

Abstract
Interruption of translation in Escherichia coli can lead to the addition of an 11-residue carboxy-terminal peptide tail to the nascent chain. This modification is mediated by SsrA RNA (also called 10Sa RNA and tmRNA) and marks the tagged polypeptide for proteolysis. Degradation in vivo of λ repressor amino-terminal domain variants bearing this carboxy-terminal SsrA peptide tag is shown here to depend on the cytoplasmic proteases ClpXP and ClpAP. Degradation in vitro of SsrA-tagged substrates was reproduced with purified components and required a substrate with a wild-type SsrA tail, the presence of both ClpP and either ClpA or ClpX, and ATP. Clp-dependent proteolysis accounts for most degradation of SsrA-tagged amino-domain substrates at 32°C, but additional proteases contribute to the degradation of some of these SsrA-tagged substrates at 39°C. The existence of multiple cytoplasmic proteases that function in SsrA quality-control surveillance suggests that the SsrA tag is designed to serve as a relatively promiscuous signal for proteolysis. Having diverse degradation systems able to recognize this tag may increase degradation capacity, permit degradation of a wide variety of different tagged proteins, or allow SsrA-tagged proteins to be degraded under different growth conditions.
Keywords: 10Sa RNA, λ repressor, Escherichia coli, intracellular proteolysis, molecular recognition, ATP-dependent degradation
Certain proteins and protein fragments in Escherichia coli are modified by carboxy-terminal addition of an 11-residue peptide tag (Tu et al. 1995). This tagging process requires functional SsrA RNA (10Sa RNA), which encodes the last 10 residues of the peptide (Tu et al. 1995) and results in rapid degradation of the tagged protein by carboxy-terminal-specific proteases (Keiler et al. 1996). SsrA-mediated tagging of proteins translated from defective messenger RNAs lacking termination codons has been demonstrated, and a model in which SsrA functions both as a tRNA and an mRNA has been proposed (Keiler et al. 1996). The 363-nucleotide SsrA RNA has sequences that form a tRNA-like structure and has been shown to be chargeable with alanine (Komine et al. 1994; Williams and Bartel 1996; Felden et al. 1997). In the model proposed by Keiler et al., when ribosomes stall at the 3′ end of the damaged message, SsrA charged with alanine binds to the ribosome like a tRNA and contributes the alanine to the idle nascent chain. Translation then switches from the mRNA to a small open reading frame (ORF) in SsrA that encodes the carboxy-terminal degradation peptide. This system provides both a method to avoid the accumulation of ribosomes stalled at the end of defective messages and a general quality-control mechanism that allows the cell to rid itself of incomplete protein fragments that might have inappropriate cellular activities. Cells devoid of SsrA RNA grow more slowly and show a certain degree of temperature sensitivity (Oh and Apirion 1991; Komine et al. 1994; Trempy et al. 1994).
The involvement of carboxy-terminal amino-acid sequences in targeting proteins for rapid degradation was recognized before the discovery of the SsrA-tagging system (Bowie and Sauer 1989; Parsell et al. 1990), and a periplasmic protease (Tsp or Prc) that degrades protein substrates in a carboxy-terminal-specific manner was purified and characterized (Silber et al. 1992). The carboxy-terminal substrate sequences recognized by Tsp are similar to those of the SsrA tag (Keiler et al. 1995; Tu et al. 1995), and Tsp is responsible for degradation of SsrA-tagged proteins that are exported to the periplasm (Keiler et al. 1996). Cytoplasmic proteins with carboxy-terminal degradation sequences, however, are still proteolyzed rapidly in cells lacking Tsp (Silber and Sauer 1994; Keiler et al. 1996), indicating that other proteases must be responsible for carboxy-terminal-specific degradation of proteins in the bacterial cytoplasm.
Essentially all cytoplasmic degradation in prokaryotes, archaea, and eukaryotes is energy-dependent. E. coli, for example, has at least five ATP-dependent proteases [Lon (La); HflB (FtsH); ClpAP; ClpXP; and ClpYQ (HslUV)] (for review, see Gottesman 1996). These enzymes appear to have distinct substrate preferences, as a mutation in a single protease gene is often sufficient to stabilize a specific unstable protein. For example, mutations in Lon lead to stabilization of the N protein of bacteriophage λ, the SulA and RcsA proteins of E. coli, and the CcdA protein of the episomal F factor. HflB appears to be responsible for degradation of the cII protein of λ and the heat-shock σ factor RpoH (Herman et al. 1993, 1995). The principal substrates for ClpYQ degradation have not yet been identified, although this two-component protease has been implicated in degradation of both Lon subtsrates and HflB substrates in vivo (Missiakas et al. 1996; Kanemori et al. 1997; Khattar 1997; W.-F. Wu and S. Gottesman, unpubl.). ClpAP and ClpXP are two-component proteases that share a common proteolytic subunit, ClpP, but have different ATPase regulatory subunits, ClpA or ClpX. Proteins stabilized by mutations in clpX but not in clpA include λ O, phage Mu repressor variants, and the stationary-phase σ factor, RpoS; clpA but not clpX mutants stabilize certain LacZ fusion proteins and the MazE protein. ClpB, an ATPase with extensive sequence similarity to ClpA, has not thus far been demonstrated to have a direct role in proteolysis but may act as a chaperone (Squires and Squires 1992).
In the studies presented here, we show that intracellular degradation of variants of the amino-terminal domain of λ repressor containing the SsrA peptide tag is dramatically reduced in cells lacking ClpP or lacking both ClpX and ClpA, and is somewhat reduced in cells lacking ClpX or ClpA only. Purified ClpXP and purified ClpAP degrade SsrA-tagged protein substrates in vitro, suggesting that these ATP-dependent enzymes are directly responsible for degradation of SsrA-tagged proteins in the bacterial cytoplasm.
Results
At sufficiently high concentrations, a protein containing the 93 amino-acid amino-terminal domain of λ cI repressor binds to the λ operators and prevents lytic growth of superinfecting phage (Sauer et al. 1979; Jordan and Pabo 1988). When degradation signals such as the SsrA tag, however, are attached to the carboxyl terminus of this fragment, proteolysis reduces the steady-state quantities of this domain to levels insufficient for function (Parsell et al. 1990; Keiler et al. 1996). Therefore, phage immunity provides an assay for the intracellular degradation of tagged amino-terminal domain derivatives.
For the work reported here, we have used three pairs of amino-domain variants (see Fig. 1). In each pair, one variant contains the protease-sensitive SsrA tag (AANDENYALAA) and the other bears a protease-resistant control tag (AANDENYALDD); the pairs differ in the linker regions that connect these carboxy-terminal tags to the amino-terminal domain. We have also used two phages, λcI−, which produces no functional λ repressor, and the weaker λc17 (see Materials and Methods), which is capable of producing some λ repressor. Wild-type strains expressing the three SsrA-tagged proteins (λN-AA, λN-L1-AA, and λN-L2-AA) were found, as expected, to be sensitive to superinfection by λcI− or λc17, whereas cells expressing the three control-tagged proteins (λN-DD, λN-L1-DD, and λN-L2-DD) were immune.
