA damage. These findings are important in evaluating the clinical impact of mutations in these proteins and how patients lacking Artemis or ATM respond to oxidative stress.
MATERIALS AND METHODS
Cell culture and reagents
Primary fibroblasts from 1BR.3 and 48BR (control), F02/385 and CJ179 (Artemis null), 495GOS and 411BR (Ligase IV hypomorphic), AT1BR and AT5BR (ATM defective) and hTERT immortalized fibroblasts, 1BRhTERT, CJ179hTERT, AT1BRhTERT and 495GOShTERT, were grown in minimal essential medium (MEM) as described previously (5). Primary fibroblasts derived from Glutathione deficient (GS−/−) and control (GS+/+) patients were a generous gift from Dr M. Harm-Ringdahl and S. Haghoost (Stockholm University, Sweden). Primary mouse embryo fibroblasts (MEFs) derived from control (ATM WT, LigIV WT), LigIV defective (LigIVY288C) and A-T (ATM−/−) mice were cultured in either 3% or 20% O2 in MEM supplemented with 15% heat inactivated foetal calf serum (FCS), penicillin–streptomycin and l-Glutamine. TBH was added to complete culture medium at concentrations specified. For longevity studies (chronic treatment) fresh TBH was added to the medium every 72 h. For repair analysis (acute treatment), TBH was added at a concentration of 10 mM. After 30 min incubation at 37°C, TBH was removed and cells allowed to repair for times indicated. The ATM inhibitor (ATMi) KU-55933 was a gift from KuDos Pharmaceuticals and was used at a final concentration of 10 µM.
Immunofluorescence
Primary γ-H2AX, α-53BP1, pKAP-1, TriMeK20-H4, α-BudR, Caspase-3 (Asp175) and α-PAR antibodies were obtained from Milipore (Watford, UK), Bethyl (Montgomery, AL, USA) Abcam (Cambridge, UK), Santa Cruz (CA, USA), Cell Signalling (Boston, MA, USA) and Enzo Life Sciences (Exeter, UK) respectively. Secondary antibodies were obtained from Dako (Glostrup, Denmark). Cells were fixed in 3% paraformaldehyde, 2% sucrose phosphate-buffered saline (PBS) for 10 min and permeabilized in 20 mM HEPES pH 7.4, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton X-100 (Sigma-Aldrich, Poole, UK) for 2 min. Primary antibody incubations were carried out for 45 min at 1:800 for γ-H2AX, 1:500 α-53BP1, 1:300 pKAP-1, 1:200 TriMeK20-H4, 1:100 α-BudR, 1:400 Caspase-3 (Asp175), 1:200 α-PAR, in PBS supplemented with 2% bovine serum fraction V albumin (BSA) (Sigma-Aldrich, Poole, UK) and followed by washing in PBS. Incubations with α-mouse TRITC and FITC or with α-rabbit Cy3 secondary antibodies (Sigma-Aldrich, Poole, UK) were performed at RT at 1:200 in 2% BSA for 45 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Poole, UK) for 10 min.
β-Galactosidase staining
Primary fibroblasts were plated onto 3 cm2 dishes and grown in 20% O2 for 7 weeks or irradiated with 10 Gy. Growth medium was removed and cells washed with PBS before fixation with 2% formaldehyde, 0.2% glutaraldehyde in PBS for 10 min RT. Cells were then washed with PBS and stained with 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 40 mM citric acid/sodium phosphate (pH 6.0), 0.15 M NaCl, 2 mM MgCl2 and 1 mg 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside powder (New England Biolabs, Hitchin, UK). Cells were incubated overnight at 37°C. After 18 h of incubation, cells were monitored for the development of blue staining under a light microscope.
Transient knockdown of protein expression
Knockdown of KAP-1 in primary human fibroblasts was achieved using Metafectene (Biotex, Planegg, Germany) mediated transfection (according to the maunfacturer’s instructions) with 100 pmol of siRNA duplexes per 2× 105 cells. siRNA duplexes were StealthTM RNAi synthesized by Invitrogen (Paisley, UK): KAP-1= CAGUGCUGCACUAGCUGUGAGGAUA (human cDNA nucleotides 450–475). For DSB repair experiments, cells were treated with 10 mM TBH for 30 min 72 h post-transfection.
DNP detection
Primary human fibroblasts were grown to conflency in 3 cm2 dishes at 20% oxygen and 1 × 106 cells harvested at either Week 0 or Week 7. Cells were lysed in 2× reducing SDS loading buffer at 95°C for 5 min and loaded onto a 7% polyacrylamide gel. Proteins were subsequently transferred to a nitrocellulose membrane and derivatized with DNPH followed by immunoblotting using α-DNP and α-HRP antibodies as instructed by the manufacturers (Cell Biolabs, San Diego, CA, USA).
