

Abstract
Expression of c-Myc sensitizes cells to a wide range of pro-apoptotic stimuli. We here show that this pro-apoptotic effect is mediated through release of mitochondrial holocytochrome c into the cytosol. First, activation of c-Myc triggers release of cytochrome c from mitochondria. This release is caspase-independent and blocked by the survival factor IGF-1. Second, c-Myc-induced apoptosis is blocked by microinjection of anticytochrome c antibody. In addition, we show that microinjection of holocytochrome c mimics the effect of c-Myc activation, sensitizing cells to DNA damage and to the CD95 pathway. Both p53 and CD95/Fas signaling have been implicated in c-Myc-induced apoptosis but neither was required for c-Myc-induced cytochrome c release. Nonetheless, inhibition of CD95 signaling in fibroblasts did prevent c-Myc-induced apoptosis, apparently by obstructing the ability of cytosolic cytochrome c to activate caspases. We conclude that c-Myc promotes apoptosis by causing the release of cytochrome c, but the ability of cytochrome c to activate apoptosis is critically dependent upon other signals.
Keywords: c-Myc, cytochrome c, apoptosis
The c-Myc protein, encoded by the c-myc proto-oncogene, is both a potent inducer of cell proliferation and of apoptosis (Askew et al. 1991; Evan et al. 1992). The pro-apoptotic property of c-Myc is shared with other mitogenic oncoproteins such as E1A (White et al. 1991) and is thought to act as a built-in restraint to the emergence of neoplastic clones within the soma (Harrington et al. 1994a; Evan and Littlewood 1998; Hueber and Evan 1998).
c-Myc resembles transcription factors of the basic helix–loop–helix leucine zipper (bHLH–LZ) family and exhibits sequence-specific DNA binding when dimerized with its partner Max. Although mutagenesis studies are consistent with the notion that c-Myc exerts its biological effects as a transcription factor, the mechanism by which c-Myc exerts its biological effects remains obscure. Regions of the protein required for induction of cell proliferation coincide with those needed for apoptosis and include all the requisite motifs characteristic of bHLH–LZ transcription factors. However, c-Myc target genes have not been well defined. In particular, it is not known whether proliferation and apoptosis are mediated by the same, overlapping, or discrete sets of genes.
Nonetheless, substantial evidence indicates that c-Myc-induced apoptosis and mitogenesis are discrete downstream programs, neither of which is necessarily dependent upon the other. Thus, activation of the molecular machinery mediating cell-cycle progression is not required for c-Myc-induced apoptosis (Rudolph et al. 1996). Furthermore, c-Myc-induced apoptosis in serum-deprived fibroblasts is inhibited by survival factors such as insulin-like growth factor 1 (IGF-1) that exert little, if any, mitogenic effect on such cells (Harrington et al. 1994b). Likewise, the apoptosis suppressor Bcl-2 inhibits c-Myc-induced apoptosis (Bissonnette et al. 1992; Fanidi et al. 1992; Wagner et al. 1993) without any measurable effect on the oncoprotein’s mitogenic activity (Fanidi et al. 1992).
One intriguing possibility is that c-Myc does not itself induce apoptosis but rather acts to sensitize cells to other pro-apoptotic insults. Indeed, c-Myc expression has been shown to sensitize cells to a wide range of mechanistically distinct insults such as serum or growth-factor deprivation (Askew et al. 1991; Evan et al. 1992), nutrient privation (Evan et al. 1992), hypoxia (Alarcon et al. 1996), p53-dependent response to genotoxic damage (Evan et al. 1992), virus infection (Cherney et al. 1994), interferons (Evan et al. 1992; Bennett et al. 1994), tumor necrosis factor (TNF) (Klefstrom et al. 1994), and CD95/Fas (Hueber et al. 1997), many of which have no obvious effect on cell proliferation. For c-Myc to act as a sensitizer to so many disparate triggers of apoptosis it must act presumably at some common node in the regulatory and effector machinery of apoptosis.
One frequent feature of apoptosis is the early translocation of holocytochrome c (hcC) from mitochondria to the cytosol. The mechanism by which this release occurs, and its relationship with other mitochondrial changes such as opening of the mitochondrial permeability transition pore and/or collapse of the inner membrane potential (for review, see Green and Reed 1998), are still obscure. In contrast, the way in which hcC activates the apoptotic machinery is reasonably well documented. Elegant experiments using cell-free systems have shown that hcC interacts with Apaf-1, a mammalian homolog of the Caenorhabditis elegans Ced4 adaptor protein (Zou et al. 1997), which then recruits and activates pro-caspase 9 (P. Li et al. 1997). This ternary complex, or ‘apoptosome’ triggers ATP-dependent autocatalytic processing of caspase 9 which, in turn, activates caspase 3 and other effector caspases.
Much evidence now favors the idea that key effectors mediating hcC release are ‘BH3 proteins’—a heterologous family of pro-apoptotic proteins that share the BH3 homology domain with Bcl-2 and probably act by interfering with Bcl-2 protective function (for review, see Kelekar and Thompson 1998). This is consistent with observations that one of the anti-apoptotic functions of Bcl-2 family members is to block hcC release (Kharbanda et al. 1997; Kluck et al. 1997; Yang et al. 1997b; for review, see Green and Reed 1998). Understanding the molecular mechanism by which Bcl-2 blocks apoptosis is of fundamental importance as it underlies the oncogenic synergy between Bcl-2 and c-Myc (Strasser et al. 1990) which arises because Bcl-2 blocks c-Myc-induced apoptosis specifically without significantly affecting c-Myc-induced proliferation (Bissonnette et al. 1992; Fanidi et al. 1992; Wagner et al. 1993). Bcl-2 family proteins are also key downstream targets of survival-signaling pathways, such as that initiated by IGF-1, which also inhibit oncogene-induced apoptosis (Harrington et al. 1994b; Evan and Littlewood 1998). Activation of the IGF-1 receptor tyrosine kinase triggers a survival-signal routing through Ras, PI3-kinase, and the serine/threonine kinase PKB/Akt (Kauffmann-Zeh et al. 1997; Kulik et al. 1997), which then phosphorylates and functionally inactivates Bad, a BH3 protein that antagonizes Bcl-2 (Datta et al. 1997; del Peso et al. 1997).
In this paper we have examined the role of cytochrome c in c-Myc-induced, CD95-dependent apoptosis of fibroblasts. Our studies reveal that a key pro-apoptotic action of c-Myc is to cause release into the cytosol of hcC, a release that is blocked by the survival factor IGF-1 but not by inhibition of CD95- or p53-signaling pathways. We also show that cytosolic hcC is, by itself, a poor inducer of apoptosis in fibroblasts that can cooperate with other triggers of apoptosis such as the p53-dependent response to X-irradiation or CD95 engagement. In fibroblasts, CD95 signaling is required for efficient activation of the apoptotic caspase machinery following c-Myc-induced cytochrome c release or microinjection of pure cytochrome c.
Results
c-Myc triggers cytochrome c release from mitochondria, which is inhibited by IGF-1 signaling
Activation of c-Myc induces apoptosis in serum-deprived Rat-1 fibroblasts (Evan et al. 1992). Because of the emerging role of cytosolic cytochrome c in apoptosis, we investigated the effect c-Myc might have on the intracellular localization of cytochrome c in low-serum conditions using Rat-1 fibroblasts that express a conditional 4-hydroxytamoxifen (OHT)-dependent c-Myc protein (Rat-1/c-MycER, Littlewood et al. 1995). Rat-1/c-MycER cells were serum-deprived for 48 hr and subcellular fractions prepared at various time points following activation of c-Myc. Cytochrome c was then assayed by immunoblotting in both the heavy membrane pellet, containing the crude mitochondrial fraction, and in the residual cytosolic supernatant. c-Myc activation had no discernible effect on the total level of cytochrome c protein in the cell (data not shown). However, within 8 hr of c-Myc activation, a significant proportion of cytochrome c had been lost from the heavy membrane fraction into the cytosol (Fig. 1A, see also Fig. 4C, below). Cytochrome c progressively accumulated in the cytosolic fraction until, by 24 hr after the onset of c-Myc activation, very little of the total cellular cytochrome c remained in the mitochondrial pellets. As a control, we also assayed the distribution of the mitochondrial inner membrane protein, cytochrome c oxidase subunit IV. As expected, the great majority of this protein fractionates with the insoluble mitochondrial fraction and this distribution was unaffected by c-Myc activation (Fig. 1B). The small, constant amount of cytochrome c oxidase IV present in the soluble fractions may arise from leakage caused by unavoidable disruption of mitochondria during fractionation.
Figure 1.