Figure 1.

Proteins used for degradation studies of SsrA-tagged proteins. Cartoon representation of the structures of hybrid proteins. The names of these proteins and of the plasmids that encode them are indicated to the left. [λ(1-93)] The 93-residue sequence of the amino-terminal, operator-binding domain of the λ cI repressor. (M2 Flag) The epitope sequence DYKDDDDK. (6His) The sequence HHHHHH. (SsrA tag AA) The sequence AANDENYALAA; (SsrA tag DD) The sequence AANDENYALDD. (trpAt) The AARLMSG sequence encoded by the stem–loop region of the trpAt transcriptional terminator. Note that because of heterogeneity in the position of SsrA tagging (Keiler et al. 1996), the λN–trpAt protein may consist of several species, some of which differ by 1 or 2 amino acids from the otherwise identical sequence of λN-L2-AA.
Immunity in protease-deficient mutants
To test for cytoplasmic proteases required for degradation of SsrA-tagged proteins, we assayed for increased λ immunity in known or suspected protease-defective strains expressing the λN-AA or λN-L1-AA proteins. No changes in immunity were observed in strains with an insertion mutation in clpB, an insertion mutation in hflA, a deletion of lon, mutations in lon and clpQ, or a temperature-sensitive mutation in hflB (data not shown). A very different result was observed in cells with defects in the ClpXP or ClpAP proteases. In strains containing insertions in the protease subunit (clpP) or regulatory subunits (clpA or clpX), expression of λN-AA or λN-L1-AA resulted in immunity to superinfection by λc17 (Fig. 2), suggesting that these proteases are involved in degradation of SsrA-tagged proteins.
Figure 2.

λN-AA and λN-L1-AA mediated immunity in clp mutants. Isogenic strains expressing λN-AA or λN-L1-AA were grown in tryptone broth with ampicillin and plated on LB–Amp plates in top agar. Ten-microliter serial dilutions of λcI− or λc17 phage were spotted, plates were incubated overnight at 32°C, and an approximate eop was calculated relative to plating on the wild-type strain SG22163 transformed with pBR322. E. coli strains: wild-type (SG22163); clpP (SG22174); clpA (SG22176); clpX SG22177); and clpAclpX (SG22178). In control experiments, the heteroimmune phage λimm21cI, whose operators are not repressed by amino-terminal domain variants of λ repressor, plated with an eop of 1 on all strains containing λN-AA or λN-L1-AA and on wild-type strains expressing λN-DD or λN-L1-DD. λcI− and λc17 plated with an eop of 10−4 or less on wild-type strains expressing λN-DD or λN-L1-DD.
Among the different clp mutants, no differences in λc17 immunity were observed, but clear differences in immunity to λcI− were evident (Fig. 2). The patterns of λcI− immunity suggest that the highest steady-state levels of SsrA-tagged amino-domain proteins are achieved when ClpP is defective or when both ClpX and ClpA are inactivated by mutation. In contrast, inactivation of ClpX alone in a strain expressing λN-AA resulted in very modest immunity to λcI−, and inactivation of ClpA alone gave partial immunity (λN-L1-AA) or no immunity (λN-AA) to λcI− (see Fig. 2 for quantitation of immunity). In control experiments, we found that expression of the control-tagged λN-DD or λN-L1-DD proteins provided full immunity [Efficiencies of plating (eop) 10−4 or lower] to λcI− and λc17, in both wild-type and clp mutants. Therefore, the Clp-mediated increases in phage immunity are dependent on the presence of λ amino-terminal domain proteins bearing a wild-type SsrA degradation tag at their carboxyl terminus. We interpret these data as suggesting that either ClpXP or ClpAP can degrade and thereby reduce the intracellular levels of the λN-AA and λN-L1-AA proteins.
Protein turnover in vivo
Degradation of λN-AA or λN-L1-AA in wild-type and protease-deficient strains was assayed directly by blocking protein synthesis and measuring the disappearance of the protein by Western blotting. Transformed cells expressing either λN-AA or λN-L1-AA were grown, induced by the addition of IPTG for 30 min, and treated with spectinomycin to block further protein synthesis. Samples were removed during the subsequent incubation and examined by Western blotting for remaining levels of λN-AA or λN-L1-AA. Figure 3A shows a Western blot of λN-AA turnover in wild-type cells, in which the half-life is <10 min, and in clpP mutant cells, in which the half life is longer than 1 hr. The initial level of λN-AA also was higher in the clpP mutant host, a result consistent with the immunity experiments. In parallel experiments, the λN-DD protein showed no detectable turnover (half life >1 hr) in either the wild-type or clpP strain (data not shown). The rapid turnover of λN-AA and slow turnover of λN-DD had been demonstrated previously in a different wild-type strain background (Keiler et al. 1996).
Figure 3.


Clp-dependent turnover in vivo of λN-AA and λN-L1-AA. (A) Western blot analysis of λN-AA in wild-type (SG22163) or clpP mutant (SG22175) cells after spectinomycin blocking of protein synthesis. Cells were grown to mid-log phase at 32°C and induced with IPTG for 30 min before addition of spectinomycin. Samples were removed at the times shown into cold TCA, precipitated, and resuspended for electrophoresis in 15% SDS–polyacrylamide gels. Gels were blotted and probed with anti-λ-repressor antibody. (B) Turnover of λN-L1-AA in wild-type and clp mutants. Experimental conditions were the same as in A except 10%–20% tricine SDS–polyacrylamide gels were used, and Western blots were probed with the M2 anti-FLAG antibody and quantified by the Eagle Eye II gel imaging system. Curves and strains are marked as for Fig. 2.
Figure 3B shows time courses for λN-L1-AA degradation in a set of isogenic clp mutants. As expected from the immunity results, degradation of λN-L1-AA is fastest in the wild-type strain that contains functional ClpAP and ClpXP. A small increase in half life was observed in the ClpA-defective strain, and relatively long half lives were observed in strains lacking ClpP, ClpA and ClpX, or ClpX. We note that in different experiments, the results with clpX mutant hosts were somewhat variable, often showing less stabilization than observed in Figure 3B; we currently have no explanation for this variability. Mutations in lon, clpB, hflA, or fhflB had no detectable effect on λN-L1-AA turnover under the conditions of these experiments (data not shown). Therefore, the high turnover rates of the λN-AA and λ N-L1-AA proteins are entirely consistent with the immunity tests. More importantly, the turnover experiments demonstrate that the increased phage immunity and increased steady-state levels of λN-AA and λN-L1-AA in ClpAP-defective or ClpXP-defective strains result mainly from decreased rates of degradation.