RESULTS
ATM and Artemis are required for the repair of ROS-induced DSBs
Firstly, we examined the induction and repair of DSBs following treatment with TBH or t-BOOH, a stable H2O2 derivative that generates ROS. To avoid the impact of replication-induced DSBs and/or the characterized role of ATM in cell cycle checkpoint arrest, we used confluence arrested primary human fibroblasts (21). Fibroblasts derived from patients lacking ATM (AT1BR), Artemis (CJ179) or harbouring a hypomorphic mutation in DNA ligase IV (495GOS) were exposed to 10 mM TBH for 30 min, which, from preliminary studies, causes a similar level of γ-H2AX foci formation to that induced by 3 Gy γ-rays (data not shown). In control experiments, we have previously shown that low doses of H2O2 fail to induce γ-H2AX foci in confluent cells despite inducing a large number of SSBs assessed by the comet assay, consistent with the notion that SSBs do not activate ATM or ATR in non-dividing cells (22). Here, we show that, like H2O2, exposure to 10 mM TBH also induces a large number of SSBs as assessed by poly(ADP-ribose) (PAR) synthesis (Supplementary Figure S1). DSB repair was monitored by enumerating the rate of loss of γ-H2AX foci (22). As shown previously, following exposure to γ-rays, ATM and Artemis fail to repair a subset (∼15%) of induced DSBs that represent those repaired with slow kinetics in control cells (5) (Figure 1A). Thus, at 2 h post IR ATM−/− (AT1BR) and Artemis−/− (CJ179) cells display a similar level of γ-H2AX foci to control cells but have elevated foci numbers at 24 h post exposure. In contrast, 495GOS (LIG4 Syndrome) shows slow repair of all DSBs due to the hypomorphic mutation which does not fully inactivate DNA ligase IV. By 72 h post 3 Gy, all three repair defective cell lines harbour a similar level of γ-H2AX foci. Surprisingly, the kinetics of γ-H2AX foci loss following exposure to 10 mM TBH are almost identical to that observed following exposure to γ-rays. Significantly, elevated unrepaired DSBs are observed in all three mutant cell lines from 24 to 72 h post treatment (Figure 1B). Similar results were also obtained after addition of TBH to control cells exposed to the ATM inhibitor, KU55933 (Supplementary Figure S2). The low number of γ-H2AX foci induced by TBH treatment and their slow rejoining in 495GOS after IR, strongly supports the notion that under these non-replicating conditions, γ-H2AX foci arising after TBH treatment represent DSBs. These findings strongly suggest that despite the difference in complexity of ROS and IR-induced DSBs, they are repaired with similar kinetics, have a similar sized slow DSB repair component and a similar requirement for ATM and Artemis for repair.
Figure 1.

Rate of DSB repair of γ-ray and TBH-induced DSBs in control and mutant cell lines. (A) Control (1BR.3), an A-T (AT1BR), an Artemis-defective (CJ179) and a DNA ligase IV-deficient (459GOS) cell line were exposed to 3 Gy γ-rays or (B) 10 mM TBH for 30 min and γ-H2AX foci enumerated at the times indicated. For TBH, Time 0 represents 30 min post TBH removal. Cells were grown to confluency prior to treatment and maintained under confluency during analysis. The results have been normalized to DSB induction at 30 min post treatment to allow comparison between lines and treatments. Similar γ-H2AX foci numbers were induced by the IR and TBH treatments in all cell lines (data now shown). Results represent the mean ± SEM of three experiments.
Primary A-T MEFs accumulate endogenously arising γ-H2AX foci under non-replicating conditions
Having shown that ATM and Artemis are required for the repair of a subset of ROS-induced DSBs following non-physiological exposure to TBH, we next examined whether DSBs can arise endogenously under non-replicating conditions (likely via ROS damage) and whether ATM and Artemis function in their repair. Previous studies have shown that MEFs are sensitive to the oxygen tension at which they are grown due to high levels of ROS and/or limited free radical scavenging capacity (10). As a result, MEFs display reduced proliferation potential when cultured at 20% compared to 3% oxygen, which is closer to the physiological oxygen tension. We examined the accumulation of γ-H2AX foci under non-replicating conditions exploiting the ability of primary MEFs (like human fibroblasts) to undergo confluency arrest. Similar experiments were previously carried out using LigIVY288C MEFs (23) and are included here for comparison (Figure 2A). Firstly, we verified that the confluency arrested MEFs were non-replicating by examining BudR incorporation (Supplementary Figure S3A). We failed to observe any cells incorporating BudR under such conditions. Accumulation of γ-H2AX foci in LigIVY288C MEFs but not control MEFs provides strong evidence that DSBs arise under these non-replicating conditions. Low passage WT and ATM−/− primary MEFs were examined in parallel (Artemis−/− primary MEFs were not available). Although WT MEFs showed no change in foci numbers/cell over the 4 week period examined, ATM−/− MEFs showed a gradual and statistically significant increase in γ-H2AX foci similar to that previously observed in LigIVY288C MEFs (Figure 2A). An examination of the distribution of γ-H2AX foci between cells showed that at Week 4 >50% of the population had increased foci numbers compared to cells at Week 0, demonstrating that the results are not caused by a minor subfraction of the population with very high foci numbers (Figure 2B). In these experiments, the MEFs were grown at 20% oxygen but similar findings were observed in cells grown at 3% oxygen suggesting that under these conditions DSB accumulation is not oxygen tension dependent (Supplementary Figure S3B). To verify that these DSBs were not reflective of apoptotic cells, we carried out caspase staining of Week 4 MEFs, and failed to observe any caspase positive cells. Treatment with 100 µM etoposide, in contrast, efficiently induced apoptosis (Supplementary Figure S3C). These findings provide the first evidence that ATM functions in the repair of endogenously induced DSBs and that the rate of DSB accumulation is similar to that observed in LigIVY288C MEFs. Further, the results show that WT cell cultures can efficiently repair any DSBs generated so that no overall DSB accumulation is observed.
