


Abstract
Genome engineering using single-stranded oligonucleotides is an efficient method for generating small chromosomal and episomal modifications in a variety of host organisms. The efficiency of this allelic replacement strategy is highly dependent on avoidance of the endogenous mismatch repair (MMR) machinery. However, global MMR inactivation generally results in significant accumulation of undesired background mutations. Here, we present a novel strategy using oligos containing chemically modified bases (2′-Fluoro-Uridine, 5-Methyl-deoxyCytidine, 2,6-Diaminopurine or Iso-deoxyGuanosine) in place of the standard T, C, A or G to avoid mismatch detection and repair, which we tested in Escherichia coli. This strategy increases transient allelic-replacement efficiencies by up to 20-fold, while maintaining a 100-fold lower background mutation level. We further show that the mismatched bases between the full length oligo and the chromosome are often not incorporated at the target site, probably due to nuclease activity at the 5′ and 3′ termini of the oligo. These results further elucidate the mechanism of oligo-mediated allelic replacement (OMAR) and enable improved methodologies for efficient, large-scale engineering of genomes.
INTRODUCTION
Oligo-mediated genomic modification proceeds via transformation of short single-stranded (ss) DNA into cells expressing ssDNA-annealing recombinase proteins, which mediate an Okazaki-like allelic-replacement event at the replication fork (1). These synthetic ssDNA oligos can be used to insert novel sequences, to produce mismatches of >30 bp, or to delete gene segments (2). This oligo-mediated allelic-replacement (OMAR) process is distinctly different from double-stranded (ds) DNA-based recombination engineering [more commonly referred to as recombineering (3–5)] in that: (i) only an easily synthesized short ss-oligonucleotide (50–90 bp) is needed, as opposed to a larger (>700 bp) selectable dsDNA cassette and (ii) the efficiency of oligo-based allelic replacement is up to 30% versus 0.01% for dsDNA recombineering (6). Furthermore, only a ssDNA binding protein is needed for OMAR (7,8). In contrast, dsDNA recombineering needs additional homologous recombination machinery such as exonucleases. This high allelic-replacement efficiency facilitates Multiplex Automated Genome Engineering (MAGE), which has been used to iterate cyclical rounds of allelic replacement to simultaneously modify many chromosomal targets in a high-throughput fashion (9,10).
Here, we define the process of oligo-mediated site-specific genomic modification as ‘allelic replacement’ based on its putative mechanism of action in Escherichia coli. It has been observed that oligos designed to target the lagging strand provide for up to 30-fold greater replacement efficiency than compared with leading strand-targeting oligos (2,11). In the Gram-negative E. coli bacteria, a single-stranded DNA binding protein derived from the λ-phage, Red-Beta, facilitates the annealing of the oligo to the homologous chromosomal sequence by stabilizing the ssDNA secondary structure, and may also protect against nuclease degradation (12). The processing and annealing of oligos to an exposed single-stranded region of the chromosome at the replication fork may show mechanistic similarities with those of Okazaki fragments. Recent studies have suggested that a full-length single-stranded intermediate may be also mediate dsDNA recombineering (13,14). However, detailed mechanisms of oligo-mediated allelic replacement still remain to be elucidated.
In addition to prokaryotic systems, the use of short oligonucleotides to generate site-specific mutations in the chromosome has been well-established for eukaryotic systems including yeast (15,16), mammalian cells (17,18) and plants (19,20). Strategies using RNA–DNA chimeric oligonucleotides (RDO) and triplex-forming oligonucleotides (TFO) have been developed (21,22) based on earlier observation that single-stranded oligodeoxynucleotides (ODN) could generate site-specific changes to the yeast chromosome (23). Chemical modifications to the ODNs such as phosphorothioate linkages (24) or base variations such as locked nucleic acid (LNA) have also been shown to improve the conversion efficiency (25). While reports of efficiencies of up to 9% have been documented (26), the conversion efficiency has generally remained 0.01–0.1% (21). These limited efficiencies are likely linked to DNA repair and homologous recombination proteins (e.g. MSH2, RAD51 and RAD54), which have been implicated in this oligo-mediated conversion process (27,28).
Previous studies showed that the endogenous mismatch repair (MMR) system in E. coli can significantly reduce the efficiency of OMAR by correcting mismatched bases introduced as a part of the synthetic oligonucleotide (29). Of the several mismatch repair pathways employed by E. coli, the methyl-directed MutHLS system specifically acts on mismatches, insertions, deletions of 1–6 nt (30,31). Mispaired bases are first detected by the MutS protein dimer at the mismatch site, which triggers MutL recruitment. Together, MutS and MutL activate the endonuclease MutH, which preferentially nicks the unmethylated strand of the hemi-methylated dGATC site of newly synthesized DNA. The excision and repair synthesis is subsequently carried out by one of several single-stranded nucleases, DNA polymerase III, ssDNA binding protein and DNA ligase. Various in vitro and in vivo studies have suggested that the correction efficiency of this MMR system varies based on the type of mismatches present (30,32,33). Costantino and Court (29) showed that the efficiency of OMAR could be increased by more than 100-fold by knocking out mutS. This approach enabled the detection of recombinants via low-throughput PCR screening (∼50–100 clones) instead of traditional antibiotic marker selections (>106 colonies). However, a ΔmutS strain suffers from a 100-fold higher background mutation rate, which causes undesired mutations to accumulate (34). Furthermore, mutator strains tend to have decreased fitness under laboratory conditions in comparison to wild-type strains, because most random mutations are deleterious (35,36).