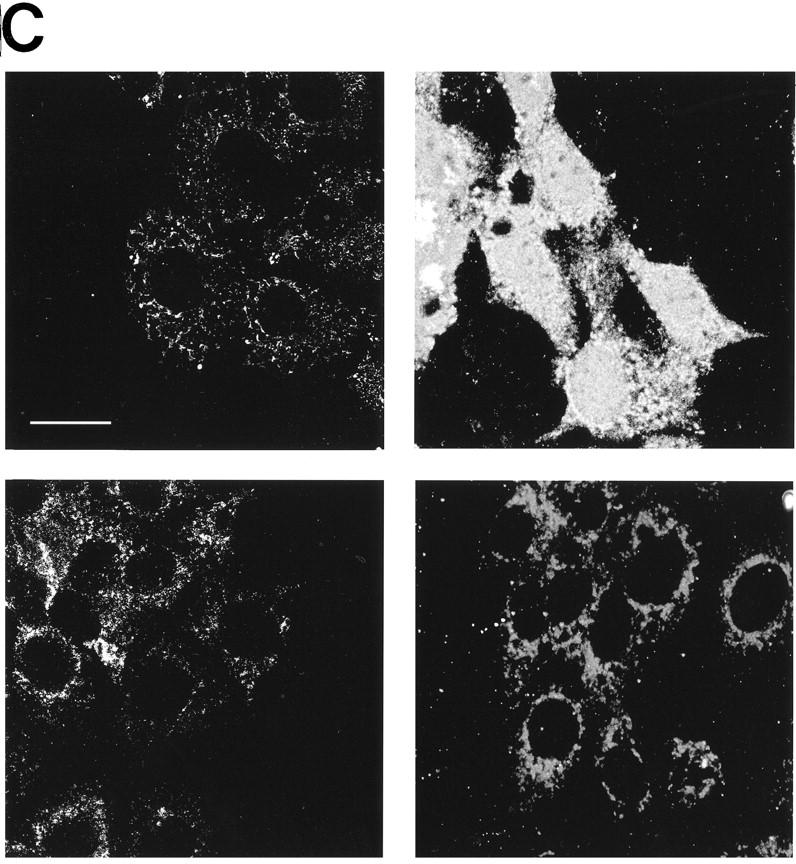

c-Myc induces cytochrome c release from mitochondria in serum-deprived fibroblasts. (A) Serum-deprived Rat-1/c-MycER fibroblasts were treated with OHT (100 nm) in the presence or absence of IGF-1. IGF-1, if present, was added at the same time as OHT. At the indicated time points, subcellular fractionation was performed as described in Materials and Methods. The heavy membrane fraction (HMF; 10 μg of protein), containing crude mitochondria, and the postmitochondrial fraction (supernatant; S; 20 μg of protein) were analyzed for cytochrome c content by SDS-PAGE and immunoblotting with anti-cytochrome c antibody. (B) Rat-1/c-MycER fibroblasts were treated for 24 hr and subcellular fractionation was performed as in A. The heavy membrane (HMF; 10 μg of protein) and postmitochondrial fractions (supernatant; S; 20 μg of protein) were analyzed by SDS-PAGE analysis and immunoblotting for cytochrome c oxidase subunit IV. (C) Serum-deprived Rat-1/cMycER fibroblasts were treated as in A for 24 hr prior to fixation, immunostaining, and confocal-laser-scanning analysis as described in Materials and Methods. (Top left) Rat-1/c-MycER fibroblasts were left untreated and stained with anti-cytochrome c antibody; (top right) Rat-1/c-MycER fibroblasts were treated with OHT for 24 hr and stained with anti-cytochrome c antibody; (bottom left) Rat-1/c-MycER fibroblasts were treated with OHT and IGF-1 for 24 hr and stained with anti-cytochrome c antibody; (bottom right) Rat-1/c-MycER fibroblasts were treated with OHT for 24 hr and stained with anti-cytochrome c oxidase subunit IV antibody. Projections of 20 Z-sections (0.5-μm steps) ranging from the bottom to the top of the cells are shown. Bar = 20 μm. (D) Serum-deprived Rat-1/c-MycΔ(106–143)ER fibroblasts were treated with OHT. Subcellular fractionation and subsequent analysis of cytochrome c content in the heavy membrane fraction were performed as described in A at the indicated time points.
Figure 4.





Cytochrome c release induced by c-Myc is neither blocked by dominant-negative mutants of p53 or FADD nor by the caspase inhibitor zVAD.fmk (A) Effect of p53min on apoptosis induced by c-Myc. (▵) Rat-1/cMycER and (○) Rat-1/c-MycER/p53min cells were serum deprived for 48 hr and c-Myc activated by the addition of OHT. The fate of the cells was followed by time-lapse videomicroscopy at one frame every 3 min and the results are expressed as cumulative cell deaths against time. (B) p53min does not inhibit c-Myc-induced cytochrome c release. Serum-deprived Rat-1/c-MycER/p53min fibroblasts were treated with OHT for the indicated time. Subcellular fractionation and immunoblot analysis of cytochrome c content in the heavy membrane fraction (HMF) and the post-mitochondrial fraction (supernatant; S) were then performed as described in Fig. 1A. (C) zVAD.fmk does not inhibit c-Myc-induced cytochrome c release. Subcellular fractionation and subsequent immunoblot analysis of serum-deprived Rat-1/c-MycER fibroblasts treated with OHT was performed as described in Fig. 1A. Where indicated, zVAD.fmk (100 μm) was added at the same time as OHT. (D) Inhibition of c-Myc-induced apoptosis in Rat-1 fibroblasts by a dominant-negative mutant of FADD. (○) Rat-1/c-MycER − OHT, (●) Rat-1/c-MycER + OHT, (▴) Rat-1/c-MycER/FADD DN − OHT, or (▵) Rat-1/c-MycER-FADD DN + OHT fibroblasts were serum-deprived and cell-death analyzed following activation of c-Myc with 100 nm OHT by phase-contrast time-lapse microscopy. (E) A dominant-negative mutant of FADD does not inhibit c-Myc-induced cytochrome c release. Serum-deprived Rat-1/c-MycER/FADD DN fibroblasts were treated with OHT for the indicated time. Subcellular fractionation and immunoblot analysis of cytochrome c content in the heavy membrane fraction (HMF) and the postmitochondrial fraction (supernatant; S) were then performed as described in Fig. 1A.
To confirm our biochemical data, we next analyzed cytochrome c subcellular localization immunocytochemically following activation of c-Myc in low serum. Cells were fixed and stained with monoclonal anti-cytochrome c antibody 24 hr after treatment with OHT. Most untreated cells (c-Myc inactive) exhibited a thread-like cytoplasmic staining consistent with a mitochondrial localization for cytochrome c (Fig. 1C, top left). In contrast, the great majority of cells expressing activated c-Myc for 24 hr exhibited a markedly more diffuse fluorescence, sometimes filling the entire cell, with no evidence of particulate mitochondrial localization (Fig. 1C, top right). Staining of analogous cells with anti-cytochrome c oxidase subunit IV antibody showed a particulate mitochondrial pattern indicating that gross mitochondrial integrity is maintained at this time (Fig. 1C, bottom right). Whereas diffuse staining of cytochrome c was observed in all dead cells, costaining with the DNA marker Hoechst 33342 showed it to be evident in a substantial proportion of cells with apparently normal nuclear morphology (data not shown; note also that cells in Fig. 1C, top right, exhibit normal cell shape). This implies that cytochrome c redistribution precedes nuclear collapse. We conclude that the release of cytochrome c into the cytosol induced by c-Myc occurs before gross apoptotic collapse and without general disruption of mitochondrial structure.
The transcriptionally inactive mutant c-MycΔ(106–143) is unable to trigger cell death in serum-deprived fibroblasts (Evan et al. 1992; Amati et al. 1993). We therefore used this mutant to determine whether c-Myc-induced release of mitochondrial cytochrome c is likely to involve a transcriptional mechanism. Rat-1 fibroblasts expressing a notionally conditional OHT-dependent form of c-MycΔ(106–143) [Rat-1/c-MycΔ(106–143)ER cells] were serum deprived, OHT was added for different periods, and the amount of cytochrome c present in the crude mitochondrial fraction was assayed. Addition of OHT exerted no effect on levels of mitochondrial cytochrome c (Fig. 1D). Thus, the transcriptionally inactive c-MycΔ(106–143) mutant does not trigger hcC release from mitochondria. Incidentally, this also provides confirmation that changes observed in cytochrome c distribution in Rat-1/c-MycER cells are not merely an artifact of OHT treatment.
c-Myc-induced apoptosis in fibroblasts is potently inhibited by the survival factor IGF-1 (Harrington et al. 1994b), probably acting via the PI3 kinase/Akt pathway (Kauffmann-Zeh et al. 1997). We therefore examined whether IGF-1 exerted any effect on c-Myc-induced hcC release from mitochondria. Levels of IGF-1 effective in preventing c-Myc-induced apoptosis and c-Myc-induced caspase activation (see Fig. 5B, below) efficiently inhibited release of cytochrome c from mitochondria and consequent accumulation in the cytosol following c-Myc activation in low serum (Fig. 1A). In contrast, IGF-1 exerted no effect on the distribution of cytochrome c oxidase subunit IV localization, which remained principally mitochondrial throughout (Fig. 1B). The effect of IGF-1 was confirmed immunocytochemically: IGF-1 suppressed onset of diffuse cytochrome c staining effectively following c-Myc activation. Cells retained a particulate subcellular localization of cytochrome c (Fig. 1C, bottom left). Taken together, these data show that c-Myc induces release of cytochrome c from mitochondria and that this is inhibited by the survival factor IGF-1.