Figure 2.

ATM−/− MEFS show decreased proliferative capacity and increased DSB formation under non-replicating and replicating conditions. (A) Early passage WT, ATM−/− and LigIVY288C primary MEFs were grown to confluency in 20% oxygen and maintained under non-replicating conditions for the times indicated. At weekly intervals γ-H2AX foci were enumerated. The results represent the mean and standard deviation of three experiments. The results for LigIVY288C MEFs have been previously published and are included for comparison (23). (B) Distribution of γ-H2AX foci in A. The number of foci scored in (A) at Week 0 and 4 is plotted as the percentage of cells with 0–2, 3–5 or >6 foci per cell. (C) Primary ATM−/− MEFs were maintained under replicating conditions. When confluent cell numbers were counted, the MEFs passaged and the number of cells re-plated recorded. Trypan blue exclusion was carried out to count only viable cells. After 50 days, MEFs grown at 3% oxygen commenced rapid growth and were considered to have immortalized. (D) At weekly intervals, γ-H2AX were enumerated. ATM−/− MEFs ceased growth after Week 2 at 20% oxygen. In contrast ATM−/− MEFs proliferated in 3% O2 but did not continue to accumulate γ-H2AX foci with time, possibly due to selective loss of cells with high unrepaired DSBs.
Primary ATM−/− MEFS show decreased proliferation potential and increased γ-H2AX foci numbers under replicating conditions
We previously showed that LigIVY288C MEFs maintained under replicating conditions have reduced proliferation capacity and elevated γ-H2AX foci numbers compared to control MEFs and that the impact was greater at 20% compared to 3% oxygen (23). Here, we examined proliferation and γ-H2AX foci formation in replicating primary ATM−/− MEFs. ATM−/− MEFs showed markedly reduced proliferation potential at 3% oxygen compared to control MEFs with a proliferative capacity similar to that previously shown by LigIVY288C MEFs (Figure 2C) (23). At 20% oxygen, ATM−/− MEFs have little proliferative capacity, displaying an impact more marked than the LigIVY288C MEFs. γ-H2AX foci accumulated at an elevated frequency compared to control MEFs under both 20% and 3% oxygen with the level of foci accumulation being greater at 20% compared to 3% oxygen (Figure 2D). Significantly, γ-H2AX foci numbers correlated with proliferative capacity, being greater in ATM−/− MEFs compared to LigIVY288C MEFs and greater at 20% compared to 3% oxygen. The γ-H2AX foci accumulating in these replicating cells could represent unrepaired DSBs or the phosphorylation of γ-H2AX at stalled replication forks via ATR activation (which likely involves the formation of single-stranded DNA regions rather than DSB formation). Furthermore, since ATM is also required for cell cycle checkpoint arrest, the enhanced levels of DNA damage could be a consequence of a failure to undergo checkpoint arrest, a failure to repair endogenously induced DSBs or the combined impact. Notwithstanding the underlying basis, these findings provide evidence that ATM−/− MEFs accumulate a greater level of damage compared to control cells and that, in contrast to the situation with non-replicating cells, this impact is more marked when MEFs are grown at 20% compared to 3% oxygen.
ATM, Artemis and DNA ligase IV prevent endogenous DSB accumulation in human fibroblasts
Primary human fibroblasts are less sensitive to oxygen tension and have enhanced proliferative capacity compared to primary MEFs (10). To examine whether endogenous DSBs arise in non-replicating human fibroblasts and to assess the role of ATM and Artemis in the repair of such lesions, early passage primary human fibroblasts defective in ATM (AT1BR) or Artemis (CJ179) or deficient in DNA ligase IV (495GOS) were grown to confluency and maintained in culture (20% oxygen) for up to 7 weeks. γ-H2AX foci were enumerated at weekly intervals. DSB accumulation was observed in all three mutant cell cultures similar to, although less dramatic than, the findings with primary MEFs consistent with the notion that MEFs incur a higher level of oxidative damage compared to human cells (Figure 3A) (10). The accumulation was slightly less marked for the LIG4 Syndrome cells (495GOS) compared to the A-T and Artemis-defective cells. The larger error bars observed in repeat experiments using the Artemis defective cell line (CJ179) limited direct comparison between cell lines, however.
Figure 3.