Protein degradation in vitro
To test directly for degradation of SsrA-tagged proteins by Clp proteases, 35S-labeled λN-L1-AA and λN-L1-DD were purified and incubated with purified ClpAP or ClpXP. As shown in Figure 4A, both Clp proteases degraded the SsrA-tagged λN-L1-AA protein efficiently but degraded the control-tagged λN-L1-DD protein at rates only 5% to 10% of that seen with λN-L1-AA (Fig. 4A). Degradation required both ClpP and either ClpX or ClpA and, in every case, required ATP (data not shown). These results demonstrate that both ClpXP and ClpAP can degrade proteins with the carboxy-terminal SsrA degradation peptide.
Figure 4.
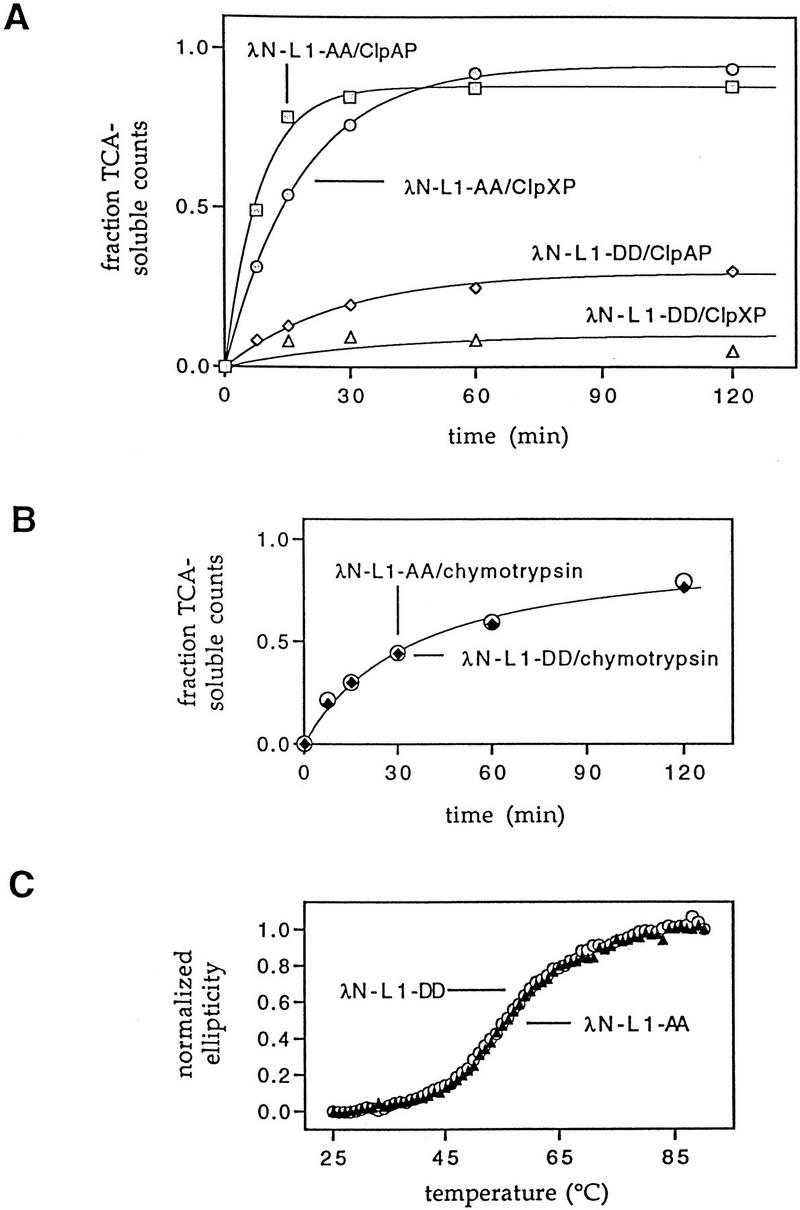
Degradation in vitro of SsrA-tagged substrates by ClpAP and ClpXP. (A) Purified 35S-labeled λN-L1-AA or λN-L1-DD proteins were incubated with purified ClpAP or ClpXP in the presence of ATP at 30°C as described in Materials and Methods. Time-dependent release of TCA soluble counts is shown. (B) 35S-Labeled λN-L1-AA or λN-L1-DD were incubated with chymotrypsin using the same experimental conditions as in A. (C) Denaturation of the λN-L1-AA and λN-L1-DD proteins monitored by changes in circular dichroism shows no difference in thermal stability. Normalized CD ellipticity was calculated as (ε25−ε)/(ε25−ε93) where e is the ellipticity at 230 nm at each temperature and ε25 and ε93 are the ellipticities at 230 nm at 25°C and 93°C, respectively.
As shown in Figure 4B, λN-L1-AA and λN-L1-DD were equally sensitive to degradation by chymotrypsin, a relatively nonspecific protease (Benyon and Bond 1989), indicating that the SsrA tag does not increase the general protease sensitivity of proteins to which it is attached. Purified λN-L1-AA and λN-L1-DD also had superimposible CD spectra (data not shown) and identical melting profiles in thermal denaturation experiments monitored by changes in circular dichroism (Fig. 4C). These data show that the susceptibility of λN-L1-AA to degradation by ClpXP and ClpAP does not result from major changes in the structure or stability of this tagged protein. Together, these results support a model in which the SsrA tag is recognized directly by ClpXP or ClpAP and argue against models in which the tag destabilizes the substrate, allowing proteases to recognize amino-acid sequences exposed in the denatured state.
Degradation after SsrA tagging in vivo
The experiments described above use amino-terminal domain variants in which the SsrA peptide tag is encoded by the genes for the λN-AA or λN-L1-AA proteins. Using the λN–trpAt system of Keiler et al. (1996), we also examined turnover in vivo of proteins in which the carboxy-terminal degradation peptide is added cotranslationally by the SsrA-mediated tagging system. As diagrammed in Figure 1, the gene for λN–trpAt encodes the amino-terminal domain and amino acids from the trpAt transcription terminator. There is no translation termination codon within this sequence. Therefore, when this message is read, translation ceases without termination and release of the growing polypeptide. Keiler et al. (1996) demonstrated that, under these circumstances, SsrA has an essential role in tagging and allows release of the stalled polypeptide. The λN-L2-AA and λN-L2-DD proteins contain the same amino acids from the trpAt terminator region but, unlike λN–trpAt, have the SsrA tag or control tag encoded in the gene (Fig. 1), and therefore are SsrA-independent.