Here, we present a strategy using oligonucleotides containing commercially available chemically modified base analogs to avoid recognition and correction by the native mismatch repair system. We also describe results suggesting that oligos are processed by nucleases at the replication fork, causing incomplete incorporation of the full-length oligo. These results will facilitate the design of improved oligos for allelic replacement in high-throughput genomic engineering applications.
MATERIALS AND METHODS
Abbreviations
Table 1 lists the abbreviations used in this article.
Table 1.
List of abbreviations used in this article
| AR | Allelic replacement |
|---|---|
| PT bases | Phosphorothioated bases |
| F-U | 2′ Fluoro dU |
| F-A | 2′ Fluoro dA |
| F-G | 2′ Fluoro dG |
| F-C | 2′ Fluoro dC |
| 5-Me-C | 5-Methyl dC |
| DiPr | 2,6-Diaminopurine |
| 2-AP | 2-Aminopurine |
| Iso-G | Iso-dG |
| Iso-C | Iso-dC |
Media, chemicals and reagents
Liquid cultures of all strains were grown in LB-min-rich media containing tryptone (10 g/l), yeast extract (5 g/l) and NaCl (5 g/l), and buffered to pH 7.45 with NaOH. Chloramphenicol (cm) and kanamycin (kan) were added to LB-agar plates (LB-min with 15 g/l agar) at concentrations of 20 µg/ml and 30 µg/ml, respectively. X-Gal (40 µg/ml) and IPTG (0.1 mM) were used on LB-min plates for functional assay of β-galactosidase activity. MacConkey–Maltose (Mac–Mal) or MacConkey–Galactose (Mac–Gal) agar plates were made by adding D-(+)-maltose monohydrate (10 g/l) or D-(+)-galactose (10 g/l), respectively, to Difco MacConkey Agar Base (40 g/l) which contains peptone (17.0 g/l), proteose peptone (3.0 g/l), bile salts no. 3 (1.5 g/l), NaCl (5.0 g/l), agar (13.5 g/l), neutral red (30 mg/l) and crystal violet (1 mg/l). Multiplex PCR kits were purchased from Qiagen (Cat #206143). PCR primers and oligonucleotides were synthesized by Integrated DNA Technologies (IDT) with standard purification for 90-bp oligonucleotides and vendor recommended purification for modified bases. Sanger DNA sequencing was performed by Agencourt Bioscience Corporation.
Oligonucleotides
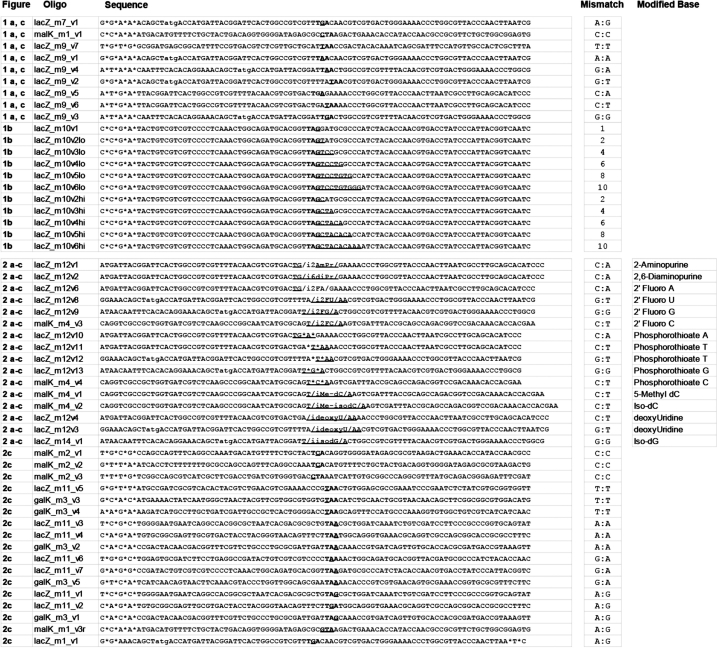
The Oligonucleotides (oligos) used in this study (listed in full in Table 2) are targeting the lagging strand of replicating DNA, meaning that they have the complementary sequence to the lagging strand.
Table 2.
A list of all oligos used in this study as well as their mismatch type or number of mismatches
 |
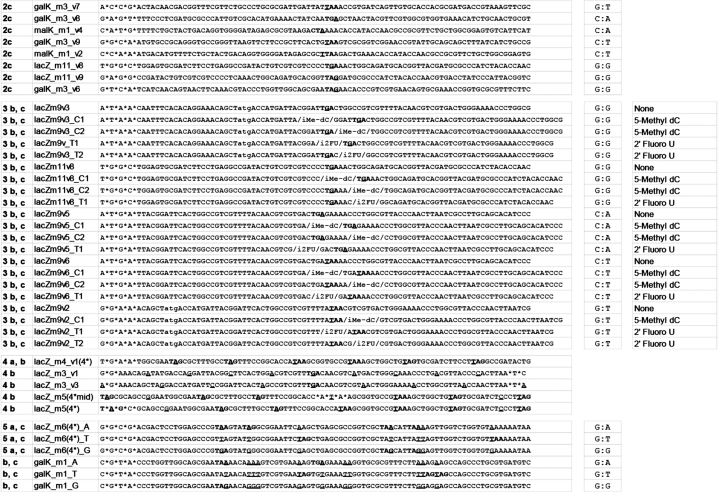 |
Oligos are organized based on the relevant figures presented.
Strains used
Escherichia coli strains EcNR1 and EcNR2 have been previously described (9). In brief, a defective Phage λ-Red construct was introduced by P1 transduction into E. coli MG1655 at the bioA location to produce EcNR1 (ΔbioA::λ-Red-bla). The relevant λ genes Redα, Redβ and Redγ are under regulation of the pL promoter and the temperature sensitive cI857 repressor. EcNR2 was made by using λ-Red homologous recombination to replace mutS with a chloramphenicol acetyltransferase (cat) cassette in EcNR1, thereby generating the ΔmutS::cat genotype. EcZS2 was made by introducing a kanamycin resistance (kan) cassette to replace the recA gene in EcNR1, generating a ΔrecA::kan genotype.