ATM, Artemis and DNA ligase IV function to prevent endogenous DSB accumulation in non-replicating primary human fibroblasts. (A) Early passage primary human fibroblasts derived from a control (1BR.3), A-T (AT1BR), Artemis-defective (CJ179) and a LIG4 Syndrome (495GOS) patient were grown to confluency and maintained under non-replicating conditions. At weekly intervals, γ-H2AX foci were enumerated. Results represent the mean ± SEM of three experiments. (B) Distribution of γ-H2AX foci in A. The foci counted in panel A at Week 0 and 7 are plotted as the percentage of cells with 0–2, 3–5 or greater than 6 foci per cell. (C) Primary human fibroblasts derived from control (48BR and GS+/+), A-T (AT5BR), Artemis-defective (CJ179), LIG4 syndrome (495GOS) and Glutathione deficient (GS−/−) patients were maintained under confluency in 20% O2. After 7 weeks, they were analysed for the presence of protein carbonyl derivatives by immunoblotting with α-DNP. The α-DNP signal was not significantly increased in any of the repair defective cells but was elevated in GS−/− cells. (D) Quantification of α-DNP immunoblots from three independent experiments. (E) Glutathione proficient (GS+/+) and deficient (GS−/−) cells were maintained under non-replicating conditions for 7 weeks. γ-H2AX foci were enumerated at weekly intervals. No increase in γ-H2AX foci accumulation was observed in the GS−/− cells. (F) Staining for the senescence marker, β-galactasidase (β-Gal), and the apoptotic marker, capase-3, was carried out at Week 7 in all three mutant cell lines. Irradiated 1BR.3 cells were used as a positive control. No β-galactasidase staining or cleaved caspase-3 was observed in any of the non-IR treated cell lines; in contrast strong straining was observed following 10 Gy IR or 100 µM etoposide. AT1BR cells at Week 7 are shown but the results are representative of all cell lines.
Examination of the distribution of γ-H2AX foci between cells showed that at Week 7 >50% of the cells had elevated foci numbers compared to cells at Week 0 (Figure 3B). Similar findings were obtained with AT5BR, another A-T cell line, F02/385, another Artemis defective cell line and 411BR, another LIG4 Syndrome cell line (Supplementary Figure S4).
Non-replicating ATM and Artemis-defective human fibroblasts do not show significantly enhanced oxidative stress or senescence
A potential explanation for these findings is that ATM−/−, Artemis−/− and DNA ligase IV-defective cells incur high levels of ROS damage due to increased oxidative stress. Indeed, previous studies have reported that ATM−/− neuronal cells may suffer oxidative stress (24). However, neuronal cells are highly metabolically active in contrast to primary fibroblasts. To gain insight into whether the patient human fibroblasts might suffer elevated oxidative damage, we examined the level of oxidized proteins using an assay that monitors protein carbonyl derivatives following pre-derivitization of the carbonyl groups with DNPH (dinitrophenylhydrazine) and immunoblotting with α-DNP (25,26). We used a primary human fibroblast lacking glutathione synthetase (GS), to verify the efficacy of our assay (Figure 3C and D) (27,28). GS is involved in the generation of GSH, which is central to several antioxidant responses. Control and mutant primary fibroblasts were maintained for 7 weeks and the level of α-DNP assessed. Although we observed a small increase in α-DNP levels in the A-T cell line after 7 weeks maintenance under non-replicating conditions, the difference was not statistically significant. No difference was observed in the Artemis and LIG4 Syndrome cells. In marked contrast, we observed a clear increase in GS−/− cells after prolonged maintenance in G0 phase. The GS−/− cells, however, did not show any increase in γ-H2AX foci numbers suggesting that they efficiently repair any DSBs induced (Figure 3E). The fact that GS−/− cells did not show enhanced protein oxidation at Week 0 suggests either that ROS damage is specifically enhanced following prolonged maintenance at G0 phase or that selection during replication prevents the accumulation of cells with high levels of oxidized proteins. Nothwithstanding this, these studies demonstrate that neither ATM nor Artemis-defective cells have dramatically increased levels of glutathione-based oxidative damage. Further, GS−/− cells, which do have increased protein oxidation, can efficiently repair any excess DSBs generated. Although, it cannot be ruled out that other forms of oxidative damage are specifically enhanced, the results provide evidence that the mutant human fibroblasts do not specifically suffer generalized oxidative stress.
An alternative possibility to explain elevated γ-H2AX foci numbers is that the repair defective cells might undergo premature senescence or apoptosis when maintained under non-replicating conditions. Staining for β-galactosidadase, a senescence marker and cleaved caspase-3, a marker of apoptosis, showed that at 7 weeks when significant unrepaired DSBs have accumulated, none of the repair defective cell lines exhibit any signs of senescence or apoptosis (Figure 3F).
Artemis and ATM-deficient cells accumulate ROS-induced DSBs at HC regions
Given our previous findings that ATM functions in the repair of HC-DSBs, our finding of a similar DSB repair defect following TBH or γ-ray exposure suggests that ROS damage induces HC-DSBs. This was not entirely expected since we predicted that HC might serve as a barrier to restrict ROS-induced DNA damage. Therefore, we sought to consolidate the notion that endogenously arising DSBs can arise in HC regions by exploiting the studies using primary MEFs, which have detectable densely staining DAPI chromocentres, similar to those observed in mouse NIH3T3 cells. Following maintenance of non-replicating WT and ATM−/− MEFs for 5 weeks at 20% oxygen, cells were processed to examine 53BP1 foci (another DSB marker that co-localizes with γ-H2AX) together with DAPI staining. Using imaging software to assess overlap between 53BP1 and densely staining DAPI chromocentres, we observed 60–70% overlap at 5 weeks compared to 30–40 % at Week 0 (Figure 4). This is similar to the overlap observed in ATM−/− human cells following IR exposure (4). As for IR-induced DSBs, the 53BP1 foci localize to the periphery of DAPI-staining chromocentres and are never located within their centre.