Figure 5.


A dominant-negative mutant of FADD confers increased resistance to microinjected cytochrome c and inhibits c-Myc-induced caspase activation. (A) (●) Rat-1/c-MycER and (○) Rat-1/c-MycER/FADD DN fibroblasts grown in 10% FCS were microinjected with 25 μm pure hcC mixed with dextran-coupled rhodamine dye and the number of injected cells then evaluated by fluorescence microscopy. Cells were incubated at 37°C and, at the indicated time points, the number of nonapoptotic cells was evaluated and expressed as percentage of the initial number of injected cells. Results presented are the mean ±s.e.m. of five independent experiments, each of ∼100 injected cells. (B) Rat-1/c-MycER and Rat-1/c-MycER/FADD DN fibroblasts were serum-deprived prior to addition of OHT (100 nm). Where indicated, IGF-1 (100 ng/ml) was added simultaneously. Twenty-four hours later, postmitochondrial fractions were prepared and assayed for DEVD cleaving activity as described in Materials and Methods. Results presented are the mean ±s.e.m. of at least three independent experiments.
Microinjected anti-cytochrome c antibodies inhibit c-Myc-induced apoptosis
To analyze the requirement for cytosolic hcC in c-Myc-induced apoptosis we asked whether introduction of blocking anti-cytochrome c antibodies would interfere with c-Myc-induced cell death in low serum. We used purified anti-cytochrome c antibody 6H2B4, which has been shown previously to inhibit activation of caspases by hcC in a cell-free system and to block death upon microinjection into rat neurons (Neame et al. 1998). Serum-deprived Rat-1/c-MycER fibroblasts were injected with either anti-cytochrome c antibody or with a control isotypic antibody. c-Myc was then activated by addition of OHT, and the rate of cell death among injected cells analyzed by phase and fluorescence microscopy at the indicated time points. As shown in Figure 2, microinjection with anti-cytochrome c antibody blocked apoptosis induced by c-Myc significantly. Thus, accumulation of cytosolic hcC is necessary for c-Myc-induced apoptosis.
Figure 2.

Microinjected anti-cytochrome c antibodies inhibit c-Myc-induced cell death. Serum-deprived Rat-1/c-MycER fibroblasts were injected with either control or anti-cytochrome c antibodies (20 mg/ml) mixed with dextran-conjugated rhodamine dye. (□) Control antibodies; (█) anti-cytochrome c antibodies. Six hours later, the number of injected cells was evaluated, OHT (100 nm) was added and the cells were then incubated at 37°C. At the indicated time points, cells were analyzed by fluorescence microscopy and the percentage of nonapoptotic cells was determined. Results are means ±s.e.m. of 4 (control antibody) and 6 (anti-cytochrome c antibody) independent experiments, each involving ∼70 cells.
Cytosolic holocytochrome c triggers apoptosis
We investigated whether increasing the levels of cytosolic hcC might be the mechanism by which c-Myc promotes apoptosis. We therefore analyzed the effect of direct introduction of hcC into the cytosol by microinjection. We used standardized conditions to microinject pure bovine hcC into the cytoplasm of Rat-1/c-MycER fibroblasts. As shown in Figure 3 (A,B), cytoplasmic microinjection of hcC at a concentration of 25 μm induces morphological changes characteristic of fibroblast apoptosis (McCarthy et al. 1997): A sudden onset of membrane blebbing, followed by cell shrinkage and rounding of the cell body, nuclear pyknosis, and chromatin condensation (data not shown).
Figure 3.
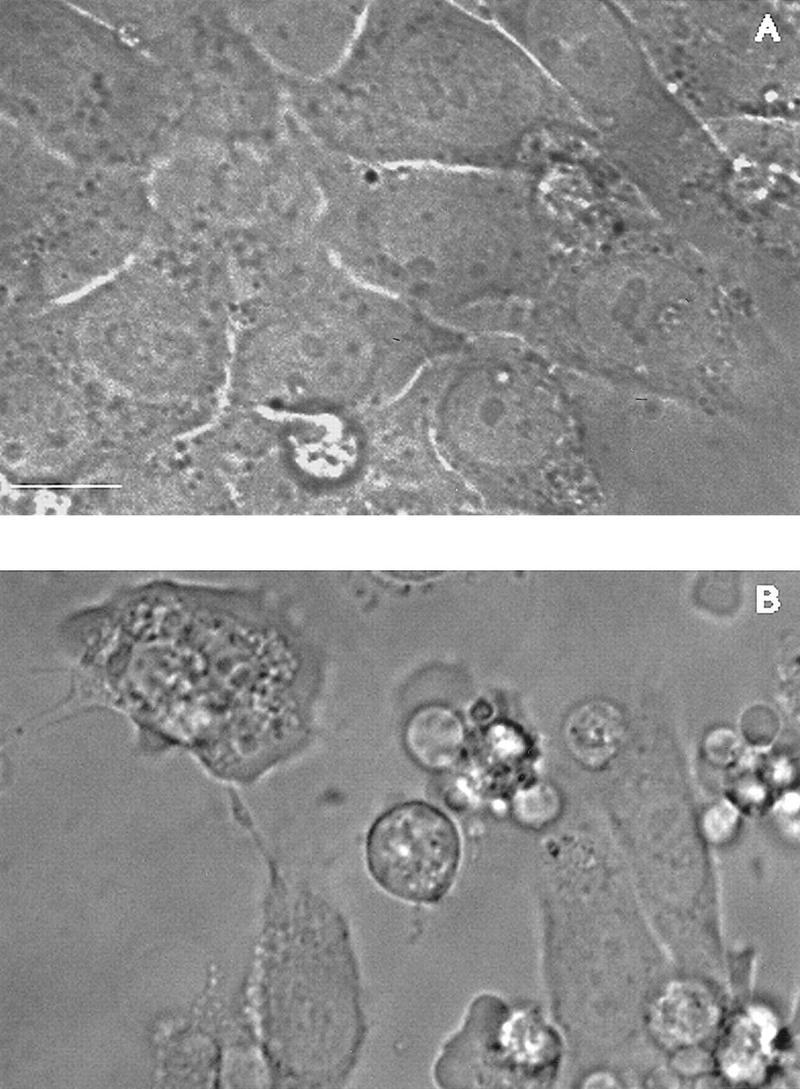



Microinjected cytochrome c induces apoptosis. (A,B) Morphology of Rat-1/c-MycER fibroblasts following cytoplasmic microinjection of cytochrome c. Rat-1/c-MycER fibroblasts grown in 10% FCS in the absence of OHT were injected with either 25 μm pure cytochrome c (B) or water (A) and incubated for 2 hr at 37°C prior to microscopic analysis. Bar = 20 μm. (C) Cytochrome c-induced apoptosis is dose dependent. Rat-1/c-MycER fibroblasts grown in 10% FCS were microinjected with pure cytochrome c (hcC) at various concentrations mixed with dextran-conjugated rhodamine dye. (█) 2μm hcC; (●) 10 μm hcC; (▴) 25 μm hcC; (▵) 25μm hcC + zVad.fmk. Injected cells were then followed by time-lapse phase and fluorescence microscopy as described in Materials and Methods. The fates of 25 cells, picked randomly from a frame containing ∼50 injected cells, were analyzed. The onset of apoptosis was scored as the start of membrane blebbing and the end point of cell death was scored as the time of cell detachment from substratum. The time between these two events is represented for each individual cell death by the length of the horizontal line. These data are representative of at least three independent experiments. Where indicated, zVAD.fmk (100 μm) was added 1 hr prior to microinjection. (D) Microinjected cytochrome c-induced apoptosis is independent of c-Myc activity. Rat-1/c-MycER fibroblasts grown in 10% FCS were microinjected with hcC (10 μm) mixed with dextran-conjugated rhodamine dye. Where indicated (□), OHT (100 nm) was added 2 hr prior to microinjection. (█) No OHT added. Microinjected cells were followed by time-lapse phase and fluorescence microscopy and scored for apoptosis as described in Materials and Methods. The number of cell deaths is expressed as a percentage of the total number of viable injected cells present in the entire frame at the beginning of the time lapse experiment (∼50 cells). Data shown are representative of at least three independent experiments. (E) IGF-1 does not protect against microinjected cytochrome c-induced apoptosis. Experiments were performed as described in D except that Rat-1/c-MycER fibroblasts serum deprived for 48 hr prior to microinjection were used. (█) IGF-1 (100 ng/ml) was added 2 hr prior to microinjection. (□) No IGF-1 added. Data shown are representative of at least three independent experiments.