Oligo-mediated allelic replacement
Electrocompetent EcNR1 and EcNR2 cells induced for allele replacement were generated as previously described (9). In brief, individual colonies from a freshly streaked overnight plate were inoculated into 3 ml LB-min aliquots and grown in a rotator drum at 300 r.p.m. at 32°C. Upon reaching OD600 of 0.7, the glass tubes were moved to a 42°C shaking water bath for 15 min to induce the expression of λ-Red proteins. Cells were then immediately chilled on ice for at least 5 min and subsequently made electrocompetent by repeated (at least twice) pelleting and resuspension in cold sterile dH2O. One microliter of cells (∼7 × 108) were finally concentrated 20-fold into 50 µl reactions containing oligos (typically ∼1–5 µM) in dH2O and electroporated with a BioRad GenePulser using a 1 mm gap cuvette at 1.8 kV, 25 µF and 200 Ω. Electroporated reactions were immediately added to 3 ml of warm LB-min media and recovered for at least 3 h prior to plating.
Colorimetric assays to determine allelic-replacement efficiencies
Allelic-replacement efficiencies were determined by using oligos (Table 2) designed to introduce an inactivating nonsense mutation in the lacZ, malK or galK gene, and then screening for the function of that gene. All oligos used were designed to be complementary to the lagging strand of the replicating chromosome. Inactivating the lacZ gene abolishes β-galactosidase activity and results in white colonies on LB-min X-Gal/IPTG agar plates, rather than the blue colonies indicative of a functional lacZ gene. Targeted inactivation of the malK gene abolishes the ability of cells to catabolize maltose and results in white colonies on the pH-indicated Mac–Mal agar plates, compared with red colonies from a functional malK gene. Similarly, inactivation of the galK gene can be screened on Mac–Gal plates. Cells are generally plated at a density of 300–500 colonies per plate, after which the allelic-replacement efficiency is calculated from the ratio of the number of mutants to total cells. In general, cells were recovered for at least 3 h after electroporation to a density of 109 cells/ml prior to plating. The 3 h recovery ensured that all replicating chromosomes [up to 8 h in rich medium (37)] have properly segregated to separate daughter cells, thereby guaranteeing clonality of each colony on the plate. Colonies produced from this procedure are always mono-colored, whereas a shorter recovery time often produced sector-colored colonies suggesting non-clonality.
RESULTS
In vivo characterization of MutS binding and mismatch repair efficiency
We first performed a detailed characterization of the ability of the E. coli mismatch repair system to correct different types of mismatches in vivo. We designed 90 bp lagging strand targeting oligos to inactivate either the lacZ or malK genes by incorporation of a premature nonsense stop codon, which could be detected by colorimetric assay. Eight oligos targeting several different locations in lacZ or malK were designed (lacZm9v1 through 7, malKm1v1), which generated each of eight possible single base pair mismatches (C:C, T:T, G:G, A:A, C:T, C:A, G:T, G:A). Allelic-replacement experiments were performed by introducing each oligo into the mutS(+) EcNR1 strain or the mutS(−) EcNR2 derivative. The efficiency of allelic replacement is defined as the number of mutant cells per total colony forming units (CFU) as determined by the ratio of either white versus total colonies on LB-X-Gal/IPTG plates for lacZ-inactivating oligos, or white versus total colonies on Mac–Mal plates for malK-inactivating oligos. Consistent with previous observations (29,30,32), we found that the efficiency of mismatch repair depended on the base pairing type (Figure 1a), with C:C mismatches very poorly corrected and G:G mismatches most efficiently corrected. Interestingly, we also observed context dependency of MMR correction, and found an oligo (lacZm7v1) that generated an A:G (chromosome:oligo) mismatch at +25 bp position of the lacZ gene at ∼20% allelic-replacement efficiency for both mutS(+) and mutS(−) strains. The lacZm7v1 oligo is different from the lacZm9v1 oligo only at the mismatched base with a G instead of an A, but has a greatly increased efficiency of 20% versus 5% in the mutS(+) strain. In contrast, the lacZm9v4 oligo, which generated a G:A mismatch, was only 3.3% efficient in the presence of mutS and 20.9% efficient in its absence. In general, mismatches containing the more common transition mutations were better corrected by MutS than transversion mutations. We further observed that oligos containing eight or more mismatches have the same allelic-replacement efficiency in both mutS(+) and mutS(−) strains (Figure 1b), consistent with previous findings (6, 31). Thus the ability of mutS to recognize the mismatch lesion is critical for MMR activation, in terms of both mismatch type and size.
Figure 1.

(a) Allelic-replacement (AR) efficiency of oligos making various mismatches with the chromosome, designated by [chromosomal base:oligo base], using E. coli strains with wild-type mutS (solid black bars, EcNR1) and ΔmutS (black bars, EcNR2). Asterisks above the figure indicate mismatches generated through transition mutations. All other mismatches are transversion mutations. Oligo lacZm7v1 used for the A:G mismatch (gray solid and open bars) is a single-nucleotide variant of oligo lacZm9v1 used to generate the A:A mismatch. Oligos used to generate each mismatch are given in Table 2. The mean and ranges of values are shown for n=2. (b) AR efficiency of oligos making mismatches of increasing size in mutS(+) (filled circle, EcNR1) and mutS(−) strains (open circle, EcNR2). Error bars are standard errors with n = 3.