Figure 4.
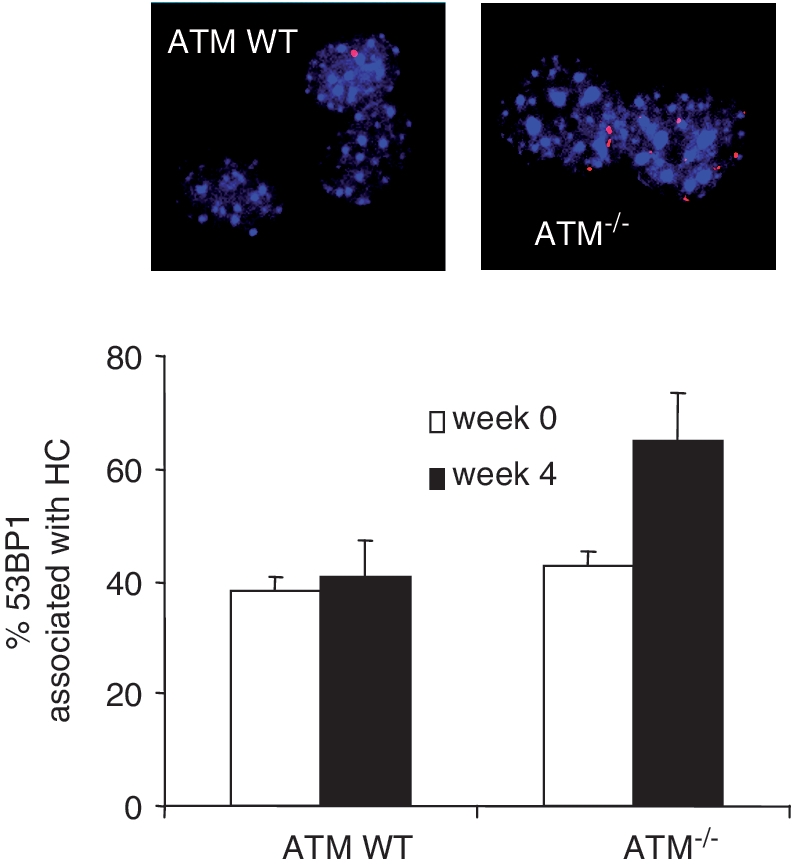
ROS induces DSBs in MEFs at HC chromocentres. At Weeks 0 and 4, ATM WT and ATM−/− MEFs were processed to allow visualization of 53BP1and DAPI chromocentres. The percent of 53BP1 foci that are associated (at the periphery) with DAPI chromocentres was scored using a Zeiss microscope and Image J software. The high overlap (40%) at Week 0 likely reflects the enhanced persistence of HC-DSBs in control and ATM−/− MEFS, which is distinct to our previous analysis of stochastically arising DSBs at 30 min post IR (4). Importantly, the DSBs that accumulate in ATM−/− cells by Week 4 have an enhanced localization to chromocentres. The actual number of γH2AX foci accumulating in these experiments was similar to that obtained in Figure 2A.
Artemis functions downstream of KAP-1 phosphorylation
Our previous studies have focused on the role of ATM in HC-DSB repair. Here, we examined the point at which Artemis functions. First, we exploited our previous finding that KAP-1 siRNA relieves the requirement for ATM for DSB repair due to the requirement for KAP-1 in generating the HC superstructure (4). We, therefore, examined whether KAP-1 siRNA similarly relieves the requirement for Artemis. hTERT immortalized cell lines were used to provide enhanced siRNA efficiency. Artemis−/− (CJ179hTERT) cells were exposed to 10 mM TBH following either control or KAP-1 siRNA and γ-H2AX foci enumerated in KAP-1 negative cells only. hTERT derivatives of WT (1BR.3), A-T (AT1BR) and a LIG4 Syndrome cell line (495GOS) were also examined. Following control siRNA, the anticipated DSB repair defect was observed at 24 h post TBH treatment in AT1BR, CJ179 and 495GOS cells (Figure 5A; consistent with Figure 1B). While siRNA KAP-1 substantially relieved the requirement for ATM for DSB repair, this was not observed in CJ179 or 495GOS cells. Similar results were also observed for IR treated CJ179hTERT cells (Supplementary Figure S5). The ability of KAP-1 siRNA to relieve DSB accumulation in ATM−/− cells after TBH substantiates the findings above that TBH induces HC-DSBs. Importantly, the failure of KAP-1 siRNA to relieve the defect in CJ179 cells demonstrates that Artemis does not directly function in promoting HC relaxation and suggests that it functions downstream of ATM-dependent KAP-1 phosphorylation.
Figure 5.