Turnover experiments for λN–trpAt and λN-L2-AA are shown in Figure 5. λN–trpAt protein, expressed in an ssrA mutant, migrated faster in the gel (as expected for untagged protein) and was proteolytically stable (Fig. 5A; see also Keiler et al. 1996). In the ssrA+ host, only about half of the λN–trpAt protein appears to be tagged, but this species was degraded rapidly in the clpP+ strain and had a longer half life in the clpP− mutant (Fig. 5A). The shorter λN–trpAt protein in the ssrA+ host appears to be untagged, based on its electrophoretic mobility, and was longer lived than the tagged protein. λN-L2-AA, which has essentially the same sequence as tagged λN–trpAt, was turned over rapidly in clpP+ cells and is significantly stabilized in the clpP− mutant (Fig. 5B). These results show that SsrA-tagged λN–trpAt and λN-L2-AA are degraded in a clpP-dependent fashion. We note, however, that SsrA-tagged λN–trpAt was degraded faster than λN-L2-AA in both clpP+ and clpP− strains (Fig. 5C), indicating that the cotranslational, SsrA-dependent tagging of λN–trpAt may result in the tagged protein becoming exposed to or made more sensitive to other proteases in addition to being substrates for ClpAP and ClpXP.
Figure 5.


Degradation of λN-trpAt and λN-L2-AA in vivo. (A) Western blots of λN–trpAt turnover following spectinomycin treatment in ssrA+ clp+ (SG22163), ssrA+ clpP::kan (SG22175), ssrA::cat clp+ (SG22183), and ssrA::cat clpP::kan (SG22184) strains. Note the presence of tagged and untagged variants of the λN-trpAt protein in ssrA+ strains. Lanes marked with an asterisk are untagged samples from the ssrA− host run on the same gel as ssrA+ samples to allow comparison of electrophoretic mobilities. Experimental procedures were the same as described in Fig. 3; 10%–20% gradient tricine gels (Novex) were used for electrophoresis. (B) Western blots of λN-L2-AA turnover. Strains and conditions were identical to those shown in A. In control experiments, λN-L2-DD expressed in the wild-type strain (SG22163) showed no significant turnover over 1 hr (data not shown). (C) Time courses of degradation of SsrA-tagged λN–trpAt protein (upper band in A) and λN-L2-AA in ssrA+ clp+ (SG22163) and ssrA+ clpP::kan (SG22175) strains. These data were obtained by scanning of the gels shown in A and B.
High-temperature effects on Clp-dependent turnover
The immunity and turnover experiments described above were performed at 32°C. We observed, however, that cells expressing λN-AA did not acquire immunity at 39°C in clpP or clpA clpX mutant strains (Fig. 6). Preliminary turnover experiments indicated that, as above, this lack of immunity reflected rapid degradation of the λN-AA protein at elevated temperatures, suggesting that proteases other than ClpAP or ClpXP must participate in degradation of this SsrA-tailed protein at 39°C. C. Herman and R. D’Ari (pers. comm.) have made similar observations and identified HflB as a protease contributing to λN-AA degradation under these conditions. Unlike λN-AA, however, we found that λN-L1-AA was still immune in clpP or clpA clpX mutant strains at high temperature (Fig. 6), suggesting that λN-L1-AA is not sensitive to HflB or proteases other than ClpXP or ClpAP at 39°C. Because both of these proteins have the same SsrA tag but only λN-AA appears to be degraded in a Clp-independent fashion at 39°C, factors in addition to the SsrA tag must influence degradation by proteases such as HflB.
Figure 6.

Phage immunity at 39°C. Immunity to λcI− of wild-type (SG22163), clpP (SG22174), and clpA clpX (SG22178) strains expressing λN-AA and λN-AA at 39°C. Except for temperature, the plating conditions were identical to those described in Fig. 2.
Discussion
SsrA-mediated addition of a peptide signal to the carboxyl terminus of incompletely translated protein fragments is a newly discovered way for bacterial cells to mark abnormal proteins for destruction (Keiler et al. 1996). Cytoplasmic proteases that recognize and degrade SsrA-tagged proteins are critical components of this quality-control system and have now been identified. Our studies show that ClpXP and ClpAP, two related protease complexes, degrade SsrA-tagged proteins in the bacterial cytoplasm. This ATP-dependent degradation of SsrA-tagged substrates can be reproduced with purified components in vitro, and requires a substrate with a wild-type SsrA tail, the presence of both ClpP and either ClpA or ClpX. Keiler et al. (1996) showed previously that periplasmic proteins containing the SsrA-tag are degraded by Tsp (Prc), a protease that does not require or use ATP. There are, therefore, at least three proteases in E. coli, which function in the SsrA quality-control surveillance system. In addition, as discussed below, there are hints that one or more additional cytoplasmic proteases may also degrade SsrA-tagged substrates. This indicates both that carboxy-terminal-specific substrate recognition is relatively common among bacterial proteases and that the SsrA quality-control system has evolved to use a substantial number of different proteolytic systems.
ClpAP and ClpXP have mini-proteasome structures composed of four stacked rings (Kessel et al. 1995; Grimaud et al. 1998), in which ClpP, the proteolytic component, forms the central portion. The crystal structure of ClpP shows a double-ring assembly of heptamers in which the active sites are located in a central proteolytic chamber with small axial entrance pores (Wang et al. 1997). It has been proposed that ClpX or ClpA, which themselves form the outer rings of the proteolytic complex, act in a chaperone-like fashion to help denature substrates and feed them into the active-site chamber of ClpP (Gottesman et al. 1997; Wang et al. 1997). This model is appealing as both ClpX and ClpA, in the absence of ClpP, have been shown to have ATP-dependent chaperone activities that catalyze the disassembly of protein oligomers (Wickner et al. 1994; Levchenko et al. 1995). The ability of ClpA and ClpX to use the energy of ATP hydrolysis to aid in protein degradation may be important to allow SsrA-tagged substrates to be degraded even when these proteins are stably folded, as is the case, for example, for the SsrA-tagged amino-terminal domain variants of λ repressor studied here.
Recognition of protein substrates by ClpXP and ClpAP is mediated by the ClpX and ClpA subunits (Gottesman et al. 1993; Wojtkowiak et al. 1993), and recent protein-dissection studies have shown that ClpX contains modular domains that bind to the carboxy-terminal peptides of several target proteins, including SsrA-tagged Arc repressor (Levchenko et al. 1997). Because both ClpA and Tsp also contain domains homologous to the substrate-binding domains of ClpX (Levchenko et al. 1997), it seems very likely that ClpXP, ClpAP, and Tsp will all recognize SsrA-tagged substrates by a common mechanism that employs independent peptide-binding domains to recognize the carboxy-terminal SsrA tag.