Mismatch repair evasion using modified bases
The E. coli MutS protein appears to be evolutionarily optimized to bind at high affinity to G:T mismatches and at low affinity to C:C mismatches, which correspond, respectively, to the most and least frequent polymerization errors during DNA replication (12). Since MutS binds poorly to the infrequently occurring C:C mismatches, it is likely that MutS will have reduced affinity to even more unusual DNA base pairing. We thus hypothesized that MutS cannot bind to some mismatches created by base pairing with non-standard nucleotides due to distortions of the DNA backbone not found in nature (33). Oligos containing these modified mismatching bases should have reduced MMR correction and increased allelic-replacement efficiency, especially for otherwise efficiently corrected mismatches (e.g. G:T, G:G). To test this hypothesis, we designed 90-mer oligos with the mismatching base containing non-standard ribose or nucleobase chemical modifications that were commercially available (Table 2) against the screenable lacZ and malK genes. The internal modified bases we explored include: (i) 2′ Fluoro bases (A, G, U, C), which have a fluorine modified ribose known to increase base pair binding affinity and confer some nuclease resistance compared with native RNA (38); (ii) Phosphorothioated bases, which contain a sulfur atom in place of a non-bridging oxygen in the phosphate backbone, which is known to increase resistance to nuclease degradation (39); (iii) 2-Aminopurine, which is a naturally fluorescent deoxyAdenosine analog that leads to reversible inactivation of the E. coli MMR when supplemented in bulk (40); (iv) 2,6-Diaminopurine, which forms three hydrogen bonds when base paired with deoxyThymidine, thus increasing binding affinity; (v) 5-Methyl-deoxyCytidine, which has increased binding affinity; (vi) Iso-deoxyGuanosine, which will base pair with 5-Methyl Iso-deoxyCytidine but not with deoxyCytidine (41), and Iso-deoxyCytidine, which will base pair with Iso-deoxyGuanosine, but not with deoxyGuanosine; and (vii) deoxyUridine, which can substitute for deoxyThymidine. Oligos were synthesized by Integrated DNA Technologies and HPLC- or PAGE-purified based on vendor recommendations. For side-by-side comparison, we also obtained oligos with standard nucleotides for each of the modified oligos. Modified or unmodified oligos were introduced into EcNR1, and the efficiency of oligo incorporation was assessed by colorimetric assay on LB-XGal/IPTG or Mac-Mal color indicator plates.
We found that oligos with certain modified bases could increase the allelic-replacement efficiency by more than 10–20-fold in a mutS(+) strain (Figure 2a). In particular, oligos containing 2′-Fluoro-deoxyUridine, 5-Methyl-deoxyCytidine, 2,6-Diaminopurine or Iso-deoxyGuanosine had the most improved allelic-replacement efficiencies. Oligos with phosphorothioated bases A, C and T at the mismatch site had an average of 5-fold improvement over non-phosphorothioated oligos. No significant improvement in allelic-replacement efficiency was observed for oligos with phosphorothioated-deoxyGuanosine, 2-Aminopurine, 2′-Fluoro-deoxyAdenosine, 2′-Fluoro-deoxyGuanosine or deoxyUridine. Using strain EcNR2, we also quantified the allelic-replacement efficiency for these modified oligos in the absence of mutS. Replacement efficiencies were further improved in this ΔmutS background (Figure 2b), suggesting that while certain modified bases could significantly evade mutS detection, some level of mutS activity was still present. Sequencing of the lacZ and malK genes from the mutant white colonies confirmed that DNA polymerase correctly replicated the modified bases. Oligos with Iso-deoxyCytidine and 2′ Fluoro-deoxyCytidine did not result in any allelic replacement, presumably due to significant disruption of DNA polymerization by these bases. Taken together, our results suggest that the intact MMR system can be easily side-stepped by careful design of the oligonucleotide sequence with the help of modified bases.
Figure 2.

(a) AR efficiency of oligos with modified bases (black bars) giving rise to different mismatch types [chromosome base : oligo base] and non-modified standard bases (white bars) characterized in a mutS(+) strain (EcNR1). Designation of modified bases are: 2′ Fluoro dU (F-U); 5-Methyl dC (5-Me-C); 2,6-Diaminopurine (DiPr); Iso-dG (Iso-G); Phosphorothioated A (PT-A); Phosphorothioated T (PT-T); Phosphorothioated T (PT-T); Phosphorothioated G (PT-G); Phosphorothioated C (PT-C); 2-Aminopurine (2-AP); 2′ Fluoro dA (F-A); 2′ Fluoro dG (F-G); deoxyUridine. Oligos with Iso-dC and 2′ Fluoro dC had AR efficiency of <0.2%, which was the detectable limit and thus were not shown. The mean and ranges of values are shown for n = 2. (b) AR efficiency of the same oligos with modified bases as in (a), tested in the mutS(+) EcNR1 strain and the mutS(−) EcNR2 strain. The mean and ranges of values are shown for n = 2. (c) White bars correspond to mismatch repair correction efficiency for different mismatch types as computed by difference in AR efficiency in mutS(−) and mutS(+) strains normalized by AR efficiency in mutS(−). Labeled black bars correspond to correction efficiency in oligos with modified bases, showing a drastic decrease compared with standard bases, (G:T with F-U, T:C with 5MeC, C:A with DiPr, G:G with Iso-G). See Table 2 for a list of oligos used. Error bars are standard errors with n = 6–15. (d) Chemical structures of the best-performing modified bases.