Artemis functions downstream of pKAP-1 foci formation. (A) KAP-1 siRNA does not relieve the requirement for Artemis for DSB repair. KAP-1 siRNA in hTERT immortalized human fibroblasts derived from a control (1BR.3), A-T (AT1BR), Artemis-defective (CJ179) and LIG4 Syndrome (495GOS) patient. Following siRNA, cells were treated with 10 mM TBH for 30 min and γ-H2AX enumerated at times indicated 30 min post treatment. Following control siRNA, the anticipated DSB repair defect at 24 h post TBH treatment was observed in control, A-T, Artemis and LIG4 syndrome cells (as in Figure 1A). siRNA KAP-1 substantially relieves the requirement for ATM. This is not observed in Artemis or LIG4 syndrome cells. Identical results were obtained with Artemis−/− and ATM inhibited−/− cells following 3 Gy IR (Supplementary Figure S2). Similar results for ATM−/− cells have been shown previously following IR exposure (4). (B) Primary human fibroblasts derived from a control (1BR.3), A-T (AT1BR) and Artemis-defective (CJ179) patient cells were examined for γ-H2AX and pKAP-1 foci following maintenance under non-replicating conditions for 7 weeks, 72 h post TBH exposure (10 mM for 30 min) or IR (3 Gy). γ-H2AX foci persisting in ATM−/− cells show no evidence of pKAP-1 foci. In contrast, the majority of γ-H2AX foci persisting in the absence of Artemis co-localize with pKAP-1 (>70%). We have not attempted to quantify the level of co-localization since, particularly in control cells, the number of residual foci is low.
Next, we examined whether KAP-1 phosphorylation occurs normally in Artemis defective cells. Previously, we reported that pKAP-1 occurs in a pan nuclear manner at early times post IR exposure and as discrete foci at later times (4,6). Further, pKAP-1 foci only form at HC-DSBs, where there is a high concentration of KAP-1 (6). We examined whether Artemis−/− cells form pKAP-1 foci after IR, TBH exposure and prolonged maintenance in G0. Under all conditions, we observed that the γ-H2AX foci persisting in the absence of ATM do not harbour pKAP-1 foci consistent with the notion that ATM has an overlapping function with DNA-PKcs in generating γ-H2AX but uniquely phosphorylates KAP-1 (Figure 5B). Strikingly, however, nearly all of the γ-H2AX foci persisting in Artemis−/− cells under all three conditions co-localize with pKAP-1 foci (∼70% co-localization). These findings consolidate the notion that Artemis functions downstream of KAP-1 phosphorylation and that unrepaired DSBs harbouring pKAP-1 persist in the absence of Artemis. Additionally, the fact that the endogenously arising DSBs co-localize with pKAP-1, which form predominantly at HC-DSBs, further adds to the conclusion above that ROS can induce DSBs within HC-DNA and thus, that HC is not a substantial barrier to endogenously arising DSBs.
DISCUSSION
ATM and Artemis are required for the repair of a subset of IR-induced DSBs, representing those that arise in HC (5). Here, we examine whether ATM and Artemis function in the repair of DSBs induced by oxidative damage or those arising endogenously. Previous studies have shown that oxidative damage activates ATM-dependent cell cycle checkpoint arrest but ATM’s role in the repair of endogenously induced DSBs has not been previously examined (15). We show that ATM and Artemis are required for the repair of DSBs that arise endogenously or following TBH treatment and provide strong evidence that they represent HC-DSBs.
To avoid the impact of replication damage and checkpoint arrest, we have primarily focused on DSB repair in non-replicating cells. We find that DSBs generated by TBH treatment or IR are repaired with similar kinetics, have a similar magnitude of a slow component and a similar requirement for ATM and Artemis for repair. This is further supported by our finding that KAP-1 siRNA bypasses the need for ATM in the repair of ROS-induced DSBs as for IR-induced DSBs and that the persisting DSBs harbour pKAP-1 foci, a feature which only arises at KAP-1 rich HC DSBs. This strongly suggests that ROS-induced DSBs arise in HC with a ratio of HC:EU DSBs being similar (1:5) to that induced by IR. This ratio is similar to the estimated ratio of HC:EU DNA (4). Previous findings have demonstrated that chromatin is a barrier to ROS-induced DNA damage. Our findings here suggest that HC does not act as a substantial additional barrier. We have previously shown that DSBs induced by neocarzinostatin (NCS) are also repaired with similar kinetics to IR-induced DSBs (4). Since neither NCS nor TBH induce the clustered damage typical of IR, these findings suggest that the complexity of damage induced by X or γ-rays does not significantly influence the overall rate of DSB repair. Interestingly, etoposide-induced DSBs are repaired more rapidly than ROS or IR-induced DSBs consistent with the notion that directly ligatable DNA ends are repaired more rapidly than ‘dirty’ DNA ends. In contrast, DSBs induced by high LET radiation, which are highly complex, are repaired more slowly. Overall, these findings are consistent with the notion that the slow component of DSB repair after X-rays represents the repair of HC DSBs. If the repair defect in ATM and Artemis defective cells after X-rays is predominantly due to the role of ATM or Artemis in HC-DSBs, this suggests that neither protein has a significant role in the repair of complex lesions induced by X or γ-rays. It should be stressed, however, that these studies do not assess the fidelity of repair.