ClpAP degrades certain β-galactosidase fusion proteins in vivo and degrades model peptides and casein in vitro (Gottesman et al. 1990; Tobias et al. 1991). Remarkably, the ClpAP-dependent degradation of certain of these β-galactosidase fusions is controlled by the identity of their amino-terminal amino acids (Tobias et al. 1991). Therefore, there is a somewhat startling difference between our results, which indicate carboxy-terminal recognition by the ClpAP enzyme, and those of Tobias et al. (1991), which suggest amino-terminal recognition. These apparently conflicting results might be reconciled if ClpAP contained a substrate-binding domain that recognized amino-terminal determinants in addition to the peptide-binding domain that apparently recognizes carboxy-terminal determinants.
ClpX recognizes the carboxy-terminal regions of several specific substrates including MuA transposase, Mu repressor, and certain virulent derivatives of Mu repressor (Levchenko et al. 1995; Laachouch et al. 1996; Vogel et al. 1996). Hence, the carboxy-terminal specificity shown here for ClpXP degradation of SsrA-tagged substrates is consonant with prior results. It is important to note, however, that the common specificity that we observe for ClpAP and ClpXP degradation of SsrA-tagged proteins contrasts with previous results, in which little or no cross-reactivity was observed for specific substrates (for review, see Gottesman 1996). It remains to be determined how the ClpA and ClpX enzymes recognize common SsrA-tagged substrates but have distinct specificities in other cases.
ClpXP and ClpAP must be responsible for most of the intracellular degradation of the λN-AA and λN-L1-AA proteins in E. coli growing at 32°C, as little residual degradation of either SsrA-tailed protein is observed in cells mutant for clpP or mutant for both clpA and clpX. Several observations, however, suggest that other cytoplasmic proteases are also capable of degrading SsrA-tagged proteins under appropriate conditions. For example, degradation of the λN-AA protein at 39°C is not fully eliminated by a clpP mutation. C. Hermann and R. D’Ari (pers. comm.) have found that HflB, a membrane-bound protease of the AAA family (Tomoyasu et al. 1995), has an important role in the high-temperature degradation of λN-AA. We note, however, that degradation of λN-L1-AA, which has the same SsrA tag as λN-AA, remains dependent on ClpP at both high and low temperatures. HflB must not, for some reason associated with the linker region of λN-L1-AA, be capable of degrading this protein efficiently. These differences might arise from linker-mediated alterations in the accessibility of the SsrA tail or possibly from binding interactions of the linker regions that sequester the associated protein from membrane-bound HflB.
The existence of multiple proteases that can function in SsrA quality-control surveillance may be biologically important in providing increased degradation capacity, in allowing degradation of virtually any incomplete translation product that is marked for degradation by this system, in allowing degradation in different cellular compartments, or in ensuring that SsrA-tagged proteins can be degraded under a wide variety of growth conditions. Functional redundancy or overlap of this sort makes biological sense for a general quality-control system but also requires that the SsrA degradation tag be recognized as a ubiquitous signal by proteases that normally have discrete substrate preferences. How this occurs is an intriguing problem in molecular recognition.
There are also clues that SsrA RNA may influence proteolysis of tagged proteins by mechanisms in addition to directing carboxy-terminal addition of the SsrA tag to incomplete proteins. For example, the shorter, presumably untagged species of the λN–trpAt protein is degraded faster in ssrA+ strains than in ssrA− strains. Moreover, the SsrA-tagged species of λN–trpAt in which the tag is added cotranslationally is degraded faster than λN-L2-AA, a protein with essentially the same sequence in which the SsrA tail is DNA encoded (Fig. 5). There are a number of ways to rationalize these results. SsrA recognition of stalled ribosomes might recruit proteases or degradation chaperones; the proteolytic susceptibility of proteins synthesized from mRNAs without stop codons might be influenced by changes in the kinetics of translation or protein folding; and/or additional proteolytic activities may be induced in ssrA+ strains.
Some of the phenotypes of ssrA mutants suggest effects beyond those caused by Clp-dependent stabilization of protein fragments. ssrA mutants grow slowly under a number of conditions (Oh and Apirion 1991; Komine et al. 1994; Trempy et al. 1994), whereas clpP mutants do not suffer the same growth problems under these conditions (S. Gottesman, unpubl.). Although this may simply indicate that degradation of some SsrA-tagged proteins requires proteases such as Tsp or HflB, it is also possible that the relief of ribosome stalling by SsrA is as important as the subsequent degradation of proteins or that tagging of particular proteins as part of their normal synthesis is important for cell growth. It is worth pointing out that ssrA mutants have been found to have a variety of as yet unexplained phenotypes including induction of a protease activity with Lon-like specificity (Kirby et al. 1994), defects in phage P22 growth (Retallack et al. 1994), and enhancement of the activities of a variety of phage and bacterial repressors (Retallack and Friedman 1995). The extent to which these phenotypes might be independent of the degradation of SsrA-tagged proteins is an important question that remains to be answered.
Materials and methods
Strains and plasmids
Hosts for experiments in vivo were a set of isogenic strains, each containing lacIQ inserted into the malP gene. The parental strain, SG21163, is derived from SG20250 (Gottesman et al. 1985); a malP::lacIQ derivative was constructed as described previously (Jubete et al. 1996). From SG21163, the various protease mutations were introduced by Pl transduction and either selection for the inserted antibiotic resistance (SG21164, clpB::kan; SG21165, clpP::cat Δlon; SG22168, hflA::kan; SG22174, clpP::cat; SG22175, clpP::kan (polar on clpX); SG22176, clpA::kan; and SG22177, clpX::kan), linkage of lon to proC (SG22186, Δlon rcsA51::kan), or linkage to a marker for hflBts [SG22166, hflB1ts linked to tet, transduced from SL93, a gift from C. Herman and R. D’Ari (Herman et al. 1993)]. To construct the clpX clpA double mutant, SG22178, we first linked zba-1091::tet [from SG1091; (Maurizi et al. 1985)] to clpX::kan and then transduced from this host to the clpA::kan strain, selecting for tetracycline resistance. Transductants were screened initially for increased immunity in the presence of the pλN–SPT plasmid and subsequently checked by back transduction of the tet resistance to a kanamycin sensitive strain and demonstration of ability to transfer the linked clpX::kan marker. Strain GL005 is lon− clpQ− rcsA− (G. Leffers, unpubl.).