We calculated the MMR correction efficiency as the difference in allelic-replacement efficiencies between the mutS(−) and mutS(+) strains normalized to the absolute mutS(−) allelic-replacement efficiency. The MMR correction efficiency was calculated for each mismatch type for both standard bases and the best performing modified bases (Figure 2c). For mismatches naturally well-recognized by mutS and corrected at near 100% efficiency by MMR (e.g. G:T or T:C), utilization of modified bases 2′-Fluoro-deoxyUridine or 5-Methyl-deoxyCytidine significantly decreased the MMR correction efficiency to levels similar to wild-type C:C mismatch correction. Furthermore, MMR-correction of C:A and G:G mismatches were more efficiently avoided using 2,6-Diaminopurine and Iso-deoxyGuanosine modified bases, respectively. Thus, 2′-Fluoro-deoxyUridine can be used as a substitute for deoxyThymidine, 5-Methyl-deoxyCytidine for deoxyCytidine, 2,6-Diaminopurine for deoxyAdenosine and Iso-deoxyGuanosine for deoxyGuanosine (Figure 2d) to induce high efficiency allelic replacement by avoidance of the native MMR machinery. The use of oligos with modified bases is especially attractive for performing allelic replacement in organisms with less well-described MMR systems or when a general mutator phenotype, such as that of ΔmutS, needs to be avoided to reduce background mutations.
Previous studies have shown that six C–C mismatches spaced every 3 bp in tandem were sufficient to decrease mutS affinity and avoid MMR correction (6). Thus, we sought to determine if modified bases could also create local distortions in the DNA structure in a manner that reduces mutS binding and MMR-correction efficiency for nearby mismatches. Modified bases 2′-Fluoro-deoxyUridine (2fU) and 5-Methyl-deoxyCytidine (5mC) were chosen because they showed the highest MMR evasion phenotype. Oligos containing easily corrected G:G, G:T, C:A and C:T mismatches within 1–5 bp from a correctly paired modified base were used for allelic replacement in mutS(+) EcNR1 and mutS(−) EcNR2 (Figure 3a). We found that in EcNR1, mismatch C:T could be much more efficiently incorporated when a 2fU base was located in close proximity (Figure 3b). However, this improved allelic-replacement efficiency was not seen with other mismatch pairs, suggesting that such a strategy might be dependent on sequence context. We have previously observed similar sequence context effects on the allelic-replacement efficiency at the target site due to inhibitory hairpin secondary structures of the oligo (9). An oligo folding energy of less than −12.5 kcal/mol significantly decreased the AR efficiency and should be avoided by redesigning the oligo homology arms with less hairpin structure. The 5mC base did not improve the efficiency of incorporation of any nearby mismatches, suggesting that the modified base did not create sufficient DNA structural distortion to affect MMR correction. The allelic-replacement efficiency was substantially higher in EcNR2 indicating that the nearby modified bases did not affect replication fidelity (Figure 3c).
Figure 3.

(a) Diagram for mismatch repair avoidance using proximal modified bases that can prevent MutS recognition and activation of the MutHLS complex. (b) Use of proximal modified bases 5-Methyl-dC (grey bars) and 2′-Fluoro-deoxyUridine (black bars) to improve the AR efficiency of different mismatch types. In a mutS(+) strain, efficiencies of oligos with 2′-Fluoro-U and 5-Methyl-dC bases within 5 bp from the mismatch are compared with oligos that do not contain modified bases (white bars). The mean and ranges of values are shown for n = 2. (c) AR efficiency for the same oligos as (b), but in a mutS(−) strain. Variation in the AR efficiency of different oligos in a mutS(−) background is attributed to the impact of oligo sequences and local structural variations of the genome on annealing and folding characteristics. The mean and ranges of values are shown for n=2.
Local processing of the oligonucleotide during allelic replacement
While mismatches can be effectively introduced through MMR evasion, the overall efficiency of allelic replacement is determined by the oligo processing at the replication fork. Previously, we and others have generally observed that mismatches located at the 3′ or 5′ terminus of the oligo could not be incorporated into the chromosome through allelic replacement (11) or double-stranded DNA recombineering (13). However, a more recent study has suggested that a single base pair mismatch located at the oligo termini could be incorporated, but at very low efficiency (<0.01%) (42). To further elucidate the basis of these observations, we performed a detailed investigation of how oligonucleotides are processed at the site of allelic replacement. To this end, we performed allelic replacement using a 90 bp oligo (lacZm4v1) that contained six single-nucleotide polymorphisms in the coding region, resulting in six non-consecutive amber stop codons. Incorporation of any of the six mutations on the single oligo would inactivate the lacZ gene, the function of which was assayed for in the ΔmutS EcNR2 strain. The oligos were introduced by allelic replacement to produce white colonies at an efficiency of ∼10%. We isolated 47 white lacZ mutants and sequenced the lacZ region targeted by the oligo (Figure 4a). Surprisingly, only 34% of the isolates contained all six amber stop mutations. More than 53% of isolates had four or five mutations while the rest had one to three mutations. In general, the mutations were grouped consecutively, where only the 5′ or 3′ terminal mutations were not incorporated. In rare instances, we observed incorporation of the two 5′ and 3′ terminal mutations, but not mutations in the middle of the oligo (e.g. isolates 5, 13 and 14). We speculate that these anomalies correspond to rare events in which two independent allelic-replacement events occurred sequentially, where the first led to incomplete incorporation at one terminus and the second led to incomplete incorporation at the other terminus. The middle sequences were therefore left unchanged. These putative multiple replacement events occur for ∼6% of isolates that have undergone allelic replacement.
Figure 4.