Our findings also provide strong evidence that DSBs arise in human cell cultures and MEFs under non-replicating conditions. Although replication is known to be a significant source of endogenous DNA breakage and to limit the lifespan of MEFs whether significant DSBs arise under non-replicating conditions has been less unclear (10). Perhaps surprisingly, the rate of DSB accumulation under non-growing conditions was not significantly affected by the oxygen tension and could not be prevented by treatment with anti-oxidants (data not shown). Although, it is difficult to verify that these represent DSBs arising from oxidative damage, we have shown that they do not represent DSBs arising from apoptosis or senescence. We have also shown previously that the accumulated DSBs in LigIV-deficient MEFs do not localize to telomeres (23). Thus, we consider it likely that they do arise from oxidative damage. The lack of correlation of their induction with oxygen tension is unclear but since DSBs likely arise with different dose response kinetics to SSBs (possibly dose squared kinetics), it is possible that the correlation with oxygen tension is masked. The strong impact of oxygen tension under replicating conditions in MEFs is consistent with previous findings (10,23). The lack of impact of anti-oxidants likely reflects the long term nature of these experiments and the ability of cells to adjust to continuous exposure to high oxidant levels. Loss of ATM under replicating conditions resulted in a marked increase in DSB accumulation, which could be a consequence of ATM’s repair or checkpoint function, or the dual roles. Indeed, under replicating conditions loss of ATM appears to have a more severe impact than the hypomorphic mutation in DNA ligase IV (compared to a similar impact under non-replicating conditions), suggesting that the checkpoint role of ATM may be significant.
A range of studies have argued that A-T cells are oxidatively stressed with increased ROS levels, impaired mitochondrial membrane potential and/or decreased scavenging capacity (29–32). Such studies have led to suggestions that elevated levels of oxidative stress may be a factor contributing to the A-T clinical phenotype (24,33). Our findings of enhanced DSB accumulation after TBH treatment cannot be attributed to high levels of oxidative damage, however, since the level of DSBs incurred by the TBH treatment was identical in control and A-T cells. This strongly suggests that ATM and Artemis function in the repair of ROS-induced DSBs. It is difficult to eliminate the possibility that elevated oxidative damage contributes to the increased DSB accumulation in A-T cells grown for prolonged periods in culture. However, the similar levels of DSBs accrued in A-T and Artemis cells suggests that this may not be the case, unless Artemis cells are similarly oxidatively stressed. Further, the DSBs that accumulate during prolonged growth are those located at HC regions suggesting that they represent DSBs that specifically require ATM for their repair and, therefore, do not represent a steady state level of stochastically induced DSBs. Our analysis of oxidized proteins in control and A-T cells further suggests that A-T cells do not incur dramatically increased protein damage. While this analysis does not exclude elevated levels of particular oxygen species, this possibility is unlikely. ROS levels may be different between cell types. Our use of human fibroblasts likely explains differences with other studies on A-T cells. Thus, we favour the notion that the elevated accrual of DSBs in A-T, Artemis defective and LIG4 Syndrome cell lines represents a failure to repair DSBs that arise at a similar level in control cells but are efficiently repaired. However, a contribution from enhanced DSB formation as a consequence of elevated oxidative stress cannot be eliminated. Nothwithstanding uncertainties about the origin of the DSBs, our findings demonstrate that ATM and Artemis function in the repair of endogenously induced DSBs that can arise under non-replicating conditions.
Finally, our findings demonstrate that Artemis functions downstream of ATM-dependent KAP-1 phosphorylation. The precise role of Artemis is still unclear but could represent the removal of secondary structures at HC sequences or processing DNA ends. The examination of Artemis function is clinically significant since, although Artemis defective patients display severe combined immunodeficiency (SCID), they currently survive to adulthood following bone marrow transplantation (34,35). Evaluating the impact of Artemis loss is, therefore, important for optimizing patient care and treatment. In addition to demonstrating a role for Artemis in endogenously arising DSB repair, our findings show that the accumulated DSBs harbour pKAP-1. Previous studies have suggested that pKAP-1 impacts upon higher order chromatin structure, promoting HC relaxation. There is also evidence that it represses the transcription of certain genes otherwise silenced within HC regions (36). The DSBs that accumulate in the absence of Artemis are thus distinct to those that accumulate in the absence of ATM, with the former but not the latter causing elevated levels of sustained pKAP-1 and hence some degree of HC-relaxation. Further studies will be required to assess the impact of this for Artemis patients. A recent study examining the long-term outcome of immunodeficiency patients following bone marrow transplantation showed that Artemis defective patients fare less well than Rag1/2 patients despite their similar type of immunodeficiency (35).