The isogenic protease-deficient strains were transformed with the plasmids shown in Figure 1. Plasmids pλN–SPT and pλN–CPT encode residues 1–93 of λ repressor (the amino-terminal domain) with the carboxy-terminal extensions AANDENYALAA and AANDENYALDD, respectively (Keiler et al. 1996). The proteins encoded by these plasmids are referred to as λN-AA and λN-DD, respectively. Plasmids pER130 and pER132 encode similar constructs but have a linker with an M2 FLAG epitope and 6 His sequence inserted between residues 1–93 and the carboxy-terminal extensions (Fig. 1); the resulting proteins are referred to as λN-L1-AA and λN-L1-DD. To construct pER130, PCR was used to amplify the carboxy-terminal portion of a λ repressor amino-terminal domain–M2–FLAG–6 His gene in plasmid pAD103 (A. Davidson, unpubl.) using primers ER27 (5′-TATAACGCGGCATTGCTAGCAAA-3′) and ER30 (5′-CCGGATCCTTAAGCTG CTAAAGCGTAGTTTTCGTCGTTTGCTGCGTGATGGTGATGATGGTGCTTGTCA-3′). The resulting product was cut with NheI and BamHI and ligated to the pAD103 NheI–BglII backbone fragment. Plasmid pER132 was constructed in the same fashion using primers ER27 and ER32 (5′-CCGGATCCTTAATCGTCTAAAGCGTAGTTTTC GTCGTTTGCTGCGTGATGGTGATGATGGTGCTTGTCA-3′). The expected structure of each construct was verified by restriction mapping and DNA sequencing.
The backbones for pPW500, pER161, and pER163 (Fig. 1) are from pAD100, a pBR322-based plasmid carrying a Ptrc promoter (trp/lacUV5 hybrid promoter), lacIQ and an Fl phage origin (Davidson and Sauer 1994). Cloning into pAD100 resulted in a change in the amino-terminal amino acid of λ repressor from Ser to Gly. This sequence change, however, does not affect repressor activity (Clarke et al. 1991). Construction of pPW500, which contains a trpAt terminator following the amino-terminal domain and directs transcription of an mRNA with no in-frame stop codons, has been described previously (see footnote 14 in Keiler et al. 1996). Addition of the AANDENYALAA carboxy-terminal sequence to the λN–trpAt protein is dependent on SsrA-mediated addition of the tag sequence in vivo, and therefore there can be untagged or SsrA-tagged versions of λN–trpAt. Plasmids pER161 and pER163 encode proteins (λN-L2-AA and λN-L2-DD, respectively) with linkers containing the same amino acids as those encoded by the trpA terminator sequence. As a result, the SsrA-tagged λN–trpAt protein and the λN-L2-AA protein should have the same or very similar amino-acid sequences (see Fig. 1 and legend). Plasmid pER161 was constructed by amplifying plasmid pAD103 with primers ER60 (5′-GCGCCATG GGCACAAAAAAGAAACCATTA-3′) and ER61 (5′-CCGGATCCTTAAGCTGCTAA AGCGTAGTTTTCGTCGTTTGCTGCTCCGGACATTAATCGTGCGGCGTGATGGTGATGATGGTTGCTT-3′). The resulting product was cut with NcoI and BamHI and ligated to the pAD100 NcoI–BglII backbone fragment. Plasmid pER163 was constructed in the same fashion using primers pER60 and ER62 (5′-CCGGATCCT- TAATCGTCTA AAGCGTAGTTTTCGTCGTTTGCTGCTCCGGACATTAATCGTGCGGCGTGATGGTGATGATGGGTGCTT-3′). The expected structure of each construct was verified by DNA sequencing.
Steady-state expression and degradation in vivo
Intracellular levels of different amino-terminal domain variants, which reflect a balance between the rates of synthesis and degradation of these proteins, were assayed by measuring immunity to superinfecting λ phage. Cells containing high levels of the amino-terminal domain of λ repressor are immune, whereas those containing low levels are sensitive to phage infection. Cells containing plasmids encoding λN-AA, λN-DD, λN-L1-AA, λN-L1-DD, λN-L2-AA, λN-L2-DD, and λN–trpAt were grown without IPTG induction in tryptone broth with vitamin B1, maltose, 0.01 m MgSO4, and ampicillin (100 μg/ml) at 32°C to late log phase. Lawns of cells were plated in top agar on LB or LB-Amp plates, spotted with dilutions of λcI−, λc17, λvir, and λimm21cI phages, and incubated at 32°C unless indicated. Efficiencies of plating are reported relative to growth on cells with vector alone or with no plasmid; both control strains gave identical results. The imm21cI phage plated with an efficiency of 1.0 on all hosts. λvir, defined as virulent by its ability to plate on normal λ lysogens, plated on all hosts, although the eop was low or plaques were smaller in some cases. λcI− is our standard for testing immunity; it fails to plate on normal λ lysogens. λc17 is a weak clear mutant and was used here because it was sensitive to intermediate levels of the amino-terminal domain of λ repressor in initial tests. λc17 carries a wild-type cI gene but has a duplication of 9 bp in the right operon, which creates a new, constitutively expressed rightward promoter that allows synthesis of λ replication proteins and leads to replication and titration of repressor (Sly and Rabideau 1969; Rosenberg et al. 1978). It seems likely that λc17 acts as a weak clear mutation in these tests because, in the presence of even low levels of endogenous repression within the cell, the positive self-regulation of PRM will lead to synthesis of wild-type cI (both stable and able to oligomerize) from the incoming c17 phage, preventing further phage growth and release of a phage burst.
Degradation of the amino-terminal domain variants in vivo was measured by Western blotting after protein synthesis was blocked by addition of spectinomycin. Cells were grown at 32°C in tryptone broth plus ampicillin (100 μg/ml) to an A600 of 0.3 and induced for 30 min with IPTG (final concentration 0.02%). A zero-time sample was removed and spectinomycin was added to a final concentration of 100 μg/ml to block further protein synthesis. One ml samples were removed periodically to tubes with 110 μl of 50% TCA. After centrifugation, pellets were washed with 80% acetone, dried, and resuspended in tricine-gel loading buffer. Equal quantities of protein extracts were separated on a 15% SDS–polyacrlamide gel or 10%–20% tricine SDS–polyacrylamide gel (see figure legends) and transferred to nitrocellulose filters. Filters were incubated with polyclonal anti-λ-repressor antibody, a gift of Jeffrey Roberts (Cornell University, Ithaca, NY), or the M2 monoclonal anti-FLAG antibody (Eastman Kodak). Immunoblots were developed using HPR-conjugated goat anti-rabbit or anti-mouse antibody, followed by enhanced chemiluminescence (Amersham). Blots were quantified by scanning films with the Eagle Eye II gel imager (Stratagene).