To study more precisely the effect of cytosolic hcC, cells were microinjected with pure hcC, at a concentration of 2–25 μm, mixed with a neutral Dextran-conjugated Rhodamine marker and followed by time-lapse phase and fluorescence microscopy. We determined that in each individual microinjected cell, the time taken to proceed from the onset of membrane blebbing to gross cell fragmentation, a crude measure of the length of each apoptotic event, was between 30 and 100 min (Fig. 3C). This interval is essentially identical to that observed in individual apoptotic cell deaths induced by c-Myc (McCarthy et al. 1997). Strikingly, once initiated, the duration of each apoptotic event was unaffected by the amount of hcC introduced into each cell (Fig. 3C). In complete contrast, the amount of hcC introduced into each cell influenced greatly the rapidity of onset of apoptosis, as judged by the start of membrane blebbing: More hcC triggered a more rapid onset. Nonetheless, even clonally identical cells microinjected with ostensibly identical amounts of the fluorescent mixture exhibited asynchronous onset of membrane blebbing (Fig. 3C).
To analyze whether hcC-induced cell death is caspase dependent, we incubated cells with the permeable caspase inhibitor benzyl oxycarbonyl-Val-Ala-Asp (O-methyl) fluoromethylketone zVAD.fmk prior to microinjection with hcC at a concentration of 25 μm and then followed them by time-lapse fluorescence microscopy. As shown in Figure 3C, (zVAD.fmk) effectively inhibits hcC-induced cell death. Indeed, many zVAD.fmk-treated, microinjected cells maintained a normal morphology throughout the course of the experiment and some even divided (data not shown). In addition, those few cells that did initiate membrane blebbing in the presence of zVAD.fmk exhibited a substantial increase in the length of the effector phase of apoptosis, characteristic of caspase inhibition in other systems (McCarthy et al. 1997). We conclude that microinjected hcC activates caspase-dependent apoptosis.
To test whether c-Myc has any effect on the efficiency with which microinjected hcC induces apoptosis, Rat-1/c-MycER cells were treated with OHT to activate c-Myc, microinjected with holo-cytochrome c at a concentration of 10 μm 2 hr later, and then followed by time-lapse fluorescence microscopy. c-Myc status was found to cause no discernible sensitization to cell death induced by microinjected hcC (Fig. 3D). A similar result was obtained even if c-Myc was activated 10 hr prior to injection or under reduced serum conditions (data not shown). We conclude that c-Myc does not sensitize cells to cytosolic hcC. This is consistent with the notion that c-Myc exerts its principle pro-apoptotic effect by causing hcC release from mitochondria.
We also analyzed the effect of IGF-1 on microinjected hcC. Rat-1/c-MycER cells were serum-deprived for 48 hr and injected with cytochrome c, either in the absence or the presence of IGF-1 at a level sufficient to block c-Myc-induced apoptosis. We observed IGF-1 exerts no discernible suppression of apoptosis induced by microinjected cytochrome c (Fig. 3E). This is consistent with IGF-1 acting to suppress c-Myc-induced apoptosis principally by blocking the release of cytochrome c from mitochondria.
c-Myc-induced release of cytochrome c does not require CD95 signaling or p53
Both p53 (Hermeking and Eick 1994; Wagner et al. 1994) and CD95 signaling (Hueber et al. 1997) have been implicated in c-Myc-induced apoptosis in fibroblasts. We therefore investigated whether either p53 or CD95 signaling was required for c-Myc-induced release of hcC.
The role of p53 was investigated by using a carboxy-terminal fragment of p53 (p53min; amino acids 302–390) which has been shown to act as an effective dominant interfering mutant of p53 function (Shaulian et al. 1992). When expressed in Rat-1 fibroblasts, p53min blocks X-ray-induced apoptosis efficiently (data not shown), consistent with its described activity as an inhibitor of wild-type p53. However, p53min expression did not inhibit c-Myc-induced apoptosis measurably (Fig. 4A). We also observed no detectable effect of p53min expression on c-Myc-induced cytochrome c release assessed by subcellular fractionation (Fig. 4B). Thus, p53 is not required for c-Myc-induced release of cytochrome c from mitochondria.
The CD95/Fas pathway activates caspases—in part through the activation of the upstream caspase 8 via interaction with CD95 and the Fas-associated death domain adaptor (FADD) within the death-induced signaling complex (DISC). Caspase activation can then lead to release of cytochrome c (Vander Heiden et al. 1997), at least in part through cleavage of the pro-apoptotic BH3 protein Bid (Li et al. 1998; Luo et al. 1998). We therefore investigated whether caspase activity is required for c-Myc to induce release of cytochrome c using the broad-spectrum caspase inhibitor zVAD.fmk, which blocks all discernible caspase-dependent processes in c-Myc-induced apoptosis (McCarthy et al. 1997). Analysis of cytochrome c localization by subcellular fractionation revealed that treatment with zVAD.fmk of serum-deprived Rat-1/c-MycER fibroblasts does not affect cytosolic accumulation of hcC induced by c-Myc (Fig. 4C). We conclude that cytochrome c release during c-Myc-induced apoptosis is caspase independent.
c-Myc-induced apoptosis in fibroblasts is inhibited profoundly by interference with the CD95/Fas signaling pathway, for example by blockading CD95 receptor ligation or by expression of a dominant interfering mutant of the FADD adaptor protein (FADD DN) (Hueber et al. 1997). To examine further the role of CD95 signaling in c-Myc-induced release of hcC we expressed FADD DN in Rat-1/c-MycER cells and examined the localization of hcC following c-Myc activation in low serum. As reported before (Hueber et al. 1997), expression of FADD DN in Rat-1/c-MycER fibroblasts (Rat-1/c-MycER/FADD DN) confers almost complete resistance to c-Myc-induced apoptosis in low serum (Fig. 4D) over the 24 hr of c-Myc activation. However, in such cells FADD DN exerted no detectable inhibitory effect on c-Myc-induced loss of hcC from mitochondria and its concomitant accumulation in the cytosol (Fig. 4E), a conclusion verified immunocytochemically (data not shown). Thus FADD DN inhibition of CD95 signaling protects cells from c-Myc-induced apoptosis but does not prevent c-Myc-induced release of mitochondrial cytochrome c.
FADD DN suppresses downstream activation of caspases by c-Myc and by cytosolic hcC
If the CD95 signaling pathway is not involved in release of hcC, how does it interfere with c-Myc-induced apoptosis? One possibility is that it acts to interfere with activation of the apoptotic program downstream of hcC release. To investigate this, Rat-1/c-MycER cells, either with or without FADD DN expression, were microinjected with 25 μm cytochrome c and the percentage of dead cells evaluated at the indicated time points using fluorescence microscopy. As shown in Figure 5A, expression of FADD DN inhibits apoptosis induced by microinjected hcC markedly. One possible reason for this is that in the presence of FADD DN, hcC is unable to activate the apoptotic caspase machinery. We therefore assayed the effect of FADD DN on generation of caspase activity directly in cytosolic extracts from cells 24 hr following activation of c-Myc in serum-deprived Rat-1/c-MycER fibroblasts using colorimetric peptide caspase substrates. c-Myc activation did not cause any measurable increase in YVAD cleaving activity, in agreement to the report by Kagaya et al. (1997). However, c-Myc activation led to a profound increase in DEVD cleaving activity that was blocked by addition of IGF-1 to the growth medium over the period of c-Myc activation (Fig. 5B). Expression of FADD DN also suppressed emergence of DEVD cleaving activity in response to c-Myc activation completely (Fig. 5B). In conclusion, our data show that although FADD DN blocks c-Myc-induced apoptosis, it has no effect on accumulation of cytochrome c in the cytosol. Rather, it acts to obstruct cytosolic cytochrome c from activating downstream caspases.
Cytosolic cytochrome c sensitizes Rat-1 cells to X irradiation and CD95 death signaling
Our data suggest that neither CD95 nor p53 participate directly in the main pro-apoptotic function of c-Myc. Rather, they raise the intriguing possibility that apoptosis may result from the cooperation between either of these two signaling pathways and c-Myc-induced hcC release. To investigate this possibility further we microinjected cells with an amount of hcC that induces little apoptosis on its own and assessed their sensitivity to both CD95 and p53-dependent killing.