In summary, we show that DSBs induced by oxidative damage are repaired with similar kinetics to IR-induced DSBs, have a similar slow component and a similar requirement for ATM and Artemis. Collectively our findings suggest that ROS-induced DSBs can arise in eu- and heterochromatin and that HC does not add a significant additional layer of protection against oxidative damage. We also show that ATM and Artemis function in the repair of endogenously induced DSBs that arise in non-replicating cells that likely represent DSBs arising from endogenously induced ROS damage. We show that Artemis functions downstream of ATM, resulting in the accumulation of DSBs that harbour pKAP-1. These findings are important for considering patient care but add further mechanistic insight into how ROS and IR-induced DSBs are repaired.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Medical Research Council (Grant number G0500897); the Association for International Cancer Research (Grant number 09-0018); the Department of Health Radiation Protection Programme (Grant number RRX116); Wellcome Research Trust (Grant number 084704/Z/08/Z). Funding for open access charge: Medical Research Council grant.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr M. Harm-Ringdahl and S. Haghoost for providing GS+/+ and GS−/− cells, and for helpful discussions. We also thank other members of the Jeggo lab for discussions/suggestions.
REFERENCES
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–714. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 3.Metzger L, Iliakis G. Kinetics of DNA double-strand break repair throughout the cell cycle as assayed by pulsed field gel electrophoresis in CHO cells. Int. J. Radiat. Biol. 1991;59:1325–1339. doi: 10.1080/09553009114551201. [DOI] [PubMed] [Google Scholar]
- 4.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 5.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Noon AT, Shibata A, Rief N, Lobrich M, Stewart GS, Jeggo PA, Goodarzi AA. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010;12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 7.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 8.Nikjoo H, O’Neill P, Wilson WE, Goodhead DT. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat. Res. 2001;156:577–583. doi: 10.1667/0033-7587(2001)156[0577:cafdts]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 9.Schmid TE, Dollinger G, Beisker W, Hable V, Greubel C, Auer S, Mittag A, Tarnok A, Friedl AA, Molls M, et al. Differences in the kinetics of gamma-H2AX fluorescence decay after exposure to low and high LET radiation. Int. J. Radiat. Biol. 2010;86:682–691. doi: 10.3109/09553001003734543. [DOI] [PubMed] [Google Scholar]
- 10.Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 2003;5:741–747. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karanjawala ZE, Murphy N, Hinton DR, Hsieh CL, Lieber MR. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr. Biol. 2002;12:397–402. doi: 10.1016/s0960-9822(02)00684-x. [DOI] [PubMed] [Google Scholar]
- 12.Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 1999;9:1501–1504. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- 13.Takao N, Li Y, Yamamoto K. Protective roles for ATM in cellular response to oxidative stress. FEBS Lett. 2000;472:133–136. doi: 10.1016/s0014-5793(00)01422-8. [DOI] [PubMed] [Google Scholar]
- 14.Tchirkov A, Lansdorp PM. Role of oxidative stress in telomere shortening in cultured fibroblasts from normal individuals and patients with ataxia-telangiectasia. Hum. Mol. Genet. 2003;12:227–232. doi: 10.1093/hmg/ddg023. [DOI] [PubMed] [Google Scholar]
- 15.Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J. Biol. Chem. 2001;276:21951–21959. doi: 10.1074/jbc.M011303200. [DOI] [PubMed] [Google Scholar]
- 16.Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, Fugmann S, Ferguson DO, Schatz DG, Sekiguchi J. Defective DNA repair and increased genomic instability in Artemis-deficient murine cells. J. Exp. Med. 2003;197:553–565. doi: 10.1084/jem.20021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX following exposure to ioninsing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 18.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 19.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 20.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 22.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. gamma H2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 23.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 24.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair. 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 25.Maity P, Bindu S, Choubey V, Alam A, Mitra K, Goyal M, Dey S, Guha M, Pal C, Bandyopadhyay U. Lansoprazole protects and heals gastric mucosa from non-steroidal anti-inflammatory drug (NSAID)-induced gastropathy by inhibiting mitochondrial as well as Fas-mediated death pathways with concurrent induction of mucosal cell renewal. J. Biol. Chem. 2008;283:14391–14401. doi: 10.1074/jbc.M800414200. [DOI] [PubMed] [Google Scholar]
- 26.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 27.Njalsson R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. 2005;62:1938–1945. doi: 10.1007/s00018-005-5163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ristoff E, Larsson A. Patients with genetic defects in the gamma-glutamyl cycle. Chem. Biol. Interact. 1998;111–112:113–121. doi: 10.1016/s0009-2797(97)00155-5. [DOI] [PubMed] [Google Scholar]
- 29.Biton S, Barzilai A, Shiloh Y. The neurological phenotype of ataxia-telangiectasia: solving a persistent puzzle. DNA Repair. 2008;7:1028–1038. doi: 10.1016/j.dnarep.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 31.Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007;16:2154–2164. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- 32.Meredith MJ, Dodson ML. Impaired gluthathione biosynthesis in cultured human ataxia-telangiectasia cells. Cancer Res. 1987;47:4576–4581. [PubMed] [Google Scholar]
- 33.Rotman G, Shiloh Y. Ataxia-telangiectasia: is ATM a sensor of oxidative damage and stress? BioEssays. 1997;19:911–917. doi: 10.1002/bies.950191011. [DOI] [PubMed] [Google Scholar]
- 34.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 35.Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, Mahlaoui N, Debre M, Casanova JL, Dal Cortivo L, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood. 2009;113:4114–4124. doi: 10.1182/blood-2008-09-177923. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem. 2007;282:36177–36189. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.