Purification of 35S-labeled substrate proteins
The λN-L1-AA and λN-L1-DD proteins were expressed in E. coli strain X90 transformed with pER130 or pER132. Cells were grown to mid-log phase at 37°C in M9 minimal medium lacking cysteine and methionine, and protein expression was induced by adding isopropyl-1-thio-β-d-galactopyranoside to 200 μg/ml. Twenty minutes after induction, [35S]methionine was added (40 μCi/ml culture). After 1 hr at 37°C, the cells were harvested by centrifugation and frozen at −80°C. Frozen cells were thawed, resuspended in Buffer A [6 m guanidine hydrochloride, 0.1 m NaH2PO4, 0.01 m Tris (pH 8.0)] containing 10 mm imidazole (40 μl/ml culture) and allowed to lyse while rocking at 4°C for 1 hr. After this time, cell debris was removed by centrifugation at 20,000g, Ni–NTA agarose (Qiagen) was added (75 μl/ml supernatant) and the samples were placed on a rocker for 15 min. The resin was washed three times with buffer A containing 10 mm imidazole, and the λN-L1-AA or λN-L1-DD proteins were eluted with buffer A containing 400 mm imidazole. Samples were dialyzed against three changes of buffer containing 50 mm HEPES/NaOH (pH 7.5), and 100 mm NaCl. The purified proteins were greater than 90% pure as assayed by SDS–polyacrylamide gel electrophoresis and staining with Coomassie blue.
Proteins
Purified ClpP and 6 His-tagged ClpX were gifts from Igor Levchenko and Tania Baker (MIT, Cambridge, MA); purified ClpA was a gift from Sue Wickner (NIH, Bethesda, MD). TLCK treated α-chymotrypsin from bovine pancreas (60 U/mg protein) was purchased from Sigma. Unlabeled λN-L1-AA or λN-L1-DD were purified by denaturing nickel NTA chromatography according to the Qiagen protocol with minor modifications.
Proteolytic assays in vitro
Proteolytic digestions were performed in a total volume of 20 μl in buffer containing 25 mm HEPES/NaOH (pH 7.5), 25 mm potassium acetate, 5 mm MgCl2, 10% glycerol, and 0.02% NP-40. Complete ClpAP or ClpXP digests also contained 5 mm ATP, 0.1 mg/ml creatine kinase, 10 mm creatine phosphate, 10 pmoles of 35S-labeled protein substrate (λN-L1-AA or λN-L1-DD), 3.5 pmoles of ClpP subunits, and 3 pmoles of ClpA or 3.5 pmoles ClpX subunits. Complete chymotrypsin digests contained 10 pmoles protein substrate and 200 ng of α-chymotrypsin. Digestion solutions were incubated at 30°C for different times, the reactions stopped by the addition of TCA to 10%, and the tubes left on ice. After a minimum of 30 min, the samples were centrifuged for 10 min at 20,000g and 4°C, and TCA soluble counts were determined by scintillation counting in Ready Safe liquid scintillation cocktail (Beckman). Control 2 hr digests lacking either ClpX or ClpA, ClpP, or ATP showed an average of 3%–4% of the total 35S counts in the supernatant after TCA precipitation. These average values were subtracted as background from the complete digestion samples containing ClpAP/ATP or ClpXP/ATP.
Circular-dichroism spectra and thermal melts
The circular-dichroism (CD) spectra of the λN-L1-AA and λN-L1-DD proteins were recorded at a concentration of 3.8 μm in 25 mm HEPES/NaOH (pH 7.5), 50 mm NaCl at 25°C using an AVIV model 60DS spectropolarimeter. Thermal melts, performed using the same buffer and protein concentrations, were monitored by changes in CD ellipticity at 230 nm. Melts were performed in 1°C increments with a 42-sec equilibration time and a 30-sec averaging time at each temperature.
Acknowledgments
We thank Ken Keiler and Patrick Waller for strains and plasmids; Igor Levchenko, Tania Baker, and Sue Wickner for purified proteins; Jeffrey Roberts for λ repressor antibodies; C. Hermann and R. D’Ari for strains and communication of unpublished results; and Tania Baker and Michael Maurizi for helpful discussions and comments on the manuscript. This work was supported in part by the intramural program of the National Cancer Institute and by National Institutes of Health grant AI-16897.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL susang@helix.nih.gov; FAX (301) 496-3875.
References
- Benyon RJ, Bond JS. Proteolytic enzymes: A practical approach. Oxford, UK: IRL Press; 1989. [Google Scholar]
- Bowie JU, Sauer RT. Identification of C-terminal extensions that protect proteins from intracellular proteolysis. J Biol Chem. 1989;264:7596–7602. [PubMed] [Google Scholar]
- Clarke ND, Beamer LJ, Goldberg HR, Berkower C, Pabo CO. The DNA binding arm of λ repressor: Critical contacts from a flexible region. Science. 1991;254:267–270. doi: 10.1126/science.254.5029.267. [DOI] [PubMed] [Google Scholar]
- Davidson AR, Sauer RT. Folded proteins occur frequently in libraries of random amino acid sequences. Proc Natl Acad Sci. 1994;91:2146–2150. doi: 10.1073/pnas.91.6.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felden B, Himeno H, Muto A, McCutcheon JP, Atkins JF, Gesteland RF. Probing the structure of the Escherichia coli 10Sa RNA (tmRNA) RNA. 1997;3:89–103. [PMC free article] [PubMed] [Google Scholar]
- Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Trisler P, Torres-Cabassa AS. Regulation of capsular polysaccharide synthesis in Escherichia coli K12: Characterization of three regulatory genes. J Bacteriol. 1985;162:1111–1119. doi: 10.1128/jb.162.3.1111-1119.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Clark WP, Maurizi MR. The ATP-dependent Clp protease of Escherichia coli: Sequence of clpA and identification of a Clp-specific substrate. J Biol Chem. 1990;265:7886–7893. [PubMed] [Google Scholar]
- Gottesman S, Clark WP, de Crecy-Lagard V, Maurizi MR. ClpX, an alternative subunit for the ATP-dependent Clp protease of Escherichia coli. J Biol Chem. 1993;268:22618–22626. [PubMed] [Google Scholar]
- Gottesman S, Wickner S, Maurizi MR. Protein quality control: Triage by chaperones and proteases. Genes & Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- Grimaud, R., M. Kessel, F. Beuron, A.C. Steven, and M.R. Maurizi. 1998. Structural and enzymatic similarities between the E. coli. ATP-dependent proteases, ClpXP and ClpAP. J. Biol. Chem. 273: (in press). [DOI] [PubMed]
- Herman C, Ogura T, Tomoyasu T, Hiraga S, Akiyama Y, Ito K, Thomas R, D’Ari R, Bouloc P. Cell growth and λ phage development controlled by the same essential Escherichia coli gene, ftsH/hflB. Proc Natl Acad Sci. 1993;90:10861–10865. doi: 10.1073/pnas.90.22.10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C, Thévenet D, D’Ari R, Bouloc P. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc Natl Acad Sci. 1995;92:3516–3520. doi: 10.1073/pnas.92.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SR, Pabo CO. Structure of the lambda complex at 2.5 A resolution: Details of the repressor-operator interactions. Science. 1988;242:893–899. doi: 10.1126/science.3187530. [DOI] [PubMed] [Google Scholar]