(a) Distribution of the mutation incorporation pattern of 47 isolated clones having undergone allele replacement, as determined by sequencing of the targeted 90 bp lacZ region. The lacZm4v1 oligo and mismatch positions are shown on the abscissa axis. A grey block indicates verified mutation incorporation at one of six mismatches. A black box indicates isolates in which a discontinuous group of incorporated mismatches was observed. Basepair conversion of each mismatch is given above the plot. The percentage of isolates containing 1–6 out of the total six possible mismatches is given on the right of the plot. (b) Pattern of incomplete incorporation of oligos covering four different targets (lacZm3v1, lacZm3v3, lacZm4v1, lacZm5v4) in a mutS(−) strain. Each black circle represents the frequency that a mutation at a given position on the 90 bp oligo is found among all sequenced isolates. Position 1 corresponds to the 5′ terminus.
The pattern of incomplete incorporation of the full length oligo was observed across different target sequences and at different loci (i.e. lacZ, galK). For oligos that do undergo allelic replacement, mutations nearest to the termini are the least frequently incorporated (Figure 4b). Only 7% of clones incorporated mismatches 6 bp from the 3′ terminus, and 52% of oligos incorporated mismatches 9 bp from the 5′ terminus. In contrast, mutations at the center of the 90 bp oligo were incorporated 97% of the time. Previously, we showed that oligos with terminal phosphorothioated (PT) bases had increased allelic-replacement efficiency, presumably due to the prevention of ssDNA-nuclease activity (9). Thus, we sought to use protective PT bases at the termini in order to determine whether the observed incomplete oligo incorporation was due to oligo truncation from cytosolic nuclease degradation or by other mechanisms. However, we found that oligos containing four PT bases at the 5′ terminus had the same incomplete incorporation pattern as oligos without PT bases or oligos with four PT bases at the center of the oligo, leading us to hypothesize that this oligo processing mechanism may be acting more generally at the replication fork by specific endonucleases. We found that oligo truncation also occurred for leading strand targeting oligos, despite these oligos having >10-fold lower allelic-replacement efficiency (data not shown). Oligo processing appears to be RecA-independent, as oligo truncation was also found using the ΔrecA strain EcZS2.
We then performed allelic-replacement experiments in which three oligo variants (lacZm6_A, lacZm6_T and lacZm6_G) that exclusively introduced non-consecutive G:G, G:T or G:A mismatches were simultaneously co-electroporated into cells for allelic replacement to screen for mutants. In this multiplexed reaction, we anticipate that the three oligo variants would incorporate competitively into the chromosome. In the ΔmutS EcNR2 strain, we found that of 93 white clones screened and sequenced, all three variants (G:G, G:T and G:A) were represented equally, with similar oligo truncation patterns seen previously (Figure 5a). However, using galK oligo variants (galKm1_A, galKm1_T and galKm1_G) in the mutS(+) EcNR1 strain, we found that 94% of the isolated strains were the G:A variant, 5% were G:T, and 1% were G:G (Figure 5b). The under-representation of G:G and G:T variants demonstrated that the MMR system in EcNR1 could very efficiently correct multiple mismatches. We also observed a stronger oligo truncation pattern at the 3′ terminus in the mutS(+) EcNR1 strain in comparison to the ΔmutS EcNR2 strain (Figure 5c). Mismatches encoded on the 90 bp oligo at positions 66–70 were found in only 43% of mutants of mutS(+) EcNR1 in comparison to 84% of mutants of ΔmutS EcNR2. We speculate that the oligo-derived mismatches here are being processed in the 3′ to 5′ direction to produce the 3′ truncation pattern.
Figure 5.

(a) Distribution of the mutation incorporation pattern of 93 isolated clones as determined by sequencing of the targeted 90 bp lacZ region using a pool of three oligos that generated either C to G, C to T or C to A mutations in a mutS(−) strain. Nucleotide position and mismatch position of the oligos (lacZm6_A, lacZm6_T, lacZm6_G) are shown on the abscissa axis. (b) Distribution of the mutation incorporation pattern of 93 isolated clones generated using oligos (galKm1_A, galKm1_T, galKm1_G) in a mutS(+) strain. (c) Comparison of incomplete oligo incorporation pattern in mutS(+) (open diamond) and mutS(−) (filled diamond) strains. Dotted line is a linear regression fit of the mutS(−) data (R2 = 0.977).
DISCUSSION
Oligo-mediated allele-replacement techniques can now reach efficiencies of up to 20–30%, with iterable cycles capable of being performed every 2–3 h (9). This high allelic-replacement efficiency relied on the removal of the native mismatch repair system through knockout of components of the MutHLS complex. However, the background mutation rate is 100 times higher in such MMR-deficient strains, which presents an undesirable limitation for high accuracy genome engineering. Recently, Sawitzke et al. (11) showed that oligos with multiple C:C mismatches making silent mutations can effectively avoid MutS recognition. Here, we presented an alternative strategy that uses oligonucleotides containing modified bases 2′-Fluoro-deoxyUridine, 5-Methyl-deoxyCytidine, 2,6-Diaminopurine or Iso-deoxyGuanosine to effectively evade MMR to improve allelic-replacement efficiency in a strain with native MMR activity. This strategy may be generally useful to improve and expand the capabilities of OMAR in other organisms. Strategies that increase the efficiency of allelic replacement while maintaining genomic stability will be essential for high-precision genome engineering. We anticipate that there are other modified nucleotide bases that can both be well-processed by the DNA replication machinery and destabilize the mismatch lesion to prevent MMR activity at the targeted site. Practical considerations for researchers using oligos with modified bases may include cost and availability of customized DNA synthesis. While we have not observed any negative effects on viability of E. coli cells transformed with the modified bases tested here, a more detailed cell viability study using these modified bases are needed, especially for eukaryotic organisms. Additional investigations are needed to determine how modified bases perform among other organisms with different MMR systems.