Rat-1/c-MycER fibroblasts are normally refractory to CD95 killing but become sensitive upon activation of c-Myc (Hueber et al. 1997). To determine whether hcC confers similar sensitization, Rat-1/c-MycER cells were microinjected with hcC plus fluorescent marker, in the absence of OHT (c-Myc inactive) and followed by time-lapse phase and fluorescence microscopy as described above. CD95 was then activated by addition of human recombinant soluble CD95 ligand (CD95Ls) as described previously (Hueber et al. 1997). This treatment by itself did not trigger detectable changes of cytochrome c localization as assessed by immunostaining (data not shown). Mock-injected cells remained largely refractory to killing by CD95Ls: After 24 hr incubation with CD95Ls <20% of mock-injected cells were dead (data not shown). In contrast, addition of CD95Ls to cells microinjected with hcC (10 μm) triggered widespread apoptosis compared with that observed in hcC-injected cells not treated with CD95Ls (Fig. 6A). Moreover, the sensitization to CD95Ls afforded by microinjected cytochrome c is dose-dependent. Cells injected with 10 μm hcC exhibited obvious sensitization within 12 hr of CD95Ls treatment, whereas cells injected with 2 μm exhibited no significant CD95Ls-dependent cell death over the same period (Fig. 6A), although a significant increase in cell death was observed over controls after 24 hr (data not shown). The dose-dependency of hcC sensitization to CD95 killing confirms that sensitization is mediated by cytochrome c and not a trivial consequence of microinjection, introduction of the fluorescent marker dye, and/or sustained illumination during time-lapse videomicroscopy. We conclude that cytosolic hcC sensitizes Rat-1 fibroblasts to CD95-induced apoptosis in a dose-dependent manner.
Figure 6.




Microinjected cytochrome c cooperates with CD95 stimulation and X-irradiation. (A) Dose-dependent sensitization to CD95 ligation by microinjected cytochrome c. Rat-1/c-MycER fibroblasts were injected with either 2 or 10 μm pure hcC mixed with dextran-conjugated rhodamine dye. Where indicated, CD95Ls (50 ng/ml) was added immediately after injection. (▵) 10 μm hcC + CD95Ls, (▴) 10 μm hcC − CD95Ls, (○) 2 μm hcC + CD95Ls, (●) 2 μm hcC − CD95Ls. Cell death was scored as described in Fig. 3D. Data shown are representative of four (10 μm hcC ± CD95Ls) and three (2 μm hcC ± CD95Ls) independent experiments, each involving ∼50 injected cells. (B) Sensitization to X-irradiation by c-Myc. Rat-1/c-MycER fibroblasts were grown in 2% FCS for 48 hr. X-ray irradiation was then performed as described in Materials and Methods. (○) OHT (100 nm) added just after irradiation; (●) OHT not added. (C) Sensitization to X-irradiation by microinjected cytochrome c. Rat-1/c-MycER fibroblasts were injected with 10 μm pure hcC mixed with dextran-conjugated rhodamine dye or with the dye alone. Where indicated, cells were exposed to X-rays, as described in Materials and Methods, immediately following microinjection. (▵) 10 μm hcC, (○) no hcC + 10 Gy, (●) 10 μm hcC + 10 Gy. Cells were then incubated at 37°C and, at the indicated time points after irradiation, the number of apoptotic cells was evaluated and expressed as percentage of the initial number of injected cells. Results presented are representative of at least five independent experiments, each involving ∼100 injected cells. (D) Sensitization to X-irradiation by microinjected cytochrome c is inhibited by p53min. Rat-1/c-MycER/p53 min fibroblasts were injected with 10 μm and exposed to X-rays as described in C. Results presented are representative of at least five independent experiments, each involving ∼100 injected cells. (▵) 10 μm hcC, (●) 10 μm hcC + 10 Gy.
We next asked if cytosolic hcC sensitizes cells to p53-dependent killing. Rat-1/c-MycER fibroblasts were X-irradiated with 10 Gy, a dose that induces little apoptosis unless c-Myc is activated (Fig. 6B). This dose has, by itself, no detectable effect on cytochrome c localization assessed by immunostaining (data not shown) and triggers little cell death in mock-injected cells (Fig. 6C). However, microinjection of Rat-1/c-MycER cells with 10 μm hcC, which by itself induces little apoptosis over 6 hr (Fig. 6C), greatly sensitizes the cells to X-ray-induced cell death (Fig. 6C). Thus, microinjected hcC, like c-Myc, sensitizes Rat-1 fibroblasts to X-ray-induced apoptosis. To verify the involvement of p53 in that process, we repeated similar experiments using Rat-1/c-MycER/p53min fibroblasts. As shown in Figure 6D, these cells did not exhibit increased resistance to hcC microinjection. This indicates that the sensitivity of Rat-1 fibroblasts to hcC microinjection is not dependent on wild-type p53. However, these cells exhibited no increased sensitivity to X-ray-induced cell death when microinjected with hcC (Fig. 6D). Thus, the cooperation between hcC microinjection and X-irradiation is dependent on p53 activation.
Taken together, our data show that cytosolic hcC shares with c-Myc the ability to sensitize fibroblasts to mechanistically distinct triggers of apoptosis, in this case signaling through the CD95 receptor and the p53-dependent response to X-irradiation.
Discussion
c-Myc induces release of hcC from mitochondria
The induction of apoptosis by oncoproteins like c-Myc and E1A is probably an important mechanism for the self limitation of potentially malignant somatic clones. c-Myc is a transcription factor thought to exert its biological effects through modulation of target genes. Unfortunately, no comprehensive characterization of c-Myc target genes has been achieved yet so it is not yet possible to determine whether the two opposing biological activities of c-Myc—cell proliferation and apoptosis—arise from regulation of different, overlapping, or identical gene targets. However, the recent finding that c-Myc (and E1A) activates p53 via p19ARF, a protein encoded by the alternate reading frame of the p16 tumor-suppressor gene (de Stanchina et al. 1998; Zindy et al. 1998), has provided one mechanism linking c-Myc with the induction of apoptosis.
One of the most intriguing aspects of c-Myc is its ability to potentiate the apoptotic effects of a wide range of mechanistically distinct cellular insults. This suggests that c-Myc acts at some common point in the control and execution of apoptosis. However, c-Myc-induced apoptosis is inhibited by Bcl-2 proteins and survival factors and dependent on CD95 signaling, implying that wherever c-Myc acts it is nonetheless still susceptible to a variety of controls and influences. Given the emerging role of cytosolic hcC as an initiator of apoptosis, we began by investigating any potential relationship between hcC release and c-Myc activation. Our analyses show, both by biochemical fractionation and by immunocytochemical localization, that c-Myc causes progressive release of hcC from mitochondria. Accumulation of cytosolic cytochrome c is appreciable within 8 hr of c-Myc activation. This timing is consistent with release of hcC resulting from the actions of a transcriptional target of c-Myc, a notion reinforced by the inactivity of the transcriptionally inactive c-Myc mutant Δ(106–143). However, there is no obvious known gene target of c-Myc that currently fits the role of modulator of cytochrome c localization.
Immunocytochemical analysis revealed significant heterogeneity in the extent of hcC redistribution in different cells following c-Myc activation. Given the reasonably synchronous entry of c-MycER cells into the cell cycle following estrogen activation, this is unlikely to reflect inherent asynchrony in the activation of the ER moiety by OHT (Eilers et al. 1991). We feel that the most likely explanation is that additional factors dictate the precise timing of cytochrome c accumulation following c-Myc activation.
There are several reasons to believe the accumulation of cytosolic hcC induced by c-Myc is an early initiating event in apoptosis rather than a mere consequence of cellular demise. First, hcC accumulation occurs in cells before they exhibit any overt apoptotic morphology. Second, it occurs independently of caspase activation and is therefore not a trivial consequence of activation of some other apoptotic pathway, such as that mediated by the CD95–FADD–caspase 8 pathway. Third, even after accumulation of cytosolic hcC, cells can be protected from its depredations, for example by blockading CD95 signaling (see below).
IGF-1 inhibits c-Myc-induced cytochrome c release
In fibroblasts, IGF-1 is a potent suppressor of c-Myc-induced apoptosis, and this raised the possibility that it acts to prevent release of hcC from mitochondria. Our studies show this to be the case. The presence of IGF-1 reduced c-Myc-induced accumulation of cytosolic hcC, mirroring its protective effects on c-Myc-induced apoptosis (Harrington et al. 1994b). In fibroblasts, an important anti-apoptotic signaling pathway activated by the IGF-1 receptor routes through PI3-kinase and the serine/threonine Akt kinase (Kauffmann-Zeh et al. 1997; Kulik et al. 1997). Akt activation is reported to prevent release of cytochrome c occurring during neuronal cell death (Neame et al. 1998). c-Myc-induced release of cytochrome c is also blocked by Bcl-2 over expression (P. Juin, A. Hueber, T. Littlewood, and G. Evan, unpubl.), consistent with the notion that Akt protects cells from apoptosis by functionally inactivating the Bcl-2 antagonist Bad (Datta et al. 1997; del Peso et al. 1997).