- Jubete Y, Maurizi MR, Gottesman S. Role of the heat shock protein DnaJ in the Lon-dependent degradation of naturally unstable proteins. J Biol Chem. 1996;271:30798–30803. doi: 10.1074/jbc.271.48.30798. [DOI] [PubMed] [Google Scholar]
- Kanemori M, Nishihara K, Yanagi H, Yura T. Synergistic roles of HslVU and other ATP-dependent proteases in controlling in vivo turnover of σ32 and abnormal proteins in Escherichia coli. J Bacteriol. 1997;179:7219–7225. doi: 10.1128/jb.179.23.7219-7225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Silver KR, Downard KM, Papayannopoulos IA, Biemann K, Sauer RT. C-terminal specific protein degradation: Activity and substrate specificity of the Tsp protease. Protein Sci. 1995;4:1507–1515. doi: 10.1002/pro.5560040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Waller PRH, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- Kessel M, Maurizi MR, Kim B, Kocsis E, Trus BL, Singh SK, Steven AC. Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26S proteasome. J Mol Biol. 1995;250:587–594. doi: 10.1006/jmbi.1995.0400. [DOI] [PubMed] [Google Scholar]
- Khattar MM. Overexpression of the hslVU operon suppresses SOS-mediated inhibition of cell division in Escherichia coli. FEBS Lett. 1997;414:402–404. doi: 10.1016/s0014-5793(97)01024-7. [DOI] [PubMed] [Google Scholar]
- Kirby JE, Trempy JE, Gottesman S. Excision of a P4-like cryptic prophage leads to Alp protease expression in Escherichia coli. J Bacteriol. 1994;176:2068–2081. doi: 10.1128/jb.176.7.2068-2081.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laachouch JE, Desmet L, Geuskens V, Grimaud R, Toussaint A. Bacteriophage Mu repressor as a target for the Escherichia coli ATP-dependent Clp protease. EMBO J. 1996;15:437–444. [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Luo L, Baker TA. Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes & Dev. 1995;9:2399–2408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA. PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- Maurizi MR, Trisler P, Gottesman S. Insertional mutagenesis of the lon gene in Escherichia coli: lon is dispensable. J Bacteriol. 1985;164:1124–1135. doi: 10.1128/jb.164.3.1124-1135.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D, Schwager F, Betton JM, Georgopoulos C, Raina S. Identification and characterization of HslV HslU (ClpQ ClpY) proteins involved in overall proteolysis of misfolded proteins in Escherichia coli. EMBO J. 1996;15:6899–6909. [PMC free article] [PubMed] [Google Scholar]
- Oh B-K, Apirion D. 10Sa RNA, a small stable RNA of Escherichia coli, is functional. Mol Gen Genet. 1991;221:52–56. doi: 10.1007/BF00264212. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Silber KR, Sauer RT. Carboxy-terminal determinants of intracellular protein degradation. Genes & Dev. 1990;4:277–286. doi: 10.1101/gad.4.2.277. [DOI] [PubMed] [Google Scholar]
- Retallack DM, Friedman DI. A role for a small stable RNA in modulating the activity of DNA-binding proteins. Cell. 1995;83:227–235. doi: 10.1016/0092-8674(95)90164-7. [DOI] [PubMed] [Google Scholar]
- Retallack DML, Thompson LL, Friedman DI. Role for 10Sa RNA in the growth of λ-P22 hybrid phage. J Bacteriol. 1994;176:2082–2089. doi: 10.1128/jb.176.7.2082-2089.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg M, Court D, Shimatake H, Brady C. The relationship between function and DNA sequence in an intercistronic regulatory region in phage λ. Nature. 1978;272:414–423. doi: 10.1038/272414a0. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Pabo CO, Meyer BJ, Ptashne M, Backman KC. The regulatory functions of the λ repressor reside in the amino terminal domain. Nature. 1979;279:396–400. doi: 10.1038/279396a0. [DOI] [PubMed] [Google Scholar]
- Silber KR, Sauer RT. Deletion of the prc (tsp) gene provides evidence for additional tail-specific proteolytic activity in Escherichia coli K-12. Mol Gen Genet. 1994;242:237–240. doi: 10.1007/BF00391018. [DOI] [PubMed] [Google Scholar]
- Silber KR, Keiler KC, Sauer RT. Tsp: A tail-specific protease that selectively degrades proteins with nonpolar C-termini. Proc Natl Acad Sci. 1992;89:295–299. doi: 10.1073/pnas.89.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly WS, Rabideau K. Mechanism of λc17cI virulence. J Mol Biol. 1969;42:385–400. doi: 10.1016/0022-2836(69)90231-9. [DOI] [PubMed] [Google Scholar]
- Squires C, Squires CL. The Clp proteins: Proteolysis regulators or molecular chaperones? J Bacteriol. 1992;174:1081–1085. doi: 10.1128/jb.174.4.1081-1085.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-End rule in bacteria. Science. 1991;254:1374–1376. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman AJ, Oppenheim AB, Yura T, Yamanaka K, Miki H, Hiraga S, Ogura T. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor σ32. EMBO J. 1995;14:2551–2560. doi: 10.1002/j.1460-2075.1995.tb07253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempy JE, Kirby JE, Gottesman S. Alp suppression of Lon: Dependence on the slpA gene. J Bacteriol. 1994;176:2061–2067. doi: 10.1128/jb.176.7.2061-2067.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu G-F, Reid GE, Zhang J-G, Moritz RL, Simpson RJ. C-terminal extension of truncated recombinant proteins in Escherichia coli with a 10Sa RNA decapeptide. J Biol Chem. 1995;270:9322–9326. doi: 10.1074/jbc.270.16.9322. [DOI] [PubMed] [Google Scholar]
- Vogel JL, Geuskens V, Desmet L, Higgins NP, Toussaint A. C-terminal deletions can suppress temperature-sensitive mutations and change dominance in the phage Mu repressor. Genetics. 1996;142:661–672. doi: 10.1093/genetics/142.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi MR. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad. 1994;91:12218–12222. doi: 10.1073/pnas.91.25.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KP, Bartel DP. Phylogenetic analysis of tmRNA secondary structure. RNA. 1996;2:1306–1310. [PMC free article] [PubMed] [Google Scholar]
- Wojtkowiak D, Georgopoulos C, Zylicz M. ClpX, a new specificity component of the ATP-dependent Escherichia coli Clp protease, is potentially involved in λ DNA replication. J Biol Chem. 1993;268:22609–22617. [PubMed] [Google Scholar]