Previously, Costantino and Court (29) had shown that using the λ-Red system in E. coli, the efficiency of targeting is 30-fold higher with the oligo that corresponds to the lagging strand in comparison to oligos that correspond to the leading strand (1). Since oligos targeting the leading strand is much less efficiently incorporated into the chromosome even in mismatch repair deficient strains (i.e. ΔmutS), the use of modified bases to avoid mismatch repair in these leading strand targeted oligos is not a practical route for making genomic changes. Instead, we recommend using lagging strand targeting ODNs with modified bases as a much more efficient approach for genome engineering.
When examining the local processing of the oligo at the target site, we found that the full-length 90 bp oligo was often not incorporated in its entirety. The center of the oligo was incorporated most frequently, with incorporation frequency decreasing toward the two termini. The 3′ and 5′ terminal bases were never observed to undergo any allelic replacement in our experiments. Exonuclease activity in both directions may account for these observations. Interestingly, we noted that tandem terminal 5′ phosphorothioated bases, which increase the overall efficiency of oligo incorporation, did not affect the frequency at which the incomplete 5′ incorporation was observed. Protection of the mismatching bases with phosphorothioated bases also showed no effect in preventing incomplete incorporation. Furthermore, oligos that targeted the leading strand also produced similar 5′ and 3′ truncations of the oligo. These observations suggest that phosphorothioated bases are effective at reducing oligo degradation by cytosolic exonucleases, but do not affect processing of the oligo at the replication fork.
The results of this work provide a general guideline for the efficient design of oligonucleotides for allelic-replacement strategies such as MAGE. To design mismatch mutations on the 90-mer oligo, we suggest placement of mutations at the center of the oligonucleotide to prevent terminal truncations from interfering with the targeted allelic replacement. We speculate that removal of endogenous nucleases may reduce oligo truncation, but might simultaneously result in other phenotypes such as decreased fitness. The oligo chew-back phenomenon observed here may be beneficial in certain applications. For example, in protein mutagenesis or sequence diversity generation using allelic replacement, a single oligo can be used to generate a heterogeneous population of cells containing multiple truncated forms of the full target oligo to produce combinatorial variants. Oligo-mediated genetic exchange has been documented in many other organisms including Gram-positive bacteria, yeast and mammalian cells (15,21,43). Improvements to the efficiency and predictability of oligo incorporation by avoiding native repair systems and overcoming nuclease degradation should be generalizable to these other organisms. Such efforts will help further develop OMAR as a general method for protein engineering, pathway optimization and strain manufacturing relevant to academic and industrial research.
FUNDING
National Science Foundation, multiple programs (SynBERC) (Grant #SA5283-11210); the Department of Energy (Genomes to Life Center) (Grant #DE-FG02-03ER6344); and the Wyss Institute for Biologically Inspired Engineering. Funding for open access charge: Wyss Institute for Biologically Inspired Engineering.
ACKNOWLEDGEMENTS
The authors thank Joshua Mosberg and Marc Lajoie for useful manuscript comments and also thank Peter Carr, Farren Isaacs, and Donald Court for helpful discussions.
REFERENCES
- 1.Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl Acad. Sci. USA. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu D, Sawitzke JA, Ellis H, Court DL. Recombineering with overlapping single-stranded DNA oligonucleotides: testing a recombination intermediate. Proc. Natl Acad. Sci. USA. 2003;100:7207–7212. doi: 10.1073/pnas.1232375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 4.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 2007;421:171–199. doi: 10.1016/S0076-6879(06)21015-2. [DOI] [PubMed] [Google Scholar]
- 7.Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- 9.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Church GM. Rapid and Multiplexed Genome Engineering and Genotyping Methods: Applications for Synthetic Biology and Protein and Pathway Engineering. Method Enzymol. 2011 doi: 10.1016/B978-0-12-385120-8.00018-8. (in press) [DOI] [PubMed] [Google Scholar]
- 11.Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL. Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J. Mol. Biol. 2011;407:45–59. doi: 10.1016/j.jmb.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erler A, Wegmann S, Elie-Caille C, Bradshaw CR, Maresca M, Seidel R, Habermann B, Muller DJ, Stewart AF. Conformational adaptability of Redbeta during DNA annealing and implications for its structural relationship with Rad52. J. Mol. Biol. 2009;391:586–598. doi: 10.1016/j.jmb.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Mosberg JA, Lajoie MJ, Church GM. Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics. 2010;186:791–799. doi: 10.1534/genetics.110.120782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maresca M, Erler A, Fu J, Friedrich A, Zhang Y, Stewart AF. Single-stranded heteroduplex intermediates in lambda Red homologous recombination. BMC Mol. Biol. 2010;11:54. doi: 10.1186/1471-2199-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brachman EE, Kmiec EB. Targeted nucleotide repair of cyc1 mutations in Saccharomyces cerevisiae directed by modified single-stranded DNA oligonucleotides. Genetics. 2003;163:527–538. doi: 10.1093/genetics/163.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto T, Moerschell RP, Wakem LP, Komar-Panicucci S, Sherman F. Strand-specificity in the transformation of yeast with synthetic oligonucleotides. Genetics. 1992;131:811–819. doi: 10.1093/genetics/131.4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole-Strauss A, Yoon K, Xiang Y, Byrne BC, Rice MC, Gryn J, Holloman WK, Kmiec EB. Correction of the mutation responsible for sickle cell anemia by an RNA-DNA oligonucleotide. Science. 1996;273:1386–1389. doi: 10.1126/science.273.5280.1386. [DOI] [PubMed] [Google Scholar]