Although IGF-1 inhibits c-Myc-induced hcC release and consequent apoptosis, it exerts no inhibitory effect on c-Myc-induced cell proliferation (Harrington et al. 1994b). This further underscores the separateness of the mitogenic and apoptotic pathways lying downstream of c-Myc and implies that release of cytochrome c is very unlikely to be a secondary effect of the proliferative function of c-Myc.
Cytosolic hcC as a mediator of c-Myc-induced cell death
Our data raise the possibility that one of c-Myc’s key pro-apoptotic actions is to trigger hcC release from mitochondria. To examine this possibility further, we asked whether cytosolic hcC is both necessary and sufficient for c-Myc-induced apoptosis.
We showed that cytosolic hcC is necessary for c-Myc-induced apoptosis by demonstrating that microinjection of cells with an anti-cytochrome c antibody, one which blocks cytochrome c pro-apoptotic activity in vitro (Neame et al. 1998), inhibits c-Myc-induced apoptosis. Whereas this does not prove that c-Myc induces apoptosis through cytochrome c, it does show that cytosolic cytochrome c is a necessary requirement for c-Myc-induced cell death. Whether other mitochondrial factors, such as the recently characterized apoptosis inducing factor (AIF) (Susin et al. 1999) are also released during c-Myc-induced apoptosis, and what involvement they might have in this process, remains to be established. We presume that microinjected anti-cytochrome c antibody exerts its effects on hcC already lost from mitochondria. The fact that it can effectively suppress apoptosis, at least over the time-scale of our experiments, implies that cells can tolerate loss of substantial amounts of hcC from their mitochondria with apparent impunity. This is consistent with other published reports indicating that cell death can be blocked by inhibiting the pro-apoptotic activity of cytosolic cytochrome c even though it is still lost from mitochondria (Rosse et al. 1998; Yoshida et al. 1998; see below).
To explore further the possibility that cytosolic hcC is responsible for c-Myc-induced apoptosis, we examined the consequences of microinjecting hcC directly into the cytosol of cells in the absence of c-Myc activation. We introduced varying amounts of hcC into cells over a range crudely estimated to bracket physiological levels of cytochrome c within our fibroblasts, as assessed using semiquantitative immunoblotting as a calibration guide (data not shown).
Microinjected hcC triggered apoptosis in a dose-dependent manner. However, time-lapse videomicroscopic inspection of the kinetics of these cell deaths revealed several intriguing features. Higher levels of microinjected hcC increased the incidence of apoptosis over time because they advanced the onset of apoptosis, as judged by the start of membrane blebbing, within the target cell population. Nonetheless, onset of apoptosis was asynchronous, even in cells ostensibly given the same amount of protein. This may indicate either that the precise point of delivery into the cell affects the apoptotic efficacy of the protein or, as described below, that other variables influence the rapidity with which hcC activates the death machinery. Once initiated, however, the kinetics of each individual apoptotic event, as determined as the time between the onset of membrane blebbing and cellular fragmentation (McCarthy et al. 1997) was unaffected by cytochrome c levels. We conclude from this that although cytochrome c initiates apoptosis, its link with the actual apoptotic program is contingent upon other factors and that hcC is not, itself, rate limiting for the effector phase of apoptosis. We found that inhibition of caspases by zVAD.fmk inhibited microinjected cytochrome c death significantly, in agreement with the notion that it acts via the formation of Apaf-1 complexes and activation of caspases. However, blocking caspase action alone did not always prove sufficient to inhibit completely hcC-induced apoptosis: Occasional caspase-blocked cells became stuck in an extended effector phase of apoptosis, characterized by a grossly protracted period of membrane blebbing similar to that observed following apoptosis induced by c-Myc (McCarthy et al. 1997). Such blebbing cells eventually die through a process that involves cell fragmentation and phagocytosis by neighboring cells (McCarthy et al. 1997). Whether such caspase-independent death is mediated through Apaf-1 or some other partner with which hcC interacts remains to be established.
Although c-Myc sensitizes cells to a wide range of mechanistically diverse triggers of apoptosis, we observed no apoptotic synergy between c-Myc and microinjected hcC. This is despite the fact that in the same cells we observed significant apoptotic synergy between microinjected hcC and both CD95 signaling and X-ray-induced DNA damage (see below). Intriguingly, we also observed apoptotic synergy between microinjected hcC and an E1AER fusion activated by OHT (P. Juin, A. Hueber, T. Littlewood, and G. Evan, unpubl.). This is consistent with a recent report (Fearnhead et al. 1998) and indicates that E1A, unlike c-Myc, exerts some of its pro-apoptotic activity downstream of hcC release. We also demonstrate that although IGF-1 protects from c-Myc sensitization to diverse apoptotic stimuli, it exerts no protective effect against microinjected hcC. These data are all consistent with the notion that c-Myc promotes sensitivity to apoptosis by triggering accumulation of cytosolic hcC and that IGF-1 inhibits c-Myc-induced apoptosis by blocking this accumulation.
Interestingly, unlike IGF-1, whole serum did appear to exert some protective effect against microinjected hcC (cf. Fig. 3, D and E), indicating that survival-signaling pathways operating downstream of hcC release may exist. Fetal calf serum is a rich mix of largely uncharacterized nutrients and growth and survival factors that provides significantly more potent suppression of c-Myc-induced apoptosis than does IGF-1 alone (Evan et al. 1992; Harrington et al. 1994b). We are currently investigating these additional factors.
The relationship between c-Myc, cytochrome c, p53, and CD95 signaling
CD95 signaling triggers caspase-dependent release of hcC (Vander Heiden et al. 1997). One obvious possible link between CD95, c-Myc, and cytochrome c is, therefore, that CD95 signaling is required for release of hcC induced by c-Myc. However, this is not the case because c-Myc-induced release of hcC is caspase-independent. It is, moreover, unaffected by the presence of a dominant interfering mutant of FADD, FADD DN. FADD DN is nevertheless very effective at protecting c-Myc-induced apoptosis in fibroblasts. We show that FADD DN blocks caspase activation subsequent to c-Myc-induced hcC release or hcC microinjection. Taken together, these data indicate that the immediate pro-apoptotic function of c-Myc in fibroblasts—release of mitochondrial hcC—does not depend on CD95 signaling, but that subsequent activation of the caspase apoptotic machinery requires cooperation between c-Myc-induced hcC release and CD95 signaling. Consistent with this, we show that microinjection of hcC recapitulates the effect of c-Myc by sensitizing fibroblasts to CD95 killing.
What might be the nature of the CD95-mediated signals that cooperate with cytosolic hcC to trigger apoptosis? In the case of CD95-induced apoptosis, the classical paradigm involves a direct connection between death receptor and apical caspase via a single intermediary adaptor molecule FADD (Boldin et al. 1996, Muzio et al. 1996). This leaves little apparent room for the involvement of mitochondria or cytochrome c. Nonetheless, several independent lines of study indicate that these two pathways interact and even cooperate. First, CD95 activation of caspase 8 triggers cleavage and consequent activation of the pro-apoptotic BH3 protein Bid, leading to cytochrome c release (Li et al. 1998; Luo et al. 1998). Second, Bcl-2 proteins, which presumably act at the level of mitochondria and/or the intracellular apoptosome, suppress CD95 killing in certain cell types (Scaffidi et al. 1998). Third, genetic ablation of Apaf-1 inhibits sensitivity to killing by CD95 in fibroblasts (Cecconi et al. 1998). Finally, overexpression of a dominant-negative mutant of pro-caspase 9 inhibits CD95 killing, presumably because it interferes with the normal activation of Apaf-1/cytochrome c complexes (Srinivasula et al. 1998).
Our data show that c-Myc promotes apoptosis by inducing the release of hcC into the cytosol. However, we also show that release of hcC is not, in itself, efficient at inducing apoptosis. Rather, as indicated in Figure 7, cells with a pool of cytosolic hcC are rendered acutely sensitive to other triggers of apoptosis. In addition to the CD95 pathway, p53 activation might provide such a trigger. Indeed, we show that cells microinjected with hcC are sensitized to X-irradiation by a p53-dependent mechanism. A dominant-negative mutant of p53, p53min, prevents this sensitization without having any effect on c-Myc-induced hcC release. A similar result was obtained with the p53 inhibitor Mdm 2 (P. Juin, A. Hueber, T. Littlewood, and G. Evan, unpubl.). Thus, our data provide functional evidence that c-Myc and p53 have distinct, cooperating pro-apoptotic mechanisms (Fig. 7). The nature of this p53-dependent signal cooperating with cytosolic hcC remains to be elucidated. However, it is clearly distinct from the CD95 signal cooperating with cytosolic hcC as FADD DN-expressing cells can be sensitized to X-ray by hcC microinjection (P. Juin, A. Hueber, T. Littlewood, and G. Evan, unpubl.).
Figure 7.

Mechanism of c-Myc-induced sensitization to apoptosis. c-Myc activation triggers release of cytochrome c into the cytosol. This release is blocked by IGF-1. Cells with increased cytosolic cytochrome c are sensitized to distinct death pathways such as that mediated by CD95 and p53, and possibly others too. Neither FADD DN nor p53min blocks c-Myc-induced cytochrome c release and so neither affects the sensitized state of a cell expressing c-Myc. See text for details.
Our data imply that in a cell in which p53 is already active, for example a cell sustaining chronic DNA damage or hypoxia, c-Myc-induced hcC release would trigger apoptosis by raising sensitivity to endogenous p53 signaling. In such a situation, c-Myc-induced apoptosis would appear to be dependent upon p53. Likewise, c-Myc-induced release of hcC would act to lower the threshold at which CD95 signaling triggers apoptosis. Rodent fibroblasts in vitro experience constitutive low-level CD95 signaling because of autocrine interaction between surface CD95L and the CD95 receptor (Hueber et al. 1997; Hueber and Evan 1998). In such cells, activation of c-Myc in low serum triggers hcC release, and this lowers the threshold at which endogenous CD95 signaling triggers apoptosis. In such a situation, c-Myc and cytochrome c-induced apoptosis would both appear to be dependent on CD95.
The fate of a cell expressing c-Myc is therefore acutely dependent on both the availability of survival factors, which dictate the extent of hcC release, and of other signaling pathways—p53, CD95, or other pathways—that determine how effectively cytosolic hcC triggers the apoptotic machinery. One intriguing possibility that emerges from this is that release of hcC into the cytosol need not be an ineluctable trigger of the apoptotic program. If cells can survive extended periods with cytosolic hcC, it may be possible to rescue them, either by blockading the apoptotic pathway downstream of cytochrome c or by providing sufficient trophic support to give the cells time to dispose of the cytosolic pool.
The identification of a physical mechanism by which c-Myc promotes apoptosis opens the way to a more detailed analysis of how tumors evade apoptosis. As most tumor cells express Myc proteins at high levels, and they are clearly not within their normal trophic environment, some agency must be acting to suppress their apoptosis. We have identified two independent mechanisms by which c-Myc-induced apoptosis might be forestalled: One by disabling the ability of c-Myc to cause efflux of hcC from mitochondria (the IGF-1 type of mechanism) and the other through ablation of downstream signals necessary for cytosolic hcC to activate the apoptotic program (the FADD DN type of mechanism). Obviously, different pharmacological approaches will be needed to correct each of these types of lesion, although the common goal will remain to reinstate the suicide machinery of the tumor cells to allow them to trigger their own demise.
Materials and methods
Reagents and antibodies
OHT was obtained from Sigma (St. Louis, MO). A 1 mm stock solution in 100% ethanol, stored in −20°C, was used. zVAD.fmk was obtained from Enzyme Systems Products (Dublin, CA). A 50 mm stock solution in DMSO was prepared and stored at −80°C. N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide (DEVDpNA) was obtained from Calbiochem Novabiochem (San Diego, CA) and a 100 mm stock solution in DMSO prepared and stored at −20°C was used. CD95Ls was obtained from Alexis Corp. (San Diego, CA). A 0.1 mg/ml stock solution was used according to the manufacturer’s instructions.
For immunocytochemical analysis of cytochrome c, purified monoclonal antibody 6H2B4 was used. The same antibody was used in microinjection experiments to incapacitate hcC; monoclonal antibody 7H8.2C12 was used to detect hcC by immunoblotting. Both antibodies were obtained from PharMingen (San Diego, CA), as was the mouse monoclonal IgG1 isotype control (clone MOPC-21) used for microinjection experiments. Purified monoclonal antibody specific for cytochrome c oxidase subunit IV (20E8-C12) was obtained from Molecular Probes (Eugene, OR). Horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents were obtained from Amersham, UK. Fluorescein-isothiocyanate (FITC)-conjugated secondary antibodies were obtained from Molecular Probes. For microinjection experiments, stock solutions of control and anti-cytochrome c antibodies were diluted fivefold in phosphate-buffered saline (PBS) and dialyzed against PBS overnight at 4°C. Antibodies were then concentrated to the required concentration using Microcon10 following the manufacturer’s instructions (Amicon, Beverly, MA). All other reagents used were obtained from Sigma.
Cell culture and cell lines
Rat-1 fibroblasts expressing the OHT conditional allele of c-Myc (Rat-1/c-MycER) and of the transcriptionally inactive mutant c-MycΔ(106–143) [Rat-1/c-MycΔ(106–143)ER] have been described previously (Littlewood et al. 1995). A Rat-1/c-MycER cell line constitutively expressing a dominant-negative mutant of FADD (Rat-1/c-MycER/FADD DN) was obtained by infection of Rat-1/c-MycER with the recombinant retrovirus pBabeHygro FDN4 containing the cDNA encoding FADD residues 80–208, followed by selection and cloning as described in Hueber et al. (1997). Expression of FADD DN protein was verified by immunoblotting. Results presented were obtained with one clone but identical results were also obtained with another independent clone and, for microinjection experiments, with a pool of the initial infected cells. Rat-1/c-MycER/p53min cells were generated by infecting Rat-1/c-MycER cells with the recombinant retrovirus pBabeHygro p53min encoding amino acids 302–390 of murine p53. Hygromycin-resistant clones were isolated and expression of p53min confirmed by immunoblotting. Results presented here are with one clone, but similar results were obtained with a further clone. Rat-1/c-MycER, Rat-1/c-MycER/FADD DN, and Rat-1/c-MycER/p53min cells were maintained in DMEM supplemented with 10% fetal calf serum (FCS). Cells were selected with puromycin at 5 μg/ml (Rat-1/c-MycER) or with puromycin at 5 μg/ml and hygromycin at 200 μg/ml (Rat-1/c-MycER/FADD DN and Rat-1/c-MycER/p53min). Following serum-deprivation for 48 hr, c-Myc was activated by addition of OHT to the growth medium at a concentration of 100 nm. When specified, IGF-1 was added to the growth medium at 100 ng/ml.
Biochemical techniques
Subcellular fractionation was performed as described in Juin et al. (1998) with minor modifications. Cells grown on 15-cm dishes were, at the indicated time points, scraped using a Teflon scraper, collected by centrifugation at 2,000g, and washed twice in PBS. Cells were then resuspended in sucrose-supplemented cell extract buffer (SCEB, 300 mm sucrose, 10 mm HEPES at pH 7.4, 50 mm KCl, 5 mm EGTA, 5 mm MgCl2, 1 mm DTT, 10 μm cytochalasin B, 1 mm PMSF), left on ice for 30 min, and then homogenized by 50 strokes in an ice-cold Dounce homogenizer. Unbroken cells and nuclei were pelleted by centrifugation for 10 min at 2000g. Mitochondria were collected from the resulting supernatant by centrifugation at 13,000g for 10 min. Both the mitochondrial pellet, resuspended in SCEB, and the postmitochondrial fraction were aliquoted, frozen in liquid nitrogen, and stored at −80°C prior to determination of protein concentration, immunoblotting, and/or assay of caspase activity.
For immunoblotting, similar amounts of proteins (10 μg for mitochondrial fractions, 20 μg for postmitochondrial fractions) were fractionated by SDS-PAGE and electroblotted onto Immobilon-P (Millipore).
To assay DEVD-cleaving activity in extracts, 50 μg of postmitochondrial fraction was incubated with 50 μm DEVD pNA in 100 μl of SCEB at 37°C. Cleavage of DEVDpNA was monitored over 2–3 hr by change in optical density at 405 nm using a spectrophometer, as described previously (Fraser and Evan 1997; Fraser et al. 1997). The amount of substrate cleaved as a function of time was determined using a standard calibration curve relative to free pNA (Sigma).
Immunocytochemistry
Cytochrome c immunostaining was performed as described in Bossy-Wetzel et al. (1998) using FITC-conjugated secondary antibodies. Serial z-axis optical sections of cells (collected at 0.5-μm intervals) were collected using a laser scanning confocal microscope (MRC1000, BioRad Labs, Hercules, CA) via a 100× Plan-Apochromatic oil objective. Images were collected within a linear range of fluorescence intensity based on values in ‘look up’ tables (Comos confocal imaging software, BioRad Labs) as described (Shima et al. 1997, 1998). Confocal images shown are two-dimensional projections of Z-series through the entire cell depth.