- 18.Igoucheva O, Alexeev V, Yoon K. Targeted gene correction by small single-stranded oligonucleotides in mammalian cells. Gene Ther. 2001;8:391–399. doi: 10.1038/sj.gt.3301414. [DOI] [PubMed] [Google Scholar]
- 19.Beetham PR, Kipp PB, Sawycky XL, Arntzen CJ, May GD. A tool for functional plant genomics: chimeric RNA/DNA oligonucleotides cause in vivo gene-specific mutations. Proc. Natl Acad. Sci. USA. 1999;96:8774–8778. doi: 10.1073/pnas.96.15.8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu T, Peterson DJ, Tagliani L, St Clair G, Baszczynski CL, Bowen B. Targeted manipulation of maize genes in vivo using chimeric RNA/DNA oligonucleotides. Proc. Natl Acad. Sci. USA. 1999;96:8768–8773. doi: 10.1073/pnas.96.15.8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igoucheva O, Alexeev V, Yoon K. Oligonucleotide-directed mutagenesis and targeted gene correction: a mechanistic point of view. Curr. Mol. Med. 2004;4:445–463. doi: 10.2174/1566524043360465. [DOI] [PubMed] [Google Scholar]
- 22.Kolb AF, Coates CJ, Kaminski JM, Summers JB, Miller AD, Segal DJ. Site-directed genome modification: nucleic acid and protein modules for targeted integration and gene correction. Trends Biotechnol. 2005;23:399–406. doi: 10.1016/j.tibtech.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Moerschell RP, Tsunasawa S, Sherman F. Transformation of yeast with synthetic oligonucleotides. Proc. Natl Acad. Sci. USA. 1988;85:524–528. doi: 10.1073/pnas.85.2.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen PA, Randøl M, Luna L, Brown T, Krauss S. Genomic sequence correction by single-stranded DNA oligonucleotides: role of DNA synthesis and chemical modifications of the oligonucleotide ends. J. Gene Med. 2005;7:1534–1544. doi: 10.1002/jgm.804. [DOI] [PubMed] [Google Scholar]
- 25.Parekh-Olmedo H, Drury M, Kmiec EB. Targeted nucleotide exchange in Saccharomyces cerevisiae directed by short oligonucleotides containing locked nucleic acids. Chem. Biol. 2002;9:1073–1084. doi: 10.1016/s1074-5521(02)00236-3. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura M, Ando Y, Nagahara S, Sano A, Ochiya T, Maeda S, Kawaji T, Ogawa M, Hirata A, Terazaki H, et al. Targeted conversion of the transthyretin gene in vitro and in vivo. Gene Ther. 2004;11:838–846. doi: 10.1038/sj.gt.3302228. [DOI] [PubMed] [Google Scholar]
- 27.Parekh-Olmedo H, Kmiec EB. Progress and prospects: targeted gene alteration (TGA) Gene Ther. 2007;14:1675–1680. doi: 10.1038/sj.gt.3303053. [DOI] [PubMed] [Google Scholar]
- 28.Radecke S, Radecke F, Peter I, Schwarz K. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J. Gene Med. 2006;8:217–228. doi: 10.1002/jgm.828. [DOI] [PubMed] [Google Scholar]
- 29.Costantino N, Court DL. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl Acad. Sci. USA. 2003;100:15748–15753. doi: 10.1073/pnas.2434959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunkel TA, Erie DA. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 31.Modrich P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991;25:229–253. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- 32.Babic I, Andrew SE, Jirik FR. MutS interaction with mismatch and alkylated base containing DNA molecules detected by optical biosensor. Mutat. Res. 1996;372:87–96. doi: 10.1016/S0027-5107(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, Lee SD, Larson ED, Drummond JT, Alani E, Hsieh P, et al. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc. Natl Acad. Sci. USA. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 35.Funchain P, Yeung A, Stewart JL, Lin R, Slupska MM, Miller JH. The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics. 2000;154:959–970. doi: 10.1093/genetics/154.3.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaaper RM, Dunn RL. Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: the nature of in vivo DNA replication errors. Proc. Natl Acad. Sci. USA. 1987;84:6220–6224. doi: 10.1073/pnas.84.17.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sergueev K, Court D, Reaves L, Austin S. E. coli cell-cycle regulation by bacteriophage lambda. J. Mol. Biol. 2002;324:297–307. doi: 10.1016/s0022-2836(02)01037-9. [DOI] [PubMed] [Google Scholar]
- 38.Pallan PS, Greene EM, Jicman PA, Pandey RK, Manoharan M, Rozners E, Egli M. Unexpected origins of the enhanced pairing affinity of 2′-fluoro-modified RNA. Nucleic Acids Res. 2011;39:3482–3495. doi: 10.1093/nar/gkq1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu XP, Liu JH. The terminal 5′ phosphate and proximate phosphorothioate promote ligation-independent cloning. Protein Sci. 2010;19:967–973. doi: 10.1002/pro.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matic I, Babic A, Radman M. 2-aminopurine allows interspecies recombination by a reversible inactivation of the Escherichia coli mismatch repair system. J. Bacteriol. 2003;185:1459–1461. doi: 10.1128/JB.185.4.1459-1461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham ML, Albalos M, Guettouche T, Friesenhahn MJ, Battersby TR. Nucleobase analogs for degenerate hybridization devised through conformational pairing analysis. Biotechniques. 2007;43 doi: 10.2144/000112602. 617–618, 620, 622 passim. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Muyrers JP, Rientjes J, Stewart AF. Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol. Biol. 2003;4:1. doi: 10.1186/1471-2199-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swingle B, Markel E, Costantino N, Bubunenko MG, Cartinhour S, Court DL. Oligonucleotide recombination in Gram-negative bacteria. Mol. Microbiol. 2010;75:138–148. doi: 10.1111/j.1365-2958.2009.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]