Microinjection experiments
Cells were seeded on glass-bottomed coverslip dishes (Matek Corp. Ashland, OR) 24–48 hr prior to injection. Cytosolic microinjection was then performed using an automated microinjection system (Zeiss AIS, Carl Zeiss, Thornwood, NY) as described previously (Pepperkok et al. 1993). Identical standardized conditions of pressure (120 hPa) and time (0.1 sec) were used for microinjection in all experiments. Cytochrome c (from bovine heart, Sigma C-3131) was prepared freshly at the indicated concentration in sterile water and mixed with dextran 40S-conjugated rhodamine isothiocyanate (Sigma; 0.4% final concentration) as a coinjection marker. Where indicated, microinjected cells were treated with CD95Ls (50 ng/ml) or subjected to 10 Gy of ionizing radiation from a Pantak model CB50 350kVp X-ray machine at 6 Gy per minute (320 kV and 10 mA). The number of viable positive cells (i.e., fluorescent cells exhibiting normal morphology) was then evaluated and their fates followed either by direct fluorescent microscopic analysis at discrete time points or by time-lapse fluorescence microscopy as described below.
Time-lapse videomicroscopy
Acquisition of time-lapse phase-contrast images and scoring of apoptotic cell death has been described previously (Evan et al. 1992; Fanidi and Evan 1994; Harrington et al. 1994b).
For time-lapse fluorescence-microscopy analysis, images of injected cells were recorded using a X20 Plan Achromatic objective on a Zeiss inverted fluorescent microscope (Axiovert 135TV) equipped with a cooled CCD camera (Photometrics CH250, 1317 × 1035 pixels, Tucson, AZ) controlled by a MacIntosh PowerPC 8600. A density filter (OD = 0.7%) was used to reduce incident light exposure to a minimum. Fluorescent images (exposure time 0.5 sec) were collected at 15-min intervals and recorded as described in Scales et al. (1997). A series of phase-contrast images was recorded simultaneously (exposure time 0.05 sec) and cumulative apoptotic cell deaths were scored as described (Evan et al. 1992). Crude determination of duration of the execution phase of apoptosis in injected cells was performed as described (McCarthy et al. 1997).
Acknowledgments
We thank Debbie Lyon and Jon Ramsden for help with time-lapse microscopy and Drs. Andrew Fraser and Rainer Pepperkok for fruitful discussion throughout this work. We would also like to thank all members of the Biochemistry of the Cell Nucleus Laboratory for their invaluable criticism and support. P.J. is a fellow of the EC TMR Programme. A.O.H. is a fellow of EMBO. We are grateful for support from MRC-LINK G9321238.EC and from EC grants BioTech BIO4-CT96-0052.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL G.Evan@icrf.icnet.uk; FAX 44 171 269 3581.
References
- Alarcon RM, Rupnow BA, Graeber TG, Knox SJ, Giaccia AJ. Modulation of c-Myc activity and apoptosis in vivo. Cancer Res. 1996;56:4315–4319. [PubMed] [Google Scholar]
- Amati B, Littlewood TD, Evan GI, Land H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993;12:5083–5087. doi: 10.1002/j.1460-2075.1993.tb06202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew D, Ashmun R, Simmons B, Cleveland J. Constitutive c-myc expression in IL-3-dependent myeloid cell line suppresses cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Bennett MR, Evan GI, Newby AC. Deregulated expression of the c-myc oncogene abolishes inhibition of proliferation of rat vascular smooth muscle cells by serum reduction, interferon-gamma, heparin, and cyclic nucleotide analogues and induces apoptosis. Circ Res. 1994;74:525–536. doi: 10.1161/01.res.74.3.525. [DOI] [PubMed] [Google Scholar]
- Bissonnette R, Echeverri F, Mahboubi A, Green D. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:552–554. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- Boldin M, Goncharov T, Goltsev Y, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Cherney BW, Bhatia K, Tosato G. A role for deregulated c-Myc expression in apoptosis of Epstein-Barr virus-immortalized B cells. Proc Natl Acad Sci. 1994;91:12967–12971. doi: 10.1073/pnas.91.26.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Eilers M, Schirm S, Bishop JM. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 1991;10:133–141. doi: 10.1002/j.1460-2075.1991.tb07929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- Evan G, Wyllie A, Gilbert C, Littlewood T, Land H, Brooks M, Waters C, Penn L, Hancock D. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;63:119–125. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fanidi A, Evan G. Applications of apoptosis: Making death pay. Trends Biotechnol. 1994;12:213–221. doi: 10.1016/0167-7799(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Fanidi A, Harrington E, Evan G. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature. 1992;359:554–556. doi: 10.1038/359554a0. [DOI] [PubMed] [Google Scholar]
- Fearnhead HO, Rodriguez J, Govek EE, Guo W, Kobayashi R, Hannon G, Lazebnik YA. Oncogene-dependent apoptosis is mediated by caspase-9. Proc Natl Acad Sci. 1998;95:13664–13669. doi: 10.1073/pnas.95.23.13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A, Evan G. Identification of a Drosophila melanogaster ICE/CED-3-related protease, drICE. EMBO J. 1997;16:2805–2813. doi: 10.1093/emboj/16.10.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A, McCarthy N, Evan G. drICE is an essential caspase required for apoptotic activity in Drosophila cells. EMBO J. 1997;16:6192–6199. doi: 10.1093/emboj/16.20.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Fanidi A, Evan GI. Oncogenes and cell death. Curr Opin Genet Dev. 1994a;4:120–129. doi: 10.1016/0959-437x(94)90100-7. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc-induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO J. 1994b;13:3286–3295. doi: 10.1002/j.1460-2075.1994.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- Hueber AO, Evan GI. Traps to catch unwary oncogenes. Trends Genet. 1998;14:364–367. doi: 10.1016/s0168-9525(98)01520-0. [DOI] [PubMed] [Google Scholar]
- Hueber A-O, Zörnig M, Lyon D, Suda T, Nagata S, Evan G. Requirement for the CD95 receptor-ligand pathway in c-Myc induced apoptosis. Science. 1997;278:1305–1309. doi: 10.1126/science.278.5341.1305. [DOI] [PubMed] [Google Scholar]
- Juin P, Pelletier M, Oliver L, Tremblais K, Gregoire M, Meflah K, Vallette FM. Induction of a caspase-3-like activity by calcium in normal cytosolic extracts triggers nuclear apoptosis in a cell-free system. J Biol Chem. 1998;273:17559–17564. doi: 10.1074/jbc.273.28.17559. [DOI] [PubMed] [Google Scholar]
- Kagaya S, Kitanaka C, Noguchi K, Mochizuki T, Sugiyama A, Asai A, Yasuhara N, Eguchi Y, Tsujimoto Y, Kuchino Y. A functional role for death proteases in s-Myc- and c-Myc-mediated apoptosis. Mol Cell Biol. 1997;17:6736–6745. doi: 10.1128/mcb.17.11.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI 3-kinase and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson C. Bcl-2 family proteins: The role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K, Bharti A, Yuan ZM, Saxena S, Weichselbaum R, Nalin C, Kufe D. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci. 1997;94:6939–6942. doi: 10.1073/pnas.94.13.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klefstrom J, Vastrik I, Saksela E, Valle J, Eilers M, Alitalo K. c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J. 1994;13:5442–5450. doi: 10.1002/j.1460-2075.1994.tb06879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber M. Antiapoptotic signaling by the insulin-like growth-factor-I receptor, phosphatidylinositol 3-kinase, and akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Littlewood T, Hancock D, Danielian P, Parker M, Evan G. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- McCarthy N, Whyte M, Gilbert C, Evan G. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan A, Kischkel F, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J, Zhang M, Gentz R, Mann M, Krammer P, Peter M, Dixit V. FLICE, a novel FADD homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/Apo-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R, Scheel J, Horstmann H, Hauri HP, Griffiths G, Kreis TE. Beta-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–82. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Rudolph B, Saffrich R, Zwicker J, Henglein B, Muller R, Ansorge W, Eilers M. Activation of cyclin-dependent kinases by Myc mediates induction of cyclin-a, but not apoptosis. EMBO J. 1996;15:3065–3076. [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Zauberman A, Ginsberg D, Oren M. Identification of a minimal transforming domain of p53: Negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol. 1992;12:5581–5592. doi: 10.1128/mcb.12.12.5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Haldar K, Pepperkok R, Watson R, Warren G. Partitioning of the Golgi apparatus during mitosis in living HeLa cells. J Cell Biol. 1997;137:1211–1228. doi: 10.1083/jcb.137.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Cabrera-Poch N, Pepperkok R, Warren G. An ordered inheritance strategy for the Golgi apparatus: Visualization of mitotic disassembly reveals a role for the mitotic spindle. J Cell Biol. 1998;141:955–966. doi: 10.1083/jcb.141.4.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes & Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- Wagner AJ, Small MB, Hay N. Myc-mediated apoptosis is blocked by ectopic expression of bcl-2. Mol Cell Biol. 1993;13:2432–2440. doi: 10.1128/mcb.13.4.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, Cipriani R, Sabbatini P, Denton A. Adenovirus E1B 19-kilodalton protein overcomes the cytotoxicity of E1A proteins. J Virol. 1991;65:2968–2978. doi: 10.1128/jvi.65.6.2968-2978.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]